

Abstract
Iron overload plays a key role in the secondary brain damage that develops after intracerebral hemorrhage (ICH). The significant increase in iron deposition is associated with the generation of reactive oxygen species (ROS), which leads to oxidative brain damage. In this study, we examined the protective effects of VK-28, a brain-permeable iron chelator, against hemoglobin toxicity in an ex vivo organotypic hippocampal slice culture (OHSC) model and in middle-aged mice subjected to an in vivo, collagenase-induced ICH model. We found that the effects of VK-28 were similar to those of deferoxamine (DFX), a well-studied iron chelator. Both decreased cell death and ROS production in OHSCs and in vivo, decreased iron-deposition and microglial activation around hematoma in vivo, and improved neurologic function. Moreover, compared with DFX, VK-28 polarized microglia to an M2-like phenotype, reduced brain water content, deceased white matter injury, improved neurobehavioral performance, and reduced overall death rate after ICH. The protection of VK-28 was confirmed in a blood-injection ICH model and in aged-male and young female mice. Our findings indicate that VK-28 is protective against iron toxicity after ICH and that, at the dosage tested, it has better efficacy and less toxicity than DFX does.
Keywords: Deferoxamine, intracerebral hemorrhage, iron chelator, microglial polarization, neurolucida
Introduction
Blood that escapes after spontaneous intracerebral hemorrhage (ICH) is toxic and contributes to secondary brain damage.1 Besides thrombin, hemoglobin is the most likely neurotoxin protein, as it is released in abundance from lysed red blood cells. As a result, microglia and infiltrating macrophages in the perihematomal zone become overloaded with iron degraded from hemoglobin (Hb)/heme.2 Subsequently, iron transferred from immune cells accumulates in neurons and reacts with hydrogen peroxide via the Fenton reaction to form highly toxic hydroxyl radicals that attack DNA, proteins, and lipid membranes, thereby disrupting cellular function.3
In preclinical in vivo ICH models induced by collagenase or blood injection, iron deposition is increased on the first day, peaks at day 3, and is still detectable on day 28 in the peri-hematoma region.4 Increasing evidence has shown that iron chelation reduces Hb/iron-induced neuronal toxicity and offers promise as a therapeutic strategy for ICH.2,5–7 The iron chelator deferoxamine (DFX) is well-studied in ICH models. It has been shown to offer neuronal protection, reduce reactive oxygen species (ROS), and improve neurologic function in both collagenase and blood models in mice, rats, and piglets2,8–10; however, it has not been shown to decrease lesion volume,2,11 brain edema and swelling,2,11 or even neurologic deficits, as reported by one group.11,12 Additionally, DFX is a large and unstable molecule, has poor cell permeability,13 and causes severe side effects, including exacerbated body weight loss in mice (200 mg/kg)2 and hypotension, pancytopenia, retinal toxicity and neurotoxicity in patients (at high doses),14–16 and, in one case report, acute respiratory distress syndrome in a child.17 Although a low dose of DFX (7–62 mg/kg per day) is being tested in a Phase ΙΙ clinical trial for ICH (www.clinicaltrials.gov), an alternative to DFX is highly desired for pre-clinical and clinical ICH studies.
VK-28 (5-[4-(2-hydroxyethyl) piperazine-1-ylmethyl]-quinoline-8-ol), a brain-permeable iron chelator, has been shown to provide significant neuroprotection and a marked reduction in iron deposition in models of Parkinson's disease,18 Alzheimer's disease,19 and amyotrophic lateral sclerosis,20 but no study has shown its effects on ICH outcomes. The goal of this study was to investigate the effects of VK-28 on short-term ICH outcomes, microglial polarization, white matter injury, long-term functional outcomes, and animal survival. We designed experiments to compare the therapeutic effects and side effects of DFX and VK-28 side by side in ex vivo organotypic hippocampal slice cultures (OHSCs) and in an in vivo ICH model. The insights gained from this study will be essential for selecting the proper iron chelators for future preclinical/clinical ICH studies.
Materials and methods
Study design
A power analysis based on our previous studies2 and pilot data indicated that eight mice/group would provide at least 80% power for detecting a 20% decrease in lesion volume at α = 0.05 (two-sided). To account for potential animal death, we used 10 mice/group. Animals that had a neurologic deficit score greater than 20 at 24 h post-surgery were euthanized under deep anesthesia.21 Animals that died or were euthanized within 24 h post-surgery were excluded from the sample size. Outlying data points were defined after assuming a normal distribution using statistical software (threshold was set as 2.0 fold of SD from the mean) and were excluded from the data set. Three or more independent experiments were performed for all ex vivo experiments. Animals and slice cultures for each group were randomized with the website www.randomization.com. Treatment, data collection, and data analysis were blinded by using different investigators or by masking sample labels.
Animals
The experimental procedures were conducted in accordance with the National Institutes of Health guidelines and were approved by the Johns Hopkins University Animal Care and Use Committee. Middle-aged male, aged male, and young female C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA, USA). In total, we used 167 middle-aged male mice (12-months-old), 30 aged male mice (18-months-old), and 30 young female mice (3-months-old). All mice were housed in a pathogen-free environment with free access to food and water on a 12-h light/dark cycle before and after surgery. Animal experiments were reported in accordance with the ARRIVE, STAIR, and RIGOR guidelines.22,23
OHSCs
OHSCs were cultured as previously described.24 Brains were rapidly removed from pups (seven- to nine-days old) and placed in ice-cold Hanks’ balanced salt solution (HBSS, with Ca2+ and Mg2+; Life Technologies, Frederick, MD) containing 25 mM HEPES (Life Technologies). The brains were cut coronally into 350 -µm-thick sections with a McIlwain tissue chopper (Ted Pella, Redding, CA). The hippocampal slices were isolated carefully and immediately plated on a hydrophilic PTFE cell culture insert with pore size of 0.4 µm (Millicell-CM, Millipore, Darmstadt, Germany) in a 6-well plate. We cultured the hippocampal slices with 1 mL culture medium containing 50% DMEM (Life Technologies), 25% HBSS, 25% heat-inactivated horse serum (Invitrogen, Grand Island, NY), 35 mM glucose (Sigma, St. Louis, MO), and 25 mM HEPES at 37℃ in a humidified incubator with 5% CO2. We changed the medium to 70% DMEM and 5% heat-inactivated horse serum the next day and then every two to three days. After 10 to 14 days of culture in vitro, the sections were placed in serum-free medium (75% DMEM) one day before treatment.
