

Abstract
The cerebral metabolic rate of oxygen (CMRO2) is reduced during apnea that yields profound hypoxia and hypercapnia. In this study, to dissociate the impact of hypoxia and hypercapnia on the reduction in CMRO2, 11 breath-hold competitors completed three apneas under: (a) normal conditions (NM), yielding severe hypercapnia and hypoxemia, (b) with prior hyperventilation (HV), yielding severe hypoxemia only, and (c) with prior 100% oxygen breathing (HX), yielding the greatest level of hypercapnia, but in the absence of hypoxemia. The CMRO2 was calculated from the product of cerebral blood flow (ultrasound) and the radial artery-jugular venous oxygen content difference (cannulation). Secondary measures included net-cerebral glucose/lactate exchange and nonoxidative metabolism. Reductions in CMRO2 were largest in the HX condition (−44 ± 15%, p < 0.05), with the most severe hypercapnia (PaCO2 = 58 ± 5 mmHg) but maintained oxygen saturation. The CMRO2 was reduced by 24 ± 27% in NM (p = 0.05), but unchanged in the HV apnea where hypercapnia was absent. A net-cerebral lactate release was observed at the end of apnea in the HV and NM condition, but not in the HX apnea (main effect p < 0.05). These novel data support hypercapnia/pH as a key mechanism mediating reductions in CMRO2 during apnea, and show that severe hypoxemia stimulates lactate release from the brain.
Keywords: Hypoxia, breath-holding, cerebral lactate release, cerebral nonoxidative metabolism, brain metabolism
Introduction
With its exceptionally high-energy demand and almost exclusive reliance on oxidative metabolism, the human brain is particularly vulnerable to reductions in oxygen availability. During prolonged apnea, as oxygen becomes limited, oxygen-conserving reflexes are paramount for prolonging cell survival, and consciousness. The mammalian dive response is hallmarked by bradycardia, reduced blood perfusion to nonvital organs (e.g. skeletal muscle), and reductions in whole-body oxidative metabolism.1 The ability to slow oxidative metabolism in turn delays the time before reaching critical levels of hypoxemia, and prolongs the apnea breaking point in elite divers.2,3 Indeed, the remarkable human apnea times of over 10 min (officially recognized world record in static apnea; 11:35 min) most certainly stem from effective oxygen conservation.
For the hypoxemic vulnerable brain, a logical protective mechanism of inadequate oxygen supply would be to reduce the metabolic rate. Yet, hypoxia alone may in fact increase, rather than decrease the cerebral metabolic rate of oxygen (CMRO2).4,5 On the other hand, during apnea that generates both extreme levels of hypoxia (partial pressure of arterial O2, PaO2 ≈ 30 mmHg) and hypercapnia (partial pressure of arterial CO2, PaCO2 ≈ 55 mmHg), we have recently demonstrated that the CMRO2 is reduced by ∼29%.6 In the same study, breathing during severe-hypercapnic hypoxia (PaCO2 ∼ 58.7 mmHg; PaO2 ∼ 38.9 mmHg) also reduced the CMRO2 (by ∼17%). However, no change in CMRO2 was observed while breathing at a similar level of hypoxia (PaO2 ∼ 38.0 mmHg), but with milder hypercapnia (PaCO2 ∼ 46.3 mmHg). It was therefore proposed that hypercapnia might determine the CMRO2 reduction during apnea. A hypercapnic reduction in CMRO2 is notionally mediated from the decreased extracellular pH, which reduces phosphofructokinase activity,7 and increases extracellular adenosine concentrations.8 However, the associated hemodynamic and autonomic differences with apnea compared to breathing—even with similar arterial blood gases9,10—make it difficult to isolate the CMRO2 reduction during extreme apnea to hypercapnia. For example, the mammalian dive reflex attending apnea may support a metabolic reduction independent of hypercapnia.11 In addition, in our previous study6 it was impossible to discern the metabolic impact of hypercapnia independent to the impact of severe hypoxia. As recently reported,12 the attending hypoxia may well offset some of the reduction in CMRO2 attributed to hypercapnia.
The primary purpose of this study was to quantify the CMRO2 under three distinct apnea paradigms that yield separate levels of hypoxemia and hypercapnia / acidosis, thereby dissociating the PO2 from PCO2. In elite apnea divers, maximal apneas were performed under: (a) normal conditions (NM) that yield extreme levels of both hypoxemia and hypercapnia; (b) prior hyperventilation (HV), yielding severe hypoxemia but limiting hypercapnia; and (c) prior hyperoxic hyperventilation (HX), thus removing the impact of hypoxemia, but generating the most severe hypercapnia. It was hypothesized that the reduction in CMRO2 near the termination of apnea would be mediated by hypercapnia, independent of the level of hypoxia.
Materials and methods
Participants
The ethical committees of the University of Split, School of Medicine, the University of British Columbia, and the University of South Wales approved the study procedures and experimentation, which conformed to Helsinki Declaration. Thirteen actively competitive and elite breath-hold divers (3 females; age 31 ± 8 years; BMI 23.0 ± 2.1 kg/m2) were recruited from the Croatian national apnea team. All participants provided informed written consent before experimentation. Years competing ranged from 2 to 15 years primarily in the discipline of dynamic (underwater laps in a pool) and static (resting while face down in water) apnea. Four of the participants were also involved with depth disciplines. Six of the subjects were world-class apnea competitors, having placed top 10 within the last 3 years in the international competition in at least one event. One subject had recently set a new official world record in dynamic apnea. All subjects were assessed by standard anthropometric and pulmonary functioning metrics, and a medical history questionnaire. Participants were free from any known respiratory and cardiovascular diseases.
Experimental design
All experimentation for a single subject was completed on a single day, at the Department of Integrative Physiology, University of Split School of Medicine. Participants arrived to the laboratory following abstinence from vigorous exercise, alcohol, and caffeine at least 24 h prior. Upon arrival to the laboratory and following initial screening, a 20-gage arterial catheter (Arrow, Markham, ON, Canada) was placed in the right radial artery, and a central venous catheter (Edwards PediaSat Oximetry Catheter, CA, USA) was placed in the right internal jugular vein and advanced towards the jugular bulb. Cannulation was completed under local anesthesia (1% lidocaine) with ultrasound guidance. Facial vein contamination was ruled out by assuring that all jugular venous SO2 recordings were below 75%. The arterial catheter was attached to an in-line waste-less sampling setup (Edwards Lifesciences VAMP, CA, USA) attached to a pressure transducer that was placed at the height of the right atrium (TruWave transducer). Following cannulation subjects were further instrumented with ECG and transcranial Doppler (see section “Measures”).
