

Abstract
Biological membrane organization mediates numerous cellular functions and has also been connected with an immense number of human diseases. However, until recently, experimental methodologies have been unable to directly visualize the nanoscale details of biological membranes, particularly in intact living cells. Numerous models explaining membrane organization have been proposed, but testing those models has required indirect methods; the desire to directly image proteins and lipids in living cell membranes is a strong motivation for the advancement of technology. The development of super-resolution microscopy has provided powerful tools for quantification of membrane organization at the level of individual proteins and lipids, and many of these tools are compatible with living cells. Previously inaccessible questions are now being addressed, and the field of membrane biology is developing rapidly. This chapter discusses how the development of super-resolution microscopy has led to fundamental advances in the field of biological membrane organization. We summarize the history and some models explaining how proteins are organized in cell membranes, and give an overview of various super-resolution techniques and methods of quantifying super-resolution data. We discuss the application of super-resolution techniques to membrane biology in general, and also with specific reference to the fields of actin and actin-binding proteins, virus infection, mitochondria, immune cell biology, and phosphoinositide signaling. Finally, we present our hopes and expectations for the future of super-resolution microscopy in the field of membrane biology.
Keywords: Actin, Cluster feedback, Domain, FPALM, Lipid, Live cell, PALM, Raft, Review, STORM
1. Introduction
1.1. Fundamentals of membrane organization
Biological membranes mediate vast numbers of cellular functions, serve as the fundamental barrier between cell exterior and interior, and spatially define most cellular organelles (Alberts, 2002). Biological membranes are composed of proteins, lipids, and other small molecules, typically arranged in two opposing monolayers (i.e., a bilayer) (Alberts, 2002). The bilayer arrangement allows hydrogen bonding between the aqueous phase (i.e., the cytoplasm on one side, and extracellular medium on the other) and hydrophilic lipid head groups, while restricting interactions between hydrophobic lipid tails and the hydrophilic cytoplasm and extracellular medium (Tanford, 1991). The lipids found in membranes include a large number (thousands) of molecular species, which can be subdivided into several general classes: phospholipids (including sphingolipids and glycerophospholipids with saturated and/or unsaturated fatty acid chains), glycolipids, and sterols (including cholesterol) (Alberts, 2002, Lehninger et al., 2013).
1.1.1. Membrane lateral organization
1.1.1.1. Singer–Nicolson fluid mosaic model
The idea of a cell membrane as a mosaic structure of globular proteins within a phospholipid bilayer was proposed in 1971 by S.J. Singer (Singer, 1971) and popularized the following year (Singer & Nicolson, 1972). This “fluid mosaic” model proposes the free lateral diffusion of membrane proteins, which assume a long-scale random distribution in the two dimensional homogenous lipid fluid phase (see Figure 1(A) ). One fundamental departure from this theory has been pivotal in shaping membrane research from the 1980s. Rather than being strewn randomly throughout a homogenous cell plasma membrane, proteins and lipids were proposed to be laterally heterogeneous, distributed in discrete patches. While researchers worldwide were embracing this view of cell membranes, to this day there remains deep division in the community: what are the driving forces behind plasma membrane heterogeneity?
Figure 1.
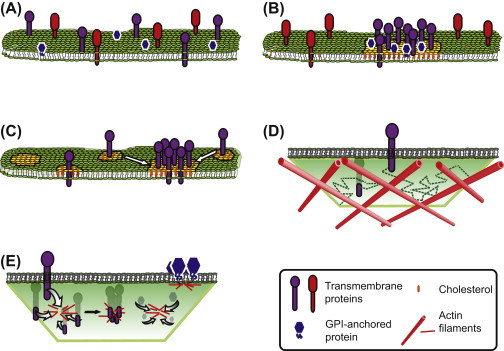
Models of cell membrane organization discussed in Section 1.1. (A) “Fluid mosaic” model. Proteins are distributed randomly through a homogenous phospholipid bilayer. (B) “Lipid raft” model. Sphingolipid and cholesterol patches are populated with proteins which have an affinity for these patches. Protein species can be raft associated or nonraft associated. (C) “Lipid shell” model. Some proteins will be targeted to self-assembled cholesterol and sphingolipid complexes which form a “lipid shell” around the protein. These “lipid shells” have an affinity for, and can coalesce with, larger lipid domains. (D) “Picket Fence” model. Transmembrane proteins are restricted in their diffusion by actin filaments (the “fence”) which appose and run parallel to the cytoplasmic leaflet of the membrane, and by other transmembrane proteins bound to these filaments (“pickets”, not shown). (E) “Active composite” model. Short actin filaments adjacent to the cytoplasmic membrane leaflet are arranged in “asters”. Transmembrane proteins and GPI-anchored proteins are advected by actin and myosin to the centers of these asters, resulting in protein nanoclustering. See Section 1.1 for more detail. Readers please note that depictions of cell membranes here do not show as much protein (relative to lipid) as would be found in actual cell membranes. (See color plate)
In the 10 years after the popular Singer–Nicolson (Singer & Nicolson, 1972) paper was published, researchers were theorizing that cell plasma membranes were organized into discrete lipid domains, and already proposing lipid–protein interactions (Moore, Lentz, & Meissner, 1978) analogous to the present day lipid shell model and boundary lipid theories (Anderson & Jacobson, 2002); researchers were also beginning to theorize that cytoskeletons could modulate lateral mobility of membrane molecules (Karnovsky, Kleinfeld, Hoover, & Klausner, 1982). The ability of glycosphingolipids to self-associate and form discrete patches (reviewed in (Thompson & Tillack, 1985)), was then understood to also encompass protein distributions. Glycosphingolipid self-association in the Golgi could form patches with which membrane proteins would combine, and these mixtures could theoretically be transported to the apical membrane, mediating the sorting of sphingolipids and proteins in polarized epithelial cells (Simons and van Meer, 1988, Simons and Wandinger-Ness, 1990). Biochemical analyses appeared to support this model—the association of (glycophosphatidyl inositol) GPI-anchored proteins along with glycosphingolipids in cell lysate insoluble detergent fractions was taken as evidence of lipid–protein complexes in native membranes (Brown & Rose, 1992). In 1997 came a popular stating of one theory of lipid–protein complexes in the cell plasma membrane: the lipid raft model (Simons & Ikonen, 1997).
1.1.1.2. The lipid raft model
This model postulates that particular subsets of lipids can self-organize, forming discrete patches within the plasma membrane (see Figure 1(B)), believed to be enriched in cholesterol, sphingolipids, and GPI-anchored proteins. The affinity of particular species of membrane proteins for these self-organizing lipids would determine their inclusion into these patches, and in doing so, determine the spatial patterning of proteins in the cell plasma membrane. Sphingolipid self-association would occur through weak interactions between their head groups. Furthermore, cholesterol helps fill gaps between lipid molecules to reduce water permeability (Finkelstein & Cass, 1967). The result (within the context of the model) is phase-separated liquid ordered (Lo) domains (i.e., “lipid rafts”) enriched in sphingolipids, cholesterol, and GPI-anchored proteins, within a surrounding liquid disordered (Ld) fluid phase enriched in unsaturated lipids and other proteins (Simons & Ikonen, 1997). The revised raft model posits that nanoscale assemblies of sphingolipids, cholesterol, and proteins are fluctuating, but can be stabilized into larger platforms important for signaling, viral infection, and membrane trafficking (Simons & Gerl, 2010). While these assemblies are now theorized as being dynamic and of variable size, the basic concept remains that preferential interactions between cholesterol, sphingolipids, and specific proteins are the central mechanism driving local heterogeneity of the cell plasma membrane (Simons & Gerl, 2010).
The lipid raft model is closely linked to the idea that membrane lipids can self-segregate into domains. In particular, separation of bilayers into Lo and Ld phases was proposed to explain membrane lateral heterogeneity in biological membranes (Brown and London, 1997, Brown and London, 1998a, Brown and London, 1998b, Simons and Ikonen, 1997). This idea is based on observations that in the absence of membrane proteins, and depending on lipid composition, temperature, and buffer conditions, artificially created lipid bilayers can separate into coexisting Lo, Ld, and/or solid (gel) phases (Honerkamp-Smith et al., 2009, Tanford, 1978, Veatch et al., 2008, Veatch et al., 2006, Veatch and Keller, 2003a, Veatch and Keller, 2003b, Veatch and Keller, 2005, Veatch et al., 2004). Giant unilamellar vesicles (GUVs) (Angelova & Dimitrov, 1986), which are composed of a single bilayer, have been used extensively as a lipid model of liquid-ordered and liquid-disordered domains (Almeida et al., 2005, Bagatolli and Gratton, 1999, Bagatolli and Gratton, 2000, Bagatolli et al., 2000, Baumgart et al., 2007, Bagatolli et al., 2003, Baumgart et al., 2003, Baumgart et al., 2007, Dietrich et al., 2001, Korlach et al., 1999, Veatch, 2007, Veatch et al., 2008, Veatch et al., 2006, Veatch and Keller, 2005). GUVs made from a mixture of 1:1:1 (unsaturated lipid:saturated lipid:cholesterol), for example, can be made to phase separate at or close to physiological temperature. In general, due to differences in fatty acid chain order and/or length, saturated and unsaturated lipids will under many conditions separate into liquid and gel phases unless sufficient sterol (cholesterol) is present to prevent demixing (Stottrup et al., 2004, Veatch and Keller, 2002, Veatch and Keller, 2003a, Veatch and Keller, 2003b). Intriguingly, disruption of interactions between the cell plasma membrane and the cortical actin cytoskeleton in a process called “blebbing” (Keller et al., 2002, Tank et al., 1982) has been shown to lead to lipid phase separation in bilayers attached to or derived from cells (Baumgart et al., 2007, Veatch et al., 2008). Thus, cell membranes can be caused to phase separate, but whether phase separation occurs in unperturbed cell membranes, and if so, under what conditions and on what length scales, is still unclear.
1.1.1.3. The lipid shell model
Anderson and Jacobson (Anderson & Jacobson, 2002) theorized that there must be some targeting motif encoded in proteins which determines inclusion into lipid domains. The lipid shell model stresses the self assembly of cholesterol–phospholipid complexes, and the differential tendency of proteins to associate with these complexes, either through direct interactions mediated by specific amino acid motifs in transmembrane domain sequences or electrostatic interactions between the head groups of phospholipids and oppositely charged amino acids in the protein. Proteins act as individual units each wrapped in a sphingolipid–cholesterol lipid shell (see Figure 1(C)); these lipid shells do not form a distinct lipid phase, but are thought to be mobile units in the membrane. However, larger scale lipid domains (such as caveolae and “lipid rafts”) are proposed to have a characteristic Lo phase, and proteins in lipid shells would have an affinity for these domains due to the compatibility of lipids in the shell and the Lo phase of the lipid domain. Interactions between shelled proteins and those already in lipid domains may influence their time of residence in the domain, as may ligand binding.
1.1.2. Proteins as organizers
The importance of the cytoskeleton in erythrocyte membrane organization has been reported since the early 1980s (Koppel, 1981, Sheetz, 1983, Tsuji and Ohnishi, 1986) and specific models explaining the dynamics of proteins in living native cell membranes, and the potential for actin to create regions in membranes within which proteins and/or lipids are confined, concentrated, or excluded (“membrane domains”), have gained wider recognition.
1.1.2.1. The picket fence model
The basic underlying principle of this theory is that actin filaments, in a very close spatial association with the cytoplasmic leaflet of the plasma membrane, provide physical barriers which effectively compartmentalize the plasma membrane (Kusumi and Sako, 1996, Sako and Kusumi, 1994). The actin-based “membrane skeleton” (MSK), or “fence”, creates compartments with diameters of 40–300 nm (Kusumi et al., 2012), and various transmembrane proteins bound to these actin filaments act as “pickets”, which span the bilayer. Short-term diffusion of membrane lipids and non-MSK-bound membrane proteins can be confined within these compartments (see Figure 1(D)), with longer term trajectories punctuated by “hop” movements between adjacent compartments (termed “hop diffusion”) (Fujiwara et al., 2002, Kusumi et al., 2012). More recently, the model has been amended to include lipid rafts and protein oligomers, such that entire assemblies of proteins and lipids together may move within a compartment, but still be confined by the picket fence (Kusumi, et al., 2012).
1.1.2.2. Active composite model
Originally used to describe the distribution of GPI-anchored proteins in native cell membranes, this theory was developed to explain indirect observations which suggested these proteins existed either as monomers or in small, immobile, dense clusters, with an approximate average of four molecules per cluster (Sharma et al., 2004). These nanoclusters were reported to be transient and continually remodeling. However, they do not join or coalesce into larger domains, and the ratio of monomers (80%) to clusters (20%) is independent of total expression levels (Goswami et al., 2008). The authors propose the cytoplasmic side of the plasma membrane is abutted by a system of “short” (although estimated as ∼250 nm in length) actin filaments arranged in “asters”, (see Figure 1(E)) which are formed by actin treadmilling and myosin contractility. Myosin contractility results in the barbed (or plus) ends of actin filaments pointing toward aster centers. In regions rich with myosin-bound actin filaments, peripheral actin filaments can be aligned with these other filaments, and pulled toward the most actin dense regions (the centers of asters). The authors posit that these forces are transmitted to (for example) GPI-anchored (exoplasmic leaflet) proteins, and proteins which bind F-actin, which are advected along with these filaments toward the centers of asters, forming nanoclusters at aster cores. Thus actin treadmilling and actin–myosin contraction would drive dynamic nanoscale clustering of proteins at the cell surface (Gowrishankar, Ghosh, Saha, Mayor, & Rao, 2012). The theoretical basis for this model is further discussed below.
1.1.2.3. Cluster feedback model
We propose the “cluster feedback” model (see Figure 2 ) whereby the reciprocal influence between two assemblies: (1) proteins and lipids in the membrane, and (2) the underlying actin cytoskeleton and associated actin binding proteins (ABPs), results in the formation of plasma membrane domains. We theorize that it is membrane-associated molecules which signal to induce local actin organization, and that these new actin platforms immediately adjacent to the membrane modulate the clustering of membrane proteins and/or lipids (Gudheti et al., 2013). This model does not discriminate between different hypotheses explaining the initial formation of membrane clusters (depicted in Figure 2(A)). We propose that when proteins are recruited into expanding membrane clusters they do so because of protein–protein interactions (either through other cluster proteins or by binding to actin filaments), or because actin filaments affect their mobility through steric interactions. The recruitment of new molecules into the cluster subsequent to local actin remodeling must, in some cases at least, contain a method by which this recruitment is differential, thereby sometimes favoring recruitment of “like” proteins into preexisting membrane clusters. This model is further discussed in Section 3.1.
Figure 2.
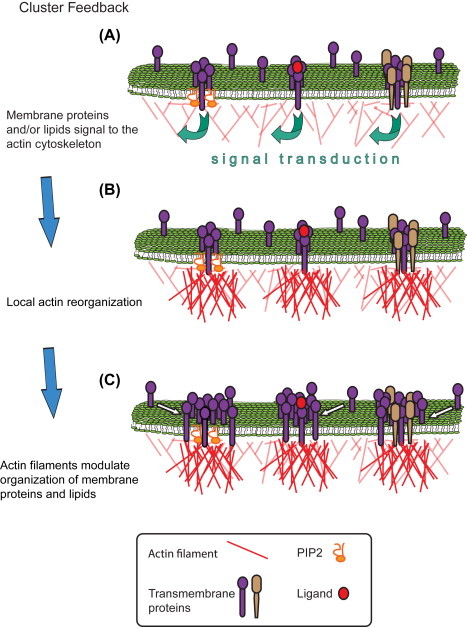
“Cluster Feedback” model of membrane organization. (A) Proteins cluster at the nanoscale in the plasma membrane. These clusters are protein–lipid (left, transmembrane protein and PIP2), mediated by ligand binding (middle), or protein–protein (right; depicted as a heterocluster; a homocluster oligomer is not shown). Of course, in clusters of any protein there exists the potential for local lipid clustering also. These protein–protein or protein–lipid clusters are collectively referred to hereafter as “membrane clusters.” In each case, signaling to the actin cytoskeleton initiated by proteins and/or lipids in membrane clusters elicits the local remodeling of actin (B), either through the recruitment and binding of actin filaments to the membrane lipids or proteins, the de novo nucleation of new filaments or branches thereof, or both. ABPs which mediate these interactions are not shown, but see Section 3.1 for more detail. The increase in actin density immediately adjacent to the membrane cluster then acts as a recruitment platform for other proteins (or lipids, not shown) diffusing in the membrane to join existing membrane clusters (C). This results in changes in the size, density, perimeter, and/or number of molecules within membrane clusters. Readers should note, the numbers of proteins depicted above are only for ease of communication—we do not hypothesize about the specific sizes (or numbers of molecules within) any of the membrane clusters here. Clusters may exist on the nano-, meso-, or micro scale; the Cluster Feedback model only predicts changes (here shown as increases) in cluster sizes from (A) to (C). See Sections 1.1 and 3.1 for more detail. (See color plate)
1.1.3. Clarifying the theories of membrane organization: the need for direct evidence
1.1.3.1. The need for improved spatial and temporal resolution
The basic goal of science is to gain knowledge through the testing of falsifiable hypotheses. In many ways and for many years, membrane research has been prevented from achieving this goal. The lack of adequate methodology has resulted in only indirect measurements being used, which has often provided insufficient evidence to discriminate between competing models. As membrane domains are widely theorized as being submicron and dynamic, their visualization with light or electron microscopy (EM) has remained indefinite. In the absence of direct images of membrane domains, the above models of membrane organization have been incompletely tested. Many experimental approaches are controversial in their ability to distinguish membrane domains. For example, cold low density detergent-insoluble fractions of lysed cell membranes have been extensively used as a determinant of lipid rafts (from (Brown & Rose, 1992) to (Recktenwald et al., 2015)), however their representation of domains in native cell membranes is tenuous and yet to be verified (Edidin, 2003, Kraft, 2013, Lichtenberg et al., 2005).