ICH model
We used ear bars and an incisor bar to hold the heads of the mice, which were anesthetized with isoflurane (4.0% for induction and 2.0% for maintenance) and ventilated with oxygen-enriched air (20%:80%) via a nose cone. Each mouse was injected in the left striatum with 0.5 µL of 0.075 U collagenase VII-S (Sigma) at the rate of 0.1 µL/min. The needle was kept in place for 5 min and then removed gradually. The stereotactic coordinates of injection were 0.6 mm anterior and 2.0 mm lateral of the bregma, and 3.0 mm in depth.25 The craniotomy was sealed with 4–0 silk sutures. To confirm the protective effect of VK-28 on ICH outcomes, we used a second ICH model in which 10 µL of autologous whole arterial blood was injected into the left striatum at a rate of 0.5 µL/min. Blood removed from the tail and was infused in two time blocks (4 µL followed by a 5-min pause and then 6 µL followed by a 10-min pause).26 For all models, rectal temperature was maintained at 37.0 ± 0.5℃ throughout experimental and recovery periods (DC Temperature Controller 40-90-8D; FHC Inc., ME). Sham-operated mice received the same treatment with needle insertion but no collagenase or blood injection.
Experimental groups
For OHSCs experiments, we used four treatment groups: vehicle, 20 µM Hb (Sigma), 20 µM Hb with 100 µM DFX (Sigma),27 and 20 µM Hb with 5 µM VK-28 (Santa Cruz Biotechnology, Dallas, TX).28
For in vivo experiments, the mice were randomly assigned to receive DFX (200 mg/kg),2 VK-28 (5 mg/kg),28 or vehicle (saline containing 5% DMSO). All drugs were administered intraperitoneally (i.p.) at 6 h after collagenase or autologous blood injection and then every 12 h for one, three, or seven consecutive days.
Propidium iodide staining
Propidium iodide (PI) (5 µg/mL, Sigma) was added to OHSCs for cell death assessment. Before OHSCs were treated with Hb or drug, images were taken under a fluorescence microscope (TE 2000-E, Nikon, Japan) at 200 ms exposure time, and the PI fluorescence intensity was recorded as P0. After 16 h of treatment, the slices were incubated with PI for 30 min and images were captured as P16. Slices were incubated for another 24 h to reach maximum death (Pmax). The fluorescence intensity was measured by Image J software (NIH, Bethesda, MD), and cell death was determined as a percentage by dividing the total number of pixels in the region of interest (ROI) by the pixels in the ROI above a threshold in the PI fluorescent image.24 Cell death was calculated as (P16-P0)/(Pmax-P0) × 100%.
Lactate dehydrogenase (LDH) activity assay
We collected the treated medium from the OHSCs and measured lactate dehydrogenase (LDH) activity by an LDH activity assay kit (Sigma). The samples were treated with substrate and assay buffer in a clear 96-well plate. Reduction of NAD to NADH was detected at 450 nm on a Spectramax M2 microplate reader (Molecular Devices LLC, Sunnyvale, CA).
ROS detection
For in vitro detection, we incubated the OHSCs for 30 min with 63 µM hydroethidine (HEt, Molecular Probes, Eugene, OR), a cell-permeable oxidative fluorescent dye, and captured images under a fluorescence microscope (Nikon Eclipse 90i) at identical exposure times. An investigator blinded to treatment group analyzed the fluorescence intensity with Image J software.
For in vivo detection, we analyzed ROS production after ICH by in situ detection of oxidized HEt.29 HEt (in DMSO) was diluted to 1 mg/mL in phosphate-buffered saline (PBS) and sonicated. At one day after ICH, mice were injected i.p. with 300 µL of HEt and euthanized 1 h later. All images were taken under a fluorescence microscope at identical exposure times, contrast settings, and intensity. An investigator blinded to treatment group analyzed the sections with Image J software.
Perls’ iron and Fluoro-Jade B staining
Ferric iron accumulation at three days after ICH was detected by 3,3′-diaminobenzidine (DAB; Vector Laboratories, Burlingame, CA)-enhanced Perls’ staining as previously described.2 Sections of brain tissue were washed with PBS and incubated in freshly prepared Perls’ solution (5% potassium ferrocyanide with 10% hydrochloric acid) for 1 h, followed by PBS washes (3 × 5 min). After DAB incubation (3 min) and hematoxylin (Sigma) counterstaining, Image J software was used to analyze iron deposition.
To examine degraded neurons in brain tissue, we used Fluoro-Jade B (FJB) staining according to a previously published description.30 The concentration of FJB was 0.0004%. Stained brain sections were examined with a fluorescence microscope at an excitation wavelength of 450–490 nm.
To quantify Perls’ iron-positive or FJB-positive cells, we selected at least three sections per mouse with similar areas of hematoma, and three fields with magnification of 200× per section. The numbers from these fields were averaged and expressed as positive cells per square millimeter for each mouse. Tissue sections were analyzed by an observer who was blinded to the experimental cohorts.
Hemorrhagic injury volume
On day 3 after ICH, we euthanized mice and collected 30 -µm rostral-caudal level cryosections spaced at 180 -µm intervals from bregma +1.32 to −1.64 mm. We stained the cryosections with Luxol fast blue (which stains myelin) and Cresyl violet (CV, which stains surviving neurons) to quantify lesion volume, including volume of the hematoma and peri-hematoma regions (characterized by neuronal loss and white matter damage); damaged areas were evaluated under a 10× objective with Image J software. The total injury volume in cubic millimeters was calculated as the sum of the damaged area multiplied by the distance between the sections (180 µm),31 and was corrected for brain swelling: corrected lesion volume = volume of non-hemorrhagic hemisphere – (volume of hemorrhagic hemisphere – lesion volume).29 We further quantified Luxor fast blue-positive areas to evaluate myelin integrity under 10× objective microscope, and used CV to quantify surviving neurons around the lesion under 40× objective.32
Brain water content measurement
To assess the global cerebral edema, we measured brain water content in different treatment groups as previously reported.2 Briefly, on day 3 after ICH, mice were euthanized by deep anesthesia. The brains were removed and divided into three parts: ipsilateral hemisphere, contralateral hemisphere, and cerebellum (an internal control). Samples were immediately weighed on an analytical balance to record the wet weight, and then dried at 100℃ for 24 h to obtain the dry weight. Brain water content was expressed as (wet weight-dry weight)/wet weight of brain tissue×100%.
Brain swelling measurement
Using the cytosections stained with Luxol fast blue and CV, we quantified brain swelling by calculating the percentage of hemispheric enlargement at three days after ICH with Image J software as described previously.2 The percentage of hemisphere enlargement was expressed as: (ipsilateral hemisphere volume/contralateral hemisphere volume) ×100%.
Spectrophotometric assay for hemoglobin content
The hemoglobin content of brains was quantified with the Hemoglobin Assay Kit (Sigma) at 24 h after collagenase injection. Briefly, mice were anesthetized and then transcardially perfused with PBS. The injured hemisphere was collected in a tube with 500 µL of ice cold PBS. The tissue was homogenized for 1 min, and then centrifuged at 16,000 g for 15 min. Fifty microliters of supernatant were transferred to flat-bottom, 96-well plates. After a 5-min incubation with reaction reagent, we measured the absorbance at 400 nm. Hemoglobin content was calculated by a standard curve.