The experimental procedure comprised three maximal apneas (see experimental procedure schematic, Figure 1). Each apnea protocol was separated by a minimum of 20 min rest before commencing the preparatory phase of the next respective apnea (see Bain et al.2 and Hoiland et al.3 for description of the preparatory phase). The order of the hyperventilation apnea (HV) and the normal apnea (NM) was counter-balanced, but due to the potentially long lasting physiological effects of hyperoxia in combination with the fatiguing factor of a prolonged hyperoxic apnea (up to 21 min), the hyperoxic apnea (HX) was always performed last. For the HX protocol, subjects hyperventilated for 15 minutes from a Douglas bag that was continually filled with 100% O2. Hyperventilation for the HV (3 min) and HX (15 min) protocol was paced with auditory feedback to achieve an end-tidal PCO2 of approximately 20 mmHg. End-tidal gases were sampled at the mouth and integrated into a calibrated gas analyzer (ADI instruments, Colorado Springs, CO, USA). The 15 min of 100% oxygen hyperventilation is used during hyperoxic apnea competitions, and was based on a stimulus that yields the longest possible apnea time and, therefore, greatest increase in PaCO2 at the end of the apnea. The apnea coach (ID) was present at all times to assure complete motivation during the maximal apneas.
Figure 1.

Timeline schematic of the experimental design. Maximal apneas were performed with no prior hyperventilation (NM), 3 min of hyperventilation (HV), and 15 min of hyperventilation on 100% oxygen (HX). Compared time points between apneas included baseline (BL), apnea onset (Onset), the first involuntary breathing movement (IBM), and immediately at apnea end (End). The “a–v draw” denotes when radial artery and jugular venous draws were taken. The order of the NM and HV apneas were randomized, but the HX apnea was always performed last. See text for details.
Measures
Involuntary breathing movements: The onset of involuntary breathing movements (IBMs) was visually assessed by the apnea coach, and verified by a plethysmography belt placed around the chest, integrated into LabChart® for offline analysis. Measures analyzed at the onset of IBMs represent approximately the half waypoint of the respective apnea.
Blood gases, oximetry, and metabolites: At each of the four time points described in Figure 1, approximately 2 mL of blood was procured from the radial artery and jugular vein into a heparinized syringe. Whole blood was immediately analyzed for PO2, PCO2, O2 saturation (SO2%), glucose (Glu), lactate (La), hemoglobin (Hg), and pH, using a commercially available cassette based analyzer (ABL90 FLEX, Radiometer, Copenhagen, Denmark).
Cardiovascular: Heart rate (HR) was obtained from the R-R intervals measured from a three-lead ECG. Mean arterial blood pressure (MAP) was measured with the pressure transducer connected to the radial catheter. Because the pressure trace is lost during blood sampling, values for MAP were taken 15 s immediately before each blood draw. Heart rate and pressure measures were integrated into PowerLab® and LabChart® software (ADInstruments) for online monitoring, and saved for offline analysis.
Cerebrovascular: Cerebral blood velocity of the middle cerebral artery (MCAv) and posterior cerebral artery (PCAv) were measured using a 2-MHz pulsed transcranial Doppler ultrasound system (Spencer Technologies, Seattle, WA). A headband fixation device (model M600 bilateral head frame, Spencer Technologies) was used to fix the probes in position. Signal quality was optimized using standardized search techniques that produce test-retest reliability of ∼3% and 2% for MCAv and PCAv, respectfully.13 Once fixed into place, probe positioning was kept constant for the entire three trials. The MCAv was insonated through the left temporal window, at a depth of approximately 1 cm distal to the MCA-anterior cerebral artery bifurcation, and the PCAv was insonated at the P1 segment through the right temporal window. The MCAv and PCAv were integrated into PowerLab® and LabChart® (ADInstruments) for online monitoring and offline analysis.
Blood flow in the right internal carotid artery (ICA) and left vertebral artery (VA) was simultaneously measured using duplex vascular ultrasound (Terason 3200, Teratech, Burlington, MA). The right ICA was on average insonated 2cm from the carotid bifurcation, while the left VA was insonated at the C5–C6 or C4–C5 space, but kept constant within participant between trials.14 The steering angle was fixed to 60 ° among all trials, and the sample volume was placed in the center of the vessel adjusted to cover the entire vascular lumen. All files were screen captured and saved as video files for offline analysis at 30 Hz using custom designed software.15 Simultaneous measures of luminal diameter and velocity over a minimum of 12 cardiac cycles were used to calculate flow. The onset of involuntary breathing movements (occurring at approximately 50% of the apnea duration) generates movement of the neck muscles, and in turn hinders reliable ICA and VA blood flow measures. As such, ICA and VA blood flow following the onset of IBMs to the end of apnea was derived from the subsequent change in MCA, for ICA flow, and PCA, for VA flow. This technique has been used previously with good agreement between measures.2,3
Calculations
Under the assumption of symmetrical blood flow of contralateral ICA and VA arteries, global cerebral blood flow (gCBF) was calculated from
Arterial content of oxygen (CaO2) and venous content of oxygen (CvO2) were calculated using the equations
where 1.36 is the affinity for oxygen to hemoglobin for a given saturation, and 0.003 is the percentage of oxygen dissolved in the blood. Values are expressed as ml of O2 per 100 mL of blood (mL·dL−1).
Cerebral delivery of oxygen (CDO2) was calculated from
The cerebral metabolic rate of oxygen (CMRO2) was calculated from
Cerebral oxygen extraction fraction was calculated from
Net cerebral glucose and lactate exchange was calculated from
where a negative value indicates a net uptake, and positive value indicates a net release. Glucose and lactate values are in mmol·mL−1 and gCBF is in mL·min−1.
The oxidative carbohydrate index (OCI) provides an estimation of oxidative versus nonoxidative metabolism, and is calculated by the equation shown below. In short, a reduction from 100% indicates the presence of nonoxidative metabolism. See Ainslie et al.16 for further details.
Statistical analysis
Values are presented as mean values ± standard deviations (SD), except in Figures 3 and 5, where means ± 95% confidence intervals are shown. Baseline measures were acquired during quite rest prior to the preparatory apneas (∼10 min before the NM and HV apnea, and ∼20 min before the HX apnea) of each respective condition. Measures were averaged over 20 s around the blood draws, except for arterial blood pressure that was averaged over 15 s immediately before each blood draw (see measurements).
Figure 3.
