

Abstract
We describe a combinatorial CRISPR interference (CRISPRi) screening platform for mapping genetic interactions in mammalian cells. We targeted 107 chromatin-regulation factors in human cells with pools of either single or double single guide RNAs (sgRNAs) to downregulate individual genes or gene pairs, respectively. relative enrichment analysis of individual sgRNAs or sgRNA pairs allowed for quantitative characterization of genetic interactions, and comparison with protein–protein-interaction data revealed a functional map of chromatin regulation.
Epistasis is a biological phenomenon in which the phenotype of one gene is modified by the presence or absence of another gene1. Analyses of large numbers of genetic interactions (GIs) can reveal how groups of functionally redundant gene products perform sophisticated functions2,3. This approach has successfully been used to uncover basic mechanistic biology and to aid in understanding the underlying causes of diseases. To date, the bulk of the available GI data in eukaryotes have been generated in yeast species4,5 and Drosophila6,7. Existing experimental platforms in mammalian cells have relied on high-throughput RNA interference (RNAi) technology8,9. Owing to substantial off-target effects10,11, the use of RNAi for GI mapping has been limited. Therefore, new complementary experimental methods for interrogating GIs in mammalian cells must be developed.
The nuclease-dead Cas9 (dCas9) enables sequence-specific transcriptional regulation through the CRISPRi or CRISPR activation (CRISPRa) approaches12,13. These methods have been demonstrated for large-scale sgRNA-guided pooled screens for repression or activation, but not for combinatorial screens14,15. To develop a CRISPRi combinatorial screening approach, we constructed a library of single-sgRNA constructs targeting 107 genes (three sgRNAs per gene, on average) involved in chromatin regulation. The library also included 41 negative-control sgRNAs that did not target sites in the human genome and five positive-control sgRNAs that targeted factors known to strongly affect cell proliferation (Supplementary Table 1). This library was then used to generate a pool of double-sgRNA constructs for simultaneous targeting of gene pairs (Supplementary Fig. 1a,b). Transducing both libraries into clonal HEK293-TetON-dCas9-KRAB cells produced two cell pools containing either single or pairwise gene perturbations (Fig. 1a). The relative representation of sgRNAs from doxycycline (Dox)-induced and uninduced samples was highly reproducible (Fig. 1b,c), as quantified by next-generation sequencing (NGS) (Supplementary Fig. 1c), and was used as a proxy for cell fitness and a quantitative phenotypic readout.
Figure 1.
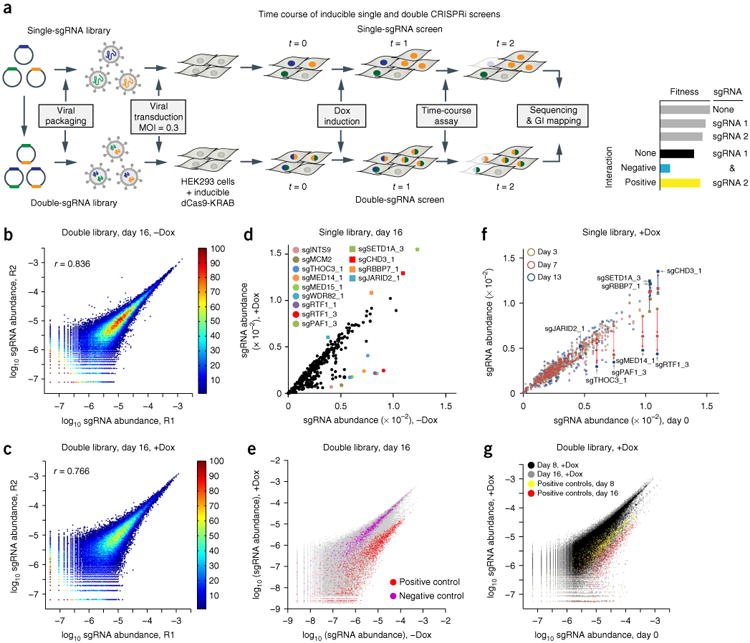
The CRISPRi screening platform for studying genetic interactions. (a) The experimental platform for single and double CRISPRi screening for GI studies. MOI, multiplicity of infection; t, time. (b,c) Characterization of independent experimental replicates for double-sgRNA libraries (R1 and R2, two independent experimental replicates). Double library without Dox (−Dox) at day 16 (b) or with Dox (+Dox) at day 16 (c). The correlation coefficient (r) for each comparison is displayed. The color bar shows the density of data. (d) Comparison of the single library with or without Dox at day 16. The colored dots show representative depleted sgRNAs, and squares show the enriched sgRNAs in the pool. (e) Comparison of the double library with or without Dox at day 16, with all data in gray and pairs containing positive (red) and negative (purple) control sgRNAs. (f) Comparison of day 0 with other time points (yellow, day 3; red, day 7; blue, day 13) in the presence of Dox for the single library. Consistent depletion and enrichment of representative sgRNAs are indicated with red and blue arrows, respectively. The comparison without Dox is shown in Supplementary Figure 4j. (g) Comparison of day 0 with other time points (black, all sgRNAs in the day 8 sample; yellow, all pairs containing positive-control sgRNAs in the day 8 sample; gray, all sgRNAs in the day 16 sample; red, all pairs containing positive-control sgRNAs in the day 16 sample) in the presence of Dox for the double library. The comparison without Dox is shown in Supplementary Figure 4k.