Neurologic function evaluation
An investigator blinded to treatment groups evaluated the mice with a neurologic deficit scoring system on days 1, 3, 7, and 28 after ICH, as described previously.21 Mice were evaluated for six tests, including body symmetry, gait, climbing, circling behavior, front limb symmetry, and compulsory circling. Each test was graded from 0 to 4, leading to a maximum deficit score of 24.21 Mice were also tested in the corner turn test at baseline and on days 1, 7, and 28 after ICH. The direction a mouse turned when facing a 30° corner was recorded in 10 trials and expressed as percentage of left turns.32
Immunofluorescence staining
Immunofluorescence was conducted as described previously.32 Brain sections were incubated overnight at 4℃ with primary antibodies including rabbit anti-Iba-1 (1:1000; Wako Chemicals, Richmond, VA, USA), mouse anti mannose receptor (CD206, 1:50; Abcam, Cambridge, MA), rat anti-CD16/32 (1:100; BD Medical Technology, Franklin Lakes, NJ), mouse anti-myelin basic protein (MBP, 1:1000; Biolegend, San Diego, CA), rabbit anti-degraded myelin basic protein (dMBP, 1:1000; Millipore, Billerica, MA), and rabbit anti-beta-amyloid precursor protein (beta-APP, 1:1000; Invitrogen). Then the sections were incubated with secondary antibodies (Alexa Fluor 488 and/or Alexa Fluor 594, 1:1000; Molecular Probes, Eugene, OR) for 1 h at room temperature. All sections were photographed under a fluorescence microscope (Nikon Eclipse TE2000-E). We acquired at least nine locations per mouse (three fields per section × three sections per mouse) for quantifications.
We quantified the number of Iba-1-positive cells around the lesion at day 3 after ICH, and counted the number of cells that co-expressed Iba-1 and CD16/32 or Iba-1 and CD206. To examine dMBP and β-APP expression, we calculated fluorescent areas by Image J software.
Microglial morphology
We analyzed the morphology of Iba-1-positive cells around the lesion one day 3 using Neurolucida software (MBF Bioscience). Cells were imaged on sections at 1.70, 0.74, and −0.34 mm from the bregma. We selected three fields per section with magnification of 200× and analyzed 105 cells in each image (we chose only microglia that displayed intact processes unobscured by background labelling or other cells).33 Cell body diameter/area, branch number and length, number of nodes and ends of microglia, and the complexity of the cells were quantified by the software.
Statistical analysis
All data are presented as mean ± SD. We used one-way or two-way analysis of variance (ANOVA) to compare differences among multiple groups followed by Bonferroni or Dunn’s post hoc analysis. All analysis was carried out with GraphPad Software (GraphPad Prism 5.0; GraphPad Software, Inc., La Jolla, CA). The criterion for statistical significance was p < 0.05.
Results
VK-28 reduces Hb-induced cell death and ROS production in OHSCs
To determine whether iron chelators inhibit Hb-induces cell death in OHSCs, we exposed cultured OHSCs obtained from C57BL/6 mice to 20 µM Hb and assessed cell death with PI staining; 16-h incubation with 20 µM Hb caused cell death in dentate gyrus and the hippocampal CA1 and CA3 regions (vehicle: 12.40 ± 3.91%; Hb: 44.45 ± 8.56%; p < 0.05; Figure 1(a) and(b)). Treating with DFX (10 µM) or VK-28 (5 µM) significantly reduced Hb-induced cell death (Hb + DFX: 14.22 ± 5.56%, Hb + VK-28: 14.88 ± 8.63%; n = 5–9 slices/group; both p < 0.01 vs. Hb; Figure 1(a) and (b)). No significant difference was seen between DFX and VK-28 treatment (p > 0.05; Figure 1(a) and (b)). To confirm this result, we collected the media of treated OHSCs and measured cellular cytotoxicity with the LDH assay. Hb increased LDH activity in the media (vehicle: 5.47 ± 0.61, Hb: 10.33 ± 1.13 milliunit/mL; n = 7 slices/group; p < 0.01; Figure 1(c)), and treating with DFX or VK-28 reduced Hb-induced LDH release (Hb + DFX: 5.77 ± 1.12, Hb + VK-28: 5.74 ± 1.01 milliunit/mL; n = 7 slices/group; both p < 0.05 vs. Hb; Figure 1(c)).
Figure 1.

VK-28 reduces Hb-induced cell death and ROS production in OHSCs. (a–c) OHSCs were treated under the conditions shown for 16 h. Slices were stained with propidium iodide (PI). Representative images of PI staining (a), the percentage of PI+ cells (b), and LDH activity (c) are shown. Vehicle, n = 5; Hb, n = 7; DFX, n = 9; VK-28, n = 8. (d–e) OHSCs were incubated with hydroethidine (HEt) for 30 min, and fluorescence intensity was measured. Representative images (d) and quantification (e) are shown. n = 5–8 slices/group. *p < 0.05, **p < 0.01 vs. vehicle; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. Hb. Results are presented as mean ± SD. One-way ANOVA followed by Dunn’s multiple comparison post-test was used. Scale bars: 1 mm.
To determine whether iron chelators are able to reverse Hb-induced ROS accumulation, we used HEt to assess cellular ROS production. Hb induced large amounts of ROS (vehicle: 0.35 ± 0.05 × 10E7, Hb: 2.89 ± 0.51 × 10E7; n = 8 slices/group; p < 0.01; Figure 1(d) and (e)); DFX and VK-28 were both able to reverse Hb-induced ROS (Hb + DFX: 0.61 ± 0.56 × 10E7, p < 0.05 vs. Hb; Hb + VK-28: 0.18 ± 0.13 × 10E7, p < 0.001 vs. Hb; n = 5-8 slices/group; Figure 1(d) and (e)).