Relying on cholesterol dependence to define raft-associated processes is also problematic. The cholesterol dependence of membrane domains is often tested by perturbing cholesterol, which is known to alter phosphatidylinositol (4,5)-bisphosphate (PIP2) availability, and in turn the organization of the actin cytoskeleton (Kwik et al., 2003), as well as other fundamental cellular processes such as endocytosis (Borroni et al., 2007). Additionally, perturbing cholesterol with cyclodextrins can result in phospholipid redistribution, and these drugs can interact with membrane proteins (Zidovetzki & Levitan, 2007). Additional hypotheses of cholesterol-mediated scaffolding of signaling proteins can also explain cholesterol dependence in lipid raft-free models of protein organization (see review in (Kraft, 2013)). Furthermore, cholesterol was found not to be enriched in membrane clusters of sphingolipids in fibroblast cells (Frisz, Klitzing, et al., 2013), and even in GUVs made from ternary lipid mixtures, the concentration of cholesterol was only modestly enriched in the Lo phase compared to the Ld phase (Veatch et al., 2006). Thus, despite its involvement in an abundance of biological functions and diseases (Maxfield and Menon, 2006, Maxfield and Tabas, 2005, Michel and Bakovic, 2007, Pichler and Riezman, 2004, Rojo et al., 2006, Saini et al., 2004, Song et al., 2014, Tabas, 2002), the roles of cholesterol in membrane lateral organization require further clarification. While the importance of cholesterol and other lipids has been heavily investigated in the field of membrane organization models, clearly membrane and cytoskeletal proteins also play a crucial role in cell membrane biology.
Many unanswered questions also remain about other models of membrane organization. Some aspects of these models are expressed as being mutually compatible. For example, a recent revision of the fluid mosaic membrane model envisages that actin fences, lipid rafts, and lipid shells (among other factors) can be congruous and all contribute to the segregation of integral membrane proteins (Nicolson, 2014). However, some aspects of these models are mutually exclusive: actin filaments in the “active composite” model are proposed as being ∼250 nm long (Gowrishankar, et al., 2012), which would presumably result in asters with diameters ∼500nm, too large to be bounded by actin membrane compartments of ≤300 nm diameter proposed by the “picket fence” model (Kusumi et al., 2012). Moreover, some tenets of these models are directly competing. For example, the “lipid shell” model hypothesizes that GPI-anchored protein clustering results from Lo phase coalescence between lipid shells and lipid domains (Anderson & Jacobson, 2002). Conversely, the “active composite” model hypothesizes that GPI-anchored protein clustering results from these proteins advecting with the movement of actin filaments toward the centers of actin asters (Gowrishankar et al., 2012). These are two very different mechanisms, each proposed to explain the same phenomenon, namely the clustering of GPI-anchored proteins.
Furthermore, the theoretical basis for some models also begs further elucidation. Actin monomers in treadmilling filaments move from the barbed to the pointed ends of filaments (Kirschner, 1980, Wegner, 1976). The “active composite” model (Gowrishankar et al., 2012) proposes a system of actin asters (see Figure 1(E)), formed by actin treadmilling and myosin contractility, with filament barbed ends pointing toward the centers of asters. If transmembrane proteins bound to F-actin are advected along with treadmilling actin filaments, and these filaments have their barbed ends pointing toward the center of an aster, then the motion of the transmembrane proteins will be toward the periphery of the aster, rather than toward the center (i.e., the treadmilling of actin will push the actin-bound transmembrane protein toward the actin filament pointed end, which is at the aster periphery). Presumably such movement would disperse transmembrane F-actin bound proteins, rather than clustering them.
The alignment of actin filaments through myosin contractility raises its own set of questions. Nonmuscle myosin II assembles into bipolar filaments that engage actin filaments of opposing polarity. These myosin II filaments can translocate oppositely oriented actin filaments, in an antiparallel manner, toward each other (Vicente-Manzanares, Ma, Adelstein, & Horwitz, 2009). In the case of the “active composite” model (see Section 1.1), myosin II is proposed to cross-link parallel actin filaments, and to translocate these actin filaments by walking along a pair of parallel actin filaments (Gowrishankar et al., 2012). Testing these and other hypotheses predicted by these models has been hampered by the inability of techniques used to directly image the structures in unperturbed living cell membranes.
1.1.3.2. Testing hypotheses about very small and very fast processes
Mounting evidence suggests that interactions between cell membranes and the actin cytoskeleton play a crucial role in controlling membrane organization. Studies of protein dynamics are highly informative, but considering the importance of the cytoskeleton, methods which can image multiple species (e.g., cytoskeletal and integral membrane proteins) simultaneously are highly desirable. Single particle tracking (SPT) can fulfill this role when combined with another microscopy method, or when adapted to allow multiple species to be tracked simultaneously (Weigel, Simon, Tamkun, & Krapf, 2011). This approach has already provided valuable information on the apparent restriction of FcεRI receptors by the actin cytoskeleton (Andrews et al., 2008), and this technique is a great tool for gathering precise information on dynamics of very small numbers of molecules. However, SPT tends to be used to quantify motions of individual particles, rather than specifically for imaging. The use of homo-Förster resonance energy transfer (homo-FRET) in describing cell surface distributions of proteins is also a powerful and dynamic method (Gowrishankar et al., 2012, Sharma et al., 2004), yet is limited in length scales to less than roughly twice the Förster radius (i.e., <10 nm) and to diffraction-limited scales (i.e., >200 nm), blocking direct observation of the size and shape of membrane domains.
Thus, methods of imaging membrane organization in living cells on length scales between 10 nm and the diffraction limited resolution of ∼200 nm are highly desirable. The desire to study the interactions between membrane components or between membrane and cytoskeletal components has further motivated the development of super-resolution imaging methods.
2. Methods
2.1. Principles of super-resolution microscopy
2.1.1. Diffraction limit
An optical imaging system, such as a microscope, is fundamentally limited in resolution due to the diffraction of light. The resolving power of a microscope is often given by the Rayleigh criterion,
where r0 is the minimum distance needed between two objects to be independently identifiable, λ is the wavelength of light emitted from a sample, and NA is the numerical aperture of the objective lens of the microscope. For practical purposes, the numerical aperture of a lens is limited to ∼1.45–1.65 and using the mean of the visible wavelengths of light (λ = 550 nm), the value of r0 is ∼200–230 nm. Although small compared to the size of a cell, this limit in resolution is substantial when compared to the sizes of proteins, viruses, and various intracellular processes. In order to resolve features below the diffraction limit, several super-resolution microscopy techniques have been developed which utilize fluorescent proteins and dyes.
2.1.2. Super-resolution imaging techniques
We here provide a brief overview of only some of a wide range of super-resolution techniques. For more in depth descriptions and comparisons of super-resolution methods, see (Coltharp and Xiao, 2012, Gould et al., 2012, Leung and Chou, 2011, Rossy et al., 2013, Schermelleh et al., 2010).
2.1.2.1. Near-field scanning optical microscopy
Near-field scanning optical microscopy (NSOM), first developed in the 1980s (Lewis et al., 1984, Pohl et al., 1984), uses a very small (subwavelength sized) aperture, placed very close to a fluorescently labeled sample to create a spot of illumination smaller than r0 (Betzig and Trautman, 1992, Betzig et al., 1991). This spot is then scanned across the sample and fluorescence intensity is recorded as a function of position to produce an image. Since the spot of illumination is very small, the emitted signal correspondingly represents a very small area of the sample and can be resolved much better than the diffraction limit would otherwise allow.
Lateral spatial resolutions of 20 nm and axial resolutions of 2–5 nm have been observed using NSOM (Oshikane et al., 2007). However, in order for the illumination spot to remain smaller than r0, the aperture must be placed, and remain, much closer to the sample than the wavelength of light used. The resolving power of NSOM is quickly reduced as the aperture is moved away from the sample which primarily restricts NSOM to the imaging of membranes, sectioned samples, or other flat surfaces.
2.1.2.2. Stimulated emission depletion microscopy
Stimulated emission depletion (STED) microscopy (Hell, 2007, Hell and Wichmann, 1994, Klar et al., 2000) takes advantage of a process known as stimulated emission to suppress the usual fluorescence of fluorophores in an illumination region, except for within a very small spot. Stimulated emission refers to the phenomenon whereby a molecule in an excited state can interact with an incident photon and emit a second in-phase photon of the same wavelength, deexciting the molecule in the process. In STED, the fluorescence of molecules is interrupted (outcompeted) by stimulated emission so that the emission of fluorescent molecules not at the precise center of the focal volume can be quenched and the effective focal volume size reduced to a few tens of nanometers, or even smaller, in either two or three dimensions (Hein et al., 2008, Meyer et al., 2008). STED has been used to image living systems (Westphal et al., 2008) and multiple fluorescent species (Meyer et al., 2008).
Typically, two lasers, often pulsed to achieve high intensity, are used in STED microscopes. An excitation laser, approximately Gaussian (actually an Airy pattern) in profile, serves to excite the fluorescent molecules of a sample. The second, a deexcitation laser, is shaped so that it reaches the sample with a toroidal profile and is timed to arrive within a few picoseconds of the excitation pulse. The intensity of the STED beam is increased to saturate the STED process until only a small spot in the sample is capable of fluorescing normally. This spot is then scanned across the sample and the emission recorded to generate an image with greatly increased resolution (Hell & Wichmann, 1994). STED microscopy has achieved better than 20 nm resolution (Gottfert et al., 2013).
The relatively high laser intensities used for STED may lead to issues with phototoxicity and excessive photobleaching (Eggeling, Widengren, Rigler, & Seidel, 1998). However, the use of photostable dyes or proteins along with optimization of the STED lasers (intensity, wavelength, and scan time) can mitigate the risk of phototoxic effects (Hotta et al., 2010).
2.1.2.3. Structured illumination microscopy
Structured Illumination Microscopy (SIM) (Gustafsson, 2000) uses a periodic pattern of bands to illuminate a fluorescently labeled sample. This style of illumination generates interference between the fluorescent probes in the sample and the illumination itself to generate moiré fringes. These interference fringes are of lower spatial frequency than the illumination bands and can be more easily resolved. The pattern is rotated and phase shifted to obtain additional information from the sample. From this additional information, and using knowledge of the original illumination profile, a high resolution image can be reconstructed (Gustafsson, 2000). SIM is capable of imaging living samples at high frame rates (Kner, Chhun, Griffis, Winoto, & Gustafsson, 2009).
Conventional SIM is capable of resolutions around 100 nm, but with the introduction of nonlinear components to the system, such as saturation of the fluorophores or use of photoswitchable probes, nonlinear SIM is capable of achieving resolutions <50 nm (Gustafsson, 2005). SIM may be sensitive to rapid changes in structure during imaging and sample-induced aberrations (Gustafsson, 2005). Since SIM requires several rotations and phase shifts of the illumination pattern for optimal resolution, it is best used in conjunction with photostable probes so that photobleaching can be minimized.
2.1.2.4. Localization microscopy: FPALM, PALM, and STORM
Fluorescence photoactivation localization microscopy (FPALM) (Hess, Girirajan, & Mason, 2006), photoactivated localization microscopy (PALM) (Betzig et al., 2006), and stochastic optical reconstruction microscopy (STORM) (Rust, Bates, & Zhuang, 2006) are imaging techniques that take advantage of the properties of photoactivatable or photoswitchable fluorescent probes to improve resolution. These probes, often fluorescent proteins or dyes, are characterized by their ability to exhibit changes to their fluorescent properties, such as transitions from an inactive dark state to an active fluorescent state, when illuminated with a particular wavelength of light (often in the blue or ultraviolet wavelength range). Two lasers are typically used in combination, an activation laser to convert inactive (nonfluorescent) molecules to an activated state and a readout laser to drive the activated molecules to emit light. Since the activation of the fluorescent probes is a stochastic process, with probability proportional to the activation laser intensity, using low activation laser intensity can cause a small subset of the total population of fluorescent molecules in the sample to be activated at any given time. This subset of activated molecules will fluoresce when excited with a readout laser and then photobleach or return to a dark state. With low density of active molecules, there is a high probability that each fluorescent molecule will be resolvable from the others. When this occurs, each molecule can be localized by finding the centroid of the image or by fitting the image with an appropriate function, such as a two-dimensional Gaussian. The process of activation, readout, and photobleaching of the fluorescent probes can be repeated until there are no more fluorescent molecules in the sample or until a desired number of molecules has been recorded (Betzig et al., 2006, Hess et al., 2006, Rust et al., 2006).
FPALM was the first of these techniques used to image living cells (Hess et al., 2007); both PALM and STORM share this capability (Jones et al., 2011, Shroff et al., 2008). Multiple fluorescent species can also be imaged (Bates et al., 2007, Bossi et al., 2008, Curthoys et al., 2013, Gunewardene et al., 2011, Shroff et al., 2007). Furthermore, these types of localization microscopy can be used to produce images in three dimensions by introducing z-dependent elements to alter the detection point spread function (PSF). This z dependency can be added in a number of ways, such as by using a cylindrical lens to induce astigmatism (Huang, Wang, Bates, & Zhuang, 2008), by simultaneous bi-plane detection (Juette et al., 2008), or by introducing strong axial dependence of the PSF using a spatial light modulator (Jia et al., 2014, Pavani et al., 2009) or 4Pi detection geometry (Shtengel et al., 2009).
The resolution of FPALM, PALM, and STORM depends largely on the number of photons detected per molecule, the background noise per pixel, the camera pixel size, and the total number of molecules localized. Using bright fluorescent probes, localization precisions <15 nm are possible in all three spatial dimensions (Jia et al., 2014).
Although these localization methods all rely on the same basic principle to achieve high resolutions, there are some subtle differences between methods. FPALM and PALM initially used fluorescent proteins (Betzig et al., 2006, Hess et al., 2006) while STORM initially used antibodies conjugated to organic dyes to label structures of interest (Rust et al., 2006). The intracellular introduction of antibodies generally requires cell membranes to be permeabilized in some manner, a consideration when STORM is used for live cell studies. However, organic dyes have the advantage of being relatively bright and photostable, especially in the presence of an oxygen scavenging system. This allows for resampling of the same fluorophore several times before photobleaching. While this increases sampling density, and potentially resolution, this resampling can result in overcounting of the number of localized molecules in the sample. Fluorescent proteins, by comparison, sometimes emit fewer photons before photobleaching. Such characteristics reduce the rates of overcounting, but fewer detected photons per molecule can also limit localization precision and, in the presence of background, the number of molecules identified. However, fluorescent proteins are expressed by the cell, eliminating the need to introduce large antibodies through the cell membrane, and they also do not require use of reducing agents in the imaging buffer. Since these localization methods rely on constructing an image from many localized point sources, labeling density and specific probe photophysics tend to be the most limiting factors.
2.1.2.5. Super-resolution optical fluctuation imaging
Super-resolution optical fluctuation imaging (SOFI) is a software-based postprocessing technique that exploits random fluctuations of fluorescence intensities of fluorophores in order to enhance resolution and reduce background in experimental image data (Dertinger, Colyer, Iyer, Weiss, & Enderlein, 2009). SOFI can be applied to many fluorescence imaging modalities provided that the fluorescent label used has two or more distinguishable states (for example, a bright and dark state), and that each molecule switches stochastically and independently of the others. To achieve enhanced resolution, a movie of a fluorescently labeled sample is acquired, and an n-th order autocumulant is calculated, effectively filtering the acquisition such that only highly correlated fluctuations remain. This results in an image with greatly reduced background and a resolution that is the square root of 2 better than the diffraction limited case. Alternatively, cross-cumulants may be used instead for even larger increases in resolution (Dertinger, Colyer, Vogel, Enderlein, & Weiss, 2010). SOFI can be used in 3D imaging applications (Dertinger, Xu, Naini, Vogel, & Weiss, 2012), with living samples (Geissbuehler et al., 2014), and has been demonstrated with organic dyes (Dertinger, Heilemann, Vogel, Sauer, & Weiss, 2010).
Sophisticated postprocessing is required to provide resolution enhancement and improvement is limited without use of high-order cumulants, which require long acquisition times. However, SOFI can be performed with relatively short acquisition times and moderate laser intensities to obtain modest resolution improvement and has the advantage of being compatible with conventional fluorophores and microscopes (Dertinger, Colyer, et al., 2012).
2.1.2.6. Universal point accumulation for imaging in nanoscale topography
Universal point accumulation for imaging in nanoscale topography (uPAINT), a generalization of PAINT (Sharonov & Hochstrasser, 2006), makes use of continuous labeling while imaging to localize and track single molecules in living samples (Giannone et al., 2010). In uPAINT, highly specific fluorescent ligands are added to the sample via solution just before imaging. The ligands bind to target proteins of interest and, with an appropriately chosen concentration, are sparse enough to be localized. Excited fluorophores will eventually photobleach but are stochastically replenished by ligands from solution. An oblique laser illumination profile is used to excite the fluorophores in order to limit the focal volume, reduce background, and prevent bleaching of out of focus molecules. uPAINT is well suited to tracking single molecules for many time steps, and multiple dye molecules can be bound to a single ligand to further increase tracking duration (up to tens of seconds). uPAINT has been used to examine up to two species simultaneously (Winckler et al., 2013). Typical localization precisions are within the range of 40–50 nm (Giannone et al., 2010). Since the fluorescent labels are added exogenously to the sample, careful optimization of ligand concentration, laser intensity, and imaging rate is essential.
2.1.2.7. Super-resolution: worth the Nobel Prize
The development of these methods has fundamentally changed the capabilities of scientific researchers, and is already leading to crucial insights in a number of fields including biomedical research, and specifically cell membrane biology. Stefan W. Hell, Eric Betzig, and W.E. Moerner were awarded the Nobel Prize in Chemistry in 2014 for their contributions toward the development of super-resolution microscopy.
2.2. Tools for quantification of membrane organization and dynamics
To be able to distinguish and refine the existing models of membrane organization, quantification of the fundamentals of membrane biology are necessary. These fundamentals relate directly to the interactions between membrane components, such as proteins and lipids. While studies of lipid phase separation can provide some insight into the behavior of lipids in the absence of proteins, methods which quantify protein–protein, lipid–lipid, and lipid–protein interactions in the (preferably living) cellular environment are essential to advancing the understanding of membrane biology (Bar-On et al., 2012, Gudheti et al., 2013, Sengupta et al., 2011). Several methods (see Section 2.1) can image the spatial distributions and time-dependence of (labeled) proteins and lipids in intact cell membranes. However, imaging is really just the first step toward understanding the processes which have led to these distributions. Quantification of the distributions of membrane components is often the next step.
