

Abstract
We investigated the effect of Rho kinase inhibition on changes in cerebral blood flow (CBF), brain injury and vascular function after ischemic stroke in spontaneously hypertensive rats (SHR). Changes in core MCA and collateral perfusion were measured by a validated laser Doppler method. Animals underwent 2 h tMCAO and 2 h reperfusion. Fasudil (0.1 mg/kg, i.v.) or vehicle was given at 30 min ischemia (n = 9/group; mean (SD)). Brain injury was determined by 2,3,5-triphenyltetrazolium chloride staining. To determine the effect of fasudil on vascular function, fasudil was given 10 min before reperfusion and parenchymal arterioles studied isolated (n = 6/group; mean(SD)). Collateral perfusion was low in vehicle-treated SHR (−8(32)%) that changed minimally with fasudil (6(24)%, p > 0.05, effect size: 0.47;95% CI−0.49–1.39). Reperfusion CBF was below baseline in vehicle (−27(26)%) and fasudil (−32(25)%, p > 0.05, effect size: 0.19; 95% CI−0.74–1.11) groups, suggesting incomplete reperfusion in both groups. Fasudil had little effect on brain injury volume (28(13)% vs. 36(7)% in vehicle, p > 0.05, effect size: 0.75; 95% CI−0.24–1.66). In isolated parenchymal arterioles, myogenic tone was similar between groups (37(6)% vs. 38(10)% in vehicle, p > 0.05, effect size: 0.09; 95% CI−1.05–1.21). There were no differences with fasudil treatment vs. vehicle in perfusion, brain injury and vascular function that may be related to the low dose that had minimal blood pressure lowering effect.
Keywords: Acute stroke, cerebral blood flow, focal ischemia, hypertension, animal models
Introduction
Chronic hypertension is a major risk factor for acute ischemic stroke.1,2 A number of clinical studies revealed at least 70% of acute stroke patients have a history of hypertension.3–5 These patients also have worse stroke outcomes compared to normotensive patients, including increased incidence of death and disability after stroke.3–5 Animal studies also showed that acute ischemic stroke caused increased neurological deficits, larger infarct and smaller penumbra in hypertensive animal models compared to their normotensive counterparts.6–9 Although the cause of worse stroke outcome is likely multifactorial, one well-established adverse effect of chronic hypertension is on cerebral vascular function and structure.10,11 For example, multiple vascular segments of the cerebral circulation have been shown to be constricted in hypertensive animals, including leptomeningeal anastomoses (LMAs) and parenchymal arterioles (PAs).12,13 Constriction of small arterioles in the brain in response to ischemia and/or reperfusion could further limit perfusion and worsen outcome of acute ischemic stroke. In addition, selective vasodilation of these vascular segments could be a therapeutic target for acute stroke patients with pre-existing hypertension.
Chronic hypertension is known to activate Rho-associated protein kinase (Rho kinase) that is expressed in many cell types, including vascular endothelial and smooth muscle cells.14–16 Rho kinase inhibits myosin phosphatase activity in vascular smooth muscle, and hence preventing de-phosphorylation of myosin light chain.17 One important consequence of Rho kinase activation is enhanced response to increased smooth muscle intracellular calcium, leading to more robust vasoconstriction.16 This Rho kinase-dependent smooth muscle calcium sensitization is an important mechanism for hypertension-induced vasoconstriction.16,18 In the cerebral circulation, Rho kinase inhibitors dilate a number of vascular segments, including the basilar artery, posterior cerebral artery, middle cerebral artery (MCA) and PA.19–23 This suggests Rho kinase is involved in basal tone regulation at almost all levels of the cerebral vasculature. Moreover, Rho kinase inhibitors including Y27632 and fasudil (also known as HA-1077) have been shown to dilate significantly more in cerebral vasculature from hypertensive than normotensive animals, further suggesting enhanced activation of Rho kinase in the cerebral circulation during hypertension.21,24 We have previously demonstrated that PAs were more constricted at early post-ischemic reperfusion due to smooth muscle calcium sensitization.19 Due to the inhibitory effect of Rho kinase on myosin phosphatase activity, we sought to determine if Rho kinase inhibition, by in vivo fasudil treatment, could increase collateral perfusion, reperfusion CBF, and reduce ischemic brain injury in hypertensive rats. We also determined if fasudil treatment in vivo would reverse the vasoconstriction of PAs after ischemia and reperfusion.