First, we assessed whether our libraries efficiently repressed gene expression. More than 75% of the sgRNAs tested were able to repress the target genes in both the single- and double-sgRNA settings, and repression efficiency was generally lower in the double-sgRNA context, in which approximately one in three sgRNAs was able to repress its respective target to less than 50% (Supplementary Fig. 2a–c). Hence, the repression efficiency achieved by CRISPRi appears to be generally lower than that achieved by RNAi8, possibly because of the effects of chromatin structure on locus accessibility and the shortcomings of current sgRNA design algorithms. We confirmed that after Dox induction, the positive-control sgRNAs resulted in a strong growth phenotype, whereas the negative-control sgRNA resulted in no detectable phenotype (Supplementary Fig. 2d).
Because the quality of the final GI data sets is highly dependent on raw-data reproducibility, we examined several aspects of our experimental setup. We optimized the experimental protocols to decrease the template switching during PCR amplification from pooled templates sharing a high degree of sequence similarity (Supplementary Fig. 3). We observed no significant bias between the sgRNA abundance before packaging the libraries into lentiviral particles and that after transduction in the HEK293-TetON-dCas9-KRAB cells, thus suggesting that our DNA packaging and transduction protocols were suitable for screening (Supplementary Fig. 4a). We confirmed that the data obtained from independent experimental replicates without (−Dox) or with (+Dox) Dox induction at different time points were highly reproducible (Fig. 1b,c and Supplementary Fig. 4b–i). Background dCas9-KRAB expression was tightly controlled to eliminate false-negative phenotypes, as verified by the absence of significant changes in sgRNA representation in −Dox samples over time (Supplementary Fig. 4j,k). Additionally, only the +Dox samples of the single and double libraries showed a depletion pattern (Supplementary Fig. 4l–o), and enrichment and depletion patterns among independent experimental replicates were highly reproducible (Supplementary Fig. 4p,q). Together, these results demonstrated that our experimental setup was able to deliver highly reproducible and reliable data.
We identified a set of single and double gene perturbations affecting cell fitness (Fig. 1d–g). Repression of a set of individual genes dramatically slowed cell proliferation (Fig. 1d); these genes included the positive controls (INTS9 and MCM2) as well as genes encoding components of the mediator complex (MED14 and MED15) and RNA polymerase II–associated factors (PAF1 and RTF1). In contrast, depletion of another set of genes, including those encoding components of the Set1/Ash2 histone methyltransferase complex (SETD1A), the Mi-2/NuRD complex (CHD3) and several histone acetyltransferase complexes (encoded by RBBP7), promoted cell proliferation (Fig. 1d). In the double-sgRNA library, pairs containing positive-control sgRNAs showed a consistent pattern of depletion, whereas the negative-control population remained mostly unchanged (Fig. 1g).
Our platform was also suitable for time-resolved analysis of genetic interactions and therefore has broader applications, including studying GI rewiring and dynamics during differentiation and development. In addition to the end-point experiments (Fig. 1d), sgRNA distribution was analyzed at intermediate time points. We observed depletion of similar sgRNA species in single (Supplementary Fig. 5a) and double (Supplementary Fig. 5b) screens, as well as consistent and evolving enrichment and depletion patterns in both single and double screens (Supplementary Fig. 5c,d). The reproducible depletion and enrichment patterns for both libraries at different time points suggested that the trend in these effects may provide another layer for identification of true-positive hits. For the single-sgRNA library in the presence of Dox, we compared sgRNA enrichment and depletion patterns at days 0, 3, 7 and 13 (Fig. 1f). The gene hits in Figure 1d showed consistent depletion (MED14, PAF1, RTF1 and THOC3) or enrichment (CHD3, SETD1A, RBBP7 and JARID2) over time. The positive controls (MCM2, GEMIN5, CENPA and INTS9) and approximately half of the 107 genes in the set exhibited consistent behavior over time, thus suggesting that perturbing these genes affects cell growth in a reproducible and time-dependent manner (Fig. 1f and Supplementary Fig. 5e). Similar results were obtained for the double library assayed at days 0, 8 and 16 (Fig. 1g and Supplementary Fig. 5f). Two pairs were chosen for validation in growth assays: MRGBP–MED6 and BRD7–LEO1. None of these four sgRNAs caused a growth phenotype when repressed individually in the pooled single screen (Supplementary Fig. 6a), whereas both pairs displayed a strong proliferation phenotype in the pooled double-sgRNA screen (Supplementary Fig. 6b) and in our validation experiments (Supplementary Fig. 6c,d).