2.2.1. Ripley's K and the pair correlation function
Several statistical techniques are commonly used to identify and quantify clustering and codistribution of labeled proteins and lipids. Ripley's K-function and the pair correlation function (PCF) test spatial heterogeneity and clustering of objects (Gudheti et al., 2013, Hess et al., 2005, Philimonenko et al., 2000, Plowman et al., 2005, Ripley, 1977, Yeomans, 2002). The K-function quantifies the number of objects (e.g., imaged proteins or lipids) of a given type found within a circle of specified radius from each other object of the same type (or a different type for co-distribution measurements) and normalizes the amplitude by the overall density such that an amplitude of zero corresponds to a random distribution (Ripley, 1977). The PCF quantifies the number of probes at a given distance from each other molecule, normalizes by the area of each ring and the average molecular density, and averages over all molecules, such that an amplitude of 1 corresponds to random. Typically, these tests are performed over a range of radii in order to determine whether there is any clustering of the probes and if so, to elucidate properties such as mean cluster radii or mean cluster density (Gudheti et al., 2013, Hess et al., 2005, Philimonenko et al., 2000, Plowman et al., 2005). There are other tests to measure spatial heterogeneity such as the Getis G statistic (Getis & Ord, 1992), which includes different normalizations and weighing parameters dependent on molecular spatial position. These differences result in increased sensitivities to certain cluster parameters (Itano et al., 2014).
The Ripley's cross-K-function (cross-K) and pair cross-correlation function (PCCF) quantify the spatial relationship between two differently labeled species in a sample. In principle, the algorithms work similarly to the K-function and PCF, by counting the measured numbers of probes of one species at a distance r from each molecule of the other species, then averaging over all molecules. These tests quantify the degree to which and the range of distances over which two labeled species colocalize (Gudheti et al., 2013, Philimonenko et al., 2000).
2.2.2. Nearest neighbor-based cluster analysis
Clustering can also be defined by nearest neighbor distance. Single-linkage cluster analysis (Sneath, 1957) defines all molecules within a user-defined distance of any other molecule in the set to be within the same cluster. This type of algorithm is useful because it precisely defines the set of molecules within each cluster, and thus the localized coordinates of those molecules can be quantified directly (Bar-On et al., 2012, Gudheti et al., 2013). The nearest neighbor distance distribution can also be used to determine the local density of the clustered probes. This information allows for comparison of cells that have been treated to disrupt or enhance clustering (Hess et al., 2005) or for comparison of nonclustered probes in different regions of the plasma membrane (Stryer, 1978, Zacharias et al., 2002). Additionally, an angular histogram (the angle between a molecule and its two nearest neighbors) reveals any preferential orientation of probes within the cluster. Orientational order can result from hexatic phases or interactions with other cellular components which may induce ordering of the proteins, such as cytoskeletal components (Hess et al., 2005).
2.2.3. Quantifying dynamics
Fluorescence microscopy techniques are capable of quantifying the dynamics of fluorescently labeled species in live cells. Tracking of single molecule trajectories over two or more consecutive frames enables calculation of molecular mobility (Manley et al., 2008). These high-density maps of short molecular trajectories provide both spatial and dynamic information, and in many ways are a high-density limit of SPT, where low densities of labeled molecules are typically used. Likewise, as single-molecule localization methods have advanced, computational methods have developed which enable larger numbers of SPT molecules in close proximity to be concurrently analyzed (Jaqaman et al., 2008). Longer molecular trajectories obtained by localization microscopy or SPT allow distinction between confined diffusion, free diffusion, active transport, and other types of motion.
Particle image correlation spectroscopy is an excellent way to quantify large numbers of (even relatively short) single molecule trajectories to test the type(s) of motion occurring in a system (Semrau & Schmidt, 2007). In addition, histograms of the turn angle (the vertex between the positions of a single molecule through three time points) of moving single molecules can reveal whether molecules have a preferred turning angle. Molecules undergoing confined diffusion (or other situations which lead to reversal of motion) are more likely to have turn angles near 180°, while freely diffusing molecules have no preferred turn angle (Gudheti et al., 2013, Hess et al., 2005).
3. Biological Applications
3.1. Membrane–actin interactions
The actin cytoskeleton is now widely recognized as being key in regulating the spatial distribution of some membrane proteins (Chichili and Rodgers, 2009, Plowman et al., 2005) and membrane lipids (Frisz et al., 2013, Head et al., 2014, Liu and Fletcher, 2006). In return, membrane proteins and lipids can locally remodel (or create new) actin filaments (see review by (Saarikangas, Zhao, & Lappalainen, 2010)). While the precise mechanisms are still under debate, instrumental in many actin-membrane interactions are actin binding proteins (ABPs), which can variously connect actin filaments with membrane proteins or the lipid membrane itself, and modulate the local nucleation, branching, cross-linking, and dissociation of actin filaments. Due to constraints of space we here discuss a small subset of these ABPs.
3.1.1. Molecular participants
3.1.1.1. ABPs: the membrane—actin connection
Long since theorized as contributing to lateral heterogeneity in plasma membranes (Edwards & Crumpton, 1991), the annexins can bind negatively charged phospholipids (Edwards and Crumpton, 1991, Jackle et al., 1994), many membrane and signaling proteins (Cornely, Rentero, Enrich, Grewal, & Gaus, 2011), and through an F-actin binding domain (Hosoya, Kobayashi, Tsukita, & Matsumura, 1992) may act as a nexus between the actin cytoskeleton and the plasma membrane (Hayes, Shao, Grieve, Levine, Bailly, & Moss, 2009). Proteins of the ezrin/radixin/moesin (ERM) family (also including ABPs with homology in their 4.1 ezrin/radixin/moesin (FERM) domains, such as talin (see review in (Critchley, 2005)) can bind F-actin, phospholipids, and a range of transmembrane proteins (including those believed to corral membrane proteins into domains such as CD317/tetherin (Rollason, Korolchuk, Hamilton, Jepson, & Banting, 2009)), and membrane-associated cytosolic proteins (see review in (Fehon, McClatchey, & Bretscher, 2010)). Some ERM proteins can also signal to other ABPs from the plasma membrane, e.g., ezrin to myosin (see review in (Manes & Viola, 2006)) to effect structural change.
3.1.1.2. Ezrin tethers, filamin traps
Viola and Gupta (2007) theorized that resting immune cell membranes contain small membrane protein clusters linked to cortical actin by ezrin. Antigen stimulation disconnects ezrin from these clusters and from actin (Gupta et al., 2006), and the theory holds that small protein clusters uncoupled from the actin cytoskeleton coalesce into large membrane domains which are then “tethered and trapped,” via rebinding to the actin cytoskeleton through the now-activated ABP filamin (Viola & Gupta, 2007). Indeed, the filamin isoform FLNa is required for actin-mediated stabilization of membrane domains at the immunological synapse (Tavano et al., 2006), and for the internalization of caveolae (Muriel et al., 2011, Sverdlov et al., 2009).
3.1.1.3. Spectrin, α-actinin, myosins (and a supervillin)
Also implicated in actin rearrangement at caveolae is the actin cross-linker α-actinin (Singleton, Dudek, Chiang, & Garcia, 2005), which is commonly isolated in detergent-resistant membranes (see review in (Chichili & Rodgers, 2009)). Among the first ABPs recognized in regulating membrane organization, spectrin family members (including α-actinin, and nonerythroid spectrin or fodrin) are able to bind F-actin and phospholipids (Davis and Bennett, 1994, Hartwig and DeSisto, 1991) and can signal to reorganize actin through various pathways (Machnicka, Czogalla, Hryniewicz-Jankowska, Boguslawska, Grochowalska, Heger, & Sikorski, 2014).
The membrane proximal actin cortex is commonly understood as being populated with myosin motors (Salbreux, Charras, & Paluch, 2012), and different myosin isoforms have been identified in DRMs (Nebl et al., 2002). Myosin II contractility may be needed for the coalescence of lipid domains along F-actin bridges (Jordan and Rodgers, 2003, Rodgers et al., 2005) and Myo1c is needed for the delivery and recycling of membrane domain-associated lipids (Brandstaetter, Kendrick-Jones, & Buss, 2012). Further connection between myosins and membrane domains may occur through binding of membrane-associated ABPs such as supervillin (Chen et al., 2003).
3.1.1.4. Arp2/3: pushing membranes around
One class of ABPs alters actin organization by initiating the formation of new actin filaments. These “nucleators” include actin-related protein 2/3 (Arp(2/3)) protein complexes, which create branched actin filament networks, and can be ultimately regulated by cooperative mechanisms including interactions with phospholipids (Derivery and Gautreau, 2010, Lebensohn and Kirschner, 2009). The proximity of these branched networks to cell membranes is central to their involvement in contexts of membrane movement—both at the whole cell level (underpinning the lamellipodial morphology (Stradal et al., 2004)) and at the level of the vesicle (aiding in endocytosis (Kaksonen, Toret, & Drubin, 2006)).
3.1.1.5. PIP2: the powerhouse
Actin filaments can be bound to membrane proteins and lipids through ABP conduits, but they can also influence the diffusion and distribution of membrane molecules through steric interactions (reviewed in (Alenghat & Golan, 2013)). Moreover, the nexus between membrane proteins and ABPs need not be direct for each to alter the spatial localization of the other. Beyond the scope of this chapter is a plethora of signaling pathways which regulate the activation of ABPs and their sorting to membrane domains, such as the Rho family GTPases (e.g., see reviews by (Bisi et al., 2013, de Curtis and Meldolesi, 2012)) and the family of Bin–amphipysin–Rvs167 (BAR) domain containing proteins (e.g., see reviews by (Aspenstrom, 2014, Frost et al., 2009)).
We instead briefly discuss some regulatory influences of the anionic membrane phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP2). By binding to PIP2, ABPs can be targeted to membranes (e.g., spectrin; see review in (Boguslawska, Machnicka, Hryniewicz-Jankowska, & Czogalla, 2014), ezrin/radixin/moesin proteins and talin (Barret et al., 2000, Hao et al., 2009, Jayasundar et al., 2012, Saltel et al., 2009) and annexins (Hayes et al., 2009, Martin-Belmonte and Mostov, 2007)). PIP2 binding is also considered a necessary step in the activation of many ABPs (Wu et al., 2014), such as ezrin/radixin/moesin proteins (Yonemura, Matsui, & Tsukita, 2002), and Arp2/3 (via WASP) (Higgs & Pollard, 2000). However, PIP2 binding may also inhibit the cross-linking of actin by filamin (Furuhashi, Inagaki, Hatano, Fukami, & Takenawa, 1992), the bundling of actin by α-actinin (Fraley et al., 2003) and the severing of actin by cofilin (van Rheenen et al., 2007). Additionally, PIP2 may be in return regulated by ABPs (e.g., talin (Di Paolo et al., 2002)). PIP2 also regulates signaling cascades upstream of ABPs and actin remodeling, and helps coordinate actin organization in a variety of contexts (see reviews by (Rocha-Perugini et al., 2014, Saarikangas et al., 2010, Sun et al., 2013)). See Section 3.4.2 for further discussion of PIP2 imaging.
We have, here, only described a small fraction of the myriad ways in which actin is bound to, and can be regulated by, membrane components. In all likelihood, there are a gross number of complicated yet precise methods by which cells can regulate how membrane proteins and lipids move and are distributed. However, some mechanisms appear crucial across multiple systems. By understanding the spatial and functional relationships between ABPs, actin, and the membrane, we can better understand exactly how cell systems are able to coordinate a staggering array of varied and distinct molecular processes at the membrane. Before assessing the future opportunities in this field, we first summarize the techniques which have led us to this point.
3.1.2. Early findings
Aside from the identification of ABPs in fractions of lysed cells (such as DRMs) to indicate their involvement in membrane domains (Jordan and Rodgers, 2003, Nebl et al., 2002, Singleton et al., 2005), classic techniques including coimmunoprecipitation and yeast two hybrid screens are used to gauge the interactions between ABPs and membrane proteins (e.g., (Sverdlov et al., 2009, Tavano et al., 2006)). Biochemical techniques such as these are useful for exploring binding relationships, but do not describe distributions of proteins (or lipids).
Diffraction-limited microscopy has been used to image membrane domains such as the immunological synapse (Tavano et al., 2006) and invadopodia (Antelmi et al., 2013), and specific inhibition of ABPs followed by widefield imaging has elucidated the importance of, for example, myosin motors in the movement of large (micrometers in length) domains into larger immunological synapses (Jordan & Rodgers, 2003).
3.1.2.1. FRET, FRAP, and FCS
Techniques such as FRAP have been extensively used to investigate the dynamics of the actin cortex (e.g., (Sund & Axelrod, 2000)), ABPs (e.g., ezrin (Coscoy et al., 2002)) or proteins moving through the plasma membrane (e.g., (Golan & Veatch, 1980)). Kenworthy et al. used FRAP very effectively to distinguish between membrane models with and without cytoskeletal interactions (Kenworthy et al., 2004), and discovered that the slow diffusion of the cholera toxin subunit B (CTxB) is due to confinement by the actin cytoskeleton (Day & Kenworthy, 2012). FRAP and SPT have been used together to identify anomalous diffusion of clustered membrane receptors, suggesting interactions with the cytoskeleton (Feder, Brust-Mascher, Slattery, Baird, & Webb, 1996). FRET has been used to investigate the coclustering of putative raft and nonraft markers (Chichili & Rodgers, 2007) and Goswami et al. were able to quantify an association between GPI-anchored protein clustering and cortical actin remodeling using homo-FRET (Goswami et al., 2008). However, neither FRET nor FRAP visualize (subdiffraction sized) cluster geometry directly, so results must be interpreted in terms of some kind of model, and FRET microscopy does not directly access clustering on length scales between ∼10 and 250 nm.
Fluorescence correlation spectroscopy (FCS) (Magde, Elson, & Webb, 1972) is a powerful method able to provide information on microsecond and millisecond molecular dynamics within a (typically) diffraction-limited observation volume of ∼0.3–1.0 μm in size (Hess & Webb, 2002). FCS uses the fluctuations in a fluorescence signal to measure molecular concentration, diffusion coefficient or transport rate, and molecular transitions between bright and dark states (Hess, Huang, Heikal, & Webb, 2002). FCS of lipid probes in cell membranes demonstrated anomalous diffusion (Schwille, Korlach, & Webb, 1999) consistent with cytoskeletal interactions or nanodomains. FCS measurements of diffusion as a function of observation volume size (Wawrezinieck, Rigneault, Marguet, & Lenne, 2005) have been very effectively used to quantify GPI-anchored protein clustering, and results also suggest that cytoskeletal interactions mediate membrane lateral organization (Lenne et al., 2006). Lasserre et al. used similar FCS measurements to quantify protein nanodomains related to activation of signaling pathways related to Akt and phosphoinositide-3 kinase, which can regulate the actin cytoskeleton (Lasserre et al., 2008). A super-resolution version of FCS which uses STED to confine the observation volume (Eggeling et al., 2009) has demonstrated that sphingolipids and GPI-anchored proteins are transiently trapped in small cholesterol-dependent complexes of <20 nm in size, while phosphoglycerolipids are not.
Conventional versions of FCS, FRAP, and FRET are, however, limited in their ability to resolve lateral heterogeneity on length scales <200 nm: FCS does not form an image at all, FRET cannot access length scales between ∼10 and 200 nm, and conventional FRAP does not image structures below the diffraction limit.
Widefield TIRF microscopy has been used to study the colocalization of various proteins with markers of caveolae (e.g., tagged filamin and caveolin-1 (Muriel et al., 2011)), and ABPs have been tracked with respect to endosomal markers and other ABPs (e.g., tagged supervillin and tagged myosin (Fang et al., 2010)). While useful in their indication of gross colocalization, direct visualization of the spatial and dynamic distributions at the nanoscale would aid in understanding how distributions of membrane-associated ABPs are spatially and functionally related to the dynamics and clustering of membrane proteins and lipids.
3.1.3. Findings using super-resolution methods
3.1.3.1. Perturbing ABP expression and watching what happens
Many groups use super-resolution microscopy to report the spatial distributions of membrane proteins and how these change with actin disruption or cholesterol depletion. Recently, membrane protein distributions have also been imaged as a function of ABP disruption, indicating the reorganization of actin in NK cells lacking the actin depolymerizing protein coronin (Mace & Orange, 2014), and alterations in the distributions of putative “raft” and “nonraft” markers in a cell-dependent manner with varying annexin (anxA6) expression levels (Alvarez-Guaita et al., 2014). Moreover, ABPs can affect motility of membrane proteins; STORM imaging has shown that the mobility of single BCRs and entire assemblies is decreased in cells lacking ezrin (Pore et al., 2013).
Combined super-resolution imaging of the ABPs themselves together with conventional transmitted light (or differential interference contrast) imaging can show the nanoscale localization of these proteins relative to diffraction limited images of membranes and other cellular structures. For example, this has been explored for spectrin (Blunk et al., 2014, Zhong et al., 2014) and α-actinin (Hou et al., 2014), just as single-species imaging of actin can inform as to nanoscale actin distributions with respect to a larger structure or a whole cell (Izeddin et al., 2011, Xu et al., 2012).
3.1.3.2. Super-resolution multicolor imaging: picturing combinations of molecules
With the advent of multicolor localization microscopy, studies directly and simultaneously imaging nanoscale actin distributions and membrane proteins, or membrane proteins and membrane-associated ABP distributions, in fixed cells, are starting to emerge. As examples, actin and lytic granules (see also Section 3.4) have been imaged together with dual-color STED in cells lacking an isoform of the ABP coronin, fuelling a novel model of granule secretion (Mace & Orange, 2014). Dual color FPALM has shown that clusters of influenza hemagglutinin (HA) can either be colocalized with, or completely exclude, the ABP cofilin (Gudheti et al., 2013). Sequential dual color PALM/dSTORM imaging has revealed a tight spatial association between the post synaptic density and Arp2/3, which is functionally reliant on the Rho GTPase Rac (Chazeau et al., 2014). Dual-color PALM has shown that actin and GPI-anchored proteins (which have extensively been used to demonstrate aspects of various models of membrane organization (see Section 1.1)), cluster on the nanoscale without appreciable colocalization, until antibody cross-linking stimulates formation of GPI-anchored protein clusters many microns in size which do associate with actin (Sengupta et al., 2011). Dual-color STED imaging of PC12 membranes has shown that mesoscale membrane clusters contain many species of protein which are further spatially organized within a single cluster, and these clusters exclude, and are strikingly bordered by, actin and spectrin (Saka et al., 2014).
3.1.3.3. Super-resolution multicolor live cell imaging: watching the dance
Live cell multispecies imaging is also now allowing extraordinary views of the dynamic interplay between membrane components and the actin cytoskeleton. As examples, FPALM has already elucidated that clusters of the influenza protein HA are associated spatially and dynamically with clusters of actin (i.e., not contained in an actin poor compartment and bounded by a fence, but rather shuffling on top of a thick actin hedge) (Gudheti et al., 2013). Exciting avenues for testing between models of membrane organization are now being realized with the ability of super-resolution microscopy to visualize the dynamics of three protein species simultaneously, in living cells, at the nanoscale. Three-color live cell FPALM experiments have already shown that HA and transferrin receptors (previously considered putative “raft” and “nonraft” markers, respectively) each form spatially segregated nano- and microscale clusters, yet both species spatially and dynamically correlate with actin clustering (Gunewardene et al., 2011).