Materials and methods
Animals
Male spontaneously hypertensive rats (SHR, Charles River, Kingston, NY, USA), aged 16–19 weeks old, were used in this study. Rats were housed in the Animal Care Facility at the University of Vermont, an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility. Rats were maintained on a 12-h light/dark cycle and allowed food and water ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Vermont and complied with the National Institutes of Health guidelines for care and use of laboratory animals. The reporting of this study was conducted in accordance with ARRIVE guidelines.
Model of transient focal ischemia
Proximal middle cerebral artery occlusion (MCAO) was performed using an intravascular filament model, as previously described.19 Briefly, animals were anesthetized with isoflurane in oxygen (1.5–2%) and mechanically ventilated to maintain blood gases within physiologic ranges (Table 1). One femoral artery catheter was placed and connected to a servo-null system (Living Systems Instrumentation, St. Albans, VT) for monitoring blood pressure. Another femoral artery catheter was placed for obtaining blood gas samples. Femoral vein catheters were inserted on both sides for chloral hydrate and treatment infusion. After instrumentation, chloral hydrate (200–300 mg/kg, i.v.) anesthesia was used in place of isoflurane because isoflurane has a potent vasodilator effect of the cerebral circulation. Toe pinch was performed and blood pressure regularly checked for depth of anesthesia. The MCA was occluded for 2 h of ischemia followed by filament removal to allow reperfusion for 2 h for CBF and brain injury experiments or for 30 min for isolated PA experiments. The different time periods of reperfusion was because our previous study found that isolated PAs were more constricted after 30 min of reperfusion.19 Animals were excluded if the drop in CBF during ischemia was <70% from baseline.
Table 1.
Body weights, mean arterial pressure and blood gases of all animals during MCAO surgeries.
| In vivo CBF experiments |
Isolated PA experiments |
|||
|---|---|---|---|---|
| SHR-vehicle | SHR-fasudil | SHR-vehicle | SHR-fasudil | |
| n | 9 | 9 | 6 | 6 |
| Weight (g) | 337 (25) | 341 (10) | 367 (31) | 348 (40) |
| Arterial pressure and blood gases | ||||
| Start | ||||
| Pressure (mm Hg) | 89 (12) | 91 (12) | 85 (15) | 84 (12) |
| pH | 7.38 (0.03) | 7.39 (0.03) | 7.44 (0.03) | 7.42 (0.02) |
| pCO2 (mm Hg) | 38.3 (3.6) | 36.8 (3.4) | 40.1 (4.5) | 41.0 (2.6) |
| pO2 (mm Hg) | 109.4 (17.0) | 100.9 (14.9) | 108.3 (9.7) | 125.8 (32.1) |
| Middle | ||||
| Pressure (mm Hg) | 86 (5) | 88 (7) | 87 (6) | 85 (4) |
| pH | 7.36 (0.03) | 7.37 (0.03) | 7.45 (0.03) | 7.44 (0.01) |
| pCO2 (mm Hg) | 40.9 (2.2) | 37.1 (3.2) | 40.9 (5.1) | 42.0 (3.1) |
| pO2 (mm Hg) | 98.8 (8.6) | 93.9 (7.5) | 108.2 (11.1) | 117.5 (25.0) |
| End | ||||
| Pressure (mm Hg) | 86 (9) | 88 (11) | 83 (11) | 88 (9) |
| pH | 7.36 (0.03) | 7.36 (0.03) | 7.46 (0.03) | 7.46 (0.01) |
| pCO2 (mm Hg) | 39.0 (3.6) | 39.7 (2.2) | 41.0 (2.9) | 41.5 (1.9) |
| pO2 (mm Hg) | 99.0 (10.6) | 95.1 (12.7) | 104.7 (7.6) | 110.5 (15.4) |
MCAO: middle cerebral artery occlusion; SHR: spontaneously hypertensive rats; CBF: cerebral blood flow; PA: parenchymal arteriole.
Measurement of changes in CBF at core and collateral MCA territories
Dual laser Doppler flowmetry (Perimed Inc., Ardmore, PA, USA) was used to measure CBF in both the MCA core ischemic and collateral territories, as described previously.25 Probe 1 was placed at +4 mm lateral of midline and −2 mm posterior of Bregma to measure changes of CBF in MCA core ischemic territory. Probe 2 was placed +3 mm lateral of midline and +2 mm anterior of Bregma to measure changes in collateral CBF. This multi-site laser Doppler measurement for collateral flow measurement during MCAO was recently validated with MRI.26 Changes in CBF were measured continuously throughout ischemia and reperfusion, and calculated and presented as percent change compared to before filament insertion or before treatment. To further assess collateral flow, discrete increased perfusion events within the collateral territory were quantified by number, duration, and magnitude (area under the curve, AUC). These events were identified from laser Doppler and blood pressure tracings during ischemia only and defined by increases in probe 2 perfusion that were largely independent of changes in blood pressure. The quantification of increased perfusion was performed by investigators who were unaware of the experimental groups, i.e. in a blinded manner.