VK-28 attenuates ROS production, iron deposition, neuronal death, brain edema, and brain swelling after ICH in vivo
To determine the initial bleeding, we measured the striatal Hb content 24 h after ICH, and found no significant difference among all groups (vehicle: 959.33 ± 227.89, DFX: 772.42 ± 363.50, VK-28: 591.58 ± 204.68 mg/dL; n = 5 mice/group; Figure S1). To investigate the effect of DFX and VK-28 on acute outcomes at 24 h after ICH, we measured ROS accumulation in situ by oxidized HEt. As expected, treatment with DFX or VK-28 at 6 h after ICH effectively decreased ROS production by approximately half compared to that of the vehicle-treated group (DFX: 0.51 ± 0.18 fold change, VK-28: 0.50 ± 0.09 fold change; n = 5–10 mice/group; Figure 2(a)). In addition, we used Perls’ staining to quantify ferric iron deposits in the perihematomal region at three days after ICH, the time of peak injury.4 Post-treatment with DFX or VK-28 significantly reduced the number of iron-positive cells compared with that in the vehicle group (vehicle: 110.50 ± 50.81, DFX: 35.53 ± 11.81, VK-28: 38.31 ± 22.83 per 200× field; both p < 0.001 vs. vehicle; n = 5 mice/group; Figure 2(b)). We further evaluated neuronal death using FJB staining. Only VK-28 treatment significantly reduced the number of FJB+ cells at three days after ICH (vehicle: 282.47 ± 29.14/mm2; DFX: 212.33 ± 35.65/mm2, p > 0.05; VK-28: 165.42 ± 17.33/mm2; p < 0.001; n = 5 mice/group; Figure 2(c)). We further confirmed the rescue effects of VK-28 and DFX using CV staining. Consistent with the results we gained from FJB staining, we found that the number of surviving neurons was significantly higher in the DFX- and VK-28-treated mice than in the vehicle-treated mice (vehicle: 67.48 ± 23.08, DFX: 147.7 ± 44.73, VK-28: 156.5 ± 40.89 per 400-x field; both p < 0.001 vs. vehicle; n = 5 mice/group; Figure 2(d)). Using the Luxol fast blue/CV staining to measure lesion volume and brain swelling at three days after ICH, we found that neither DFX nor VK-28 affected lesion volume (vehicle: 6.30 ± 2.06 mm3, DFX: 7.64 ± 2.26 mm3, VK-28: 6.44 ± 4.81 mm3; both p > 0.05 vs. vehicle; n = 5 mice/group; Figure 2(e)). DFX did not affect brain swelling, whereas VK-28 treatment significantly decreased brain swelling in the ipsilateral striatum (vehicle: 117.4 ± 4.40%, DFX: 115.4 ± 8.56%, VK-28: 105.6 ± 3.65%; p < 0.05 (VK-28 vs. vehicle); n = 5 mice/group; Figure 2(f)). DFX did not alter brain water content of the ipsilateral hemisphere, contralateral hemisphere, or cerebellum among groups, but VK-28 treatment significantly decreased brain water content in the ipsilateral striatum at three days after ICH (vehicle: 81.61 ± 0.39%; DFX: 82.24 ± 1.12%, p > 0.05; VK-28: 80.12 ± 0.41%; p < 0.01; n = 6 mice/group; Figure 2(g)).
Figure 2.

VK-28 attenuates ROS production, iron deposition, neuronal degeneration, lesion volume, and brain edema after ICH in vivo. Middle-aged male C57BL/6 mice underwent ICH by collagenase injection. DFX, VK-28, or vehicle (5% DMSO in saline) was administered i.p. at 6 h post-ICH and then daily until sacrifice. (a) HEt was injected i.p. 1 h before sacrifice. Representative images show HEt fluorescence intensity one day after ICH, and quantification shows the intensity as fold change of that in the vehicle group. Vehicle: n = 10, DFX: n = 5, VK-28: n = 5. (b) Perls’ staining and quantification of iron+ cells at three days after ICH. n = 5 mice/group. (c) Fluoro-Jade B (FJB) was used to stain degenerating neurons at day 3. Representative images and quantification are shown. n = 5 mice/group. (d) Cresyl violet (CV) staining and quantification of surviving neurons. n = 5 mice/group. (e) CV/Luxol fast blue staining was performed on post-ICH day 3, and lesion volume (corrected for brain swilling) was calculated. n = 5 mice/group. (f) The percentage of brain swelling was measured at three days after ICH. n = 5 mice/group. (g) The percentage of brain water content was measured at three days after ICH. n = 6 mice/group. (h) Field selection for ICH brain. *p < 0.05, **p < 0.01, ***p < 0.001 vs. vehicle group; N.S., not significant. Results are presented as mean ± SD. One-way ANOVA followed by Dunn’s multiple comparison post-test was used. Scale bars: (a–c) 100 µm; (d) 50 µm; (e) 1 mm.
We further confirmed the effect of VK-28 on brain water content in middle-aged (12-month-old) male mice subjected to an autologous blood ICH model. As expected, VK-28 administration reduced brain water content in the ipsilateral striatum at three days after ICH (vehicle: 81.29 ± 0.27%, VK-28: 80.12 ± 0.50%; p < 0.001; n = 6 mice/group; Figure S2(a)). Additionally, VK-28 significantly decreased brain water content on day 3 after collagenase-induced ICH in aged (18-month-old) male mice (vehicle: 81.58 ± 0.40%, VK-28: 80.08 ± 0.47%; p < 0.001; n = 5 mice/group; Figure S2(b)) and young female (vehicle: 80.44 ± 0.32%; VK-28: 79.94 ± 0.32%; p < 0.05; n = 5 mice/group; Figure S2(c)).
VK-28 improves neurologic function and corner turn preference after ICH in vivo
Mice treated with VK-28 exhibited significantly less neurologic deficit than did vehicle-treated mice on days 3, 7, and 28 (all p < 0.001 vs. vehicle; Figure 3(a)), whereas DFX-treated mice showed improved neurologic function only on days 3 (p < 0.01) and 7 (p < 0.05). Neurologic scores did not differ significantly in either group from those of vehicle-treated mice on day 1. Notably, long-term recovery was significantly better in the VK-28 group, as neurologic deficit scores were significantly lower in that group than in the DFX-treated group on day 28 (p < 0.05; Figure 3(a)).
Figure 3.
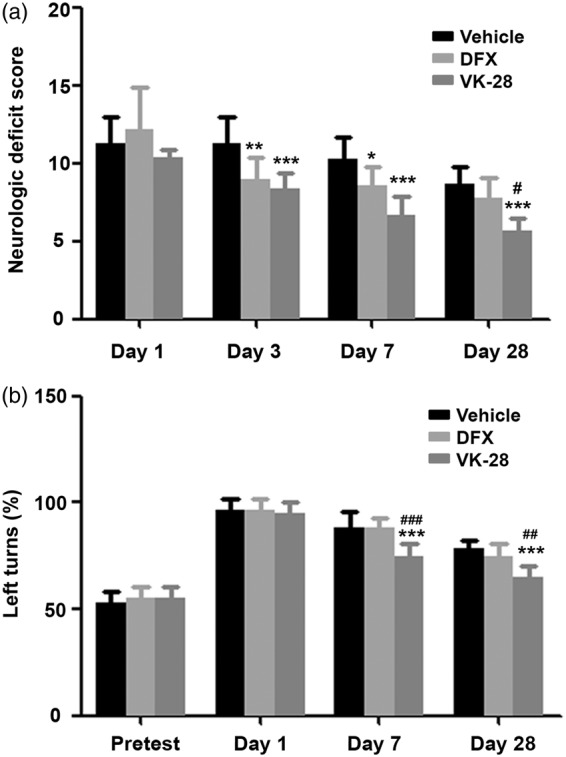
VK-28 improves neurologic function and corner turn preferences after ICH in vivo. Middle-aged male C57BL/6 mice underwent ICH by collagenase injection. DFX, VK-28, or vehicle was administered i.p. at 6 h after ICH and then daily until sacrifice. (a) Neurologic deficit score of ICH mice at days 1, 3, 7, and 28. Vehicle: n = 16, DFX: n = 10, VK-28: n = 6. (b) Mouse behavior in the corner turn test was assessed before ICH and on day 1, 7, and 28 after ICH. n = 6 mice/group *p < 0.05, **p < 0.01, ***p < 0.001 vs. vehicle group at the same time point; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. DFX group at the same time point. Results are presented as mean ± SD. Two-way ANOVA followed by Bonferroni post-test was used.