3.1.4. Proposed model: Cluster feedback
3.1.4.1. The cluster feedback model: two-way communication between actin and the membrane
The insights into membrane organization afforded by super-resolution microscopy, along with observations from decades of membrane research provide the basis for our cluster feedback model (see Section 1.1), whereby the relationship between (1) membrane proteins and lipids (including PIP2), and (2) actin and ABPs is bidirectional. The differential regulation of ABPs by membrane-associated molecules can induce the local formation and reorganization of actin filaments. These actin filaments can then work to locally alter the distribution and diffusion of membrane molecules, creating clusters of proteins and lipids in membrane domains. This model builds on ideas published in a range of membrane protein clustering contexts (Chichili and Rodgers, 2009, Gowrishankar et al., 2012, Gudheti et al., 2013, Jaumouille et al., 2014, Viola and Gupta, 2007, van Zanten et al., 2009). Experimental evidence for this model has come from observations that (1) increasing actin stability can increase coclustering of membrane proteins (e.g., (Chichili and Rodgers, 2007, Gudheti et al., 2013)), (2) ABPs are differentially localized with respect to clusters of membrane proteins (Chazeau et al., 2014, Gudheti et al., 2013, Saka et al., 2014), and (3) altering ABP expression can be sufficient to modulate membrane protein mobility (Pore et al., 2013) and clustering (e.g., (Alvarez-Guaita et al., 2014, Chazeau et al., 2014). Feedback has been demonstrated by, for example, work showing that Fcγ receptor activation can influence actin polymerization, which in turn affects Fcγ receptor mobility (Jaumouille et al., 2014), and in the T-cell activated remodeling of F-actin, which in turn mediates the organization of plasma-membrane bound signaling proteins (see review in (Kumari, Curado, Mayya, & Dustin, 2014)). Upstream regulators of ABPs have also been measured with respect to membrane protein organization, such as the elegant dual color NSOM study demonstrating that ligand binding of integrins induces clustering of, and colocalization with, GPI-anchored proteins (van Zanten et al., 2009). We hope to see more studies using super-resolution microscopy to directly image actin, membrane proteins, and the ABPs themselves, so that we may further test hypotheses of membrane protein—actin/ABP feedback predicted by the cluster feedback model.
3.1.4.2. Outlook
While various studies highlight the importance of ABPs in regulating protein distributions at the cell membrane, many ABPs can elicit change through multiple pathways. Similarly, actin can contribute to many processes which alter spatial organization of membrane proteins (e.g., endo- and exocytosis). Single species imaging of membrane proteins with or without ABPS (or actin disrupting drugs) can test for effects on membrane protein distribution, but cannot easily determine how. Better understanding of mechanisms can come from simultaneous imaging of the membrane protein and the ABP (and/or actin) being investigated. Research into other membrane platforms, such as focal adhesions, has been aided by super-resolution microscopy elucidating (for example) movement of single talin molecules with respect to β-integrins (Rossier et al., 2012) and the organization in three dimensions of multiple ABPs with respect to the membrane (Kanchanawong et al., 2010).
Despite the benefits that super-resolution microscopy has afforded membrane research, many competing models of how ABPs and actin influence distributions of membrane proteins and lipids are yet to be tested. The development of photoactivatable lipid tags (e.g. Mizuno et al., 2011) has already allowed exciting super-resolution imaging of membrane lipids (Abe et al., 2012, Honigmann et al., 2013), yet many opportunities for multichannel lipid/actin/membrane imaging remain relatively unexplored. Studies which visualize and analyze the three-dimensional organization of the membrane and actin cytoskeleton will continue to be extremely fruitful. We are now in a golden age of microscopy, where dreams of visualizing the precise dynamic steps of cellular function, including the previously enigmatic process of organizing membrane domains and the associated actin cytoskeleton, are now becoming reality. Studies combining super-resolution techniques will allow us to finally see, in three dimensions and real time, how many different players dance together to organize cellular membranes.
3.2. Virus infection
Viruses are responsible worldwide for significant illness across many species, and are able to induce changes in the organization of the plasma membrane to facilitate infection, replication, budding, release, and evasion of the host immune system (Manes, del Real, & Martinez, 2003). Exceptionally high resolution views of virus infection have been obtained with EM. For example, images of whole influenza virus (Ruigrok, Krijgsman, de Ronde-Verloop, & de Jong, 1985) and in particular the influenza fusion protein hemagglutinin (HA) (Hess et al., 2005) have been obtained and quantified by EM. While not yet rivaling the resolution obtained by EM, super-resolution microscopy is far better suited for imaging living, dynamic systems. Coupled with advances in temporal resolution (Huang et al., 2013, Nelson et al., 2014), super-resolution imaging gives investigators the tools capable of answering more questions about pathogen interactions with host cell membranes. Because this is a highly researched field, were here limit discussion to a small subset of studies only.
3.2.1. Super-resolution microscopy: viruses meet their match
Even though direct optical imaging of membrane domains with diffraction limited techniques is not always possible, many properties of viral assembly and function can be uncovered through indirect imaging methods. FRET microscopy has reported the association between viral proteins and putative lipid raft markers (Engel et al., 2010), and FRET has been quite useful in quantifying clustering of many membrane proteins on length scales <10 nm (Karpova et al., 2003, Kenworthy, 2001, Kenworthy, 2005, Kenworthy and Edidin, 1998, Kenworthy et al., 2000). FRAP (Elson, Schlessinger, Koppel, Axelrod, & Webb, 1976) has yielded insight into mechanisms of diffusion of viral membrane proteins at the cell membrane (Kenworthy et al., 2004), and nuclear magnetic resonance (NMR) has revealed how lipid phase changes as a function of temperature help protect viral stability (Polozov, Bezrukov, Gawrisch, & Zimmerberg, 2008). However, FRET is insensitive to length scales from ∼10 to 200 nm, NMR does not provide an image, and FRAP does not give direct information about spatial organization below the diffraction limit. Using super-resolution imaging, investigators have been able to directly quantify shapes, sizes, and densities of membrane protein distributions, as well as the degree of spatial overlap between different species of proteins at the nanoscale (Gould et al., 2008, Gudheti et al., 2013, Gunewardene et al., 2011, Hess et al., 2007, Shroff et al., 2007). These capabilities have allowed researchers to answer previously inaccessible questions about viral assembly, viral protein trafficking, and viral interactions with host cell components, which help build our understanding of the infection process, and have the potential to reveal new anti-viral drug targets.
3.2.2. Influenza virus hemagglutinin: the versatile membrane protein hijacking your cells
The influenza virus is responsible for tens of thousands of deaths annually. Influenza can use host cell proteins to aid in infection, and mass spectrometry has shown that a number of host cell proteins are also preferentially incorporated into influenza virus released from infected cells (Shaw, Stone, Colangelo, Gulcicek, & Palese, 2008), leading to the question of how these associations occur. Some answers may be found with the influenza membrane protein hemagglutinin (HA), which is crucial in many steps of viral infection. HA binds sialic-acid containing cell surface receptors (Skehel & Wiley, 2000); HA catalyzes membrane fusion necessary for viral entry (Biswas et al., 2008, Chernomordik et al., 1998, Skehel and Wiley, 2000, Skehel and Wiley, 2002, Wiley and Skehel, 1987); and clustering of HA in the viral membrane is crucial for fusion to be accomplished (Bentz, 2000, Chernomordik et al., 1998, Chernomordik et al., 1997, Kozlov and Chernomordik, 1998, Kumar et al., 2001, Takeda et al., 2003). HA assembles with other viral components before budding (Nayak, Hui, & Barman, 2004), and in the late 1990s, biochemical experiments were able to show that the influenza virus buds from areas of the host membrane where viral components including HA and certain cell lipids are concentrated (Scheiffele et al., 1999, Scheiffele et al., 1997). HA dynamics have been investigated with SPT, which helped elucidate the HA-dependent mechanism by which viral RNA traverses the nuclear envelope (Babcock et al., 2004, Lakadamyali et al., 2003), and FRAP experiments measured the diffusion coefficient of HA ∼0.1 μm2/s, and suggested an immobile fraction (∼25%) of HA (Kenworthy et al., 2004). While these experiments have greatly helped shape our understanding of influenza infection, they have not fully clarified the nanoscale organization of viral and host cell components during infection.
Recently, super-resolution experiments have made substantial advances in the understanding of several aspects of the influenza virus life cycle. Super-resolution microscopy has also been used to determine the spatial distribution of the host cell protein CD81 (tetraspanin), which is recruited to assembling influenza viruses, and is concentrated at the growing tip and budding neck of progeny viruses (He et al., 2013). CD81 can control the progression of membrane protein distributions in, for example, immunological synapse formation (Rocha-Perugini et al., 2013), and form complexes with a number of signaling proteins and other master regulators such as the integrins (Berditchevski, 2001). Tetraspanin redistribution by influenza may be one method by which the virus is able to reorganize other host cell membrane proteins on the surface of budding virions.
3.2.3. Influenza hemagglutinin and host cell actin: an unhealthy relationship?
One other host cell protein which is greatly exploited by influenza, and viruses in general, is actin (Radtke, Dohner, & Sodeik, 2006). While it was shown that HA clusters (which are necessary precursors of viral budding (Scheiffele et al., 1997)) can persist over timescales of at least tens of seconds (Hess et al., 2007), high-speed FPALM showed that fluctuations in area, perimeter, and shape of these clusters can occur on timescales as short as 0.1 s (Nelson et al., 2014) leading to the question of how clusters are able to persist. Multicolor FPALM imaging in live cells has shown that HA mobility decreases with increased cortical actin density. Along with the discovery of two distinct populations of HA, one with low (non-zero) mobility and confined motion on 100–200 nm length scales (Gudheti et al., 2013), these findings suggest that local actin is influencing the dynamics of this membrane protein. Colocalization of HA clusters with actin clusters and the increase in HA cluster size upon treatment with actin-stabilizing jasplakinolide treatment do not seem consistent with a picket-fence description (Gudheti et al., 2013). Moreover, the intriguing nanoscale differential anti- and colocalization of HA and ABPs, including cofilin (which is strikingly excluded from some HA clusters, yet strongly colocalizes with others), suggests the relationship is more involved than HA molecules simply being confined between actin fences (Gudheti et al., 2013). Rather, these insights made possible by super-resolution microscopy suggest a dynamic “cluster feedback” between membrane HA and the underlying actin cytoskeleton (see Section 1.1 and Figure 2). Understanding these HA organizing mechanisms could be vital in identifying novel antiviral drug treatments, and understanding the HA/actin/ABP interplay may illuminate cellular processes which are used to organize the distributions of many other membrane proteins.
3.2.4. Role of Gag in HIV life cycle
The U.S. Centers for Disease Control (CDC) estimates that the Human Immunodeficiency Virus (HIV) infects tens of thousands of people in the United States each year. Understanding of the molecular dynamics involved in HIV infection and replication is critical for developing future medical treatments for infected individuals, as well as for preventing infection. Formation of the HIV immature capsid (and in turn budding, release, and maturation of the virus) depends on formation of a polyprotein assembly of the HIV protein group-specific antigen (Gag) (Lingappa, Reed, Tanaka, Chutiraka, & Robinson, 2014). Live cell sptPALM was used to compare the dynamic behaviors of Gag and the vesicular stomatitis virus (VSV) G protein (Manley et al., 2008). This allowed researchers to build dense “trajectory maps” of many different proteins to help understand the dynamic behavior of individual proteins, and entire populations and assemblies, in the plasma membrane. While the distributions and mobility of Gag and VSV-G proteins differed greatly, they were both found to be consistent with results obtained through diffraction-limited techniques (Jouvenet et al., 2006, Kenworthy et al., 2004). Using STORM, researchers revealed that Gag recruits and corrals the HIV viral envelope protein (Env) into large immobile clusters on the plasma membrane, in a process dependent on the Env cytoplasmic tail (Roy, Chan, Lambele, & Thali, 2013). STED has also elucidated this relationship, revealing that clustering of Env on viral envelopes changed as a function of viral maturity, and that this clustering required the Gag-interacting Env tail (Chojnacki et al., 2012). Using sptPALM and PALM images, micro-RNA overexpression was shown to reduce Gag mobility, and also reduce Gag cluster size and density (Chen et al., 2014), which could in turn affect the clustering of HIV-1 Env and overall infectivity of the virus.
Multicolor super-resolution studies indicate that Gag colocalizes with a variety of host cell transmembrane proteins by interacting with basic motifs within their cytoplasmic tails (Grover, Veatch, & Ono, 2015). Correlative iPALM/EM images and multicolor 3D super-resolution imaging have beautifully shown host cell endosomal sorting complexes required for transport (ESCRT) machinery bundled up inside the Gag lattice of budding virus particles (Van Engelenburg et al., 2014), indicating that after hijacking ESCRT to help bud the host cell membrane, HIV virions swallow ESCRT whole.
3.2.5. Outlook
Ground-breaking progress in our understanding of virus infection has already emerged from the use of super-resolution microscopy. In particular, the manipulation of host membrane protein organization by viruses has been shown, at the nanoscale, to be imperative for some steps in viral infection. These microscopy methods could also be employed to help understand currently unknown mechanisms in past and rising health threats such as pox viruses (e.g., smallpox and vaccinia), coronaviruses (e.g., SARS), and filoviruses (e.g., Ebola and Marburg viruses). There are at least six membrane proteins known to be associated with the currently unclear process of virion formation in the vaccinia virus (Liu, Cooper, Howley, & Hayball, 2014). In filoviruses, the proteins responsible for viral assembly and virion production have been identified, but the mechanisms of interaction with the host cell remain unclear (Stahelin, 2014). Understanding the interplay between host and viral membrane protein organization at the nanoscale with super-resolution microscopy will undoubtedly continue to rapidly improve our understanding of virus infection, and so aid in the development of targeted and efficacious antiviral therapies. Super-resolution microscopy has also been used to study the defenses mounted by cells upon viral infection (see Section 3.4.1 for more detail on immune cells). FPALM imaging of zebrafish cells showed that snakehead rhabdovirus (SHRV) infection resulted in downregulated caveolin expression, which in turn dispersed the clustering of a zebrafish type I interferon receptor (IFN-R) homolog, the clustering of which was crucial for the innate immune response (Gabor et al., 2013). Exciting recent developments now show that FPALM can be used in vivo to image membrane structures within living zebrafish (Gabor, Kim, Kim, & Hess, 2015), suggesting that changes in membrane organization during viral infection, and many other possibilities, can now be investigated in vivo.
Many super-resolution viral studies have focused on assembly and related processes near the membrane. While these experiments provide invaluable information about the viral life cycle, they explore only a part of the full story. More research into the organization and dynamics of the virion envelope during binding, entry, and uncoating, could provide additional insights and help identify new antiviral drug targets. As many entire virions can be smaller than the diffraction limit, questions relating to the organization of host cell and viral proteins within the virion itself require the ability to image nanoscale structure. Super-resolution can see at the nano- and virus-scale, and we can now resolve host cell proteins (and lipids) in living cells as they are commandeered by the virus. These capabilities are well suited for understanding infection, so we can better develop methods to combat it. We look forward to seeing in real time at the nanoscale exactly how viruses use, abuse, and steal our membranes and associated proteins for their own infective purposes.
3.3. Applications to mitochondria
Mitochondrial dysfunction has been causally linked to a number of degenerative diseases including Huntington's disease, Alzheimer's disease, Parkinson's disease, and Amyotrophic Lateral Sclerosis (ALS), among others (Burte et al., 2014, Palomo and Manfredi, 2014). Though dysfunction appears in a number of ways, it is often associated with changes in mitochondrial morphology and may be accompanied by altered regulation of mitochondrial fission and fusion proteins (Johri & Beal, 2012).
3.3.1. Mitochondrial fission: membrane contacts and protein helices
Mitochondrial fission in mammals is supported by the mitochondrial outer membrane (MOM) protein mitochondrial fission factor (Mff), which localizes to sites of fission (Otera et al., 2010). Mff is the MOM receptor essential for recruiting dynamin-related protein 1 (Drp1, or the yeast homolog Dnm1) to sites of fission (Otera & Mihara, 2011), where it has long been believed that self-assembling Drp1 oligomers form constrictive helices which sever mitochondria (Labrousse, Zappaterra, Rube, & van der Bliek, 1999).
Conventional microscopy images of dividing mitochondria indicated that these Drp1 sites were smaller than the diffraction limit (e.g., (Labrousse et al., 1999)), while EM studies showed Drp1 (or Dnm1) formed tubular membrane clusters (Yoon, Pitts, & McNiven, 2001), and also helices ∼100–130 nm in diameter at sites of extreme mitochondrial constriction and division (Ingerman et al., 2005). Recent multicolor PALM imaging has illustrated that Drp1 helix size reduces significantly as mitochondria are constricted and fission progresses (Rosenbloom et al., 2014). These helices represent functional membrane-associated Drp1 clusters. However, fission is also spatially correlated with sites of interaction between mitochondria and endoplasmic reticulum (ER).
EM and conventional fluorescence microscopy have shown that mitochondria maintain numerous contacts with the ER which can mediate mitochondrial constriction prior to Drp1 recruitment and mitochondrial division (Friedman et al., 2011). Super-resolution imaging (using a modified SIM geometry) in living astrocytes visualized these ER-mitochondrial contacts, which comprised fine tubules of ER (∼100 nm in width) wound around mitochondria (Brunstein, Wicker, Herault, Heintzmann, & Oheim, 2013), in agreement with multicolor STORM imaging in living kidney epithelial cells which showed ER tubules (average widths 80–130 nm) were often found at sites of mitochondrial division (Shim et al., 2012).
3.3.2. Mitochondrial membranes within membranes: organization on many levels
Mitochondria themselves contain multiple membrane structures. Mitochondrial DNA is packaged into nucleoids, and the relative alignment of nucleoids to cristae (convolutions of the inner mitochondrial membrane (IMM)) is thought to have functional impacts, since mtDNA encodes for oxidative phosphorylation system proteins which require coordinated membrane insertion (van den Heuvel & Smeitink, 2001). STED imaging indicated nucleoids were of a uniform size and shape and were themselves clustered (Kukat et al., 2011); however, PALM imaging indicated a great variability in nucleoid sizes and shapes, and suggested physical interactions between nucleoids and the IMM (Brown, Tkachuk, et al., 2011). Subsequent correlation of EM and iPALM images has illustrated the three-dimensional interplay of nucleoids and the IMM: nucleoids can intertwine with cristae, or seemingly directly contact cristae tips or sides (Kopek, Shtengel, Xu, Clayton, & Hess, 2012).