We used a manually curated set of protein complexes and pathways involved in chromatin regulation (Supplementary Table 2) to create a GI map depicting the observed genetic interactions among different functional modules (Fig. 2a). GIs were scored (Online Methods and Supplementary Fig. 7) with the S-score framework16, and 95 significant negative (S score < −1) and 178 significant positive (S score >2.5) genetic interactions were identified (Fig. 2a and Supplementary Data Set 1). To establish genetic relationships among modules, groups of GIs were further collapsed into interaction bundles (Fig. 2b and Supplementary Methods). We observed conserved positive GIs between the CNOT and PAF1C modules8 and negative interactions between the CNOT module and the MLL2/3 module, a tumor-suppressive coactivator of p53 (ref. 17) and enhancer of ligand-dependent transcriptional activation by nuclear hormone receptors18.
Figure 2.
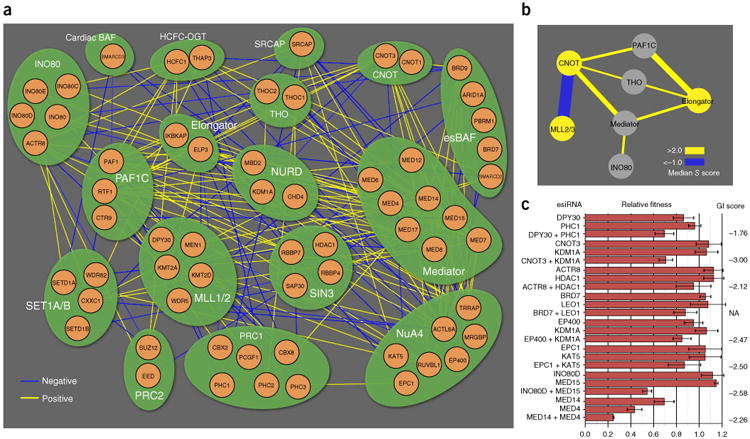
Module map of chromatin-related genes, based on a curated set of the indicated protein complexes. (a) Network representation of significant negative (blue) and positive (yellow) genetic interactions. (b) Intermodule connectivity map with negative (blue) and positive (yellow) edges. Edge thickness indicates interaction magnitude. (c) Validation of select GIs with esiRNA for eight interaction pairs. Calculated S scores (with the exception of BRD7 + LEO1, which was removed during the data-filtering process but was predicted to be negative) (Supplementary Fig. 6b,d) from the pooled double-CRISPRi screen are shown at right. Error bars, s.d. Number of technical replicates (n) = 3 for CNOT3 + KDM1A, BRD7 + LEO1, LEO1, EPC1 + KAT5, KAT5, INO80D + MED15, INO80D, MED14 + MED4 and MED4; n = 4 for all other measurements; NA, S score not measured.
We validated eight of the negative genetic interactions uncovered by our CRISPRi screen (Fig. 2c) by using an orthogonal silencing reagent, endonuclease-prepared interfering RNA (esiRNA)19. Treatment of cells with individual esiRNA had little or no effect on proliferation, whereas pairwise treatment resulted in detectable growth defects (Fig. 2c). We also compared our GI map with a previously published chromatin-centric map generated by combinatorial esiRNA8 in mouse fibroblasts and observed nine conserved genetic interactions (Supplementary Table 3). These data verified that our CRISPRi platform is able to recapitulate known interactions and to identify new genetic interactions in mammalian cells.
In addition to GI mapping, our platform can be used for mapping chemogenetic interactions. We performed a screen by using the drug rapamycin and the single-sgRNA library, and observed both protective and sensitizing effects (Supplementary Fig. 8). Null alleles of the orthologs of seven of the hits have previously been shown to cause rapamycin resistance in yeast (Supplementary Table 4), four of which displayed decreased expression after drug treatment in mammalian cells (Supplementary Table 5), thus suggesting that these genes function downstream of the mTOR signaling pathway.