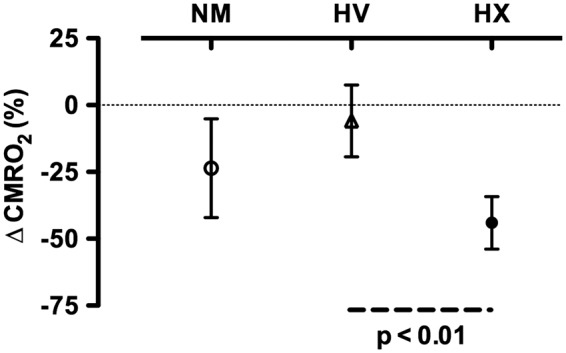
Percent change in the cerebral metabolic rate of oxygen (CMRO2) from baseline to the end the normal apnea (NM), apnea following 5 min of hyperventilation (HV), and apnea following 15 min of 100% O2 hyperventilation (HX). The reduction in CMRO2 was significantly larger in the HX compared to the HV apnea. Error bars denote 95% confidence intervals.
Figure 5.
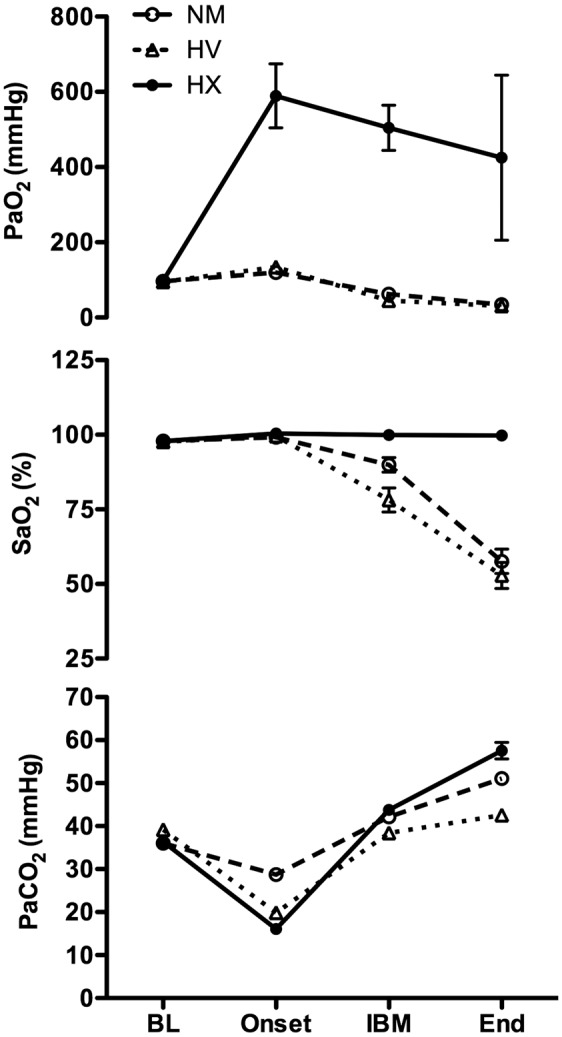
Arterial partial pressure of O2 (PaO2; top panel), arterial oxygen saturation (SaO2; middle panel), and the partial pressure of arterial CO2 (PaCO2; bottom panel) for all three apneas at baseline (BL), the apnea onset (Onset), the first involuntary breathing movement (IBM), and immediately at the termination of apnea (End). Error bars denote 95% confidence intervals. By design, there was a significant main effect of condition, time, and time*condition for all variables. There were no differences at baseline for any variable between conditions. See text for exact post hoc statistical analysis.
After testing the primary outcome variable for normality using repeated Shapiro–Wilks W tests, statistical analysis was performed using a two-way repeated measures analysis of variance (ANOVA) using the factors of three conditions (NM, HV, HX) and four time points (BL, Onset, IBM, End). The Huynh–Feldt correction was applied when sphericity was not met. When appropriate, post hoc analysis was performed using a two-tailed repeated measures Student's t-test. When a significant condition*time interaction was observed, the delta from baseline only was compared between conditions (nine comparisons). When a significant main effect of time was observed, post hoc comparisons were made to baseline only (three comparisons per condition). Correction for multiple comparisons was made using a Bonferroni adjustment. Correlation analysis was performed using a simple linear regression. Cohen's d (d) was calculated for effect size of the primary outcome variable (CMRO2). Significance was determined at an alpha of 0.05.
Results
Data from two participants were omitted due to technical difficulties with procuring blood samples. Analyzed sample size was in turn based on n = 11. The mean apnea times (range) were: NM trial 5:10 min (3:37–6:49); HV trial 5:38 min (3:50–7:11); and HX trial 16:11 min (9:39–21:02).
Cerebral metabolism
Individual values for CMRO2 are presented in Figure 2. Mean values, statistical main effects, and within trial post hoc time effects for CMRO2, OCI, CDO2, O2 extraction, net glucose uptake, and net lactate release are presented in Table 1. The end apnea CMRO2 compared to within-trial baseline was reduced by 24 ± 27% during the NM trial (p = 0.05, d = 1.25), unchanged during the HV trial (−6 ± 20%, p > 0.05), and reduced by 44 ± 15% (p < 0.05, d = 3.05) during the HX trial. The reduction in CMRO2 from baseline to end apnea during the HX trial was greater compared to the HV trial (p < 0.05, d = 2.24), but statistically similar to the NM trial (p > 0.05). The reduction in CMRO2 from baseline to end apnea was not statistically different between the NM and HV trial (p > 0.05; see Figure 3). The reduction in CMRO2 amongst all apneas significantly correlated with the end apnea PaCO2 (r2 = 0.32) and similarly arterial pH (r2 = 0.35; see Figure 4).
Figure 2.