Treatment with Rho kinase inhibitor fasudil
To investigate the effect of Rho kinase inhibition on changes in collateral perfusion, reperfusion CBF and brain injury (n = 9), fasudil (0.1 mg/kg) or vehicle (lactated Ringer solution) was infused intravenously via femoral catheter at 30 min of ischemia. Animals were randomly assigned to either treatment or vehicle group. In another set of experiments, the effect of Rho kinase inhibition on PA reactivity to pressure and vasoactive agents (n = 6) after ischemia and reperfusion, fasudil (0.1 mg/kg) or vehicle was infused 10 min prior to reperfusion after which PAs were dissected and studied isolated and pressurized. We chose 0.1 mg/kg fasudil for infusion because pilot studies showed that this dose decreased systemic pressure minimally, by 4(8)%, whereas higher doses of 0.3 and 3 mg/kg decreased pressure by 15(12)% (n = 7) and 43% (n = 1), respectively.
Determination of acute brain injury and edema
Rats were quickly decapitated under deep isoflurane anesthesia after 2 h of reperfusion and the brain removed. Brains were sliced to 2 mm coronal sections and incubated for 30 min at 37℃ in 2% 2,3,5-triphenyltetrazolium chloride (TTC) in phosphate-buffered saline. Brain sections were then fixed in 3.7% formalin in PBS at 4℃ for 45 min, followed by imaging using a digital scanner. Brain injury was identified as white (unstained) area of each section and measured with ImageJ software (NIH, Bethesda, MD, USA). Edema was measured and calculated by subtracting the area of contralateral from the ipsilateral hemisphere. Volume of acute brain injury was calculated by summing the edema-corrected brain injury areas of the sections for each brain by investigators blinded to the groups.
Determination of reactivity of isolated PAs to pressure and vasoactive agents
Following 30 min of reperfusion, rats (n = 6 in each group) were quickly decapitated under isoflurane anesthesia. Brains were removed and placed in cold, oxygenated physiologic saline solution (PSS). PAs, which branched perpendicularly off the MCA and penetrated into the brain tissue, were dissected from the M2 region of the ipsilateral MCA, mounted onto glass cannulas in an arteriograph chamber and secured with silk suture, as previously described.19 PAs were pressurized to 40 mm Hg and equilibrated for 1 h to allow spontaneous development of myogenic tone. After the equilibration period, myogenic reactivity was assessed with stepwise increases in intravascular pressure up to 80 mm Hg, and lumen diameter recorded once stable. Intraluminal pressure was returned to 40 mm Hg for the remaining experiment. A pharmacological approach was used to investigate the effect of fasudil on PA smooth muscle reactivity after ischemia and reperfusion. Constriction to thromboxane mimetic U46619 was evaluated by cumulatively adding U46619 (10−9–10−6 M) to the bath. After washout with PSS, reactivity to L-type calcium channel blocker diltiazem (10−9–10−6 M) was tested. At the conclusion of the experiment, passive structural inner diameter of PAs was determined in calcium-free PSS from 5 to 100 mm Hg for calculation of myogenic tone and percent reactivity of vasoactive agents.
Drugs and solutions
Fasudil (Sigma, St. Louis, MO, USA) was dissolved in sterile-lactated Ringer solution prior to animal treatment. All isolated vessel experiments were performed using a bicarbonate-based Ringer’s PSS, the ionic composition of which was (mmol/L): NaCl 119.0, NaHCO3 24.0, KCl 4.7, KH2PO4 1.18, MgSO4 × 7H2O 1.17, CaCl2 1.6, EDTA 0.026, and glucose 5.5. Zero calcium PSS was the same PSS composition without CaCl2 and addition of EGTA 0.05 mmol/L. PSS was made each week and stored without glucose at 4℃. Glucose was added to the PSS prior to each experiment. PSS was aerated with 5% CO2, 10% O2 and balanced N2 to maintain pH. Diltiazem was purchased from MP Biomedicals (Santa Ana, CA, USA) and made up weekly to a 1 mmol/L stock. U46619 was purchased from Sigma and aliquots of 10 mmol/L stocks were stored frozen at −20℃. TTC and formalin were purchased from Sigma.