Our approach for mapping genetic interactions in mammalian cells by using CRISPRi presents several advantages as compared with RNAi-based methods. CRISPRi knockdown is more specific14, thus raising fewer concerns about off-target effects in the same cells. In comparison to Cas9-mediated gene knockout, CRISPRi also offers several advantages. Cas9-mediated knockout allows complete loss of function to be attained, and knockouts may be heterogeneous among alleles; in contrast, CRISPRi-based gene knockdown leads to homogeneous partial loss of function20. Additionally, by changing the transcription-effector part of dCas9-KRAB into a transcriptional activator (CRISPRa)14, the approach can be potentially exploited for gain-of-function screening, an application not possible with RNAi or Cas9-mediated gene editing. Furthermore, combining CRISPRi and CRISPRa into the same cells may allow for simultaneously activating one gene while repressing another, thus greatly expanding the types of epistatic screens that can be performed. These important considerations should be taken into account in choosing a method for screening higher-order genetic interactions.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Online Methods
Step-by-step protocol
A protocol for the CRISPRi interaction screen is available as Supplementary Note 1.
Construct design
The cloning vectors used in this study were constructed through standard molecular cloning techniques, including PCR, restriction-enzyme digestion and DNA ligation. Custom oligonucleotides were purchased from Integrated DNA Technologies (IDT). The Escherichia coli strain DH5α was used for the transformation, which was followed by selection with 100 μg/ml of carbenicillin or 50 μg/ml of kanamycin. DNA was extracted and purified with Plasmid Miniprep or Midiprep Kits (Macherey-Nagel). Sequences of the constructs were verified with Quintarabio's DNA-sequencing service.
The dCas9-KRAB and sgRNA-expression constructs have previously been described21,22. The SpeI and SalI sites were removed in the sgRNA-expression construct. The single-sgRNA-expression constructs were cloned as previously described, with minor modifications (primer sequences in Supplementary Table 6; cloning strategy in Supplementary Fig. 1a). The PCR products and the lentiviral mouse U6 (mU6) promoter–based sgRNA-expression vector were digested with BstXI and XhoI and ligated to produce the final construct. The single-sgRNA-expression vector had unique SpeI and SalI sites introduced to enable the insertion of the mU6-sgRNA-expression cassettes to construct higher-order sgRNA-expression vectors.
To construct a lentiviral vector for mU6-driven expression of pairs of sgRNAs, mU6-sgRNA-expression cassettes were prepared from digestion of the storage vector with XbaI and XhoI enzymes, and were ligated into the target single-sgRNA-expression vector backbone, by using the compatible sticky ends generated by digestion of the target single-sgRNA-expression vector with SpeI and SalI enzymes (Supplementary Fig. 1b).
Single-sgRNA-library preparation
We designed a library of 358 sgRNAs including a set of 107 genes encoding epigenetic regulators (approximately 1–3 sgRNAs per gene) by using the top prediction hits from the CRISPR-ERA algorithm23. The library also included 41 nontargeting negative-control sgRNAs (NC_1 through NC_41) and five positive controls targeting genes (MCM2, GEMIN5, CENPA, INTS9 and POLR1D) strongly affecting cell proliferation. We excluded any sgRNAs containing XbaI, XhoI, SpeI and SalI restriction sites, which were used for double-sgRNA-library construction. We synthesized individual oli-gonucleotides encoding sgRNAs in a 96-well format, pooled and constructed the single-sgRNA-expression vectors individually, by ligating the oligonucleotides into a common sgRNA lentiviral vector with BstXI and XhoI sites. After sequencing validation, we manually mixed 358 sgRNA constructs in equal amounts for the single-sgRNA screens and double-sgRNA-library construction. The sgRNA sequence and corresponding genes are listed in Supplementary Table 1.
Combinatorial sgRNA-library preparation
To generate the pooled storage-vector library, equal amounts of the 358 single-sgRNA-expression vectors were pooled. Pooled lentiviral vector libraries containing combinatorial gRNAs were constructed with the strategy outlined in Supplementary Figure 1b. Briefly, the pooled mU6-sgRNA inserts were excised in a one-pot digestion of the pooled storage-vector library with XbaI and XhoI. The destination lentiviral vectors were digested with SpeI and SalI. The digested inserts and vectors were ligated via their compatible ends (i.e., XbaI–SpeI and XhoI–SalI) to create the pooled double-sgRNA library (358 × 358 = 128,164 total combinations) in the lentiviral vector. The lentiviral sgRNA-library pools were prepared in DH5α ultracompetent cells (Agilent Technologies) and purified with a Plasmid Midi Kit (Macherey-Nagel). The representation of each of the double-sgRNA constructs was then quantified by NGS with the oligonucleotides listed in Supplementary Table 6.
Lentivirus production and transduction
Lentiviral particles were produced by packaging in HEK293T cells, as previously described, with minor modifications21,22. Briefly, HEK293T cells were transfected with standard packaging vectors with Mirus TransIT-LT1 transfection reagent (Mirus MIR 2300), according to the manufacturer's instructions. Viral supernatant was harvested 48–72 h after transfection and either filtered through a 0.45-μm syringe filter or snap frozen.