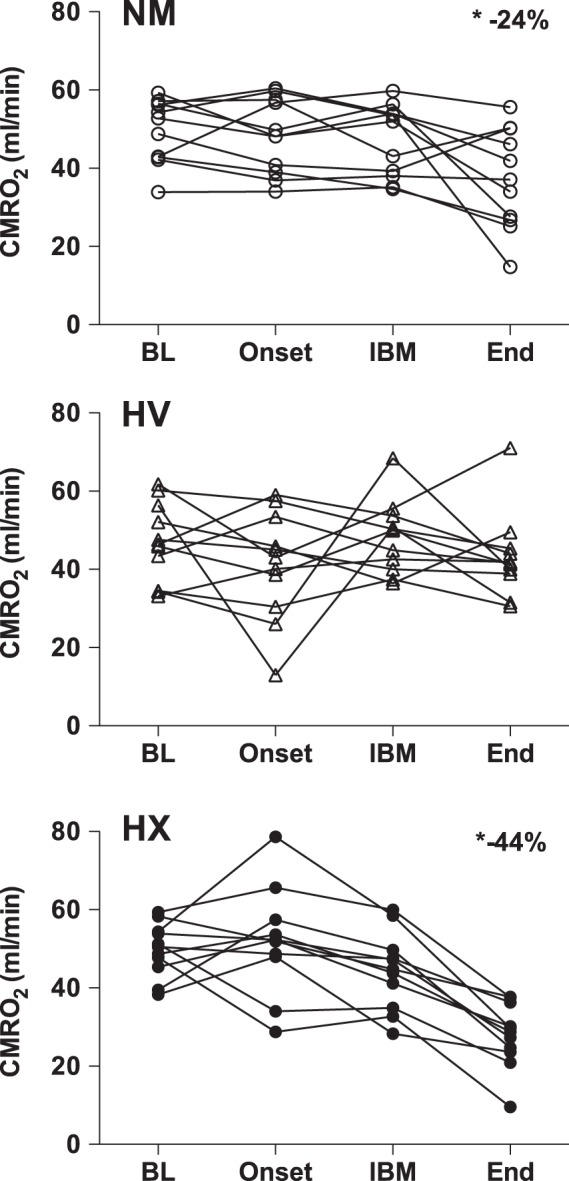
Individual data of the cerebral metabolic rate of oxygen (CMRO2) at baseline (BL), the onset of apnea (Onset), the first involuntary breathing movement (IBM), and immediately at apnea end (End), for the normal apnea (NM; top panel), apnea following 5 min of hyperventilation (HV; middle panel), and apnea following 15 min of 100% O2 hyperventilation (HX; bottom panel). Compared to baseline, there was an average ∼24% reduction in CMRO2 at the end of the NM apnea, no significant change at the end of the HV apnea, and an ∼44% reduction at the end of the HX apnea.
Table 1.
Mean ± SD of the cerebral metabolic rate of oxygen (CMRO2), oxidative carbohydrate index (OCI), O2 extraction fraction, and the net cerebral glucose uptake and lactate release, at baseline (BL), apnea onset, first involuntary breathing movement (IBM), and the end of a normal apnea (NM), apnea with prior hyperventilation (HV), and apnea with prior 100% oxygen hyperventilation (HX).
| 3 × 4 ANOVA |
||||||||
|---|---|---|---|---|---|---|---|---|
| BL | Onset | IBM | End | Condition | Time | Interaction | ||
| CMRO2 (mL·min−1) | NM | 50 ± 8 | 48 ± 10 | 47 ± 9 | 37 ± 13* | p > 0.05 | p < 0.05 | p < 0.05 |
| HV | 47 ± 9 | 41 ± 14 | 48 ± 9 | 43 ± 11 | ||||
| HX | 50 ± 7 | 52 ± 13 | 44 ± 10 | 28 ± 8* | ||||
| OCI (%) | NM | 101 ± 22 | 95 ± 11 | 122 ± 65 | 97 ± 39 | p > 0.05 | p > 0.05 | p > 0.05 |
| HV | 111 ± 32 | 72 ± 24 | 101 ± 36 | 119 ± 71 | ||||
| HX | 100 ± 16 | 81 ± 9 | 110 ± 34 | 63 ± 33 | ||||
| O2 Extraction (%) | NM | 43 ± 7 | 49 ± 8 | 32 ± 6* | 31 ± 10 | p < 0.05 | p < 0.05 | p < 0.05 |
| HV | 38 ± 6 | 50 ± 17 | 36 ± 3 | 42 ± 8 | ||||
| HX | 40 ± 7 | 52 ± 19 | 21 ± 5* | 11 ± 4* | ||||
| Net Glu uptake (mmol·min−1) | NM | 0.38 ± 0.08 | 0.39 ± 0.09 | 0.34 ± 0.13 | 0.32 ± 0.11 | p > 0.05 | p > 0.05 | p > 0.05 |
| HV | 0.35 ± 0.15 | 0.43 ± 0.12 | 0.40 ± 0.18 | 0.32 ± 0.11 | ||||
| HX | 0.38 ± 0.13 | 0.43 ± 0.23 | 0.33 ± 0.17 | 0.37 ± 0.19 | ||||
| Net La release (mmol·min−1) | NM | 0.02 ± 0.04 | 0.03 ± 0.05 | 0.04 ± 0.06 | 0.14 ± 0.21 | p > 0.05 | p = 0.05 | p < 0.05 |
| HV | 0.02 ± 0.03 | 0.09 ± 0.07 | 0.07 ± 0.08 | 0.15 ± 0.12* | ||||
| HX | 0.02 ± 0.06 | 0.12 ± 0.08 | −0.01 ± 0.06 | 0.00 ± 0.10 | ||||
Denotes significant change from baseline within trial. See text for individual differences where a main effect of condition and interaction is observed.
Figure 4.

Linear regression of the percent change in the cerebral metabolic rate of oxygen (CMRO2) from baseline to the end of apnea over the end apnea partial pressure of arterial CO2 (PaCO2), (left panel), and arterial pH (right panel). Regression analysis includes apneas from all three conditions. When analyzed separately, the correlations for end apnea pH over the change in CMRO2 are: r2 = 0.71 for the NM apnea (p < 0.05), r2 = 0.01 for the HV apnea (p > 0.05), and r2 = 0.23 (opposite direction) for the HX apnea (p > 0.05). These separate analyses may indicate a nonlinear impact of pH on the CMRO2.
There were no significant main effects of OCI, or net glucose uptake across the brain; however, there was a significant interaction (p < 0.05) in net lactate release across the brain. Post hoc tests revealed no significant differences in the change in lactate release between conditions, but the change from baseline to end apnea trended to be larger in the HV trial compared to the HX trial (p = 0.06). Allowing post hoc comparisons at p = 0.05, there was an increased cerebral lactate release at the end of apnea compared to baseline in the HV trial (p < 0.05). The cerebral lactate release in the NM trial followed similar (albeit nonsignificant) change patterns as the HV trial, but there was no change in net cerebral lactate release in the HX trial (Table 1). Similarly to our previous study,6 there were only very modest changes in arterial lactate from baseline to the end of apnea in all three conditions (NM: + 0.1 ± 0.2 mmol·L−1, HV: + 0.3 ± 0.2 mmol·L−1, HX: + 0.1 ± 0.2 mmol·L−1).