Data calculations and statistical analysis
The number of animals used in each experiment was justified by statistical power calculation based on previous similar studies.25,27 Power calculation was based on difference between means of the two groups and standard deviation of brain injury volume and myogenic tone of isolated PAs. Probability of type I error (α) was set at 0.05 and that of type II error (β) was set at 0.2. Therefore, statistical power (1−β) of each experiment was at least 80%. For brain injury volume statistical power calculation, difference between means was 9.94, standard deviation was 7.30 and the test was one-tailed. For myogenic tone statistical power calculation, effect size was 12.30, standard deviation was 7.49 and the test was also one-tailed. Results are presented as mean (SD). Myogenic tone was calculated as a percent decrease in diameter from the fully relaxed diameter in calcium-free PSS by the equation: (1−(ϕtone/ϕpassive)) ×100%; where ϕtone is the inner diameter of the vessel with tone and ϕpassive is the inner diameter of the vessel in calcium-free PSS. Percent constriction to U46619 was calculated as a percent change in diameter from baseline by the equation: [1−(ϕdrug/ϕbaseline)] × 100%, where ϕdrug is the diameter of vessel after drug exposure, and ϕbaseline is the diameter before giving drug. Percent reactivity to diltiazem was calculated from the equation [(ϕdose−ϕbaseline)/(ϕpassive−ϕbaseline)] × 100%, where ϕdose is the diameter at a specific concentration of drug. Mann–Whitney or t-test was used to determine statistical differences between groups where appropriate. Repeated measures one-way analysis of variance (ANOVA) was used for changes of CBF versus baseline. Effect size was calculated using Cohen’s d. Differences were considered significant at P < 0.05.
Excluded animals
A total of seven animals were excluded in this study, two in CBF and brain injury experiments and five in isolated PA experiments. All but one excluded animal had a drop of CBF that was <70%. The remaining excluded animal had a malformed brain.
Results
Effect of fasudil on collateral perfusion
Collateral perfusion remained largely unchanged or negative throughout 90 min of post-treatment period in vehicle-treated SHR (Figure 1(a)). Fasudil modestly increased collateral perfusion starting at 60 min of ischemia but this was not statistically significant (p = 0.198 at 60 min). To further evaluate the change in collateral perfusion, we determined the number, duration, and magnitude of discrete collateral perfusion increases from the laser Doppler tracings. Within the 90 min of post-treatment period, fasudil did not increase the number (Figure 1(b)), duration (Figure 1(c)) and magnitude (Figure 1(d)) of increased collateral perfusion events.
Figure 1.

Effect of fasudil on changes in collateral perfusion during ischemia. (a) Graph showing changes in collateral perfusion calculated as percent change from prior to treatment (30 min after filament occlusion). Changes in collateral perfusion was minimal and mostly negative in vehicle-treated SHR. Fasudil did not significantly increased collateral perfusion. Graphs showing (b) number, (c) duration, and (d) magnitude of increased collateral perfusion events in vehicle or fasudil-treated SHR. Fasudil treatment did not increase these discrete events.
Effect of fasudil on reperfusion CBF
Figure 2 shows the change in CBF at core MCA region over the entire ischemia and reperfusion duration. The drop in CBF during filament occlusion was sustained and consistent throughout ischemia in both groups of animals, suggesting severity of stroke was similar (Figure 2). After removal of the filament, CBF was lower than baseline in both groups, demonstrating incomplete reperfusion. The decrease in CBF during reperfusion was not significantly different at any time points in the fasudil group when compared to the controls.
Figure 2.
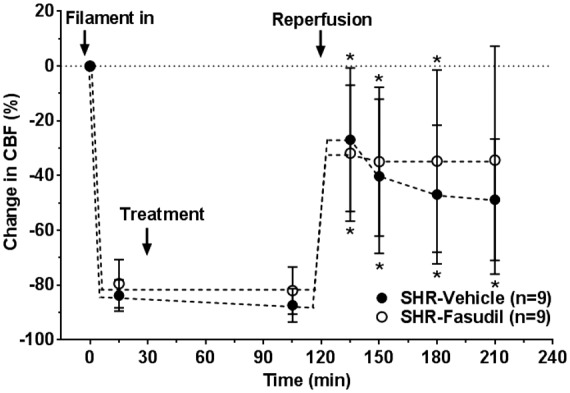
Effect of fasudil on changes in ischemic and reperfusion CBF in core MCA territory. Decrease in CBF in response to filament insertion was similar between groups and sustained throughout ischemia. During reperfusion, CBF was lower than baseline in both groups and fasudil had no significant effect. *p < 0.05 vs. baseline using repeated measures ANOVA.