Cell culture
HEK293T and HEK293 cells were cultured in DMEM supplemented with 10% FBS, 100 units/ml streptomycin and 100 mg/ml penicillin at 37 °C, with 5% CO2. To generate the Dox-inducible CRISPRi HEK293-dCas9-KRAB cell line, cells were transduced with lentiviral constructs expressing dCas9-KRAB from the TRE3G promoter and rtTA. Pure polyclonal populations of the CRISPRi cell line were treated with doxycycline and sorted by flow cytometry with a BD FACS Aria2 instrument on the basis of mCherry expression. Clones displaying high mCherry expression after Dox induction and undetectable mCherry expression in the absence of Dox were selected for further characterization.
Growth competition assay
The target cells were transduced with the packaged libraries in the presence of 8 μg/ml polybrene (Sigma) at a multiplicity of infection (MOI) of approximately 0.3 (corresponding to an infection efficiency of 30–40%) to ensure single-copy integration in most infected cells. After puromycin selection (2 μg/ml for 3 d) cells were harvested at 0, 9 or 16 d for single-sgRNA-library screening and at 0, 8 or 16 d for double-sgRNA screening.
For the rapamycin-resistance screen, after puromycin selection, cells were treated with either DMSO (vehicle) or rapamycin (Selleck Chemicals, 100 nM in DMSO). Growth medium was replaced with fresh medium every 2 d. Cells were harvested at 20 d.
After the cell samples were collected, genomic DNA was isolated with a QIAamp DNA Blood Maxi Kit (Qiagen), according to the manufacturer's protocol. The cassette encoding a single sgRNA or sgRNA pair was amplified by PCR (Supplementary Fig. 1c), and relative sgRNA abundance was determined by NGS with the Illumina MiSeq platform for single-sgRNA-library screens or with the Illumina HiSeq-2500 platform for double-sgRNA-library screens. The method to sequence the sgRNA regions of the constructs is shown in Supplementary Figure 1, and the custom primers used are listed in Supplementary Table 6. Two independent replicate experiments of each screen were performed. In the single-sgRNA screen, we were able to detect 349 out of the 358 in the sgRNA library.
Optimization of sgRNA- and NGS-library preparation
sgRNA-library preparation and cloning as well as NGS-library preparation relied on several PCR amplification steps. Individual fragments within the pool that share substantial sequence similarity may cause template switching during PCR amplification, thus leading to generation of incorrect products (Supplementary Figs. 1c and 3a). Template switching occurs because of enzyme processivity issues (for example, premature substrate release by DNA polymerase), thus leading to the creation of partially amplified products that may act as primers in later PCR amplification cycles. Depending on the length of the partial products (for example, the point at which the enzyme releases the substrate), hybrid DNA molecules composed of parts of several different DNA fragments may be generated, thereby resulting in skewed readout and contributing to experimental noise. As result, we systematically optimized the PCR conditions.
To quantify the extent of template switching, we constructed a single-sgRNA library in which each sgRNA was paired to a unique barcode sequence (Supplementary Fig. 3a, left), thus allowing for identification of each construct by sequencing two regions together: the sgRNA and the respective barcode (Supplementary Fig. 3a, left, black boxes). This methodology allowed for precise quantification of the fraction of correct versus template-switched products after a PCR amplification step. The PCR conditions were then adjusted to maximize the overall representation of the correct products over the template-switched products and were used to generate the double-sgRNA library. Whereas the double-sgRNA library used in this paper did not contain barcode sequences, and each double construct was identified by sequencing of the two sgRNAs (Supplementary Fig. 1), the incorporation of a barcode in the starting single-sgRNA library made it possible to easily analyze higher-order combinatorial sgRNA libraries by sequencing only the barcode cluster region (Supplementary Fig. 3a, right, black box).
Several different parameters including template-DNA input, PCR cycle number and DNA polymerase were tested (Supplementary Fig. 3b) and were found to influence the outcomes of the reactions. Three different DNA polymerases were evaluated: Phusion High-Fidelity DNA Polymerase (NEB), LongAmp Taq DNA Polymerase (NEB) and Kapa-HiFi DNA Polymerase (Kapa Biosystems). Our results clearly indicated that decreasing the number of PCR cycles as well as the template-DNA input led to a marked improvement in the fraction of correct fragments in the resulting pool. We observed a clear benefit of using LongAmp or Kapa-HiFi polymerases, rather than the Phusion enzyme, with higher input-DNA amounts. When 100 pg template was used, the template-switched products decreased to less than 5% with LongAMP after 19 cycles. These reaction conditions were used to prepare the deep-sequencing sample library in subsequent experiments.