The maintenance of the highly convoluted and distinctive IMM requires protein subunits of the mitochondrial inner membrane organizing system (MINOS) (Zerbes et al., 2012). Very little was known about the distribution of these proteins within mammalian mitochondria, although EM indicated their enrichment at cristae junctions of budding yeast mitochondria (Harner et al., 2011, Rabl et al., 2009).
Multicolor STED imaging of mitochondrial membranes and MINOS units in human cell lines revealed a highly ordered distribution of the subunit mitofilin, which consistently formed clusters at cristae junctions. Mitofilin was highly colocalized with MINOS subunits MINOS1 and CHCHD3, in a distinct, regular (“rail-like”) pattern of clusters within or adjacent to the IMM at the rim of mitochondria, indicating a coordinated targeting and organization of MINOS with respect to the mitochondrial membrane (Jans et al., 2013).
This super-resolution work has illustrated the compartmentalization of mitochondrial membranes. FPALM tracking of individual mitochondrial membrane proteins (Tom20 and ATP synthase) in living cells further illustrated their altered mobility within (and transitions between) microcompartments, and these domains were present on both the IMM and MOM (Appelhans et al., 2012).
3.3.3. Outlook
Super-resolution imaging of mitochondria has already allowed the quantification of membrane–membrane interfaces in two and three dimensions, and the distributions of membrane bound and associated proteins. This heterogeneity appears to be imperative to specific organelle processes (such as mitochondrial fission and oxidative phosphorylation). Furthermore, it has revealed that mitochondrial membranes are partitioned into distinct microcompartments, within which the movement of membrane proteins is restricted. Further investigation into the distributions of proteins at the nanoscale will inform as to the processes which allow the highly ordered physical arrangements of convoluted mitochondrial membranes in the meso- and microscale. Applicable to a range of diseases in which mitochondria are involved, studies such as these also enlighten us as to the functional organization of these fundamental miniature energy conversion factories.
3.4. Signaling
3.4.1. Immune system function
Immune cell biology is at the cutting edge of emerging technologies. Super-resolution microscopy has afforded some unprecedented advances in cellular immunology, and its use has resulted in true paradigm shifts in the field.
B-Cells, T-Cells, natural killer (NK) cells, and mast cells have all been used as systems to investigate the recruitment and reorganization of membrane proteins in response to activating molecules. The nanometer localization precision provided by super-resolution techniques has enabled new and previously impossible observations of fundamental processes, such as the organization of receptors in the immunological synapse (IS), and how actin is organized with respect to the IS and lytic granules. Because this field is vast and diverse, we have limited discussion to a small number of super-resolution studies.
3.4.1.1. Mast cell signaling: interplay of molecular dynamics, clustering, and signaling
Mast cells are populated with FcεRI, a high affinity membrane receptor for antigen-specific IgE. When multivalent antigen cross-links these IgE–FcεRI complexes, mast cells are stimulated to release preformed granules containing immune system mediators (see review in Siraganian, 2003). EM imaging has demonstrated that antigen cross-linking induces FcεRI–IgE complex aggregation in fixed cells (Stump et al., 1988, Veatch et al., 2012). In live cells, antigen cross-linking induces altered dynamics as demonstrated with SPT (Spendier, Lidke, Lidke, & Thomas, 2012), and with nonimaging methods (e.g., FCS and FRAP with stationary spot illumination) (Larson et al., 2005, Menon et al., 1986) but super-resolution imaging provides a means of directly imaging the protein dynamics and clustering, and their context as a function of time in living cells.
The kinetics following antigen cross-linking of IgE–FcεRI complexes was measured in living rat basophilic leukemia (RBL)-2H3 mast cells at the nanoscale with STORM, first reported in 2013 (Shelby, Holowka, Baird, & Veatch, 2013). Importantly, this allowed the imaging of a distinct time series in receptor organization. Prior to antigen addition, IgE-bound FcεRI receptors are uniformly distributed throughout the membrane. Within 2 minutes of antigen addition, the mobility of receptors slows dramatically and the receptors themselves are laterally confined. It is not until 5 minutes post antigen addition that receptors organize into ∼70 nm clusters, which are too small to resolve using conventional microscopy. Furthermore, the mobility of single receptors after antigen addition was highly dependent on the surrounding receptor density, even before strong clustering occurred. Single receptors reversibly associated with small and slowly moving receptor clusters soon after antigen addition, and before calcium mobilization, suggesting initial signaling promoted these molecular associations. Antigen-mediated clustering could be reversed; addition of an antigen-competitive monovalent ligand restored the uniform distribution of receptors in nonantigen-treated cells. This elegant and robust study is a fine example of the profound biological insight which can be gained from carefully quantified super-resolution data.
3.4.1.2. NK cells: pass a lytic granule cannonball through the eye of a needle
One long-standing question has faced immunity research for years: how can NK cells pass a cannonball through the eye of a needle? NK cells (and cytotoxic T cells) can directly eliminate virus-infected or tumor cells through specialized secretory lysosomes known as lytic granules (Bryceson et al., 2011, Lieberman, 2003), with diameters around 250 nm (Brown, Oddos, et al., 2011). Confocal studies suggested granules were secreted through a large (many microns in diameter) central clearance in an otherwise hyperdense actin barrier (Orange et al., 2003). Recent super-resolution imaging of NK cells showed actin filament accumulation at the IS upon NK cell stimulation, which is not centrally cleared as previously thought ((SIM; Brown, Oddos, et al., 2011; STED; Rak et al., 2011)). Rather, the holes in the actin mesh (only present upon cell activation) are only large enough to accommodate a single granule (Brown et al., 2012, Brown et al., 2011, Rak et al., 2011). Two-color STED showed the close spatial association of actin and granules adjacent to the IS (Mace & Orange, 2012a), and that the actin ABP coronin aided actin hole formation, facilitating granule secretion through the otherwise dense actin cortex (Mace & Orange, 2014). So, the lytic granule cannonball can pass through, not because of a large barrier clearance, but because the eye of the dense actin mesh needle locally expands to accommodate it. This fundamental change in ideas is due to the insight made possible by imaging advances: “The use of super-resolution imaging was critical to the identification of the granule-sized clearances formed in response to activating signal” (Mace & Orange, 2012b).
3.4.1.3. T-cell signaling: are clusters pre-formed in the cell?
We here present only a subset of super-resolution studies investigating T-cell membrane organization; for further discussion, see recent reviews (Garcia-Parajo et al., 2014, Rossy et al., 2013, Sherman et al., 2013).
When T-Cells engage peptides on the surfaces of antigen-presenting cells, subunits of plasma membrane-bound T-Cell receptors (TCR) become phosphorylated by the Src family kinase Lck or Fyn, and in turn recruit the Syk family kinase, ζ-associated protein-70 (ZAP-70). ZAP-70 and Lck mediate the phosphorylation of the scaffolding proteins linker for activation of T-cells (LAT) and SH2 domain-containing leukocyte protein-76 (SLP76) (for review see (Le Floc'h & Huse, 2015)). These multimolecular signaling complexes (along with other associated effectors and adaptors) can further group into microclusters, and these complexes and microclusters drive signal amplification and underpin T-cell activation (for review see (Sherman et al., 2013)). As such, the spatial distributions and dynamics of these proteins, and TCR-dependent signaling microclusters, are of tremendous interest. Because many aspects of their organization span length scales smaller than the diffraction-limited resolution, immense advances in T-cell membrane organization research have been made with super-resolution microscopy.
In activated T-cells, imaging of phosphorylated LAT microclusters with single color dSTORM (Owen et al., 2010) and of ZAP-70 with single-color PALM (Hsu & Baumgart, 2011) showed microclusters of each were in fact comprised of many smaller clusters, unresolvable in conventional TIRF images (Hsu and Baumgart, 2011, Owen et al., 2010). In resting T-cells, plasma membrane-bound TCR and LAT were shown with single color PALM to precluster into independent “protein islands.” Following T-cell activation, these islands appeared to concatenate, forming TCR and LAT microclusters, respectively (Lillemeier et al., 2010). Further use of dual color PALM also indicated that TCR and ZAP-70 resided in separate clusters which mixed upon T-cell activation (Sherman et al., 2011). Surprisingly, the same study indicated that clusters of LAT and TCR decreased in their spatial overlap upon T-cell activation (Sherman et al., 2011). Similarly, dual-color PALM imaging of ZAP-70 and another of its substrates, SLP-76, indicated that ZAP-70 and SLP-76 clusters segregated as a function of time after activation (Hsu & Baumgart, 2011). Debate ensued. How were T-cells regulating the precise clustering, mixing, and segregation of signaling proteins in response to activation?
Through the use of super-resolution microscopy, a story has begun to emerge. Single-color PALM revealed that following activation, T-cell plasma membranes contained dramatic increases in the numbers of clusters of LAT (Hsu and Baumgart, 2011, Williamson et al., 2011), SLP-76, and ZAP-70 (Hsu & Baumgart, 2011). Dual-color dSTORM and PALM imaging suggested that T-cell activation involved docking of subsynaptic vesicles and subsequent localized delivery of LAT molecules to plasma membrane activation sites, forming new, immobile LAT clusters (Williamson et al., 2011). Dual-color PALM showed SLP-76 accumulation at the rims of LAT clusters in activated T-cells (Sherman et al., 2011), and dual color dSTORM-TIRF imaging showed that upon T-cell activation, phosphorylated (p)LAT formed vesicular fusion-dependent “signaling nanoterritories,” elongated pLAT nanoclusters adjacent to (within 20 nm), but spatially segregated from clusters of phosphorylated SLP-76 (Soares et al., 2013). Conversely, pLAT that was preclustered in the plasma membranes of resting T-cells (in morphologically distinct circular nanoclusters) rarely associated with pSLP76 (Soares et al., 2013). These results suggest that two different populations of LAT exist; with (1) activation induced fusion of vesicular LAT to the plasma membrane predominantly contributing to signaling complexes, and (2) preexisting plasma membrane-bound LAT clusters remaining relatively inert with respect to signal transduction.
The roles of the actin cytoskeleton in microcluster arrangement have also been studied using super-resolution techniques. PALM imaging has shown the distributions of SLP76 proximal to, but at the peripheries of LAT clusters, were abolished with actin disruption (Sherman et al., 2011), and actin depolymerization severely impaired TCR and LAT microcluster structure, resulting in deformed clusters with reduced circularity (Hsu & Baumgart, 2011). In resting T-cells, single color PALM imaging has indicated that actin may work to physically segregate “protein islands,” as depolymerization induced the formation of large TCR and large LAT aggregations (Lillemeier et al., 2010).
Super-resolution microscopy continues to allow visualization of previously unresolvable molecular organizations, providing data which is invaluable in advancing the understanding of dynamic nanoscale clustering crucial to T-cell function. Moreover, results from these studies have far-reaching implications for understanding general molecular processes which guide membrane protein clustering and the potential influences of various pools of protein on the organization at the plasma membrane.
3.4.1.4. B-cell signaling: nanoscale changes can direct global signaling
B-cell plasma membranes undergo considerable reorganization during antigen recognition, and the formation of signaling-competent microclusters of B-cell receptors (BCRs) is mediated by dynamic modulation of the actin cytoskeleton by the ezrin/radixin/moesin family of ABPs (Harwood and Batista, 2010, Treanor et al., 2011). Imaging by tracking single molecules with STORM has shown that individual BCRs, and BCR microclusters, each exhibit reduced mobility in ezrin deficient cells (Pore et al., 2013). Imaging with dSTORM showed for the first time that BCR isotypes IgM and IgG, and the co-factor CD19, are differentially assembled into preformed clusters on the naive B-cell plasma membrane (Mattila et al., 2013). Antigen activation induces a local coalescence of nanoclusters of BCR and the cofactor CD19, with the membrane spanning molecular organizer CD81 (tetraspanin) determining reorganization. Mattila et al. propose that CD19 is sequestered from BCR nanoclusters by CD81, and B-cell activation induces actin remodeling which allows CD19 to converge with BCRs and elicit cell signaling (Mattila et al., 2013). The alterations in the clustering of these proteins occur at the nanoscale, without alterations to their global distributions. These studies highlight the insights allowed by super-resolution in understanding immune processes which are otherwise invisible (see review by (Grove, 2014)).
3.4.1.5. Outlook
The studies presented here have demonstrated dramatic advances made in understanding functional membrane domains at the nanoscale, and interactions between these domains and cellular components such as the actin cytoskeleton. The super-resolution tools available are not just allowing visualization of structures with better detail; they have enabled numerous discoveries which have led to fundamental changes in the understanding of these systems.
3.4.2. Phosphoinositides and associated pathways
3.4.2.1. Phosphoinositides: in many places and doing many things
Phosphoinositides are phosphorylated phosphatidylinositols commonly found in the cytoplasmic (inner) leaflet of the cell membrane (Balla, 2013). Mammals produce seven different types of phosphoinositide, each of which is found in a different domain of the membrane, and each of which is differentiated by the location of the phosphorylation of the inositol ring (Di Paolo & De Camilli, 2006). Phosphoinositides are involved in, and regulate, an immense number of cellular events, including cell signaling, membrane transport and regulation, cytokinesis, and organelle distinction (Di Paolo & De Camilli, 2006). They can do this by directly binding to proteins, by activating proteins upstream of structural change, and by mediating a number of signaling pathways. One particularly important phosphoinositide is phosphatidylinositol (4,5)-biphosphate (PIP2), which is also one of the more prevalent subspecies (Liu & Bankaitis, 2010). Of particular interest in membrane organization research, PIP2 has the potential to be a master regulator, as it can function to remodel the actin cytoskeleton in many different ways (see Section 3.1 for more detail), and may be a functional conduit between membrane proteins, lipids, and membrane-associated actin. Though these lipids are essential for cellular functionality, data suggest that PIP2-mediated processes are altered by nanoscale spatial distributions not resolvable by conventional microscopy. Super-resolution microscopy has allowed nanoscale investigation to begin.
3.4.2.2. Imaging PIP2 with super-resolution microscopy
STED imaging of a PIP2-binding pleckstrin-homology (PH) domain construct has indicated that PIP2 forms circular and linear clusters of approximately 73 nm in size in PC12 plasma membrane sheets (van den Bogaart et al., 2011). This is in agreement with cluster sizes of PIP2 imaged with dSTORM of antibody-labeled PIP2 (Wang & Richards, 2012). Simultaneous dual-color STED revealed, stunningly, that PIP2 was almost never found in the same clusters as phosphatidylinositol (3,4,5)-trisphosphate (PIP3), and that PIP3 clusters (∼100 nm size) were significantly larger than PIP2 clusters (Wang & Richards, 2012).
PIP2 has also been shown to affect the nanoscale organization of membrane components. For example, PIP2 depletion from PC12 cells was shown to reduce the clustering of syntaxin-1A, a protein which normally clusters at sites of synaptic vesicle exocytosis (van den Bogaart et al., 2011), and STED-FCS has indicated that the PIP2 phosphatase synaptotagmin binds selectively to mobile syntaxin membrane clusters, estimated to contain PIP2 (Honigmann et al., 2013). Multispecies PALM/dSTORM imaging in fixed cells has shown that clusters of PIP2 (a cytoplasmic leaflet lipid) tightly colocalize with, and are dependent on, sphingomyelin (an exoplasmic leaflet lipid). This may be a result of localized PIP2 production, as further dual-color imaging showed that the PIP2-producing kinase, phosphatidylinositol 4-phosphate 5-kinase (PIP5Kβ, located at the cytoplasmic leaflet), also colocalizes with sphingomyelin clusters (Abe et al., 2012).
These collective images of PIP2 in membrane sheets and whole cell plasma membranes have yielded data on PIP2 distributions, in fixed cells, with resolutions of ∼30–70 nm. The direct imaging of PIP2 and other lipid species at the nanoscale in living cells is a tantalizing prospect. Due to the compatibility of super-resolution microscopy with live cell imaging, much can be learned about biological membranes; for example, how are PIP2 and PIP3 molecules spatially segregated? Are molecular motions confined within clusters, or do certain molecules just spend longer dwell times within? Localization microscopy can provide single-molecule information such as high-density molecular trajectories (Gudheti et al., 2013, Manley et al., 2008) within the context of other membrane components, information which can be invaluable for quantification of membrane organization and dynamics to address a wide range of interesting questions.
Interactions between PIP2 and other lipids, PIP2 and membrane proteins, and PIP2 and the actin cytoskeleton remain greatly underexplored using super-resolution imaging methods. The exciting developments we have described are just part of the story, and these studies demonstrate that a multitude of fundamental processes can (and hopefully will) be better understood by using super-resolution imaging of phosphoinositides.
3.5. Future directions
3.5.1. Technical considerations
For localization microscopy itself, improving time resolution is still an important goal, especially for live cell imaging. Biological processes occur not just on the minute, second, and millisecond timescale, but movements of lipids and proteins can be exceptionally fast, and methods which can image on submillisecond timescales will yield valuable and previously invisible insights into how cells are really organized in time.
As is frequently the case in fluorescence methods, the properties of fluorescent probes themselves can be limiting, and can restrict the speed at which fast images can be acquired. Ideally, probes would emit very large numbers of photons (i.e., >105) before switching to a dark state or bleaching, they would have minimal blinking on millisecond and microsecond timescales (as this can lead to erroneously increased estimates of molecule number), and probes would not disrupt biological function. Of course, increased numbers of detected photons can in principle improve localization precision, although many other factors are involved at length scales below 20 nm, such as localization errors due to emission dipole orientation effects (Enderlein, Toprak, & Selvin, 2006), camera pixel heterogeneities (Pertsinidis, Zhang, & Chu, 2010), and sample/stage drift (Mlodzianoski et al., 2011). However, to gain substantial increases in imaging speeds, using probes with high peak emission rates (i.e., 106 photons detected per molecule per second), may be the most important (and perhaps overlooked) property. While organic dyes can emit very large numbers of photons, they are still limited in emission rate by blinking, flicker, and saturation. Furthermore, very little data on maximum emission rate per molecule is available—most studies focus on number of detected photons, measured extinction coefficient and quantum yield, which do not address emission rate at saturation. To optimize use and choice of probes, there is considerable need for systematic photophysical studies of probes used for localization microscopy, such as those being carried out by Markus Sauer (van de Linde & Sauer, 2014), Carlos Bustamante (Lee, Shin, Lee, & Bustamante, 2012), Dominique Bourgeois (Avilov et al., 2014), and others.