Effect of fasudil on brain injury and edema formation
Figure 3(a) shows representative images of TTC-stained brain coronal sections where red color represents healthy tissue, while white represents injured brain tissue. Fasudil treatment did not significantly affect acute brain injury volume (Figure 3(b)) and edema formation (Figure 3(c)).
Figure 3.

Effect of fasudil on acute brain injury volume and edema formation. (a) Representative images of rat brain coronal sections showing acute brain injury (white) after TTC staining in each group. Graphs showing (b) brain injury volume and (c) edema formation in vehicle or fasudil-treated SHR. Fasudil treatment had no significant effect on brain injury and edema.
Effect of fasudil on reactivity of isolated PAs
Constricted PAs after ischemia and reperfusion, caused by smooth muscle calcium sensitization, may be a mechanism for incomplete reperfusion.19 Therefore, we sought to determine if fasudil treatment 10 min before reperfusion could prevent vasoconstriction of PAs. Figure 4 shows the reactivity of PAs to pressure and vasoactive agents after 2 h of ischemia and 30 min of reperfusion. Active diameter of PAs was not significantly different at all pressures studied in the fasudil group versus controls (Figure 4(a)). The percent tone was also similar between the groups (Figure 4(b)). In addition, we tested reactivity of PAs to U46619 and diltiazem. Constriction to U46619 (Figure 4(c)) and dilation to diltiazem (Figure 4(d)) were similar in PAs from vehicle and fasudil-treated animals.
Figure 4.

Effect of fasudil on reactivity of PAs to pressure and vasoactive agents. (a) Graph showing active inner diameters of PAs in response to pressure. Fasudil treatment did not affect inner diameter of PAs at any pressure studied. (b) Graph showing percent tone of PAs at different pressures. Fasudil had no significant effect on percent tone of PAs. Graphs showing (c) percent constriction to U-46619 and (d) percent reactivity to diltiazem. Fasudil had no effect on reactivity to U46619 or diltiazem.
Discussion
Rho kinase is activated and leads to vasoconstriction in cerebral vasculature during chronic hypertension.21 Previous studies have shown hypertension worsens stroke outcome that may be related to the constricted state of the cerebral vasculature. For example, vasoconstriction of LMAs could significantly limit collateral perfusion from the ACA during ischemia, leading to small penumbra and large infarct seen in hypertensive animals.13 In addition, constriction of PAs could markedly limit reperfusion to the brain parenchyma that could also worsen stroke outcome. Therefore, we sought to determine if Rho kinase inhibitor fasudil would prevent constriction of these specific vascular segments in the brain, leading to improved collateral and reperfusion CBF. We found that fasudil, at a low dose that had minimal effects on blood pressure, had no significant effect on collateral flow, reperfusion, acute brain injury, or edema. In agreement with the in vivo study, isolated vessel experiments revealed that the same dose of fasudil did not significantly prevent vasoconstriction of PAs.
In addition to chronic hypertension, Rho kinase is also activated in neurons after ischemic brain injury.28 The neuroprotective effect of Rho kinase inhibition by fasudil has been demonstrated and likely involves multiple mechanisms. For example, fasudil exerts its neuroprotective effect by directly inhibiting neuronal cell death pathways.28,29 Alternatively, the neuroprotective effect of fasudil may be secondary to its vascular protective effect, through endothelial nitric oxide-dependent vasodilation and increase in CBF.30,31 These previous studies were performed in either cell culture or normotensive animals. Despite previous studies showing promising vascular protecting effects, fasudil did not show any effect in hypertensive rats. It is possible that in an effort to limit the blood pressure lowering effect, this dose of fasudil was insufficient to inhibit Rho kinase that is highly activated in hypertension. This low dose of fasudil was also unlikely to have direct neuroprotective effect because previous studies that showed the efficacy of fasudil used was 1 to 10 mg/kg.30,32,33 That this low dose of fasudil was insufficient to inhibit Rho kinase in SHR is further supported by the finding that blood pressure was not decreased compared to vehicle-treated animals. While the blood pressure lowering effect of chloral hydrate was substantial (Table 1), it was not further lowered by fasudil, suggesting Rho kinase inhibition was too low to impact blood pressure.