Quantitative PCR to measure gene silencing at the mRNA level
Cells were harvested, and total RNA was isolated with an RNeasy Kit (Qiagen), according to the manufacturer's instructions. RNA was converted to cDNA with an iScript cDNA Synthesis Kit according to the manufacturer's instructions (Bio-Rad). Quantitative PCR reactions were prepared with a 2× master mix according to the manufacturer's instructions (Bio-Rad). Reactions were run on CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The human ribosomal-protein-encoding gene RPL19 or GAPDH was used as a control. Primer sequences for qPCR are listed in Supplementary Table 7.
Experimental validation of growth phenotypes
For the cell-growth validation experiments, the lentiviral pools of single- or double-sgRNA constructs were transduced into HEK293-dCas9-KRAB cells, and cells were selected with 2 μg/ml puromycin to remove the uninfected cells. The cell population was split into two 3 d later, and one half was treated with Dox (0.5 μg/ml) to induce dCas9-KRAB expression, whereas the other half was left untreated and served as a control. Cell viability was measured with XTT assays (Biotium) according to the manufacturer's protocol. 2,000 to 10,000 cells were plated into 96-well tissue culture plates for the growth assay and were grown for an additional 4 d (to day 7). For each well of the 96-well plates, 30 μl of XTT solution was added to the 100-μl cell cultures at the time points indicated. Cells were incubated for 6 h at 37 °C with 5% CO2. Measurement of the absorbance of the samples was performed with a spectro-photometer at a wavelength of 450–500 nm, and the background absorbance measurements were performed at a wavelength of 630–690 nm. The normalized absorbance values were obtained by subtraction of the background absorbance from the signal absorbance. For each sample, at least three independent experimental replicates were measured. The error bars represent the s.d. of the independent experimental replicates for both growth assays.
Orthogonal validation of genetic interactions with combinatorial esiRNA
Endonuclease-prepared interfering RNA (esiRNA) was generated as previously described8, with the primers listed in Supplementary Table 8. HEK293 cells were reverse-transfected with esiRNA with RNAiMAX and seeded in optical-bottom microplates. To transfect equivalent amounts of total esiRNA for both single and double treatments, single-knockdown cells were transfected with both esiRNA targeting the coding gene plus nontargeting esiRNA against eGFP. Cells were fixed and stained concurrently with a final concentration of 2% formaldehyde in phosphate-buffered saline with 1 μg/ml Hoechst 33342. Imaging and cell counting (with a nuclear mask) were performed with a CellInsight automated microscope (Thermo Scientific).
Raw data and statistics
All raw data presented here are available in Supplementary Data Sets 1–3, and per-figure mapping can be found in Supplementary Note 2. Error bars, unless otherwise noted, represent s.d. The sample numbers (n) used in individual figures are as follows and can also be found in Supplementary Data Set 3: Figure 2c, n varied from 3 to 4 (as shown in Supplementary Data Set 3); Supplementary Figure 2a, n = 2 for single- and n = 2 or 3 for double-sgRNA experiments; Supplementary Figure 2b, n = 2 for single-sgRNA experiments, and n varied between 2 and 5 for double-sgRNA experiments (as shown in Supplementary Data Set 3); Supplementary Figure 2c, box plot, n = 29 for singles, and n = 90 for doubles; Supplementary Figure 2d, n = 3; Supplementary Figure 3, n = 315, and P values were calculated with an unpaired two-sample two-tailed t test; Supplementary Figure 6c,d, for the growth curves (left), n = 3, and for the qPCR quantification (right), n = 2. Statistical methods relating to computation of the genetic-interaction scores are described in the section below.
Data processing and analysis
Raw count data can be found in Supplementary Data Sets 1 and 2. sgRNA frequencies were calculated on the basis of the ratio of the raw counts for each single- or double-sgRNA construct and the sum of the counts for all observed constructs in the experiment. The ratio of each sgRNA-pair frequency in the +Dox and −Dox samples was used as the phenotypic readout. Lower ratios indicated depletion of a particular sgRNA combination within the pool, whereas higher ratios were interpreted as enrichments.
For the rapamycin-resistance screen, to isolate the fitness effects associated with rapamycin treatment alone, the uninduced (−Dox) sgRNA frequency in the presence of rapamycin was normalized to the frequency of the vehicle-only (DMSO), uninduced (−Dox) sample. The specific effects of the combined sgRNA and rapamycin treatment were evaluated by normalizing the induced (+Dox) rapamycin-treated sample to the vehicle-only (DMSO) induced (+Dox) sample.
