

ABSTRACT
KRAS is frequently mutated in a variety of cancers including lung cancer. Whereas the mitogen-activated protein kinase (MAPK) is a well-known effector pathway of KRAS, blocking this pathway with MEK inhibitors is relatively ineffective. One major contributor to limited efficacy is attributed to the reactivation of MAPK signal following MEK inhibition by multiple feedback mechanisms. In a recent study, we have identified that epithelial-to-mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling following MEK inhibition in KRAS mutant lung cancer. In epithelial-like cells, this feedback was mediated by ERBB3. In contrast, in mesenchymal-like cells, the feedback was attributed to the fibroblast growth factor receptor 1 (FGFR1) pathway. FGFR1 was dominantly expressed in mesenchymal-like cells: suppression of SPRY proteins by MEK inhibition relieved negative feedback control of basal FGFR-FRS2 function, resulting in reactivation of MAPK signaling via FGFR1. Therapeutically, the combination of MEK inhibitor trametinib with an FGFR inhibitor induced tumor regressions in tumor xenografts derived from mesenchymal-like KRAS mutant cancer cell lines as well as a patient derived xenograft model with a representative mesenchymal phenotype. Collectively, feedback activation of MAPK by FGFR1 signaling mitigates the effect of MEK inhibitor in mesenchymal-like KRAS mutant lung tumors, and combinations of clinically available FGFR1 inhibitors and MAPK inhibitors constitute a therapeutic approach to treat these cancers effectively.
KEYWORDS: epithelial-to-mesenchymal transition, feedback, KRAS, lung cancer, MEK inhibitor
KRAS is the most frequently mutated gene in cancer including lung adenocarcinoma in which 15 to 25% of patient harbor KRAS mutations. Mutations in KRAS impair the intrinsic GTPase activity of KRAS, causing it to accumulate in a constitutively active GTP-bound state.1,2 In contrast to the successful development of ATP-competitive small molecule inhibitors blocking mutant EGFR and translocated ALK, the identification of GTP competitive inhibitors has been more difficult. This is primarily because KRAS binds much at high picomolar affinities to GTP/GDP.
Identifying alternative ways to target KRAS mutant cancers, like in other cancers driven by currently undruggable driver oncogenes,3 have been attempted.1,2 Among them, targeting the mitogen-activated protein kinase (MAPK), the best characterized downstream pathway of KRAS, has been explored. However, MEK inhibitor monotherapy demonstrates only modest efficacy in vitro and in vivo due to 2 primary reasons.4,5 The first reason is inhibition of MEK and suppression of ERK activity relieves negative feedback from ERK at multiple levels of MAPK signaling. Initially, ERK inhibition results in upregulation of RAF and MEK activities by dephosphorylating inhibitory phosphorylation sites on these proteins. In addition, ERK induces transcription of negative feedback genes including Sprouty family (SPRYs) and dual-specificity phosphatases (DUSPs). While DUSPs bind to and inactivate ERK by dephosphorylating residues required for catalytic activity of ERK, SPRY functions more upstream of MAPK signaling by disrupting SOS1 interaction with GRB2. The second reason MEK inhibitor monotherapy is ineffective is inhibition of MEK induces rewiring of kinase signaling networks, which results in reactivation of ERK and induction of other pathways including phosphoinositide 3-kinase (PI3K)-AKT; these changes occur within 24 hours in cell culture experiments.
Mechanistically, MEK inhibition leads to feedback activation of ERBB3 signaling via activated ERK phosphorylation of an inhibitory threonine 669 residue in the conserved juxtamembrane (JM) domains of EGFR and HER2.6 Moreover, MAPK inhibition downregulates transcription factor c-MYC, which relieves transcriptional repression of multiple receptor tyrosine kinases (RTKs) and has been shown to activate PI3K and MAPK signaling.7 To overcome feedback activation of MAPK signaling, several combinatorial approaches have been proposed to treat KRAS mutant cancers.8 However, since multiple mechanisms are involved in the feedback activation of MAPK signaling, it remains unclear how we can decide which regimen would be chosen to treat each cancer.