As expected, there was a significant main effect of condition, time, and condition*time interaction in O2 extraction (see Table 1 for time effects). There were no post hoc condition effects for O2 extraction between NM and HV. In contrast, the O2 extraction was lower in the HX trial compared to the HV and NM trial at the IBM onset and at the end of apnea (p all < 0.05).
Blood gases and pH
Arterial oxygen saturation (top panel) and PCO2 (bottom panel) are depicted in Figure 5. By design, there was a significant main effect of condition, time, and condition*time interaction in both SaO2 and PaCO2 (p all < 0.05). There were no post hoc condition differences at baseline for both SaO2 and PaCO2. Other than baseline, the SaO2 during the HX trial was elevated at all time points compared to both the HV and NM trials (p all < 0.05). Although the SaO2 was lower at the IBM during the HV trial (78 ± 9%) compared to the NM trial (90 ± 5%) (p < 0.05), there was no difference in SaO2 at the end of apnea (p > 0.05) between NM and HV trials. At the end of apnea, the PaCO2 during the HX trial was higher compared to the HV (p < 0.05) and NM trial (p < 0.05), and was higher in the NM trial compared to the HV trial (p < 0.05). At the onset, the PaCO2 was lower in the HX trial compared to both the NM and HV trials, and was lower in the HV compared to the NM trial (p all < 0.05). At the IBM, the PaCO2 was similar between the HX and NM trial, but reduced in the HV compared to HX and NM trials (p both <0.05). As expected, the pH inversely followed the changes in PaCO2 and (most importantly) was reduced by a greater extent at the end of apnea in the HX trial (7.28 ± 0.04) compared to the NM trial (7.36 ± 0.02) and HV (7.41 ± 0.03) trial (p both <0.05). The reduction in pH was larger in the NM trial compared to the HV trial (p < 0.05).
Cardiovascular, cerebral blood flow, and cerebral O2 delivery
Cardiovascular (HR and MAP), gCBF and CDO2 mean values, statistical main effects, and within trial post hoc time effects are shown in Table 2. In all cardiovascular and cerebrovascular variables, there were no post hoc condition effects at baseline. Although there was no main effect of condition for gCBF, the reduction from baseline at the onset was lower in the NM trial compared to the HV trial and HX trial (p both <0.05). No other between-condition differences were observed in gCBF. As expected the CDO2 was significantly elevated in the HX trial at the IBM and end of the apnea compared to the HV and NM trials (p both <0.05). The CDO2 was also reduced at the onset in the HV trial compared to the NM trial (p < 0.05). At the onset and at the end of apnea, MAP was lower in the HX compared to the NM trial (p both <0.05). The HR was higher at the onset in the HV and the HX trial, compared to the NM trial, and was higher at the IBM during the NM trial compared to the HX trial (p all < 0.05). No other between-condition differences in HR or MAP were observed.
Table 2.
Mean ± SD of the global cerebral blood flow (gCBF), cerebral oxygen delivery (CDO2), mean arterial pressure (MAP), and heart rate (HR) at baseline (BL), apnea onset, first involuntary breathing movement (IBM), and the end of a normal apnea (NM), apnea with prior hyperventilation (HV), and apnea with prior 100% oxygen hyperventilation (HX).
| 3 × 4 ANOVA |
||||||||
|---|---|---|---|---|---|---|---|---|
| BL | Onset | IBM | End | Condition | Time | Interaction | ||
| gCBF (mL·min−1) | NM | 635 ± 84 | 522 ± 85* | 909 ± 104* | 1155 ± 147* | p > 0.05 | p < 0.05 | p < 0.05 |
| HV | 718 ± 162 | 429 ± 76* | 911 ± 233* | 1151 ± 259* | ||||
| HX | 720 ± 121 | 429 ± 104* | 1016 ± 218* | 1295 ± 310* | ||||
| CDO2 (mL·min−1) | NM | 117 ± 18 | 100 ± 17* | 153 ± 19* | 124 ± 22 | p < 0.05 | p < 0.05 | p < 0.05 |
| HV | 130 ± 31 | 81 ± 13* | 128 ± 25 | 106 ± 27 | ||||
| HX | 129 ± 26 | 84 ± 14* | 206 ± 51* | 260 ± 78* | ||||
| MAP (mmHg) | NM | 101 ± 8 | 105 ± 11* | 123 ± 11* | 160 ± 16* | p < 0.05 | p < 0.05 | p > 0.05 |
| HV | 97 ± 6 | 99 ± 7 | 120 ± 9* | 147 ± 16* | ||||
| HX | 98 ± 6 | 99 ± 8 | 121 ± 9* | 150 ± 14* | ||||
| HR (bpm) | NM | 65 ± 7 | 82 ± 13* | 76 ± 12* | 60 ± 13 | p < 0.05 | p < 0.05 | p < 0.05 |
| HV | 67 ± 10 | 97 ± 15* | 74 ± 13 | 64 ± 14 | ||||
| HX | 67 ± 11 | 95 ± 19* | 67 ± 11 | 72 ± 12 | ||||
Denotes significant change from baseline within trial. See text for individual differences where a main effect of condition and interaction is observed.
Discussion
This study first confirms our previous finding6 of a reduced CMRO2 at the termination of a prolonged apnea that yields severe levels of hypoxemia (PaO2 ∼ 30 mmHg) and hypercapnia (PaCO2 ∼ 55 mmHg). We extend these findings by establishing that the hypercapnia, and resultant acidosis, during apnea is obligatory for reducing the CMRO2. This is demonstrated by the largest reduction in CMRO2 during the hyperoxic apnea (HX) that generated the most severe hypercapnia (PaCO2 ∼ 60 mmHg), without the confounding influence of hypoxia; whereas the apnea generating no hypercapnia but severe hypoxia (HV) yielded no changes in CMRO2. Moreover, with all three apneas combined, the end apnea PaCO2 and arterial pH significantly correlated with the reduction in CMRO2 (Figure 4). A secondary finding was a net cerebral lactate release at the end of apnea compared to baseline in the HV apnea and to a lesser extent (nonsignificant) in the NM apnea, with no change in the oxidative carbohydrate index (OCI). In contrast, there was no increase in net cerebral lactate release during the HX trial. These subset findings may indicate a role for hypoxia in net lactate release from the human brain.