Improvements in imaging speed have been enabled by multiemitter fitting algorithms (Cox and Jones, 2013, Cox et al., 2012, Holden et al., 2011, Huang et al., 2013), although analysis times may increase significantly when such algorithms are used. Development of increasingly efficient algorithms for larger numbers of spatially overlapping PSFs is of great interest, and can enable live cell imaging with speeds fivefold faster or more, especially when coupled to use of labels with high emission rates (Huang et al., 2013). In addition, high-speed cameras capable of imaging the action are also crucial, and recent developments in sCMOS and EMCCD sensor technology and electronics for readout have helped the field tremendously. We look forward to the ongoing technological advances in sensor development which allow for faster acquisition, and so facilitate greater temporal resolution.
The issue of photodamage needs to be explored further. As all interactions between light and a biological sample can potentially cause disruption of biological function, super-resolution techniques (including both STED and localization-based methods) seem to be particularly susceptible; the high intensities often used are expected to cause more damage, and the short length scales typically of interest are also more likely to manifest changes (however subtle). Notwithstanding, there is very little consensus in the field related to the intensity or dose thresholds for photodamage. Currently it seems that each group has their own method for testing for possible photodamage, and there seems not to be any standardization, making generalizations extremely difficult. Thus there is considerable need for systematic exploration of this crucial issue. Once better understanding of the most common damage mechanisms has been established, methods can be developed to mitigate photodamage while still allowing nanoscale structures to be imaged.
The use of super-resolution localization microscopy to count numbers of molecules is currently possible, but difficult, largely due to the need to control for several complicating factors. These factors include fluorophore blinking (Lee et al., 2012), inefficient fluorescent protein folding/activation (Durisic, Laparra-Cuervo, Sandoval-Alvarez, Borbely, & Lakadamyali, 2014), molecules lost to thresholds and excitation/activation polarization effects (whereby certain populations are excited or activated more efficiently because their transition dipoles are oriented along the direction of the incident laser electric field), and potential complications when both exogenous and endogenous molecular populations are present. Current work in the field is helping to overcome these challenges, so it is likely that in the near future, localization microscopy will become used more widely for quantification of molecular numbers, information which is not directly accessible by a number of other techniques. Moreover, discovery and engineering of new methods to tag cellular molecules (e.g., reductions in the size of tags, whether fluorescent proteins or antibodies) will bring us closer to the prospect of imaging with minimal disruption to cellular function.
Additionally, as a larger variety of photoactivatable probes becomes available, the prospect of simultaneously imaging large numbers of different species comes into reach. Simultaneous multicolor imaging can be limited by the spectra of probes used to tag them. New tags (e.g., new species of photoactivatable proteins, or newly engineered organic dyes) are emerging which are less overlapping in their emission spectra. Also emerging are new ways to reliably identify tagged species when there may be spectral overlap (including ratiometric analyses, but also the development of color recognition in sensor systems). Together these advances bring us toward simultaneous super-resolution imaging of five or more species in living cells. Some day soon, we really will be able to see, in real time, all of the dancers about which we hypothesize.
3.5.2. Biological considerations
The field of cell biology now has some truly excellent tools to understand membrane organization. We are fortunate indeed to be able to directly image nanoscale membrane clusters in living cells! As has been forecasted by the Nobel Committee in 2014, the power of super-resolution microscopy is only going to grow as time progresses.
Perhaps one of the highest priorities will be to put the current models to the test, with the aim of generating some consensus in the field. Of course, this requires cooperation between those posing models and those designing experiments; generation of testable, falsifiable hypotheses must remain of utmost importance, even as the complexity of models increases. While many models propose a starting and ending point in a dynamic process, the steps and dynamics which occur between those points can now be visualized directly. More attention should be spent on these “intermediate points,” which can strongly govern how a biological system approaches a given outcome, even when multiple models predict the same final outcome. For example, very little has been done in the way of super-resolution timelapse imaging of the a priori formation of membrane protein clusters, or their subsequent dispersal.
In the direction of refining membrane models to improve our understanding of cell membranes, it will almost certainly be useful to further investigate the coupling between signaling and membrane organization, especially considering the potential for two-way interactions, such as the way membrane proteins can “talk to” actin through ABPs or through certain lipids, and so cause changes in actin organization, which can in turn cause changes in the membrane protein clusters themselves.
Some models of membrane organization consider the bilayer as a two-dimensional plane, and interactions with other components such as the actin cytoskeleton as perturbations to the two-dimensional energy landscape. While this can be useful, there is much more to be learned from studying the three-dimensional structure of the membrane and its interactions with the actin cytoskeleton. Not only can this strategy be helpful in discerning differences between models of how membrane proteins are regulated in their spatial distributions, but also it can allow a fuller understanding of the richness of cellular functions and interactions which are possible in such systems. For example, considering the actin cytoskeleton as its native three-dimensional self allows for ABP coupling between actin, membrane proteins, lipids, lateral organization, and three-dimensional membrane topology. That is to say, both proteins and lipids are clearly important. Likewise, as many cellular organelles have three-dimensional structures and complex membrane topologies, it is clear that we must approach such systems three-dimensionally, while also further investigating how lateral organization (i.e., domains) can occur within the membrane. Thus, there are a number of challenges to solve as we endeavor to understand membranes. Just a decade ago, many of these challenges seemed insurmountable. Yet, with the capabilities of super-resolution microscopy, a fundamentally better understanding of biological membranes is within reach.
Authors' Note
Due to space constraints, we have not included a section on imaging of membranes in neurons. However, we believe the field is deserving of a dedicated chapter! While some super-resolution studies of neurons are mentioned here, we direct readers to recent reviews (MacGillavry and Blanpied, 2013, Maglione and Sigrist, 2013, Okabe, 2012, Sigrist and Sabatini, 2012).
Acknowledgments
The authors thank Julie Gosse, Greg Innes, Mary Kraft, Joshua Zimmerberg, and Markus Sauer for crucial discussions, and Patricia Byard for administrative assistance. This work was funded by NIH R15 GM094713, NIH R15 ES024593, NIH R01 AR054170, Maine Technology Institute MTAF 1106 and 2061, the University of Maine Office of the Vice President for Research, and the Maine Economic Improvement Fund.
References
- Abe M., Makino A., Hullin-Matsuda F., Kamijo K., Ohno-Iwashita Y., Hanada K. A role for sphingomyelin-rich lipid domains in the accumulation of phosphatidylinositol-4,5-bisphosphate to the cleavage furrow during cytokinesis. Molecular and Cellular Biology. 2012;32(8):1396–1407. doi: 10.1128/MCB.06113-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B. 4th ed. Garland Science; New York: 2002. Molecular biology of the cell. [Google Scholar]
- Alenghat F.J., Golan D.E. Membrane protein dynamics and functional implications in mammalian cells. Current Topics in Membranes. 2013;72:89–120. doi: 10.1016/B978-0-12-417027-8.00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida P.F.F., Pokorny A., Hinderliter A. Thermodynamics of membrane domains. Biochimica et Biophysica Acta–Biomembranes. 2005;1720(1–2):1–13. doi: 10.1016/j.bbamem.2005.12.004. (Review) [DOI] [PubMed] [Google Scholar]
- Alvarez-Guaita A., Vila de Muga S., Owen D.M., Williamson D., Magenau A., Garcia-Melero A. Evidence for Annexin A6-dependent plasma membrane remodelling of lipid domains. British Journal Pharmacology. 2014;19(10):13022. doi: 10.1111/bph.13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R.G.W., Jacobson K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 2002;296(5574):1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]
- Andrews N.L., Lidke K.A., Pfeiffer J.R., Burns A.R., Wilson B.S., Oliver J.M. Actin restricts FcepsilonRI diffusion and facilitates antigen-induced receptor immobilization. Nature Cell Biology. 2008;10(8):955–963. doi: 10.1038/ncb1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova M.I., Dimitrov D.S. Liposome electroformation. Faraday Discuss. 1986;81:303–311. [Google Scholar]
- Antelmi E., Cardone R.A., Greco M.R., Rubino R., Di Sole F., Martino N.A. Beta1 integrin binding phosphorylates ezrin at T567 to activate a lipid raft signalsome driving invadopodia activity and invasion. PLoS One. 2013;8(9) doi: 10.1371/journal.pone.0075113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelhans T., Richter C.P., Wilkens V., Hess S.T., Piehler J., Busch K.B. Nanoscale organization of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Letters. 2012;12(2):610–616. doi: 10.1021/nl203343a. [DOI] [PubMed] [Google Scholar]
- Aspenstrom P. BAR domain proteins regulate Rho GTPase signaling. Small GTPases. 2014;5(2):1–8. doi: 10.4161/sgtp.28580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilov S., Berardozzi R., Gunewardene M.S., Adam V., Hess S.T., Bourgeois D. In cellulo evaluation of phototransformation quantum yields in fluorescent proteins used as markers for single-molecule localization microscopy. PLoS One. 2014;9(6):e98362. doi: 10.1371/journal.pone.0098362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock H.P., Chen C., Zhuang X. Using single-particle tracking to study nuclear trafficking of viral genes. Biophysical Journal. 2004;87(4):2749–2758. doi: 10.1529/biophysj.104.042234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatolli L.A., Gratton E. Two-photon fluorescence microscopy observation of shape changes at the phase transition in phospholipid giant unilamellar vesicles. Biophysical Journal. 1999;77(4):2090–2101. doi: 10.1016/S0006-3495(99)77050-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatolli L.A., Gratton E. A correlation between lipid domain shape and binary phospholipid mixture composition in free standing bilayers: a two-photon fluorescence microscopy study. Biophysical Journal. 2000;79(1):434–447. doi: 10.1016/S0006-3495(00)76305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatolli L., Gratton E., Khan T.K., Chong P.L.G. Two-photon fluorescence microscopy studies of bipolar tetraether giant liposomes from thermoacidophilic archaebacteria Sulfolobus acidocaldarius. Biophysical Journal. 2000;79(1):416–425. doi: 10.1016/S0006-3495(00)76303-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatolli L.A., Sanchez S.A., Hazlett T., Gratton E. Giant vesicles, Laurdan, and two-photon fluorescence microscopy: evidence of lipid lateral separation in bilayers. Biophotonics, Pt A. 2003;360:481–500. doi: 10.1016/s0076-6879(03)60124-2. San Diego: Academic Press Inc. [DOI] [PubMed] [Google Scholar]
- Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiological Reviews. 2013;93(3):1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On D., Wolter S., van de Linde S., Heilemann M., Nudelman G., Nachliel E. Super-resolution imaging reveals the internal architecture of nano-sized syntaxin clusters. The Journal of Biological Chemistry. 2012;287(32):27158–27167. doi: 10.1074/jbc.M112.353250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barret C., Roy C., Montcourrier P., Mangeat P., Niggli V. Mutagenesis of the phosphatidylinositol 4,5-bisphosphate (PIP(2)) binding site in the NH(2)-terminal domain of ezrin correlates with its altered cellular distribution. The Journal of Cell Biology. 2000;151(5):1067–1080. doi: 10.1083/jcb.151.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M., Huang B., Dempsey G.T., Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317(5845):1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T., Hammond A.T., Sengupta P., Hess S.T., Holowka D.A., Baird B.A. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proceedings of the National Academy of Sciences USA. 2007;104(9):3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T., Hess S.T., Webb W.W. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 2003;425(6960):821–824. doi: 10.1038/nature02013. [DOI] [PubMed] [Google Scholar]
- Baumgart T., Hunt G., Farkas E.R., Webb W.W., Feigenson G.W. Fluorescence probe partitioning between L-o/L-d phases in lipid membranes. Biochimica et Biophysica Acta–Biomembranes. 2007;1768(9):2182–2194. doi: 10.1016/j.bbamem.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz J. Minimal aggregate size and minimal fusion unit for the first fusion pore of influenza hemagglutinin-mediated membrane fusion. Biophysical Journal. 2000;78(1):227–245. doi: 10.1016/S0006-3495(00)76587-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berditchevski F. Complexes of tetraspanins with integrins: more than meets the eye. Journal of Cell Science. 2001;114(Pt 23):4143–4151. doi: 10.1242/jcs.114.23.4143. [DOI] [PubMed] [Google Scholar]
- Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S., Bonifacino J.S. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Betzig E., Trautman J.K. Near-field optics: microscopy, spectroscopy, and surface modification beyond the diffraction limit. Science. 1992;257(5067):189–195. doi: 10.1126/science.257.5067.189. [DOI] [PubMed] [Google Scholar]
- Betzig E., Trautman J.K., Harris T.D., Weiner J.S., Kostelak R.L. Breaking the diffraction barrier – optical microscopy on a nanometric scale. Science. 1991;251(5000):1468–1470. doi: 10.1126/science.251.5000.1468. [DOI] [PubMed] [Google Scholar]
- Bisi S., Disanza A., Malinverno C., Frittoli E., Palamidessi A., Scita G. Membrane and actin dynamics interplay at lamellipodia leading edge. Current Opinion in Cell Biology. 2013;25(5):565–573. doi: 10.1016/j.ceb.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Biswas S., Yin S.R., Blank P.S., Zimmerberg J. Cholesterol promotes hemifusion and pore widening in membrane fusion induced by influenza hemagglutinin. The Journal of General Physiology. 2008;131(5):503–513. doi: 10.1085/jgp.200709932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunk A.D., Akbergenova Y., Cho R.W., Lee J., Walldorf U., Xu K. Postsynaptic actin regulates active zone spacing and glutamate receptor apposition at the Drosophila neuromuscular junction. Molecular and Cellular Neuroscience. 2014;61:241–254. doi: 10.1016/j.mcn.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G., Meyenberg K., Risselada H.J., Amin H., Willig K.I., Hubrich B.E. Membrane protein sequestering by ionic protein-lipid interactions. Nature. 2011;479(7374):552–555. doi: 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguslawska D.M., Machnicka B., Hryniewicz-Jankowska A., Czogalla A. Spectrin and phospholipids – the current picture of their fascinating interplay. Cellular and Molecular Biology Letters. 2014;19(1):158–179. doi: 10.2478/s11658-014-0185-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni V., Baier C.J., Lang T., Bonini I., White M.M., Garbus I. Cholesterol depletion activates rapid internalization of submicron-sized acetylcholine receptor domains at the cell membrane. Molecular Membrane Biology. 2007;24(1):1–15. doi: 10.1080/09687860600903387. [DOI] [PubMed] [Google Scholar]
- Bossi M., Folling J., Belov V.N., Boyarskiy V.P., Medda R., Egner A. Multicolor far-field fluorescence nanoscopy through isolated detection of distinct molecular species. Nano Letters. 2008;8(8):2463–2468. doi: 10.1021/nl801471d. [DOI] [PubMed] [Google Scholar]
- Brandstaetter H., Kendrick-Jones J., Buss F. Myo1c regulates lipid raft recycling to control cell spreading, migration and Salmonella invasion. Journal of Cell Science. 2012;125(Pt 8):1991–2003. doi: 10.1242/jcs.097212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A.C., Dobbie I.M., Alakoskela J.M., Davis I., Davis D.M. Super-resolution imaging of remodeled synaptic actin reveals different synergies between NK cell receptors and integrins. Blood. 2012;120(18):3729–3740. doi: 10.1182/blood-2012-05-429977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.A., London E. Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes? Biochemical and Biophysical Research Communications. 1997;240(1):1–7. doi: 10.1006/bbrc.1997.7575. [DOI] [PubMed] [Google Scholar]
- Brown D.A., London E. Functions of lipid rafts in biological membranes. Annual Review of Cell and Developmental Biology. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. (Review) [DOI] [PubMed] [Google Scholar]
- Brown D.A., London E. Structure and origin of ordered lipid domains in biological membranes. The Journal of Membrane Biology. 1998;164(2):103–114. doi: 10.1007/s002329900397. (Review) [DOI] [PubMed] [Google Scholar]
- Brown A.C., Oddos S., Dobbie I.M., Alakoskela J.M., Parton R.M., Eissmann P. Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy. PLoS Biology. 2011;9(9):13. doi: 10.1371/journal.pbio.1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.A., Rose J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68(3):533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Brown T.A., Tkachuk A.N., Shtengel G., Kopek B.G., Bogenhagen D.F., Hess H.F. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Molecular and Cellular Biology. 2011;31(24):4994–5010. doi: 10.1128/MCB.05694-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunstein M., Wicker K., Herault K., Heintzmann R., Oheim M. Full-field dual-color 100-nm super-resolution imaging reveals organization and dynamics of mitochondrial and ER networks. Optics Express. 2013;21(22):26162–26173. doi: 10.1364/OE.21.026162. [DOI] [PubMed] [Google Scholar]
- Bryceson Y.T., Chiang S.C., Darmanin S., Fauriat C., Schlums H., Theorell J. Molecular mechanisms of natural killer cell activation. Journal of Innate Immunity. 2011;3(3):216–226. doi: 10.1159/000325265. [DOI] [PubMed] [Google Scholar]
- Burte F., Carelli V., Chinnery P.F., Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nature Reviews Neurology. 2014;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- Chazeau A., Mehidi A., Nair D., Gautier J.J., Leduc C., Chamma I. Nanoscale segregation of actin nucleation and elongation factors determines dendritic spine protrusion. EMBO Journal. 2014;33(23):2745–2764. doi: 10.15252/embj.201488837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A.K., Sengupta P., Waki K., Van Engelenburg S.B., Ochiya T., Ablan S.D. MicroRNA binding to the HIV-1 Gag protein inhibits Gag assembly and virus production. Proceedings of the National Academy of Sciences USA. 2014;111(26):E2676–E2683. doi: 10.1073/pnas.1408037111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Takizawa N., Crowley J.L., Oh S.W., Gatto C.L., Kambara T. F-actin and myosin II binding domains in supervillin. The Journal of Biological Chemistry. 2003;278(46):46094–46106. doi: 10.1074/jbc.M305311200. [DOI] [PubMed] [Google Scholar]