In a recent report, we have identified a mechanism that should aid in developing biomarker-directed combinations using MEK inhibitors in KRAS mutant lung cancers.9 In KRAS mutant lung cancer cell lines, as expected, rebound activation of ERK and upregulation of AKT signaling were observed following treatment with MEK inhibitors trametinib and selumetinib. Immunoprecipitation of p85, the regulatory subunit of PI3K, revealed that activation of AKT was mediated by ERBB3 activation. Concomitant inhibition of MEK with ERBB3 by a pan-ERBB inhibitor afatinib negated ERK reactivation and upregulation of AKT, leading to cell death in vitro and tumor regressions in vivo. The effectiveness of afatinib with trametinib against KRAS mutant cancer cell lines was consistent with a previous report.10
However, feedback activation of ERK and AKT signaling was also observed in ERBB3 non-expressed cells. Using bioinformatic analyses,we have identified a positively correlated relationship between expression of ERBB3 and epithelial markers such as E-cadherin in KRAS mutant lung cancer cell lines. Induction of epithelial to mesenchymal transition (EMT) by chronic TGF-β1 treatment in an ERBB3 positive epithelial-like KRAS mutant lung cancer cell line identified that E-cadherin low/vimentin positive mesenchymal-like KRAS mutant cancer cells lose ERBB3 expression, instead dominantly express FGFR1 protein. Importantly, while feedback activation is mediated by ERBB3 in epithelial-like KRAS mutant cancer cell lines, the FGFR1-FRS2 pathway plays a critical role in the feedback reactivation of MAPK and upregulation of AKT signaling in mesenchymal-like KRAS mutant cancer cell lines. This feedback is attributed to downregulation of SPRY4 protein expression following treatment with MEK inhibitor, which relieves suppression of basal FGFR-FRS2 function, leading to reactivation of MAPK signaling and upregulation of AKT signaling in the presence of FGFR1. In mesenchymal-like KRAS mutant lung cancer cell lines, knockdown of FGFR1 or addition of FGFR inhibitor negated feedback activation of ERK and upregulation of AKT signaling following trametinib treatment. Therapeutically, the combination of trametinib with FGFR inhibitor induced robust apoptosis in vitro and tumor regressions in vivo and a patient derived xenograft model with a representative mesenchymal phenotype identified by the expression of E-cadherin and vimentin.
These findings indicate that MEK inhibition induces distinct feedback activation of RTKs based on EMT status in KRAS mutant lung cancer: ERBB3 in epithelial-like and FGFR1 in mesenchymal-like cells, respectively (Fig. 1). Combination of MEK inhibitor with corresponding RTK inhibitors overcame adaptive resistance to MEK inhibitor. Incorporating a companion diagnostic of EMT with standard genotyping of lung cancer may help to generate more effective treatment options for patients. That is, in KRAS mutant lung cancer, defining EMT status could enable patients to access biomarker-directed, MEK inhibitor based combinations. The companion diagnostic could include immunohistochemistry for representative EMT markers, E-cadherin and ERBB3 as epithelial markers, and vimintin and FGFR1 as mesenchymal markers (Fig. 2). Because the exact criteria for identifying EMT has not been established yet,11 some other biomarkers of EMT such as ZEB1, Snail, or N-cadherin may be incorporated to optimize patient selection. In addition, given that patient tumors likely consist of both epithelial and mesenchymal cells, optimization of scoring system how tumor is categorized epithelial or mesenchymal is needed. Furthermore, this intratumor heterogeneity likely limits the efficacy of the treatment strategy. Combination against epithelial or mesenchymal tumor may result in transient tumor regressions, to be led by the growth of cells with the other phenotype that cause resistance. The sequential treatment of ERBB3/MEK inhibition and FGFR1/MEK inhibition might be a strategy to delay the emergence of drug-resistant clones.
Figure 1.