For corner turn tests, all groups showed equal preference for right and left turns at baseline and a significant preference for left turns at 1 day post-ICH (p > 0.05; Figure 3(b)). DFX did not affect the ICH-induced turn preference, but VK-28 significantly corrected the preference on day 7 (p < 0.001 vs. both vehicle and DFX) and day 28 (p < 0.001 vs. vehicle, p < 0.01 vs. DFX) after ICH (day 7: vehicle: 88.33 ± 7.53%, DFX: 88.33 ± 4.08%, VK-28: 75.00 ± 5.48%; day 28: vehicle: 78.33 ± 4.08%, DFX: 75.00 ± 5.48%, VK-28: 65.00 ±5.48%; n = 6 mice/group; Figure 3(b)).
VK-28 also significantly reduced neurologic deficits on days 3, 7, and 28 after ICH and corrected corner turn preference on day 28 after ICH in aged (18-month-old) male mice (n = 5 mice/group; Figure S3(a)) and young (3-month-old) female mice (n = 5 mice/group; Figure S3(b)).
VK-28 treatment decreases microglial activation and polarization after ICH
Microglia regulate iron homeostasis by sequestering it within ferritin in the brain, but iron overload influences the physiologic properties of microglia.34 To investigate the effects of VK-28 on microglial activation after ICH, we performed immunostaining of Iba-1, a microglia/macrophage cell marker, and quantified the number of activated Iba-1-positive microglia/macrophages around the lesion at three days after ICH. We found that, compared with the number in the vehicle group (178.8 ± 39.65), the number of activated Iba-1-positive microglia/macrophages in the perihematomal region was significantly less in the DFX group (132.5 ± 27.24, p < 0.05) and VK-28 group (132.6 ± 24.5, p < 0.01; n = 5 mice/group; Figure 4(a)). We also observed that the morphology of microglia in the VK-28 treatment group differed from that in the vehicle and DFX treatment groups. Neurolucida software analysis showed that cell body area at three days post-ICH was significantly smaller in the VK-28 group (80.45 ± 25.86 µm2; p < 0.05 vs. vehicle) than in the vehicle group (99.62 ± 33.71 µm2) or DFX group (104.7 ± 31.9 µm2, Figure 4(b)). However, cell body area in the VK-28 group was still significantly larger than that in the sham group (42.15 ± 16.16 µm2, p < 0.05). No difference was detected between the DFX and vehicle groups (p > 0.05; Figure 4(b)). We did not detect differences in the number of dendrites, notes and ends, length of dendrites, and the complexity of the cells between the vehicle and drug-treated groups (p > 0.05; Figure 4(b)).
Figure 4.

VK-28 treatment decreases microglial activation and M1-like polarization after ICH in vivo. Middle-aged male C57BL/6 mice underwent ICH by collagenase injection. DFX, VK-28, or vehicle was administered i.p. at 6 h after ICH and then daily until sacrifice. (a) Immunostaining of Iba-1 reveals microglia and recruited macrophage around the lesion at three days post-ICH. Quantification of activated Iba-1+ cells is shown. *p < 0.05, **p < 0.01 vs. vehicle. n = 5 mice/group. (b) Neurolucida software was used to analyze the morphology using Iba-1 staining. Representative images show the morphology of recruited microglia at three days after ICH. Quantifications show cell body area (µm2), number of dendrites, number of notes, number of ends, length of dendrites (µm), mean length of dendrites (µm), and complexity of the cells. ***p < 0.001 vs. Sham; #p < 0.05 vs. vehicle; ††p < 0.01 vs. DFX. Sham: n = 20, Vehicle: n = 47, DFX: n = 73, VK-28: n = 45 cells. (c) Representative images of cells immunostained for Iba-1 and M1 marker CD16/32 at three days after ICH. Quantification shows the percentage of Iba-1+/CD16/32+ cells. The white dashed line marks the hematoma boundary. *p < 0.05, **p < 0.01 vs. vehicle. n = 5 mice/group. (d) Representative images of cells immunostained for Iba-1 and M2 marker CD206 at three days after ICH. Quantification shows the percentage of Iba-1+/CD206+ cells. The white dashed line marks the hematoma boundary. *p < 0.05 vs. vehicle. Results are presented as mean ± SD. One-way ANOVA followed by Dunn’s multiple comparison post-test was used. Scale bars: (a, c, d) 100 µm; (b) 10 µm.
To assess the effects of DFX and VK-28 on microglial polarization after ICH, we performed immunofluorescence staining for Iba-1 and CD16/32 (M1 marker) or CD206 (M2 marker). We found that both DFX and VK-28 decreased the percentage of Iba-1+/CD16/32+ cells (vehicle: 87.07 ± 2.44%; DFX: 77.58 ± 4.25%, p < 0.05; VK-28: 73.78 ± 8.87%, p < 0.01; n = 5 mice/group; Figure 4(c)). Surprisingly, only VK-28 increased the percentage of Iba-1 +/CD206+ cells (vehicle: 12.45 ± 1.88%; DFX: 13.56 ± 2.76%, p > 0.05; VK-28: 19.89 ± 4.49%, p < 0.05; n = 5 mice/group; Figure 4(d)).
VK-28 attenuates white matter damage after ICH
On the third day after ICH, we labeled dMBP to detect damaged myelin and β-APP to mark damaged axons. We observed that degraded myelin was distributed in the core and at the edge of the hematoma after ICH, whereas damaged axons surrounded the hematoma edge (Figure 5(a) and (b)). DFX did not significantly affect dMBP or β-APP expression, but VK-28 effectively reduced the percentage of area with dMBP from 5.77 ± 2.31% per 200× area (with vehicle treatment) to 3.78 ± 1.30% per 200 × area (p < 0.05; Figure 5(a)) and the percentage of area with β-APP expression from 16.38 ± 8.79% per 200× area to 4.54 ± 3.37% per 200 × area (p < 0.001; n = 5 mice/group; Figure 5(b)). We also immunostained tissue with Luxol fast blue (vehicle: 62.07 ± 29.73%, DFX: 39.07 ± 12.08%, VK-28: 52.03 ± 21.35%) and MBP (vehicle: 60.09 ± 17.55%, DFX: 79.81 ± 3.41%, VK-28: 80.08 ± 8.02%) to assess white matter damage. However, we detected no differences between groups (n = 5 mice/group; all p > 0.05; Figure S4(a) and (b)).
Figure 5.

VK-28 attenuates white matter damage after ICH in vivo. Middle-aged male C57BL/6 mice underwent ICH by collagenase injection. DFX, VK-28, or vehicle was administered i.p. at 6 h after ICH and then daily until sacrifice. (a) Representative images show immunostaining of degraded myelin basic protein (dMBP). The percentage of positively stained area was quantified and is shown to the right. (b) Representative images show immunostaining of beta-amyloid precursor protein (β-APP). The percentage of positively stained area was quantified and is shown to the right. n = 5 mice/group. *p < 0.05, ***p < 0.001 vs. vehicle. Dashed lines represent the boundary of hematoma. Results are presented as mean ± SD. One-way ANOVA followed by Dunn’s multiple comparison post-test was used. Scale bars: 100 µm.