- Chernomordik L.V., Frolov V.A., Leikina E., Bronk P., Zimmerberg J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. The Journal of Cell Biology. 1998;140(6):1369–1382. doi: 10.1083/jcb.140.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L.V., Leikina E., Frolov V., Bronk P., Zimmerberg J. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. The Journal of Cell Biology. 1997;136(1):81–93. doi: 10.1083/jcb.136.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichili G.R., Rodgers W. Clustering of membrane raft proteins by the actin cytoskeleton. The Journal of Biological Chemistry. 2007;282(50):36682–36691. doi: 10.1074/jbc.M702959200. [DOI] [PubMed] [Google Scholar]
- Chichili G., Rodgers W. Cytoskeleton–membrane interactions in membrane raft structure. Cellular and Molecular Life Sciences. 2009;66(14):2319–2328. doi: 10.1007/s00018-009-0022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnacki J., Staudt T., Glass B., Bingen P., Engelhardt J., Anders M. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science. 2012;338(6106):524–528. doi: 10.1126/science.1226359. [DOI] [PubMed] [Google Scholar]
- Coltharp C., Xiao J. Superresolution microscopy for microbiology. Cellular Microbiology. 2012;14(12):1808–1818. doi: 10.1111/cmi.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornely R., Rentero C., Enrich C., Grewal T., Gaus K. Annexin A6 is an organizer of membrane microdomains to regulate receptor localization and signalling. IUBMB Life. 2011;63(11):1009–1017. doi: 10.1002/iub.540. [DOI] [PubMed] [Google Scholar]
- Coscoy S., Waharte F., Gautreau A., Martin M., Louvard D., Mangeat P. Molecular analysis of microscopic ezrin dynamics by two-photon FRAP. Proceedings of the National Academy of Sciences USA. 2002;99(20):12813–12818. doi: 10.1073/pnas.192084599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox S., Jones G.E. Imaging cells at the nanoscale. The International Journal of Biochemistry and Cell Biology. 2013;45(8):1669–1678. doi: 10.1016/j.biocel.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Cox S., Rosten E., Monypenny J., Jovanovic-Talisman T., Burnette D.T., Lippincott-Schwartz J. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nature Methods. 2012;9(2):195–200. doi: 10.1038/nmeth.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley D.R. Genetic, biochemical and structural approaches to talin function. Biochemical Society Transactions. 2005;33(Pt 6):1308–1312. doi: 10.1042/BST0331308. [DOI] [PubMed] [Google Scholar]
- Curthoys N.M., Mlodzianoski M.J., Kim D., Hess S.T. Simultaneous multicolor imaging of biological structures with fluorescence photoactivation localization microscopy. Journal of Visualized Experiments. 2013;9(82):50680. doi: 10.3791/50680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Curtis I., Meldolesi J. Cell surface dynamics – how Rho GTPases orchestrate the interplay between the plasma membrane and the cortical cytoskeleton. Journal of Cell Science. 2012;125(Pt 19):4435–4444. doi: 10.1242/jcs.108266. [DOI] [PubMed] [Google Scholar]
- Davis L.H., Bennett V. Identification of two regions of beta G spectrin that bind to distinct sites in brain membranes. The Journal of Biological Chemistry. 1994;269(6):4409–4416. [PubMed] [Google Scholar]
- Day C.A., Kenworthy A.K. Mechanisms underlying the confined diffusion of cholera toxin B-subunit in intact cell membranes. PLoS One. 2012;7(4):e34923. doi: 10.1371/journal.pone.0034923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derivery E., Gautreau A. Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. Bioessays. 2010;32(2):119–131. doi: 10.1002/bies.200900123. [DOI] [PubMed] [Google Scholar]
- Dertinger T., Colyer R., Iyer G., Weiss S., Enderlein J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI) Proceedings of the National Academy of Sciences USA. 2009;106(52):22287–22292. doi: 10.1073/pnas.0907866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertinger T., Colyer R., Vogel R., Enderlein J., Weiss S. Achieving increased resolution and more pixels with Superresolution Optical Fluctuation Imaging (SOFI) Optics Express. 2010;18(18):18875–18885. doi: 10.1364/OE.18.018875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertinger T., Colyer R., Vogel R., Heilemann M., Sauer M., Enderlein J. Superresolution optical fluctuation imaging (SOFI) Advances in Experimental Medicine and Biology. 2012;733:17–21. doi: 10.1007/978-94-007-2555-3_2. [DOI] [PubMed] [Google Scholar]
- Dertinger T., Heilemann M., Vogel R., Sauer M., Weiss S. Superresolution optical fluctuation imaging with organic dyes. Angewandte Chemie International Edition in English. 2010;49(49):9441–9443. doi: 10.1002/anie.201004138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertinger T., Xu J., Naini O.F., Vogel R., Weiss S. SOFI-based 3D superresolution sectioning with a widefield microscope. Optical Nanoscopy. 2012;1(2):2. doi: 10.1186/2192-2853-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G., De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443(7112):651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Di Paolo G., Pellegrini L., Letinic K., Cestra G., Zoncu R., Voronov S. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420(6911):85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- Dietrich C., Bagatolli L.A., Volovyk Z.N., Thompson N.L., Levi M., Jacobson K. Lipid rafts reconstituted in model membranes. Biophysical Journal. 2001;80(3):1417–1428. doi: 10.1016/S0006-3495(01)76114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durisic N., Laparra-Cuervo L., Sandoval-Alvarez A., Borbely J.S., Lakadamyali M. Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nature Methods. 2014;11(2):156–162. doi: 10.1038/nmeth.2784. [DOI] [PubMed] [Google Scholar]
- Edidin M. The state of lipid rafts: from model membranes to cells. Annual Review of Biophysics and Biomolecular Structure. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- Edwards H.C., Crumpton M.J. Ca(2+)-dependent phospholipid and arachidonic acid binding by the placental annexins VI and IV. European Journal of Biochemistry. 1991;198(1):121–129. doi: 10.1111/j.1432-1033.1991.tb15994.x. [DOI] [PubMed] [Google Scholar]
- Eggeling C., Ringemann C., Medda R., Schwarzmann G., Sandhoff K., Polyakova S. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457(7233):1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- Eggeling C., Widengren J., Rigler R., Seidel C.A. Photobleaching of fluorescent dyes under conditions used for single-molecule detection: evidence of two-step photolysis. Analytical Chemistry. 1998;70(13):2651–2659. doi: 10.1021/ac980027p. [DOI] [PubMed] [Google Scholar]
- Elson E.L., Schlessinger J., Koppel D.E., Axelrod D., Webb W.W. Measurement of lateral transport on cell surfaces. Progress in Clinical and Biological Research. 1976;9:137–147. [PubMed] [Google Scholar]
- Enderlein J., Toprak E., Selvin P.R. Polarization effect on position accuracy of fluorophore localization. Optics Express. 2006;14(18):8111–8120. doi: 10.1364/oe.14.008111. [DOI] [PubMed] [Google Scholar]
- Engel S., Scolari S., Thaa B., Krebs N., Korte T., Herrmann A. FLIM-FRET and FRAP reveal association of influenza virus haemagglutinin with membrane rafts. Biochemical Journal. 2010;425(3):567–573. doi: 10.1042/BJ20091388. [DOI] [PubMed] [Google Scholar]
- Fang Z., Takizawa N., Wilson K.A., Smith T.C., Delprato A., Davidson M.W. The membrane-associated protein, supervillin, accelerates F-actin-dependent rapid integrin recycling and cell motility. Traffic. 2010;11(6):782–799. doi: 10.1111/j.1600-0854.2010.01062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder T.J., Brust-Mascher I., Slattery J.P., Baird B., Webb W.W. Constrained diffusion or immobile fraction on cell surfaces: a new interpretation. Biophysical Journal. 1996;70(6):2767–2773. doi: 10.1016/S0006-3495(96)79846-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehon R.G., McClatchey A.I., Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nature Reviews Molecular Cell Biology. 2010;11(4):276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein A., Cass A. Effect of cholesterol on the water permeability of thin lipid membranes. Nature. 1967;216(5116):717–718. doi: 10.1038/216717a0. [DOI] [PubMed] [Google Scholar]
- Fraley T.S., Tran T.C., Corgan A.M., Nash C.A., Hao J., Critchley D.R. Phosphoinositide binding inhibits alpha-actinin bundling activity. The Journal of Biological Chemistry. 2003;278(26):24039–24045. doi: 10.1074/jbc.M213288200. [DOI] [PubMed] [Google Scholar]
- Friedman J.R., Lackner L.L., West M., DiBenedetto J.R., Nunnari J., Voeltz G.K. ER tubules mark sites of mitochondrial division. Science. 2011;334(6054):358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisz J.F., Klitzing H.A., Lou K., Hutcheon I.D., Weber P.K., Zimmerberg J. Sphingolipid domains in the plasma membranes of fibroblasts are not enriched with cholesterol. The Journal of Biological Chemistry. 2013;288(23):16855–16861. doi: 10.1074/jbc.M113.473207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisz J.F., Lou K., Klitzing H.A., Hanafin W.P., Lizunov V., Wilson R.L. Direct chemical evidence for sphingolipid domains in the plasma membranes of fibroblasts. Proceedings of the National Academy of Sciences USA. 2013;110(8):E613–E622. doi: 10.1073/pnas.1216585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost A., Unger V.M., De Camilli P. The BAR domain superfamily: membrane-molding macromolecules. Cell. 2009;137(2):191–196. doi: 10.1016/j.cell.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T., Ritchie K., Murakoshi H., Jacobson K., Kusumi A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. The Journal of Cell Biology. 2002;157(6):1071–1081. doi: 10.1083/jcb.200202050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi K., Inagaki M., Hatano S., Fukami K., Takenawa T. Inositol phospholipid-induced suppression of F-actin-gelating activity of smooth muscle filamin. Biochemical and Biophysical Research Communications. 1992;184(3):1261–1265. doi: 10.1016/s0006-291x(05)80018-x. [DOI] [PubMed] [Google Scholar]
- Gabor K.A., Kim D., Kim C.H., Hess S.T. Nanoscale imaging of caveolin-1 membrane domains in vivo. PLoS One. 2015;10(2):e0117225. doi: 10.1371/journal.pone.0117225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabor K.A., Stevens C.R., Pietraszewski M.J., Gould T.J., Shim J., Yoder J.A. Super resolution microscopy reveals that caveolin-1 is required for spatial organization of CRFB1 and subsequent antiviral signaling in zebrafish. PLoS One. 2013;8(7):e68759. doi: 10.1371/journal.pone.0068759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Parajo M.F., Cambi A., Torreno-Pina J.A., Thompson N., Jacobson K. Nanoclustering as a dominant feature of plasma membrane organization. Journal of Cell Science. 2014;127(23):4995–5005. doi: 10.1242/jcs.146340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissbuehler S., Sharipov A., Godinat A., Bocchio N.L., Sandoz P.A., Huss A. Live-cell multiplane three-dimensional super-resolution optical fluctuation imaging. Nature Communications. 2014;5:5830. doi: 10.1038/ncomms6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getis A., Ord J.K. The analysis of spatial association by use of distance statistics. Geographical Analysis. 1992;24(3):189–206. [Google Scholar]
- Giannone G., Hosy E., Levet F., Constals A., Schulze K., Sobolevsky A.I. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophysical Journal. 2010;99(4):1303–1310. doi: 10.1016/j.bpj.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan D.E., Veatch W. Lateral mobility of band 3 in the human erythrocyte membrane studied by fluorescence photobleaching recovery: evidence for control by cytoskeletal interactions. Proceedings of the National Academy of Sciences USA. 1980;77(5):2537–2541. doi: 10.1073/pnas.77.5.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami D., Gowrishankar K., Bilgrami S., Ghosh S., Raghupathy R., Chadda R. Nanoclusters of GPI-anchored proteins are formed by cortical actin-driven activity. Cell. 2008;135(6):1085–1097. doi: 10.1016/j.cell.2008.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfert F., Wurm C.A., Mueller V., Berning S., Cordes V.C., Honigmann A. Coaligned dual-channel STED nanoscopy and molecular diffusion analysis at 20 nm resolution. Biophysical Journal. 2013;105(1):L01–L03. doi: 10.1016/j.bpj.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould T.J., Gunewardene M.S., Gudheti M.V., Verkhusha V.V., Yin S.-R., Gosse J.A. Nanoscale imaging of molecular positions and anisotropies. Nature Methods. 2008;5(12):1027–1030. doi: 10.1038/nmeth.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould T.J., Hess S.T., Bewersdorf J. Optical nanoscopy: from acquisition to analysis. Annual Review of Biomedical Engineering. 2012;14:231–254. doi: 10.1146/annurev-bioeng-071811-150025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar K., Ghosh S., Saha S.C.R., Mayor S., Rao M. Active remodeling of cortical actin regulates spatiotemporal organization of cell surface molecules. Cell. 2012;149(6):1353–1367. doi: 10.1016/j.cell.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Grove J. Super-resolution microscopy: a virus' eye view of the cell. Viruses. 2014;6(3):1365–1378. doi: 10.3390/v6031365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover J.R., Veatch S.L., Ono A. Basic motifs target PSGL-1, CD43, and CD44 to plasma membrane sites where HIV-1 assembles. Journal of Virology. 2015;89(1):454–467. doi: 10.1128/JVI.02178-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudheti M.V., Curthoys N.M., Gould T.J., Kim D., Gunewardene M.S., Gabor K.A. Actin mediates the nanoscale membrane organization of the clustered membrane protein influenza hemagglutinin. Biophysical Journal. 2013;104(10):2182–2192. doi: 10.1016/j.bpj.2013.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunewardene M.S., Subach F.V., Gould T.J., Penoncello G.P., Gudheti M.V., Verkhusha V.V. Super-resolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophysical Journal. 2011;101(6):1522–1528. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N., Wollscheid B., Watts J.D., Scheer B., Aebersold R., DeFranco A.L. Quantitative proteomic analysis of B cell lipid rafts reveals that ezrin regulates antigen receptor-mediated lipid raft dynamics. Nature Immunology. 2006;7(6):625–633. doi: 10.1038/ni1337. [DOI] [PubMed] [Google Scholar]
- Gustafsson M.G. Surpassing the lateral resolution limit by factor of two using structured illumination microscopy. Journal of Microscopy. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Gustafsson M.G. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proceedings of the National Academy of Sciences USA. 2005;102(37):13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J.J., Liu Y., Kruhlak M., Debell K.E., Rellahan B.L., Shaw S. Phospholipase C-mediated hydrolysis of PIP2 releases ERM proteins from lymphocyte membrane. The Journal of Cell Biology. 2009;184(3):451–462. doi: 10.1083/jcb.200807047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harner M., Korner C., Walther D., Mokranjac D., Kaesmacher J., Welsch U. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO Journal. 2011;30(21):4356–4370. doi: 10.1038/emboj.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig J.H., DeSisto M. The cytoskeleton of the resting human blood platelet: structure of the membrane skeleton and its attachment to actin filaments. The Journal of Cell Biology. 1991;112(3):407–425. doi: 10.1083/jcb.112.3.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood N.E., Batista F.D. Early events in B cell activation. Annual Review of Immunology. 2010;28:185–210. doi: 10.1146/annurev-immunol-030409-101216. [DOI] [PubMed] [Google Scholar]
- Hayes M.J., Shao D.M., Grieve A., Levine T., Bailly M., Moss S.E. Annexin A2 at the interface between F-actin and membranes enriched in phosphatidylinositol 4,5,-bisphosphate. Biochimica et Biophysica Acta. 2009;6(95):29. doi: 10.1016/j.bbamcr.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Sun E., Bujny M.V., Kim D., Davidson M.W., Zhuang X. Dual function of CD81 in influenza virus uncoating and budding. PLoS Pathogens. 2013;9(10):e1003701. doi: 10.1371/journal.ppat.1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B.P., Patel H.H., Insel P.A. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochimica et Biophysica Acta. 2014;2:532–545. doi: 10.1016/j.bbamem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein B., Willig K.I., Hell S.W. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proceedings of the National Academy of Sciences USA. 2008;105(38):14271–14276. doi: 10.1073/pnas.0807705105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell S.W. Far-field optical nanoscopy. Science. 2007;316(5828):1153–1158. doi: 10.1126/science.1137395. (Review) [DOI] [PubMed] [Google Scholar]
- Hell S.W., Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Optics Letters. 1994;19(11):780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- Hess S.T., Girirajan T.P., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophysical Journal. 2006;91(11):4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess S.T., Gould T.J., Gudheti M.V., Maas S.A., Mills K.D., Zimmerberg J. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proceedings of the National Academy of Sciences USA. 2007;104(44):17370–17375. doi: 10.1073/pnas.0708066104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess S.T., Huang S., Heikal A.A., Webb W.W. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 2002;41(3):697–705. doi: 10.1021/bi0118512. [DOI] [PubMed] [Google Scholar]
- Hess S.T., Kumar M., Verma A., Farrington J., Kenworthy A., Zimmerberg J. Quantitative electron microscopy and fluorescence spectroscopy of the membrane distribution of influenza hemagglutinin. The Journal of Cell Biology. 2005;169(6):965–976. doi: 10.1083/jcb.200412058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess S.T., Webb W.W. Focal volume optics and experimental artifacts in confocal fluorescence correlation spectroscopy. Biophysical Journal. 2002;83(4):2300–2317. doi: 10.1016/S0006-3495(02)73990-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel L., Smeitink J. The oxidative phosphorylation (OXPHOS) system: nuclear genes and human genetic diseases. Bioessays. 2001;23(6):518–525. doi: 10.1002/bies.1071. [DOI] [PubMed] [Google Scholar]
- Higgs H.N., Pollard T.D. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. The Journal of Cell Biology. 2000;150(6):1311–1320. doi: 10.1083/jcb.150.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden S.J., Uphoff S., Kapanidis A.N. DAOSTORM: an algorithm for high- density super-resolution microscopy. Nature Methods. 2011;8(4):279–280. doi: 10.1038/nmeth0411-279. [DOI] [PubMed] [Google Scholar]
- Honerkamp-Smith A.R., Veatch S.L., Keller S.L. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochimica et Biophysica Acta–Biomembranes. 2009;1788(1):53–63. doi: 10.1016/j.bbamem.2008.09.010. (Review) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigmann A., van den Bogaart G., Iraheta E., Risselada H.J., Milovanovic D., Mueller V. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nature Structural and Molecular Biology. 2013;20(6):679–686. doi: 10.1038/nsmb.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya H., Kobayashi R., Tsukita S., Matsumura F. Ca(2+)-regulated actin and phospholipid binding protein (68 kD-protein) from bovine liver: identification as a homologue for annexin VI and intracellular localization. Cell Motility Cytoskeleton. 1992;22(3):200–210. doi: 10.1002/cm.970220307. [DOI] [PubMed] [Google Scholar]