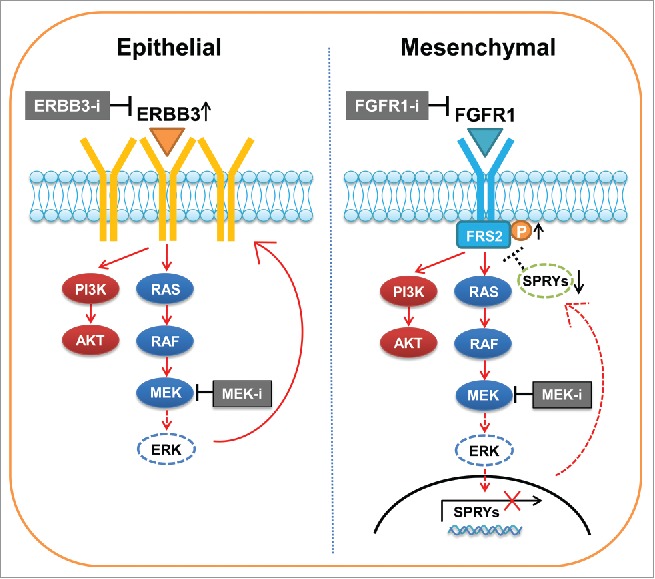
MEK inhibition induces distinct feedback activation of RTKs based on EMT status in KRAS mutant lung cancer. In epithelial-like tumor cells, MEK inhibition upregulates expression and phosphorylation of ERBB3 that re-activates MAPK and upregulates PI3K-AKT signaling. In contrast, in mesenchymal-like tumor cells, MEK inhibition suppresses sprouty proteins (SPRYs), leading to activate FRS2 phosphorylation by relieving negative feedback to FGFR1-FRS2. Combination of MEK inhibitor with ERBB3 inhibitor (ERBB3-i) or FGFR1 inhibitor (FGFR1-i) treatment exerts synergistic effect by suppressing the corresponding RTKs.
Figure 2.
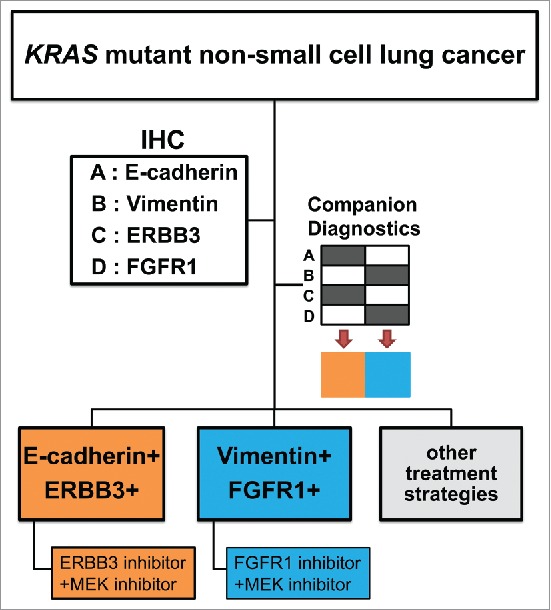
Flow chart to select combinatorial therapies stratified by expression of EMT markers. KRAS mutant lung cancer is determined whether tumor cells have epithelial or mesenchymal phenotype according to the expression of representative EMT markers, E-cadherin, vimentin, ERBB3, and FGFR1. When ERBB3 and E-cadherin are positive, the patients are treated with combination of MEK inhibitor and ERBB3 inhibitor. On the other hand, when FGFR1 and vimentin are positive, combination of the MEK inhibitor with FGFR1 inhibitors is chosen. Because not all tumors can be defined either subtype, more markers or other treatment strategies against mixed phenotype are needed to be identified. IHC; immunohistochemistry.
A mesenchymal cell phenotype had been traditionally thought to be associated with tumor metastasis by enhancing migratory capacity, invasiveness, and upregulation of extracellular matrix components.12 Intriguingly, recent findings indicate that EMT may be dispensible for metastasis, however mesenchymal cells do acquire cancer stem cell-like and chemoresistant properties.13,14 Therefore, the combination of FGFR inhibitor with MEK inhibitor might be applicable to treat residual tumor following conventional chemotherapy or chemo-refractory tumors.