VK-28 decreases mortality rate after ICH
We recorded mortality rate for 28 days after ICH; 6 of 78 vehicle-treated mice (7.69%) and 2 of 36 VK-28-treated mice (5.56%) died in that time period (Figure S5(a)). In contrast, DFX produced severe death after ICH, with a mortality rate of 33.96% (18 of 53 mice; Figure S5(a)).
We also recorded mouse body weight before and after the ICH procedure. Compared to the baseline, all mice had lost weight on day 1, with no difference between groups. However, on day 3, mice in the vehicle and VK-28 groups had begun to recover weight, whereas those in the DFX group continued to lose weight. At that time point, weight loss was significantly greater in the DFX group (8.31 ± 5.11%) than in the vehicle group (3.29%) or VK-28 group (2.10 ± 3.36%). Weight loss did not differ between the vehicle- and VK-28-treated groups (p > 0.05; Figure S5(b)).
Discussion
Iron toxicity contributes to secondary neuronal death after ICH,1 but managing iron deposition has been shown to improve ICH outcomes in pre-clinical studies.2,5–7 Iron chelation also is showing promising results in clinical trials (www.clinicaltrials.gov). DFX is a well-studied compound that chelates cellular iron effectively and improves outcomes after experimental ICH35 and in other disease models36,37 related to abnormal iron accumulation. Although several groups have reported that DFX reduces brain edema after the blood injection ICH model in rats and piglets,8,9 these results were not confirmed by another group.11,12 In a systematic review of the effect of DFX in ICH animal models, Cui et al.35 concluded that treatment window and model type (blood infusion or collagenase infusion), but not the dosage, age of the animal, or treatment method (intramuscular or intraperitoneal injection) influenced the effect of DFX on brain water content.35 They found that early post-treatment with DFX at 2 or 4 h after ICH reduced brain water content and that DFX was more effective in blood injection models than in collagenase injection models.
Ours is the first study to test VK-28 in ICH animal models and the first to compare the effects of DFX and VK-28 side by side. The collagenase and blood models are the two most commonly used animal models to mimic clinical ICH; however, neither can fully reflect the disease. The collagenase model mimics an acute cerebrovascular rupture and blood–brain barrier (BBB) breakdown. It imitates early neurologic deterioration38 and hematoma expansion39 with increased intracranial pressure,40 similar to that present in ICH patients.41 The autologous blood model mimics a single large bleed, and is used to investigate blood toxicity and mechanisms of injury, but it lacks the underlying vascular pathology and is not ideal for long-term functional studies.5,42–44 Therefore, we commonly use both models to confirm our major findings,21,29,30 as suggested by RIGOR and STAIR criteria.22,23 We did not find a strong effect of DFX on brain water content in our collagenase-induced ICH model, but we found that VK-28 was able to reduce brain water content in the ipsilateral striatum. Although we did not detect statistically significant differences in injury volume among vehicle, DFX, and VK-28 treated groups, we observed a trend toward increased lesion volume after DFX treatment. VK-28 did not appear to alter brain injury volume. Cao et al.45 found that DFX treatment attenuated the process of hematoma resolution by reducing member attack complex formation and inhibiting CD47 loss in the clot in a piglet model of ICH. We did not investigate the hematoma clearance by VK-28 or DFX treatment in our study. However, DFX treatment may result in delayed hematoma clearance, which might underlie the slightly larger lesion volume seen at day 3 post-ICH.
Tough it is important to evaluate the effect of treatment on injury volume in animal models, behavior changes are a more vital metric clinically. Like other studies have shown, DFX treatment improved short-term outcomes.35 We found that DFX improved neurologic function on days 3 and 7 compared to that in the vehicle group, but VK-28 offered improvements even up to day 28 post-ICH. Additionally, DFX had no effect on ICH-induced corner turn preference changes, but VK-28 was able to reduce the left-turn preference on days 7 and 28 post-ICH. Thus, at the dosages tested, VK-28 might be more effective than DFX at reducing neurologic deficit. In addition to assessing the effects on behavioral outcomes, determining the side effect profile of a potential treatment is also important. It has been reported that DFX can lead to hypotension, pancytopenia, retinal toxicity, and neurotoxicity clinicall.14-16,46 Few studies have evaluated side effects of DFX in preclinical animal models. We and others have found that DFX increases body weight loss and death rate after ICH.2,47 In this study, we did not observe increased mortality or weight loss after ICH in the VK-28 group as we did in the DFX group, making VK-28 a more promising compound.
Another difference we observed between DFX and VK-28 was the effect on microglial activation and polarization. Microglia are recruited to the peri-hematoma region and activated immediately after ICH.48 Activated microglia and infiltrating macrophages clean up hematoma and dead cell debris; however, when these innate immune cells become over-activated, they release large amounts of inflammatory factors that cause secondary damage to neuronal cells.48 DFX and VK-28 both decreased the number of activated microglia/macrophages in the peri-hematoma region compared with that in the vehicle group. Additionally, they deceased CD16/32 on Iba-1-positive cells. The decrease in number of activated microglia/macrophages and number of M1-like microglia/macrophages suggests that DFX and VK-28 suppressed inflammation after ICH. Such activity may represent another mechanism by which DFX and VK-28 are able to rescue degenerating neurons and improve neurologic function. Furthermore, we observed that VK-28, but not DFX, decreased the cell body area and increased CD206 expression on Iba-1-positive cells. This finding suggests that VK-28 polarized microglia/macrophages toward an M2-like phenotype, which contributes to hematoma clearance and brain healing. Indeed, Kroner et al.49 reported that iron and TNF-α polarize microglia toward an M1-like phenotype. In our study, we believe that intracellular iron chelation was able to polarize microglia/macrophages to an M2-like phenotype, and that M2 polarization might have been greater with VK-28 treatment because the effective concentration of VK-28 in the peri-hematoma region could have been higher than that of DFX.
White matter injury after ICH31 contributes to long-term complications, including motor and sensory deficits, and possibly even depression and cognitive dysfunction. However, few studies have looked into white matter injury after ICH, especially in mouse models.21 We reported that, although neither DFX nor VK-28 changed Luxol fast blue staining or MBP immunostaining, VK-28 did decrease the expression of axon/myelin acute damage markers β-APP and dMBP. Thus, VK-28 might be able to diminish white matter injury at later time points. Previous studies have shown that DFX decreases white matter injury in rats and piglets after the blood injection ICH model.8,47 However, we did not observe this effect in our collagenase model. One explanation might be that the protective effects of DFX are animal model-dependent, as it was reported to be able to decrease lesion volume in the former, but not the latter.35 The Fe2+ binding affinity is inversely proportional to the half maximal dequenching concentration [M]1/2, and the [M]1/2 value of DFX is larger than that of VK-28, which means that VK-28 has better Fe2+ binding affinity.50 The biggest advantage of VK-28 is that it can penetrate the intact BBB, whereas DFX cannot. Thus, VK-28 may target the brain at a lower systemic concentration and be more suitable for use in clinic. Another explanation would be that the collagenase injection model induces more severe damage to the BBB, and more damage to the striatum, substantia nigra, white matter, and cortex than the blood injection model at an equal hematoma volume.43,51 DFX failed to decrease neuronal death, brain swelling, and edema, and failed to decrease the expression of white matter damage-related markers in the acute phase of the collagenase injection model, perhaps because this model can cause more severe white matter damage than the blood injection model.