PAs, isolated from normotensive rats, had increased myogenic tone in response to early post-ischemic reperfusion that was associated with smooth muscle calcium sensitization.19 In this study, we sought to determine if fasudil treatment would reduce myogenic tone of PAs after MCAO. We chose fasudil because it inhibits calcium sensitization of vascular smooth muscle that was shown to be a mechanism of PA constriction.19 In agreement with the in vivo CBF measurement of reperfusion, fasudil treatment did not significantly reduce myogenic tone of PAs after ischemia and reperfusion. In addition, PA smooth muscle reactivity to pressure and receptor-mediated vasoconstriction were not affected by fasudil, as suggested by similar response to increasing pressure and U46619, respectively. In addition, sensitivity to L-type calcium channel inhibition was also not affected by fasudil as evaluated by reactivity to diltiazem. Together, the fasudil treatment used in the current study did not appear to prevent vasoconstriction and affect smooth muscle function of PAs after ischemia and reperfusion during chronic hypertension.
Fasudil has a powerful effect on systemic blood pressure, particularly in hypertensive animals.34,35 Reduced systemic blood pressure is associated with poor outcome of acute ischemic stroke, probably because of further decrease in cerebral perfusion pressure and increased risk of infarction.3,36 Because the brain during stroke has impaired cerebral autoregulation, any reduction in systemic blood pressure would be accompanied by a similar reduction in cerebral blood flow to the ischemic brain and increase both the ischemic penumbra and size of brain infarction with acute stroke. Therefore, we determined a dose of fasudil that did not significantly impact systemic blood pressure. The reported effect of fasudil in age-matched male SHR is variable, with decreased systemic blood pressure of ∼35% at 8 mg/kg versus only ∼10% at a higher 10 mg/kg.34,37 The effect of fasudil on CBF was not reported in these studies; therefore, we determined the dose of fasudil in a pilot study. Our preliminary study showed that 3 mg/kg, a dose that has been shown previously to have neuroprotective effects in normotensive rats,32 decreased blood pressure by more than 40% in anesthetized SHR. Blood pressure was decreased by ∼15% at a 10-fold lower dose of 0.3 mg/kg. Thus, we selected a dose of 0.1 mg/kg in this study for two reasons. First, this dose decreased systemic blood pressure minimally, by only ∼4%. Second, a previous study showed that 0.1 mg/kg significantly increased cortical CBF by ∼20% and was not further increased by a higher dose of 0.3 mg/kg.38 These results suggested that CBF could be increased by an appropriate dose of fasudil without marked impact on systemic blood pressure. However, we found that fasudil did not significantly increase perfusion during ischemia and reperfusion, possibly because this dose was not sufficient to inhibit Rho kinase that is highly activated in cerebral vasculature of hypertensive animals.21 It is worth noting that higher doses of fasudil may indeed provide neuroprotection in SHR if the blood pressure lowering effect could be prevented.
We have considered several commercially available Rho kinase inhibitors. We chose fasudil for several reasons. First, it is a widely used, potent (IC50 1 µM) and selective (IC50 100-fold higher for protein kinase C) Rho kinase inhibitor.39 Second, it is clinically relevant that fasudil has been approved for treatment for cerebral vasospasm after subarachnoid hemorrhage in several countries and showed efficacy in clinical trial for pulmonary arterial hypertension.40,41 Third, fasudil is as potent as another widely studied and structurally unrelated Rho kinase inhibitor Y27632 on dilating cerebral vasculature.21 Lastly, fasudil has been shown to have neuroprotective effects in animal models of acute ischemic stroke.32,33 Therefore, it appeared that fasudil was an appropriate choice of Rho kinase inhibitor for this study. However, we did not find that fasudil was efficacious in treating ischemic stroke in SHR.
In conclusion, low dose fasudil treatment in vivo had no beneficial effect on changes in collateral perfusion, reperfusion CBF, or brain injury after acute ischemic stroke in hypertensive rats. This may be partly due to insufficient fasudil dosing that had no vasodilatory effect in the cerebral vasculature. Higher doses of fasudil may be effective at limiting brain injury if the blood pressure lowering effect could be prevented.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke Grant R01 NS093289, the Totman Medical Research Trust, Cardiovascular Research Institute of Vermont.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
MJC conceived and designed the study and analyzed data; SLC designed the study and performed experiments, analyzed data, compiled figures and wrote the manuscript.