Since reactivation of ERK signal has limited the effectiveness of MEK inhibitors in KRAS mutant cancers, development of ERK inhibitors have been pursued.15-17 The combination of the ERK inhibitor VTX-11e with the MEK inhibitor PD0325901 prevented RAF-dependent reactivation of MAPK signaling and induced apoptosis in NRAS mutant melanoma cells.15 However, RTK mediated activation of pathways other than MAPK signaling may limit the efficacy of vertical, multi-step inhibition of the MAPK pathway. We have shown that apoptosis induced by FGFR inhibitor with trametinib was significantly correlated with that of induced by PI3K inhibitor with trametinib in mesenchymal-like KRAS mutant lung cancer cell lines. In line with this, PI3K-AKT-mTOR signaling was identified a key driver of resistance to another ERK inhibitor SCH772984 by reverse-phase protein array (RPPA) based pathway activation mapping analysis between sensitive and resistant cells to the drug.17 However, surprisingly, neither of upregulation of AKT phosphorylation nor induction of apoptosis by combining PI3K inhibitor was observed in SCH772984 resistant cell lines.17 Further studies are needed whether suppression of rebound ERK activation is the only cause of enhanced efficacy by combining MEK inhibitor with FGFR1 or ERBB3 inhibition.
Whereas direct inhibition of mutant KRAS has been challenging, small molecules that bind directly to mutant KRAS and inhibit interaction with effector proteins have been identified.18,19 However, because MAPK signal is a critical effector pathway, suppression of mutant KRAS could often leads to feedback RTK activation. For instance, it has been demonstrated that knockdown of activated RAS hyperactivate upstream pathways such as EGFR.20 Furthermore, while KRAS G12C specific inhibitors prevent its activation by binding to the GDP-bound oncoprotein, potentiation of nucleotide exchange by RTK activation reduces the sensitivity of the drug by lowering the levels of GDP-bound KRAS available for drug binding. In line with this, MEK or AKT inhibitors attenuated the antiproliferative effect by relieving negative feedback to RTK activation.18 Combination of KRAS and RTK inhibitors enhanced the antitumor activity of KRAS G12C specific inhibitors. These results suggest that our model may also help to select inhibitor of receptors dominantly responsible for activation of RAS nucleotide exchange. Notably, recent report showed that rigosertib bound to the RAS-binding domains (RBDs) of multiple RAS effectors and downregulated signaling such as RAF, Ral-GDS, and PI3K.21 This may suggest that the effect of rigosertib may not be attenuated by feedback activation of RTKs because the drug disrupts the association of RAS with multiple effector proteins, however, it remains to be determined.
Lastly, it is intriguing to determine whether these findings are also applicable to cancers of other organs because KRAS mutation is common in other cancers especially colorectal cancer and pancreatic cancer. In KRAS mutant lung and pancreatic cancers, KRAS knockdown experiment identified 2 classes of cells-KRAS-dependent cells (in which KRAS knockdown induced apoptosis) and KRAS-independent cells.22 Expression of epithelial markers was highly associated with KRAS dependency, whereas EMT was correlated with KRAS independence, regardless of the presence of a KRAS mutation. In contrast, the relationship between KRAS dependency and expression of epithelial markers was not evident among KRAS mutant colorectal cancers.23 These evidences suggest that EMT status may define the feedback activation of RTKs following MEK inhibition in pancreatic cancer, while colorectal cancer likely has lineage-specific feedback mechanisms.
In summary, our data demonstrated MEK inhibition leads to distinct activation of RTKs in KRAS mutant lung cancers depending on the epithelial or mesenchymal state of the cancer. While inhibitors targeting ERBB3, FGFR1, or MEK are available, feasibility of each combinatorial therapy will be the key issue in future clinical trials.
Disclosure of potential conflicts of interest
H. Ebi has received a commercial research grant from AstraZeneca.
Acknowledgments
We thank Dr. Anthony C. Faber for suggestions and comments.
Funding
This work was supported by Grant-in-Aid from the Japan Agency for Medical Research and Development (Project for Cancer Research and Therapeutics Evolution (P-CREATE 16cm0106513h0001)) and Grant-in-Aid for Scientific Research (KAKENHI 16K07164) to HE.