In clinical trials of ICH, clot removal is a very promising procedure. Although open surgery trials of hematoma evacuation (STICH I and II) have not shown expected results,52 two randomized minimally invasive surgery (MIS) trials are recruiting patients in order to compare standard medical management to early surgical hematoma evacuation using MIS (NCT02880878 and NCT02331719, www.clinicaltrials.gov). One clinical trial that combined MIS with rt-PA administration (MISTIE II) has been completed successfully.53,54 A Phase III clinical trial that began in 2013, MISTIE-III (NCT01827046, www.clinicaltrials.gov), is still ongoing. In our study, we showed that VK-28 might offer effective treatment without significant side effects in an ICH mouse. Based on our findings, we believe that VK-28 could be administered safely to patients following MIS with or without rt-PA treatment.
Our study had several limitations. First, to fully compare the effectiveness of VK-28 and DFX is, we need to conduct dose response experiments and a comparative pharmacodynamic study; in this study, we tested only one commonly used dosage of each drug. We also did not compare the therapeutic window, although 6-h delayed treatment is validated for most treatments. Second, we examined the acute effects of VK-28 on days 1 and 3 post-ICH only for ROS accumulation, iron overload, neuronal death, lesion volume, microglia/macrophage activation, and acute phase of white matter injury markers; it is also important to examine other inflammatory responses such as infiltration of macrophages, neutrophils, and T cells at different time points. Third, we have found previously that ferroptotic cell death might contribute to ICH injury55 in addition to other forms of cell death such as necrosis and apoptosis. It would be interesting to investigate which forms of cell death can be rescued by iron chelators. Lastly, we only evaluated long-term effects on neurologic deficits and corner turn preference; other behaviors such as depression/anxiety, learning, and memory should be evaluated in the future.
In conclusion, both VK-28 and DFX are protective after ICH in mice, but VK-28 appears to be more effective and safer than DFX at the dosage tested. The insights we gained from this study are valuable, and provide another alternative compound for treatment of ICH in the future.
Supplementary Material
Acknowledgments
We thank Claire Levine, MS, ELS, for assistance with manuscript preparation.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the National Institutes of Health (R01 NS078026 and R01 AT007317 to JW) and the American Heart Association (Grant-in-Aid, 13GRNT15730001 to JW; Postdoctoral Fellowship Awards 16POST29640010 to QL, 15POST25090114 to XL, and 14POST20140003 to XH) and a “Stimulating and Advancing ACCM Research (StAAR)” grant from the Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
QL, JW, and JW designed the experiments and wrote the manuscript; QL, JW, XL, XH, ZW, and JW collected and analyzed the data.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Xiong XY, Wang J, Qian ZM, et al. Iron and intracerebral hemorrhage: from mechanism to translation. Transl Stroke Res 2014; 5: 429–441. [DOI] [PubMed] [Google Scholar]
- 2.Wu H, Wu T, Xu X, et al. Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J Cereb Blood Flow Metab 2011; 31: 1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salvador GA. Iron in neuronal function and dysfunction. Biofactors 2010; 36: 103–110. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura T, Keep RF, Hua Y, et al. Iron-induced oxidative brain injury after experimental intracerebral hemorrhage. Acta Neurochir Suppl 2006; 96: 194–198. [DOI] [PubMed] [Google Scholar]
- 5.Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol 2010; 92: 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selim M. Deferoxamine mesylate: a new hope for intracerebral hemorrhage: from bench to clinical trials. Stroke 2009; 40: S90–S91. [DOI] [PubMed] [Google Scholar]
- 7.Jaremko KM, Chen-Roetling J, Chen L, et al. Accelerated hemolysis and neurotoxicity in neuron-glia-blood clot co-cultures. J Neurochem 2010; 114: 1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu Y, Hua Y, Keep RF, et al. Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke 2009; 40: 2241–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okauchi M, Hua Y, Keep RF, et al. Deferoxamine treatment for intracerebral hemorrhage in aged rats: therapeutic time window and optimal duration. Stroke 2010; 41: 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Tang J, Tan L, et al. Intracerebral hematoma contributes to hydrocephalus after intraventricular hemorrhage via aggravating iron accumulation. Stroke 2015; 46: 2902–2908. [DOI] [PubMed] [Google Scholar]
- 11.Warkentin LM, Auriat AM, Wowk S, et al. Failure of deferoxamine, an iron chelator, to improve outcome after collagenase-induced intracerebral hemorrhage in rats. Brain Res 2010; 1309: 95–103. [DOI] [PubMed] [Google Scholar]
- 12.Auriat AM, Silasi G, Wei Z, et al. Ferric iron chelation lowers brain iron levels after intracerebral hemorrhage in rats but does not improve outcome. Exp Neurol 2012; 234: 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kress GJ, Dineley KE, Reynolds IJ. The relationship between intracellular free iron and cell injury in cultured neurons, astrocytes, and oligodendrocytes. J Neurosci 2002; 22: 5848–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kontoghiorghes GJ. Comparative efficacy and toxicity of desferrioxamine, deferiprone and other iron and aluminium chelating drugs. Toxicol Lett 1995; 80: 1–18. [DOI] [PubMed] [Google Scholar]
- 15.Porter JB, Huehns ER. The toxic effects of desferrioxamine. Baillieres Clin Haematol 1989; 2: 459–474. [DOI] [PubMed] [Google Scholar]
- 16.Lai TY, Lee GK, Chan WM, et al. Rapid development of severe toxic retinopathy associated with continuous intravenous deferoxamine infusion. Br J Ophthalmol 2006; 90: 243–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atas B, Caksen H, Tuncer O, et al. Acute respiratory distress syndrome due to overdose desferrioxamine: report of a child. Med J Malaysia 2005; 60: 91–93. [PubMed] [Google Scholar]
- 18.Youdim MB, Stephenson G, Ben Shachar D. Ironing iron out in Parkinson's disease and other neurodegenerative diseases with iron chelators: a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Ann N Y Acad Sci 2004; 1012: 306–325. [DOI] [PubMed] [Google Scholar]
- 19.Bandyopadhyay S, Huang X, Cho H, et al. Metal specificity of an iron-responsive element in Alzheimer's APP mRNA 5'untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J Neural Transm Suppl 2006; 71: 237–247. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q, Zhang X, Chen S, et al. Prevention of motor neuron degeneration by novel iron chelators in SOD1(G93A) transgenic mice of amyotrophic lateral sclerosis. Neurodegener Dis 2011; 8: 310–321. [DOI] [PubMed] [Google Scholar]
- 21.Zhao X, Wu T, Chang CF, et al. Toxic role of prostaglandin E2 receptor EP1 after intracerebral hemorrhage in mice. Brain Behav Immun 2015; 46: 293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albers GW, Goldstein LB, Hess DC, et al. Stroke Treatment Academic Industry Roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke 2011; 42: 2645–2650. [DOI] [PubMed] [Google Scholar]