For the double-sgRNA screens, each double-sgRNA construct has two positions (p1 and p2) that a given sgRNA can occupy, and for the majority of sgRNAs we were able to detect pairs containing a particular sgRNA in both p1 and p2. To address whether sgRNAs expressed from p1 and p2 behave in a similar fashion, we used single-sgRNA phenotype estimates. For each individual sgRNA, we computed the median phenotype strength for all pairs containing that sgRNA in positions p1 and p2. The phenotype estimates for p1 and p2 were in very good agreement (Supplementary Fig. 7a), thus suggesting that no significant positional biases were introduced, and the sgRNA behavior was largely independent on its position within the double-sgRNA construct.
A total of seven replicate data sets (2 independent experiments and 5 technical replicates) were analyzed. Raw counts extracted from the NGS data displayed a skewed distribution for both single-sgRNA (Supplementary Fig. 7b) and double-sgRNA (Supplementary Fig. 7c) libraries and spanned more than four orders of magnitude. This skewed distribution, together with the limited sampling from the high-complexity double-sgRNA pool, contributed to a substantial degree of variability among independent experimental replicates associated with data in the low-count regime (Supplementary Fig. 7d).
To derive highly reproducible genetic-interaction scores, highly variable data in the low-count region were removed with a cutoff determined by analyzing the internal data consistency for each sequencing run. The reasoning was as follows: a successful experiment would produce a data set in which the relative abundance (RA) of each sgRNA pair sgRNA_A–sgRNA_B (RAA–B) would be highly similar and thus independent of the position (Supplementary Fig. 7a) of the individual sgRNAs within the construct (for example, RAA–B ≈ RAB–A). This relationship across the entire data set was captured by the Pearson correlation coefficient for raw data, CCAB–BA (raw), which depended on the raw count cutoff for the −Dox sample (RCCDox−). This cutoff was applied for the −Dox sample, because it closely resembled the initial state of the pool (Supplementary Fig. 3k). For each experimental replicate, an RCCDox− was chosen such that the remaining data had CCAB–BA (raw) ≥ 0.7 (Supplementary Fig. 7e).
Genetic-interaction scores were computed with the S-score metric16. Because the RCCDox− filtering tended to produce sparser data, a second filtering step was applied to remove data points with fewer than three replicate measurements (Supplementary Fig. 7f). These initial S scores were used to compute final scores for each gene by collapsing the scores from all sgRNAs targeting this gene with the following procedure:
For each sgRNA_X, a Pearson correlation coefficient (CCx:[1 … x … n] – [1 … x … N]:x) was computed with S scores for pairs in which sgRNA_X was in position p1 or p2 in the construct combined with all other individual sgRNAs in the set (1 … X … N). This procedure identifies sgRNAs displaying consistent and reproducible behavior.
- For each gene, CCx:[1 … x … n] – [1 … x … n]:x values were ranked, and one or more sgRNAs were selected on the basis of the following criteria:
- sgRNAs with CCx:[1 … x … N] – [1 … x … N]:x greater than a set threshold T1 were selected and included in further analysis.
- If no sgRNAs met criterion a, a single sgRNA with the highest positive CCx:[1 … x … n] – [1 … x … N]:x above a set threshold T2 was selected.
- If CCx:[1 … x … n] – [1 … x … N]x could not be calculated (e.g., because of missing data), then all sgRNAs containing a fraction of valid (e.g., not missing) data above a set threshold T3 were selected.
- If none of criteria a–c were met, no sgRNA was selected, and the respective gene was not included in the downstream analysis
Values for T1 and T2 were empirically determined and set to 0.4 and 0.1, respectively, by sampling the combinatorial space of T1 and T2 and maximizing the overall Pearson correlation for S scores (CCAB–BA (S scores), computed as for CCAB–BA (raw) but with S scores instead of raw counts) and the number of genes for the resulting collapsed data set (Supplementary Fig. 7g). T3 was set to 0.2; for example, only profiles for which at least 20% of valid data was present were considered.
This procedure yielded a data set displaying CCAB–BA (S score) ≥ 0.4, a value comparable to those previously obtained in yeast and mammalian systems with the same scoring metric2,4,8,24–27. Final S scores were computed by averaging this collapsed data and were used for building the network in Figure 2.
Functional-module connectivity analysis
Functional modules used in this study were manually curated from the literature (Supplementary Table 2). S-score cutoffs in Figure 2a were chosen on the basis of confidence curves, as previously described16. Intermodule bundles in Figure 2b were computed as the median of the magnitude of all intermodule interactions.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files. Source code for the data analysis pipeline is available upon request.