Influence of hypercapnia on the CMRO2
The majority of literature, including findings from this study, now supports a role for hypercapnia / pH on the CMRO2 in humans17,18—discrepant findings are discussed in Yablonskiy.19 Depressed cortical activity is also shown with CO2 inhalation,8,20,21 lending support for reductions in CMRO2. The hypercapnic suppression of CMRO2 is described by three related mechanisms. First, hypercapnia / extracellular acidosis increases adenosine concentrations, which activate adenosine A1 receptors8 and inhibit excitatory glutamatergic neurotransmission. Adenosine A1R activation may also directly attenuate mitochondrial metabolism during episodes of oxygen deprivation.22 Second, hypercapnia / acidosis may reduce phosphofructokinase activity, evidenced from an accumulation of fructose-6 phosphate and glucose-6 phosphate in un-anesthetized rat cerebral cortices following CO2 inhalation.7 Any reduction in PFK activity, and therefore glycolytic flux, will correspondingly reduce the CMRO2 provided that other noncarbohydrate substrates (e.g. amino acids) are not oxidized. This is a fair assumption given that glucose and lactate irrefutably provide the primary metabolite for cerebral metabolism in humans,23,24 barring ketone body use during extended hypoglycemia.25 Finally, hypercapnia may depress the CMRO2 via a reduction in cerebral temperature resulting from the increased CBF (dependent on the arterial blood temperature, and local brain metabolism). A temperature effect, however, is likely small. For example, assuming that the increased CBF caused a ∼0.25 ℃ drop in global brain temperature,26 a liberal Q10 temperature coefficient of 3 (Bain et al.27 for review) would theoretically equate to a metabolic reduction of only ∼5%. In summary, although we cannot delineate any one mechanism with our data, it is likely that increased adenosine, inhibited PFK activity, and to a lesser extent a reduced cerebral temperature from elevated CBF each contributed to the reduction in CMRO2 from hypercapnia.
Influence of hypoxia on the CMRO2
Hypoxia may increase the CMRO24,5,12 and in turn counter some of the metabolic depression from hypercapnia during apnea. Using MRI, Peng et al.,12 recently quantified the opposing facet of hypoxia and hypercapnia on the CMRO2 in humans. Here, Peng et al. reported that hypercapnia (5% CO2 inhalation) reduced the CMRO2 by ∼8%, hypoxia (13% O2, balanced with N2) increased the CMRO2 by ∼17%, but the combination of the two (5% CO2 and 13% O2, balanced with N2) had no effect on the CMRO2. It was thus concluded that the divergent metabolic influences of hypercapnia and hypoxia equated to a net oxidative metabolic balance. The ostensible mechanism(s) for increased CMRO2 in hypoxia remain undefined, but is suggested to involve decreased mitochondrial efficiency for ATP production,28 and perhaps also from increased sympathetic activity acting at the neuronal level. On the other hand, an increased CMRO2 during fixed FiO2 hypoxia may simply be a consequence of the concomitant hyperventilation-induced hypocapnia. For example when PaCO2 is kept eucapnic, hypoxia has no effect on the CMRO2.16
With respect to this study, the largest decrease in CMRO2 during the hyperoxic apnea may theoretically manifest in part from the absence of hypoxia, rather than simply from the largest level of hypercapnia. Nevertheless, as evidenced from our previous study,6 and the NM apnea, it is clear that the impact of hypercapnia, at least in the setting of prolonged apnea, outweighs any influence from hypoxia on the CMRO2. Moreover, there was no increase in CMRO2 at the end of the HV apnea, despite profound hypoxia (PaO2 ∼ 31 mmHg) with minimal hypercapnia (PaCO2 ∼ 43 mmHg). We therefore suggest that the impact of hypoxia on CMRO2, if any, is negligible.
Influence of hyperoxia on the CMRO2
Like hypoxia, hyperoxia has also been suggested to impact the CMRO2,4,29 although in the opposite direction, i.e. to decrease it. However, this is not a universal finding, with other studies showing no change.16,30 Aside from differences in measurement technique, it is difficult to speculate on the discrepancies between studies. In the present study, 15 min of hyperoxic breathing did not depress the CMRO2 although it did become more variable (see Onset on Figure 2, bottom panel). The variability may stem from an interaction between the 15 min of hypocapnia and hyperoxia. With obvious implications in critical care (Llitjos et al.31 for review), it is clear that more research is required to determine the cerebral metabolic impact of hyperoxia.
Cerebral lactate release and nonoxidative metabolism
There was a main effect of time and a time*condition interaction in net cerebral lactate release. Here, net lactate release was increased in the HV apnea and to a lesser extent in the NM apnea, but not in the HX apnea (Table 1). These findings suggest that the hypoxia elicited a net cerebral lactate release. Such findings are broadly comparable to Overgaard et al.,32 who reported that cerebral lactate release was increased from ∼0.05 to 0.09 mmol·min−1 with resting normoxia compared to hypoxia (PaO2 ∼ 35 mmHg).
The net lactate release from the brain may be explained by astrocyte glycogenolysis.24,33 That is, glycogen contained within the astrocytes, acting as the principle fuel storage of the brain, is converted to lactate as potential oxidative fuel for the neurons.33,34 Overgaard et al. attribute cerebral lactate release (and thus astrocyte glycogenolysis) in hypoxic exercise to a coupling of nonoxidative and oxidative metabolism. In contrast, our data indicate that the net lactate release was independent of the CMRO2 and nonoxidative metabolism, as determined by the oxidative carbohydrate index. In the present study, it may be that astrocyte glycogenolysis was initiated from noradrenaline,35,36 which we have previously shown to increase by ∼500% during apneas that generate extreme hypoxia.6 Ultimately, the fate of cerebral substrate (lactate) use is still poorly understood, even with approaches employing the use of isotope labeling and PET.37 The notion of cerebral glycogenolysis should in turn be held with caution.
Although we report reduction in the CMRO2, we are left to speculate on actual brain activity. Had brain activity been maintained, metabolic activity must have been sourced anaerobically. This may be the case during the HX apnea, where the OCI trended to decline (nonsignificant perhaps reflecting low statistical power), suggesting increased anaerobic carbohydrate metabolism. However, this was not observed in the NM or HV apnea. That glucose uptake did not change in the NM trial, despite reductions in the CMRO2 (Table 1), may indicate increased brain glucose storage, or fatty acid (e.g. ketones) metabolism. More study is ultimately required to clarify substrate usage and brain activity during prolonged apnea.
Cerebral oxygen-conserving reflexes associated with the dive reflex?