- 23.Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res 2013; 4: 279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han X, Lan X, Li Q, et al. Inhibition of prostaglandin E2 receptor EP3 mitigates thrombin-induced brain injury. J Cereb Blood Flow Metab 2016; 36: 1059–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu W, Gao Y, Chang CF, et al. Mouse models of intracerebral hemorrhage in ventricle, cortex, and hippocampus by injections of autologous blood or collagenase. PLoS One 2014; 9: e97423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Fields J, Dore S. The development of an improved preclinical mouse model of intracerebral hemorrhage using double infusion of autologous whole blood. Brain Res 2008; 1222: 214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sah R, Schwartz-Bloom RD. Optical imaging reveals elevated intracellular chloride in hippocampal pyramidal neurons after oxidative stress. J Neurosci 1999; 19: 9209–9217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu W, Xie W, Pan T, et al. Prevention and restoration of lactacystin-induced nigrostriatal dopamine neuron degeneration by novel brain-permeable iron chelators. FASEB J 2007; 21: 3835–3844. [DOI] [PubMed] [Google Scholar]
- 29.Wu H, Wu T, Hua W, et al. PGE2 receptor agonist misoprostol protects brain against intracerebral hemorrhage in mice. Neurobiol Aging 2015; 36: 1439–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu H, Wu T, Han X, et al. Cerebroprotection by the neuronal PGE2 receptor EP2 after intracerebral hemorrhage in middle-aged mice. J Cereb Blood Flow Metab 2017; 37: 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H, Wu T, Li M, et al. Efficacy of the lipid-soluble iron chelator 2,2'-dipyridyl against hemorrhagic brain injury. Neurobiol Dis 2012; 45: 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng T, Wang W, Li Q, et al. Cerebroprotection of flavanol (-)-epicatechin after traumatic brain injury via Nrf2-dependent and -independent pathways. Free Radic Biol Med 2016; 92: 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kongsui R, Beynon SB, Johnson SJ, et al. Quantitative assessment of microglial morphology and density reveals remarkable consistency in the distribution and morphology of cells within the healthy prefrontal cortex of the rat. J Neuroinflammation 2014; 11: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rathnasamy G, Ling EA, Kaur C. Consequences of iron accumulation in microglia and its implications in neuropathological conditions. CNS Neurol Disord Drug Targets 2013; 12: 785–798. [DOI] [PubMed] [Google Scholar]
- 35.Cui HJ, He HY, Yang AL, et al. Efficacy of deferoxamine in animal models of intracerebral hemorrhage: a systematic review and stratified meta-analysis. PLoS One 2015; 10: e0127256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu ZQ, Jia Y, Chen G. Possible involvement of cathepsin B/D and caspase-3 in deferoxamine-related neuroprotection of early brain injury after subarachnoid haemorrhage in rats. Neuropathol Appl Neurobiol 2014; 40: 270–283. [DOI] [PubMed] [Google Scholar]
- 37.Kostopanagiotou GG, Kalimeris KA, Arkadopoulos NP, et al. Desferrioxamine attenuates minor lung injury following surgical acute liver failure. Eur Respir J 2009; 33: 1429–1436. [DOI] [PubMed] [Google Scholar]
- 38.Specogna AV, Turin TC, Patten SB, et al. Factors associated with early deterioration after spontaneous intracerebral hemorrhage: a systematic review and meta-analysis. PLoS One 2014; 9: e96743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M, Hong X, Chang CF, et al. Simultaneous detection and separation of hyperacute intracerebral hemorrhage and cerebral ischemia using amide proton transfer MRI. Magn Reson Med 2015; 74: 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiploylee C, Colbourne F. Intracranial pressure measured in freely moving rats for days after intracerebral hemorrhage. Exp Neurol 2014; 255: 49–55. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Li Q, Wang Z, et al. Multimodality MRI assessment of grey and white matter injury and blood-brain barrier disruption after intracerebral haemorrhage in mice. Sci Rep 2017; 7: 40358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manaenko A, Chen H, Zhang JH, et al. Comparison of different preclinical models of intracerebral hemorrhage. Acta Neurochir Suppl 2011; 111: 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacLellan CL, Silasi G, Poon CC, et al. Intracerebral hemorrhage models in rat: comparing collagenase to blood infusion. J Cereb Blood Flow Metab 2008; 28: 516–525. [DOI] [PubMed] [Google Scholar]
- 44.MacLellan CL, Paquette R, Colbourne F. A critical appraisal of experimental intracerebral hemorrhage research. J Cereb Blood Flow Metab 2012; 32: 612–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao S, Zheng M, Hua Y, et al. Hematoma changes during clot resolution after experimental intracerebral hemorrhage. Stroke 2016; 47: 1626–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blake DR, Winyard P, Lunec J, et al. Cerebral and ocular toxicity induced by desferrioxamine. Q J Med 1985; 56: 345–355. [PubMed] [Google Scholar]
- 47.Ni W, Okauchi M, Hatakeyama T, et al. Deferoxamine reduces intracerebral hemorrhage-induced white matter damage in aged rats. Exp Neurol 2015; 272: 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Z, Zhang Z, Lu H, et al. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol Neurobiol 2017; 54: 1874–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kroner A, Greenhalgh AD, Zarruk JG, et al. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014; 83: 1098–1116. [DOI] [PubMed] [Google Scholar]
- 50.Zheng H, Weiner LM, Bar-Am O, et al. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer's, Parkinson's, and other neurodegenerative diseases. Bioorg Med Chem 2005; 13: 773–783. [DOI] [PubMed] [Google Scholar]
- 51.Barratt HE, Lanman TA, Carmichael ST. Mouse intracerebral hemorrhage models produce different degrees of initial and delayed damage, axonal sprouting, and recovery. J Cereb Blood Flow Metab 2014; 34: 1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fiorella D, Arthur A, Bain M, et al. minimally invasive surgery for intracerebral and intraventricular hemorrhage: rationale, review of existing data and emerging technologies. Stroke 2016; 47: 1399–1406. [DOI] [PubMed] [Google Scholar]
- 53.Sonni S, Lioutas VA, Selim MH. New avenues for treatment of intracranial hemorrhage. Curr Treat Options Cardiovasc Med 2014; 16: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mould WA, Carhuapoma JR, Muschelli J, et al. Minimally invasive surgery plus recombinant tissue-type plasminogen activator for intracerebral hemorrhage evacuation decreases perihematomal edema. Stroke 2013; 44: 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang CF, Cho S, Wang J. (-)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann Clin Transl Neurol 2014; 1: 258–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.