References
- 1.Wolf PA, D'Agostino RB, Belanger AJ, et al. Probability of stroke: a risk profile from the Framingham study. Stroke 1991; 22: 312–318. [DOI] [PubMed] [Google Scholar]
- 2.O'Donnell MJ, Xavier D, Liu L, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the Interstroke study): a case-control study. Lancet 2010; 376: 112–123. [DOI] [PubMed] [Google Scholar]
- 3.Leonardi-Bee J, Bath PM, Phillips SJ, et al. Blood pressure and clinical outcomes in the international stroke trial. Stroke 2002; 33: 1315–1320. [DOI] [PubMed] [Google Scholar]
- 4.Paciaroni M, Agnelli G, Corea F, et al. Early hemorrhagic transformation of brain infarction: rate, predictive factors, and influence on clinical outcome: results of a prospective multicenter study. Stroke 2008; 39: 2249–2256. [DOI] [PubMed] [Google Scholar]
- 5.Willmot M, Leonardi-Bee J, Bath PM. High blood pressure in acute stroke and subsequent outcome: a systematic review. Hypertension 2004; 43: 18–24. [DOI] [PubMed] [Google Scholar]
- 6.Letourneur A, Roussel S, Toutain J, et al. Impact of genetic and renovascular chronic arterial hypertension on the acute spatiotemporal evolution of the ischemic penumbra: a sequential study with MRI in the rat. J Cereb Blood Flow Metab 2011; 31: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCabe C, Gallagher L, Gsell W, et al. Differences in the evolution of the ischemic penumbra in stroke-prone spontaneously hypertensive and Wistar-Kyoto rats. Stroke 2009; 40: 3864–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ventura NM, Peterson NT, Tse MY, et al. Molecular adaptations in vasoactive systems during acute stroke in salt-induced hypertension. Mol Cell Biochem 2015; 399: 39–47. [DOI] [PubMed] [Google Scholar]
- 9.Kang BT, Leoni RF, Silva AC. Impaired CBF regulation and high CBF threshold contribute to the increased sensitivity of spontaneously hypertensive rats to cerebral ischemia. Neuroscience 2014; 269: 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumbach GL, Heistad DD. Cerebral circulation in chronic arterial hypertension. Hypertension 1988; 12: 89–95. [DOI] [PubMed] [Google Scholar]
- 11.Mayhan WG, Faraci FM, Heistad DD. Impairment of endothelium-dependent responses of cerebral arterioles in chronic hypertension. Am J Physiol 1987; 253: H1435–H1440. [DOI] [PubMed] [Google Scholar]
- 12.Sweet JG, Chan SL, Cipolla MJ. Effect of hypertension and carotid occlusion on brain parenchymal arteriole structure and reactivity. J Appl Physiol 2015; 119: 817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan SL, Sweet JG, Bishop N, et al. Pial collateral reactivity during hypertension and aging: understanding the function of collaterals for stroke therapy. Stroke 2016; 47: 1618–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eto M, Kozai T, Cosentino F, et al. Statin prevents tissue factor expression in human endothelial cells: role of Rho/Rho-kinase and Akt pathways. Circulation 2002; 105: 1756–1759. [DOI] [PubMed] [Google Scholar]
- 15.Masumoto A, Hirooka Y, Shimokawa H, et al. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 2001; 38: 1307–1310. [DOI] [PubMed] [Google Scholar]
- 16.Uehata M, Ishizaki T, Satoh H, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997; 389: 990–994. [DOI] [PubMed] [Google Scholar]
- 17.Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996; 273: 245–248. [DOI] [PubMed] [Google Scholar]
- 18.Seko T, Ito M, Kureishi Y, et al. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res 2003; 92: 411–418. [DOI] [PubMed] [Google Scholar]
- 19.Cipolla MJ, Chan SL, Sweet J, et al. Postischemic reperfusion causes smooth muscle calcium sensitization and vasoconstriction of parenchymal arterioles. Stroke 2014; 45: 2425–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gokina NI, Park KM, McElroy-Yaggy K, et al. Effects of Rho kinase inhibition on cerebral artery myogenic tone and reactivity. J Appl Physiol 2005; 98: 1940–1948. [DOI] [PubMed] [Google Scholar]
- 21.Chrissobolis S, Sobey CG. Evidence that Rho-kinase activity contributes to cerebral vascular tone in vivo and is enhanced during chronic hypertension: comparison with protein kinase C. Circ Res 2001; 88: 774–779. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Brayden JE. Rho kinase activity governs arteriolar myogenic depolarization. J Cereb Blood Flow Metab 2017; 37: 140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagaud G, Gaudreault N, Moore ED, et al. Pressure-dependent myogenic constriction of cerebral arteries occurs independently of voltage-dependent activation. Am J Physiol Heart Circ Physiol 2002; 283: H2187–H2195. [DOI] [PubMed] [Google Scholar]