Supplementary Material
Acknowledgments
The authors thank the members of the laboratories of L.S.Q. and N.J.K. for advice and helpful discussions, and the Stanford Functional Genomics Facility and UCSD Sequencing Facility for technical support. We thank D. Zhao from the laboratory of L.S.Q. for advice and help in cloning the sgRNA library. L.S.Q. acknowledges support from the NIH Office of the Director (OD), the National Institute of Dental & Craniofacial Research (NIDCR), the Department of Defense Breast Cancer Research Breakthrough Program, the Pew Charitable Trusts and the Alfred P. Sloan Foundation. D.D. and A.R. acknowledge support from the Quantitative Bioscience Institute (QBI). M.C., N.J.K. and L.S.Q. acknowledge support from the J. David Gladstone Biofulcrum Program. This work was supported by DP5 OD017887, NIH R01 DA036858, NIH U01EB021240, DOD W81XWH-17-1-0018, a Pew Scholar Fellowship and an Alfred P. Sloan Fellowship to L.S.Q., as well as by NIH grants P50 GM082250, U19 AI106754, P01 HL089707, R01 GM084279, U19 AI118610 and R01 AI120694 to N.J.K.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Author Contributions: L.S.Q. and N.J.K. conceived the project. L.S.Q., N.J.K., D.D. and A.R. designed the experiments; L.S.Q. and D.D. designed the sgRNA library; D.D. constructed the sgRNA libraries and performed library screens; D.D. and A.R. performed sample preparation and sequencing; A.R. analyzed the sequencing data, curated the data sets and scored the data; D.D., S.-H.C. and M.C. performed the CRISPRi validation experiments; D.E.G. performed the esiRNA validation experiments; M.S. contributed to analysis and figure generation; J.P.S., T.I. and P.M. provided technical advice; L.S.Q., N.J.K. and A.R. wrote the paper.
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Beltrao P, Cagney G, Krogan NJ. Cell. 2010;141:739–745. doi: 10.1016/j.cell.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins SR, et al. Nature. 2007;446:806–810. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 3.Kelley R, Ideker T. Nat Biotechnol. 2005;23:561–566. doi: 10.1038/nbt1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roguev A, et al. Science. 2008;322:405–410. doi: 10.1126/science.1162609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costanzo M, et al. Science. 2016;353:aaf1420. [Google Scholar]
- 6.Fischer B, et al. eLife. 2015;4:e05464. [Google Scholar]
- 7.Horn T, et al. Nat Methods. 2011;8:341–346. doi: 10.1038/nmeth.1581. [DOI] [PubMed] [Google Scholar]
- 8.Roguev A, et al. Nat Methods. 2013;10:432–437. doi: 10.1038/nmeth.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laufer C, Fischer B, Billmann M, Huber W, Boutros M. Nat Methods. 2013;10:427–431. doi: 10.1038/nmeth.2436. [DOI] [PubMed] [Google Scholar]
- 10.Hannon GJ. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 11.Morgens DW, Deans RM, Li A, Bassik MC. Nat Biotechnol. 2016;34:634–636. doi: 10.1038/nbt.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilbert LA, et al. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilton IB, et al. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilbert LA, et al. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konermann S, et al. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins SR, Schuldiner M, Krogan NJ, Weissman JS. Genome Biol. 2006;7:R63. doi: 10.1186/gb-2006-7-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J, et al. Proc Natl Acad Sci USA. 2009;106:8513–8518. doi: 10.1073/pnas.0902873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim DH, Lee J, Lee B, Lee JW. Mol Endocrinol. 2009;23:1556–1562. doi: 10.1210/me.2009-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kittler R, Heninger AK, Franke K, Habermann B, Buchholz F. Nat Methods. 2005;2:779–784. doi: 10.1038/nmeth1005-779. [DOI] [PubMed] [Google Scholar]
- 20.Mandegar MA, et al. Cell Stem Cell. 2016;18:541–553. doi: 10.1016/j.stem.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du D, Qi LS. Cold Spring Harb Protoc. 2016;2016 doi: 10.1101/pdb.prot090175. http://dx.doi.org/10.1101/pdb.prot090175. [DOI] [PubMed] [Google Scholar]
- 22.Du D, Qi LS. Cold Spring Harb Protoc. 2016;2016 doi: 10.1101/pdb.prot090175. https://dx.doi.org/10.1101/pdb.top086835. [DOI] [PubMed] [Google Scholar]
- 23.Liu H, et al. Bioinformatics. 2015;31:3676–3678. doi: 10.1093/bioinformatics/btv423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilmes GM, et al. Mol Cell. 2008;32:735–746. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiedler D, et al. Cell. 2009;136:952–963. doi: 10.1016/j.cell.2008.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aguilar PS, et al. Nat Struct Mol Biol. 2010;17:901–908. doi: 10.1038/nsmb.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Surma MA, et al. Mol Cell. 2013;51:519–530. doi: 10.1016/j.molcel.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information files. Source code for the data analysis pipeline is available upon request.