The cerebral metabolic changes reported from this study must be interpreted within the context of a dry (out of water) apnea. As discussed in our previous study,6 it is possible that trigeminal nerve activation with cold-water immersion prompts cerebral oxygen-conserving reflexes beyond those demonstrated from the hypercapnia, i.e. from the trigeminocardiac reflex.11 It is long accepted that trigeminal nerve activation is a potent autonomic conduit of the mammalian dive reflex, in large part responsible for the bradycardia11. This undoubtedly promotes oxygen conservation at the level of whole-body metabolism3; however, direct evidence for the trigeminocardiac reflex and reductions in CMRO2 is lacking, or to the best of our knowledge, absent. Rather, one function of the trigeminocardiac reflex is to increase CBF without altering CMRO2 (for review see Schaller38). The disproportionately rapid delivery of oxygen and glucose to the brain relative to its metabolic rate from trigeminal nerve activation may provide a protective means to assure adequate cerebral fuel delivery.11 Although we show an elevated cerebral oxygen delivery near the midpoints of a typical prolonged apnea in this study, consistent with our previous studies,6,9 it is probably the consequence of the hypercapnia. Not surprisingly, when hypercapnia does not develop, i.e. during the HV apnea, the CDO2 is never elevated above baseline. Nevertheless, despite lack of current evidence for a reduction in CMRO2 attributable to a dive / oxygen conserving reflex, a logical future study is to test the cerebral metabolic reduction to apnea with and without facial and/or whole-body water immersion.
Calculations of cerebral metabolism
To calculate the CMRO2, we employed the Fick principle with the measures of internal jugular vein and radial artery PO2, SO2, and Hg, and simultaneous measures of cerebral blood flow using duplex ultrasound of the ICA and VA. The advantage of this invasive approach compared to other techniques is its high temporal resolution. Moreover, it does not require radioactive tracers (e.g. with PET), nor does it require relying on MRI sequencing techniques that have yet to be fully validated. The major disadvantage of this technique is that we are limited to estimations of global CMRO2 from cerebral drainage solely from the jugular vein, and neglect the ∼10% of blood draining through the vertebral veins. The findings from this study must therefore be interpreted within the parameters measured, and our estimates of global CMRO2 should not be compared with specific brain regions, particularly the deeper brain regions that channel the vertebral veins.
Implications and conclusion
Describing the role of extreme acidosis and hypoxia, alone and in combination, on the cerebral metabolic functioning have implications in a myriad of clinical settings including sepsis, cardiorespiratory disease, and neurological disorders. Indeed, it is clinically well recognized that hypocapnia can incite seizures in epileptic patients, and hypercapnia can suppress them.39 In relation to extreme apnea, one subject developed seizure like symptoms (unresponsive with mild convulsions) lasting approximately 10 s at the termination of the HV apnea, but not the NM or HX apnea. In this subject the CMRO2 was notably elevated by 15% at the termination of the HV apnea, compared to baseline (see Figure 2). Although this subject was only mildly hypocapnic when the convulsions occurred at the end of the apnea (PaCO2 = 36.5 mmHg vs. 43.8 mmHg at baseline), it may be that hypercapnia in the NM apnea prevented the hypoxic induced seizure. In this case, the hypercapnic depression of metabolic activity is entirely protective. In apnea competition, profound hyperventilation (PaCO2 at ∼20 mmHg) is usually avoided to prevent shallow water blackout, notionally attributed to the reduction in breathing sensation and eventual onset of critical hypoxemia, especially upon ascent. In the ultra-elite (n = 6 in the present study), profound hyperventilation also proffers no benefit to maximal apnea time with facial immersion; in fact it will often decrease it (personal correspondence with the Croatian national apnea coach). The changes in CMRO2 ascribed to hypercapnia may help describe both the occurrence of shallow water blackout, and the reduction in ultra-elite maximal apnea time following hyperventilation.
In summary, our data provides novel evidence for a reduction in CMRO2 at the termination of a prolonged apnea in humans that, within the parameters of the present study, can be ascribed primarily to hypercapnia / acidosis. The metabolic depressing influence of hypercapnia outweighs any increase in CMRO2 that is potentially evoked from hypoxia. Moreover, we show that severe apnea induced hypoxia provokes a net release of lactate from the brain. Together, these findings have implications for the better understanding of brain survival during periods of not only voluntary apnea but also during loss of consciousness, and potentially to the myriad of clinical situations where arterial acidosis occurs. Of course, it remains undetermined whether the findings from this study are unique to the population tested (i.e. an adaptive trait of trained breath-hold divers), or can be extended to the general, nondiving, population.
Acknowledgments
The authors would like to specially acknowledge the apnea divers from the Croatia National Apnea team for their participation.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by a Canadian Research Chair and NSERC Discovery grant held by Prof Ainslie. Drs Dujic, Barak, and Ainslie were also funded through the Croatian Science Foundation (IP-2014-09-1937). Mr Bain was funded through a postgraduate NSERC scholarship.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
ARB, PNA, ID, and OB conception and design of research; ARB, PNA, OFA, RLH, ID, TM, DMB, AS, DKD, ZD, and DBM performed experiments; ARB analyzed data; ARB, PNA, OFA, RLH, ID, TM, DMB, AS, DKD, ZD, and DBM interpreted results of experiments; ARB prepared figures; ARB drafted the article; ARB, PNA, OFA, RLH, ID, TM, DMB, AS, DKD, ZD, and DBM edited / revised the article; and approved the final version of the article.