- 24.Jarajapu YP, Knot HJ. Relative contribution of Rho kinase and protein kinase C to myogenic tone in rat cerebral arteries in hypertension. Am J Physiol Heart Circ Physiol 2005; 289: H1917–H1922. [DOI] [PubMed] [Google Scholar]
- 25.Cipolla MJ, Linfante I, Abuchowski A, et al. Pharmacologically increasing collateral perfusion during acute stroke using a carboxyhemoglobin gas transfer agent (Sanguinate™) in spontaneously hypertensive rats. J Cereb Blood Flow Metab. Epub ahead of print 24 April 2017. DOI: 10.1177/0271678X17705567. [DOI] [PMC free article] [PubMed]
- 26.Cuccione E, Versace A, Cho TH, et al. Multi-site laser doppler flowmetry for assessing collateral flow in experimental ischemic stroke: validation of outcome prediction with acute mri. J Cereb Blood Flow Metab 2017; 37: 2159–2170. [DOI] [PMC free article] [PubMed]
- 27.Cipolla MJ, Sweet JG, Chan SL. Effect of hypertension and peroxynitrite decomposition with FeTMPyP on CBF and stroke outcome. J Cereb Blood Flow Metab 2017; 37: 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamashita K, Kotani Y, Nakajima Y, et al. Fasudil, a Rho kinase (ROCK) inhibitor, protects against ischemic neuronal damage in vitro and in vivo by acting directly on neurons. Brain Res 2007; 1154: 215–224. [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Li J, Hu H, et al. Rho-kinase inhibitor, fasudil, prevents neuronal apoptosis via the Akt activation and Pten inactivation in the ischemic penumbra of rat brain. Cell Mol Neurobiol 2012; 32: 1187–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yagita Y, Kitagawa K, Sasaki T, et al. Rho-kinase activation in endothelial cells contributes to expansion of infarction after focal cerebral ischemia. J Neurosci Res 2007; 85: 2460–2469. [DOI] [PubMed] [Google Scholar]
- 31.Rikitake Y, Kim HH, Huang Z, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005; 36: 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satoh S, Ikegaki I, Suzuki Y, et al. Neuroprotective properties of a protein kinase inhibitor against ischaemia-induced neuronal damage in rats and gerbils. Br J Pharmacol 1996; 118: 1592–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toshima Y, Satoh S, Ikegaki I, et al. A new model of cerebral microthrombosis in rats and the neuroprotective effect of a Rho-kinase inhibitor. Stroke 2000; 31: 2245–2250. [DOI] [PubMed] [Google Scholar]
- 34.Behuliak M, Vavrinova A, Bencze M, et al. Ontogenetic changes in contribution of calcium sensitization and calcium entry to blood pressure maintenance of Wistar-Kyoto and spontaneously hypertensive rats. J Hypertens 2015; 33: 2443–2454. [DOI] [PubMed] [Google Scholar]
- 35.Okamura N, Saito M, Mori A, et al. Vasodilator effects of fasudil, a Rho-kinase inhibitor, on retinal arterioles in stroke-prone spontaneously hypertensive rats. J Ocul Pharmacol Ther 2007; 23: 207–212. [DOI] [PubMed] [Google Scholar]
- 36.Castillo J, Leira R, Garcia MM, et al. Blood pressure decrease during the acute phase of ischemic stroke is associated with brain injury and poor stroke outcome. Stroke 2004; 35: 520–526. [DOI] [PubMed] [Google Scholar]
- 37.Tsounapi P, Saito M, Kitatani K, et al. Fasudil improves the endothelial dysfunction in the aorta of spontaneously hypertensive rats. Eur J Pharmacol 2012; 691: 182–189. [DOI] [PubMed] [Google Scholar]
- 38.Satoh S, Utsunomiya T, Tsurui K, et al. Pharmacological profile of hydroxy fasudil as a selective Rho kinase inhibitor on ischemic brain damage. Life Sci 2001; 69: 1441–1453. [DOI] [PubMed] [Google Scholar]
- 39.Shimokawa H, Seto M, Katsumata N, et al. Rho-kinase-mediated pathway induces enhanced myosin light chain phosphorylations in a swine model of coronary artery spasm. Cardiovasc Res 1999; 43: 1029–1039. [DOI] [PubMed] [Google Scholar]
- 40.Tachibana E, Harada T, Shibuya M, et al. Intra-arterial infusion of fasudil hydrochloride for treating vasospasm following subarachnoid haemorrhage. Acta Neurochir 1999; 141: 13–19. [DOI] [PubMed] [Google Scholar]
- 41.Fukumoto Y, Matoba T, Ito A, et al. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 2005; 91: 391–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.