References
- [1].Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 2014; 13(11):828-51; PMID:25323927; https://doi.org/ 10.1038/nrd4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell 2014; 25(3):272-81; PMID:24651010; https://doi.org/ 10.1016/j.ccr.2014.02.017 [DOI] [PubMed] [Google Scholar]
- [3].Ham J, Costa C, Sano R, Lochmann TL, Sennott EM, Patel NU, Dastur A, Gomez-Caraballo M, Krytska K, Hata AN, et al.. Exploitation of the apoptosis-primed state of MYCN-amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell 2016; 29(2):159-72; PMID:26859456; https://doi.org/ 10.1016/j.ccell.2016.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer 2015; 15(10):577-92; PMID:26399658; https://doi.org/ 10.1038/nrc4000 [DOI] [PubMed] [Google Scholar]
- [5].Pratilas CA, Solit DB. Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin Cancer Res 2010; 16(13):3329-34; PMID:20472680; https://doi.org/ 10.1158/1078-0432.CCR-09-3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, Engelman JA. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res 2012; 72(13):3228-37; PMID:22552284; https://doi.org/ 10.1158/0008-5472.CAN-11-3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al.. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012; 149(2):307-21; PMID:22500798; https://doi.org/ 10.1016/j.cell.2012.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Russo M, Di Nicolantonio F, Bardelli A. Climbing RAS, the everest of oncogenes. Cancer Discov 2014; 4(1):19-21; PMID:24402942; https://doi.org/ 10.1158/2159-8290.CD-13-0906 [DOI] [PubMed] [Google Scholar]
- [9].Kitai H, Ebi H, Tomida S, Floros KV, Kotani H, Adachi Y, Oizumi S, Nishimura M, Faber AC, Yano S. Epithelial-to-mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling induced by MEK inhibition in KRAS mutant lung cancer. Cancer Discov 2016; 6(7):754-69; PMID:27154822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, Cottino F, Prahallad A, Grernrum W, Tzani A, Schlicker A, et al.. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep 2014; 7(1):86-93; PMID:24685132; https://doi.org/ 10.1016/j.celrep.2014.02.045 [DOI] [PubMed] [Google Scholar]
- [11].Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest 2009; 119(6):1429-37; PMID:19487819; https://doi.org/ 10.1172/JCI36183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119(6):1420-8; PMID:19487818; https://doi.org/ 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015; 527(7579):525-30; PMID:26560028; https://doi.org/ 10.1038/nature16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al.. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015; 527(7579):472-6; PMID:26560033; https://doi.org/ 10.1038/nature15748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rebecca VW, Alicea GM, Paraiso KH, Lawrence H, Gibney GT, Smalley KS. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS-mutant melanoma. Pigment Cell Melanoma Res 2014; 27(6):1154-8; PMID:25130256; https://doi.org/ 10.1111/pcmr.12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, et al.. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 2013; 3(7):742-50; PMID:23614898; https://doi.org/ 10.1158/2159-8290.CD-13-0070 [DOI] [PubMed] [Google Scholar]
- [17].Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, et al.. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell 2016; 29(1):75-89; PMID:26725216; https://doi.org/ 10.1016/j.ccell.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016; 351(6273):604-8; PMID:26841430; https://doi.org/ 10.1126/science.aad6204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, et al.. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov 2016; 6(3):316-29; PMID:26739882; https://doi.org/ 10.1158/2159-8290.CD-15-1105 [DOI] [PubMed] [Google Scholar]
- [20].Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov 2013; 3(1):112-23; PMID:23103856; https://doi.org/ 10.1158/2159-8290.CD-12-0231 [DOI] [PubMed] [Google Scholar]
- [21].Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, Gupta YK, Reddy MV, Ueno L, Hart JR, et al.. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell 2016; 165(3):643-55; PMID:27104980; https://doi.org/ 10.1016/j.cell.2016.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J. A gene expression signature associated with “K-Ras addiction’ reveals regulators of EMT and tumor cell survival. Cancer Cell 2009; 15(6):489-500; PMID:19477428; https://doi.org/ 10.1016/j.ccr.2009.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, Haber DA, Settleman J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 2012; 148(4):639-50; PMID:22341439; https://doi.org/ 10.1016/j.cell.2011.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]