References
- 1.Butler PJ, Jones DR. Physiology of diving of birds and mammals. Physiol Rev 1997; 77: 837–899. [DOI] [PubMed] [Google Scholar]
- 2.Bain AR, Dujic Z, Hoiland RL, et al. Peripheral chemoreflex inhibition with low-dose dopamine: New insight into mechanisms of extreme apnea. Am J Physiol Regul Integr Comp Physiol 2015. DOI: ajpregu 00271 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoiland RL, Ainslie PN, Bain AR, et al. Beta 1-blockade increases maximal apnea duration in elite breath hold divers. J Appl Physiol (1985) 2016. DOI: jap 00127 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu F, Liu P, Pascual JM, et al. Effect of hypoxia and hyperoxia on cerebral blood flow, blood oxygenation, and oxidative metabolism. J Cereb Blood Flow Metab 2012; 32: 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vestergaard MB, Lindberg U, Aachmann-Andersen NJ, et al. Acute hypoxia increases the cerebral metabolic rate - a magnetic resonance imaging study. J Cereb Blood Flow Metab 2016; 36: 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bain AR, Ainslie PN, Hoiland RL, et al. Cerebral oxidative metabolism is decreased with extreme apnea in humans; impact of hypercapnia. J Physiol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Folbergrova J, Norberg K, Quistorff B, et al. Carbohydrate and amino acid metabolism in rat cerebral cortex in moderate and extreme hypercapnia. J Neurochem 1975; 25: 457–462. [DOI] [PubMed] [Google Scholar]
- 8.Dulla CG, Dobelis P, Pearson T, et al. Adenosine and ATP link PCO2 to cortical excitability via pH. Neuron 2005; 48: 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willie CK, Ainslie PN, Drvis I, et al. Regulation of brain blood flow and oxygen delivery in elite breath-hold divers. J Cereb Blood Flow Metab 2015; 35: 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steinback CD, Breskovic T, Frances M, et al. Ventilatory restraint of sympathetic activity during chemoreflex stress. Am J Physiol Regul Integr Comp Physiol 2010; 299: R1407–1414. [DOI] [PubMed] [Google Scholar]
- 11.Lemaitre F, Chowdhury T, Schaller B. The trigeminocardiac reflex - a comparison with the diving reflex in humans. Arch Med Sci 2015; 11: 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng SL, Ravi H, Sheng M, et al. Searching for a truly “iso-metabolic” gas challenge in physiological MRI. J Cereb Blood Flow Metab 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willie CK, Colino FL, Bailey DM, et al. Utility of transcranial Doppler ultrasound for the integrative assessment of cerebrovascular function. J Neurosci Meth 2011; 196: 221–237. [DOI] [PubMed] [Google Scholar]
- 14.Thomas KN, Lewis NC, Hill BG, et al. Technical recommendations for the use of carotid duplex ultrasound for the assessment of extracranial blood flow. Am J Physiol Regul Integr Comp Physiol 2015; 309: R707–720. [DOI] [PubMed] [Google Scholar]
- 15.Woodman RJ, Playford DA, Watts GF, et al. Improved analysis of brachial artery ultrasound using a novel edge-detection software system. J Appl Physiol (1985) 2001; 91: 929–937. [DOI] [PubMed] [Google Scholar]
- 16.Ainslie PN, Shaw AD, Smith KJ, et al. Stability of cerebral metabolism and substrate availability in humans during hypoxia and hyperoxia. Clin Sci (Lond) 2014; 126: 661–670. [DOI] [PubMed] [Google Scholar]
- 17.Chen JJ, Pike GB. Global cerebral oxidative metabolism during hypercapnia and hypocapnia in humans: Implications for BOLD fMRI. J Cereb Blood Flow Metab 2010; 30: 1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu F, Uh J, Brier MR, et al. The influence of carbon dioxide on brain activity and metabolism in conscious humans. J Cereb Blood Flow Metab 2011; 31: 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yablonskiy D.A. Cerebral metabolic rate in hypercapnia: Controversy continues. J Cereb Blood Flow Metab 2011; 31: 1502–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zappe AC, Uludag K, Oeltermann A, et al. The influence of moderate hypercapnia on neural activity in the anesthetized nonhuman primate. Cereb Cortex 2008; 18: 2666–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thesen T, Leontiev O, Song T, et al. Depression of cortical activity in humans by mild hypercapnia. Hum Brain Mapp 2012; 33: 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duarte JM, Cunha RA, Carvalho RA. Adenosine A(1) receptors control the metabolic recovery after hypoxia in rat hippocampal slices. J Neurochem 2016; 136: 947–957. [DOI] [PubMed] [Google Scholar]
- 23.Rasmussen P, Nyberg N, Jaroszewski JW, et al. Brain nonoxidative carbohydrate consumption is not explained by export of an unknown carbon source: Evaluation of the arterial and jugular venous metabolome. J Cereb Blood Flow Metab 2010; 30: 1240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia 2007; 55: 1263–1271. [DOI] [PubMed] [Google Scholar]
- 25.Morris AA. Cerebral ketone body metabolism. J Inherit Metab Dis 2005; 28: 109–121. [DOI] [PubMed] [Google Scholar]
- 26.Hayward JN, Baker MA. Role of cerebral arterial blood in the regulation of brain temperature in the monkey. Am J Physiol 1968; 215: 389–403. [DOI] [PubMed] [Google Scholar]
- 27.Bain AR, Nybo L, Ainslie PN. Cerebral vascular control and metabolism in heat stress. Compr Physiol 2015; 5: 1–36. [DOI] [PubMed] [Google Scholar]
- 28.Solaini G, Baracca A, Lenaz G, et al. Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys Acta 2010; 1797: 1171–1177. [DOI] [PubMed] [Google Scholar]
- 29.Rockswold SB, Rockswold GL, Zaun DA, et al. A prospective, randomized clinical trial to compare the effect of hyperbaric to normobaric hyperoxia on cerebral metabolism, intracranial pressure, and oxygen toxicity in severe traumatic brain injury. J Neurosurg 2010; 112: 1080–1094. [DOI] [PubMed] [Google Scholar]
- 30.Diringer MN, Aiyagari V, Zazulia AR, et al. Effect of hyperoxia on cerebral metabolic rate for oxygen measured using positron emission tomography in patients with acute severe head injury. J Neurosurg 2007; 106: 526–529. [DOI] [PubMed] [Google Scholar]
- 31.Llitjos JF, Mira JP, Duranteau J, et al. Hyperoxia toxicity after cardiac arrest: What is the evidence? Ann Intens Care 2016; 6: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Overgaard M, Rasmussen P, Bohm AM, et al. Hypoxia and exercise provoke both lactate release and lactate oxidation by the human brain. FASEB J 2012; 26: 3012–3020. [DOI] [PubMed] [Google Scholar]
- 33.Dienel GA, Cruz NF. Contributions of glycogen to astrocytic energetics during brain activation. Metab Brain Dis 2015; 30: 281–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown AM. Brain glycogen re-awakened. J Neurochem 2004; 89: 537–552. [DOI] [PubMed] [Google Scholar]
- 35.Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol 1995; 45: 347–360. [DOI] [PubMed] [Google Scholar]
- 36.Matsui T, Soya S, Okamoto M, et al. Brain glycogen decreases during prolonged exercise. J Physiol 2011; 589: 3383–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dienel GA. Brain lactate metabolism: The discoveries and the controversies. J Cereb Blood Flow Metab 2012; 32: 1107–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaller B. Trigeminocardiac reflex. A clinical phenomenon or a new physiological entity? J Neurol 2004; 251: 658–665. [DOI] [PubMed] [Google Scholar]
- 39.Yang XF, Shi XY, Ju J, et al. 5% CO(2) inhalation suppresses hyperventilation-induced absence seizures in children. Epilepsy Res 2014; 108: 345–348. [DOI] [PubMed] [Google Scholar]