- Hotta J., Fron E., Dedecker P., Janssen K.P., Li C., Mullen K. Spectroscopic rationale for efficient stimulated-emission depletion microscopy fluorophores. Journal of American Chemical Society. 2010;132(14):5021–5023. doi: 10.1021/ja100079w. [DOI] [PubMed] [Google Scholar]
- Hou Y., Crossman D.J., Rajagopal V., Baddeley D., Jayasinghe I., Soeller C. Super-resolution fluorescence imaging to study cardiac biophysics: alpha-actinin distribution and Z-disk topologies in optically thick cardiac tissue slices. Progress in Biophysics and Molecular Biology. 2014;115(2–3):328–339. doi: 10.1016/j.pbiomolbio.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Hsu C.J., Baumgart T. Spatial association of signaling proteins and F-actin effects on cluster assembly analyzed via photoactivation localization microscopy in T cells. PLoS One. 2011;6(8):24. doi: 10.1371/journal.pone.0023586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F., Hartwich T.M., Rivera-Molina F.E., Lin Y., Duim W.C., Long J.J. Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nature Methods. 2013;10(7):653–658. doi: 10.1038/nmeth.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B., Wang W.Q., Bates M., Zhuang X.W. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319(5864):810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingerman E., Perkins E.M., Marino M., Mears J.A., McCaffery J.M., Hinshaw J.E. Dnm1 forms spirals that are structurally tailored to fit mitochondria. The Journal of Cell Biology. 2005;170(7):1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itano M.S., Graus M.S., Pehlke C., Wester M.J., Liu P., Lidke K.A. Super-resolution imaging of C-type lectin spatial rearrangement within the dendritic cell plasma membrane at fungal microbe contact sites. Frontiers in Physics. 2014;2:46. doi: 10.3389/fphy.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeddin I., Specht C.G., Lelek M., Darzacq X., Triller A., Zimmer C. Super-resolution dynamic imaging of dendritic spines using a low-affinity photoconvertible actin probe. PLoS One. 2011;6(1):0015611. doi: 10.1371/journal.pone.0015611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackle S., Beisiegel U., Rinninger F., Buck F., Grigoleit A., Block A. Annexin VI, a marker protein of hepatocytic endosomes. The Journal of Biological Chemistry. 1994;269(2):1026–1032. [PubMed] [Google Scholar]
- Jans D.C., Wurm C.A., Riedel D., Wenzel D., Stagge F., Deckers M. STED super-resolution microscopy reveals an array of MINOS clusters along human mitochondria. Proceedings of the National Academy of Sciences USA. 2013;110(22):8936–8941. doi: 10.1073/pnas.1301820110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K., Loerke D., Mettlen M., Kuwata H., Grinstein S., Schmid S.L. Robust single-particle tracking in live-cell time-lapse sequences. Nature Methods. 2008;5(8):695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaumouille V., Farkash Y., Jaqaman K., Das R., Lowell C.A., Grinstein S. Actin cytoskeleton reorganization by Syk regulates Fcgamma receptor responsiveness by increasing its lateral mobility and clustering. Developmental Cell. 2014;29(5):534–546. doi: 10.1016/j.devcel.2014.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasundar J.J., Ju J.H., He L., Liu D., Meilleur F., Zhao J. Open conformation of ezrin bound to phosphatidylinositol 4,5-bisphosphate and to F-actin revealed by neutron scattering. The Journal of Biological Chemistry. 2012;287(44):37119–37133. doi: 10.1074/jbc.M112.380972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S., Vaughan J.C., Zhuang X. Isotropic 3D super-resolution imaging with a self-bending point spread function. Nature Photonics. 2014;8:302–306. doi: 10.1038/nphoton.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A., Beal M.F. Mitochondrial dysfunction in neurodegenerative diseases. Journal of Pharmacology and Experimental Therapeutics. 2012;342(3):619–630. doi: 10.1124/jpet.112.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.A., Shim S.H., He J., Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nature Methods. 2011;8(6):499–508. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan S., Rodgers W. T cell glycolipid-enriched membrane domains are constitutively assembled as membrane patches that translocate to immune synapses. The Journal of Immunology. 2003;171(1):78–87. doi: 10.4049/jimmunol.171.1.78. [DOI] [PubMed] [Google Scholar]
- Jouvenet N., Neil S.J., Bess C., Johnson M.C., Virgen C.A., Simon S.M. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biology. 2006;4(12):e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette M.F., Gould T.J., Lessard M.D., Mlodzianoski M.J., Nagpure B.S., Bennett B.T. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nature Methods. 2008;5(6):527–529. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- Kaksonen M., Toret C.P., Drubin D.G. Harnessing actin dynamics for clathrin-mediated endocytosis. Nature Reviews Molecular Cell Biology. 2006;7(6):404–414. doi: 10.1038/nrm1940. [DOI] [PubMed] [Google Scholar]
- Kanchanawong P., Shtengel G., Pasapera A.M., Ramko E.B., Davidson M.W., Hess H.F. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468(7323):580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnovsky M.J., Kleinfeld A.M., Hoover R.L., Klausner R.D. The concept of lipid domains in membranes. The Journal of Cell Biology. 1982;94(1):1–6. doi: 10.1083/jcb.94.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova T.S., Baumann C.T., He L., Wu X., Grammer A., Lipsky P. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. Journal of Microscopy–Oxford. 2003;209:56–70. doi: 10.1046/j.1365-2818.2003.01100.x. [DOI] [PubMed] [Google Scholar]
- Keller H., Rentsch P., Hagmann J. Differences in cortical actin structure and dynamics document that different types of blebs are formed by distinct mechanisms. Experimental Cell Research. 2002;277(2):161–172. doi: 10.1006/excr.2002.5552. [DOI] [PubMed] [Google Scholar]
- Kenworthy A.K. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24(3):289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- Kenworthy A.K. Fleeting glimpses of lipid rafts: how biophysics is being used to track them. Journal of Investigative Medicine. 2005;53(6):312–317. doi: 10.2310/6650.2005.53608. [DOI] [PubMed] [Google Scholar]
- Kenworthy A.K., Edidin M. Distribution of a glycosylphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of <100 angstrom using imaging fluorescence resonance energy transfer. The Journal of Cell Biology. 1998;142(1):69–84. doi: 10.1083/jcb.142.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy A.K., Nichols B.J., Remmert C.L., Hendrix G.M., Kumar M., Zimmerberg J. Dynamics of putative raft-associated proteins at the cell surface. The Journal of Cell Biology. 2004;165(5):735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy A.K., Petranova N., Edidin M. High-resolution FRET microscopy of cholera toxin B-subunit and GPI-anchored proteins in cell plasma membranes. Molecular Biology of the Cell. 2000;11(5):1645–1655. doi: 10.1091/mbc.11.5.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner M.W. Implications of treadmilling for the stability and polarity of actin and tubulin polymers in vivo. The Journal of Cell Biology. 1980;86(1):330–334. doi: 10.1083/jcb.86.1.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar T.A., Jakobs S., Dyba M., Egner A., Hell S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proceedings of the National Academy of Sciences USA. 2000;97(15):8206–8210. doi: 10.1073/pnas.97.15.8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kner P., Chhun B.B., Griffis E.R., Winoto L., Gustafsson M.G. Super-resolution video microscopy of live cells by structured illumination. Nature Methods. 2009;6(5):339–342. doi: 10.1038/nmeth.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopek B.G., Shtengel G., Xu C.S., Clayton D.A., Hess H.F. Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proceedings of the National Academy of Sciences USA. 2012;109(16):6136–6141. doi: 10.1073/pnas.1121558109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel D.E. Association dynamics and lateral transport in biological membranes. Journal of Supramolecular Structure and Cellular Biochemistry. 1981;17(1):61–67. doi: 10.1002/jsscb.380170107. [DOI] [PubMed] [Google Scholar]
- Korlach J., Schwille P., Webb W.W., Feigenson G.W. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proceedings of the National Academy of Sciences USA. 1999;96(15):8461–8466. doi: 10.1073/pnas.96.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov M.M., Chernomordik L.V. A mechanism of protein-mediated fusion: coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophysical Journal. 1998;75(3):1384–1396. doi: 10.1016/S0006-3495(98)74056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft M.L. Plasma membrane organization and function: moving past lipid rafts. Molecular Biology of the Cell. 2013;24(18):2765–2768. doi: 10.1091/mbc.E13-03-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukat C., Wurm C.A., Spahr H., Falkenberg M., Larsson N.G., Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proceedings of the National Academy of Sciences USA. 2011;108(33):13534–13539. doi: 10.1073/pnas.1109263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M., Kenworthy A.K., Roth M.G., Zimmerberg J. Raft association plays an important role in influenza hemagglutinin-mediated fusion. Molecular Biology of the Cell. 2001;12:74A. (Meeting abstract) [Google Scholar]
- Kumari S., Curado S., Mayya V., Dustin M.L. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochimica et Biophysica Acta. 2014;2:546–556. doi: 10.1016/j.bbamem.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumi A., Fujiwara T.K., Chadda R., Xie M., Tsunoyama T.A., Kalay Z. Dynamic organizing principles of the plasma membrane that regulate signal transduction: commemorating the fortieth anniversary of Singer and Nicolson's fluid-mosaic model. Annual Review of Cell and Developmental Biology. 2012;28:215–250. doi: 10.1146/annurev-cellbio-100809-151736. [DOI] [PubMed] [Google Scholar]
- Kusumi A., Sako Y. Cell surface organization by the membrane skeleton. Current Opinion in Cell Biology. 1996;8(4):566–574. doi: 10.1016/s0955-0674(96)80036-6. [DOI] [PubMed] [Google Scholar]
- Kwik J., Boyle S., Fooksman D., Margolis L., Sheetz M.P., Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proceedings of the National Academy of Sciences USA. 2003;100(24):13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrousse A.M., Zappaterra M.D., Rube D.A., van der Bliek A.M. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Molecular Cell. 1999;4(5):815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- Lakadamyali M., Rust M.J., Babcock H.P., Zhuang X.W. Visualizing infection of individual influenza viruses. Proceedings of the National Academy of Sciences USA. 2003;100(16):9280–9285. doi: 10.1073/pnas.0832269100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson D.R., Gosse J.A., Holowka D.A., Baird B.A., Webb W.W. Temporally resolved interactions between antigen-stimulated IgE receptors and Lyn kinase on living cells. The Journal of Cell Biology. 2005;171(3):527–536. doi: 10.1083/jcb.200503110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasserre R., Guo X.J., Conchonaud F., Hamon Y., Hawchar O., Bernard A.M. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nature Chemical Biology. 2008;4(9):538–547. doi: 10.1038/nchembio.103. [DOI] [PubMed] [Google Scholar]
- Le Floc'h A., Huse M. Molecular mechanisms and functional implications of polarized actin remodeling at the T cell immunological synapse. Cellular and Molecular Life Sciences. 2015;72(3):537–556. doi: 10.1007/s00018-014-1760-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensohn A.M., Kirschner M.W. Activation of the WAVE complex by coincident signals controls actin assembly. Molecular Cell. 2009;36(3):512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Shin J.Y., Lee A., Bustamante C. Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM) Proceedings of the National Academy of Sciences USA. 2012;109(43):17436–17441. doi: 10.1073/pnas.1215175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehninger A.L., Nelson D.L., Cox M.M. 6th ed. W.H. Freeman; New York: 2013. Principles of biochemistry. [Google Scholar]
- Lenne P.F., Wawrezinieck L., Conchonaud F., Wurtz O., Boned A., Guo X.J. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO Journal. 2006;25(14):3245–3256. doi: 10.1038/sj.emboj.7601214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung B.O., Chou K.C. Review of super-resolution fluorescence microscopy for biology. Applied Spectroscopy. 2011;65(9):967–980. doi: 10.1366/11-06398. [DOI] [PubMed] [Google Scholar]
- Lewis A., Isaacson M., Harootunian A., Muray A. Development of a 500 Å spatial resolution light microscope: I. Light is efficiently transmitted through λ/16 diameter apertures. Ultramicroscopy. 1984;13(3):227–231. [Google Scholar]
- Lichtenberg D., Goni F.M., Heerklotz H. Detergent-resistant membranes should not be identified with membrane rafts. Trends in Biochemical Sciences. 2005;30(8):430–436. doi: 10.1016/j.tibs.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nature Reviews Immunology. 2003;3(5):361–370. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- Lillemeier B.F., Mortelmaier M.A., Forstner M.B., Huppa J.B., Groves J.T., Davis M.M. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nature Immunology. 2010;11(1):90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Linde S., Sauer M. How to switch a fluorophore: from undesired blinking to controlled photoswitching. Chemical Society Reviews. 2014;43(4):1076–1087. doi: 10.1039/c3cs60195a. [DOI] [PubMed] [Google Scholar]
- Lingappa J.R., Reed J.C., Tanaka M., Chutiraka K., Robinson B.A. How HIV-1 Gag assembles in cells: putting together pieces of the puzzle. Virus Research. 2014;193:89–107. doi: 10.1016/j.virusres.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Bankaitis V.A. Phosphoinositide phosphatases in cell biology and disease. Progress in Lipid Research. 2010;49(3):201–217. doi: 10.1016/j.plipres.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Cooper T., Howley P.M., Hayball J.D. From crescent to mature virion: vaccinia virus assembly and maturation. Viruses. 2014;6(10):3787–3808. doi: 10.3390/v6103787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A.P., Fletcher D.A. Actin polymerization serves as a membrane domain switch in model lipid bilayers. Biophysical Journal. 2006;91(11):4064–4070. doi: 10.1529/biophysj.106.090852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace E.M., Orange J.S. Dual channel STED nanoscopy of lytic granules on actin filaments in natural killer cells. Communicative and Integrative Biology. 2012;5(2):184–186. doi: 10.4161/cib.18818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace E.M., Orange J.S. New views of the human NK cell immunological synapse: recent advances enabled by super- and high-resolution imaging techniques. Frontiers in Immunology. 2012;3:421. doi: 10.3389/fimmu.2012.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace E.M., Orange J.S. Lytic immune synapse function requires filamentous actin deconstruction by Coronin 1A. Proceedings of the National Academy of Sciences USA. 2014;111(18):6708–6713. doi: 10.1073/pnas.1314975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGillavry H.D., Blanpied T.A. Single-molecule tracking photoactivated localization microscopy to map nano-scale structure and dynamics in living spines. Current Protocols in Neuroscience. 2013;2(220):1–2. doi: 10.1002/0471142301.ns0220s65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka B., Czogalla A., Hryniewicz-Jankowska A., Boguslawska D.M., Grochowalska R., Heger E. Spectrins: a structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochimica et Biophysica Acta–Biomembranes. 2014;1838(2):620–634. doi: 10.1016/j.bbamem.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Magde D., Elson E.L., Webb W.W. Thermodynamic fluctuations in a reacting system—measurement by fluorescence correlation spectroscopy. Physical Review Letters. 1972;29:705–708. [Google Scholar]
- Maglione M., Sigrist S.J. Seeing the forest tree by tree: super-resolution light microscopy meets the neurosciences. Nature Neuroscience. 2013;16(7):790–797. doi: 10.1038/nn.3403. [DOI] [PubMed] [Google Scholar]
- Manes S., del Real G., Martinez C. Pathogens: raft hijackers. Nature Reviews Immunology. 2003;3:557–568. doi: 10.1038/nri1129. [DOI] [PubMed] [Google Scholar]
- Manes S., Viola A. Lipid rafts in lymphocyte activation and migration. Molecular Membrane Biology. 2006;23(1):59–69. doi: 10.1080/09687860500430069. [DOI] [PubMed] [Google Scholar]
- Manley S., Gillette J.M., Patterson G.H., Shroff H., Hess H.F., Betzig E. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nature Methods. 2008;5(2):155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- Martin-Belmonte F., Mostov K. Phosphoinositides control epithelial development. Cell Cycle. 2007;6(16):1957–1961. doi: 10.4161/cc.6.16.4583. [DOI] [PubMed] [Google Scholar]
- Mattila P.K., Feest C., Depoil D., Treanor B., Montaner B., Otipoby K.L. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity. 2013;38(3):461–474. doi: 10.1016/j.immuni.2012.11.019. [DOI] [PubMed] [Google Scholar]
- Maxfield F.R., Menon A.K. Intracellular sterol transport and distribution. Current Opinion in Cell Biology. 2006;18(4):379–385. doi: 10.1016/j.ceb.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Maxfield F.R., Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438(7068):612–621. doi: 10.1038/nature04399. (Review) [DOI] [PubMed] [Google Scholar]
- Menon A.K., Holowka D., Webb W.W., Baird B. Cross-linking of receptor-bound IgE to aggregates larger than dimers leads to rapid immobilization. The Journal of Cell Biology. 1986;102(2):541–550. doi: 10.1083/jcb.102.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer L., Wildanger D., Medda R., Punge A., Rizzoli S.O., Donnert G. Dual-color STED microscopy at 30-nm focal-plane resolution. Small. 2008;4(8):1095–1100. doi: 10.1002/smll.200800055. [DOI] [PubMed] [Google Scholar]
- Michel V., Bakovic M. Lipid rafts in health and disease. Biology of the Cell. 2007;99(3):129–140. doi: 10.1042/BC20060051. (Review) [DOI] [PubMed] [Google Scholar]
- Mizuno H., Abe M., Dedecker P., Makino A., Rocha S., Ohno-Iwashita Y. Fluorescent probes for superresolution imaging of lipid domains on the plasma membrane. Chemical Science. 2011;2(8):1548–1553. doi: 10.1039/C1SC00169H. [DOI] [Google Scholar]
- Mlodzianoski M.J., Schreiner J.M., Callahan S.P., Smolkova K., Dlaskova A., Santorova J. Sample drift correction in 3D fluorescence photoactivation localization microscopy. Optics Express. 2011;19(16):15009–15019. doi: 10.1364/OE.19.015009. [DOI] [PubMed] [Google Scholar]
- Moore B.M., Lentz B.R., Meissner G. Effects of sarcoplasmic reticulum Ca2+-ATPase on phospholipid bilayer fluidity: boundary lipid. Biochemistry. 1978;17(24):5248–5255. doi: 10.1021/bi00617a026. [DOI] [PubMed] [Google Scholar]
- Muriel O., Echarri A., Hellriegel C., Pavon D.M., Beccari L., Del Pozo M.A. Phosphorylated filamin A regulates actin-linked caveolae dynamics. Journal of Cell Science. 2011;124(Pt 16):2763–2776. doi: 10.1242/jcs.080804. [DOI] [PubMed] [Google Scholar]
- Nayak D.P., Hui E.K., Barman S. Assembly and budding of influenza virus. Virus Research. 2004;106(2):147–165. doi: 10.1016/j.virusres.2004.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebl T., Pestonjamasp K.N., Leszyk J.D., Crowley J.L., Oh S.W., Luna E.J. Proteomic analysis of a detergent-resistant membrane skeleton from neutrophil plasma membranes. The Journal of Biological Chemistry. 2002;277(45):43399–43409. doi: 10.1074/jbc.M205386200. [DOI] [PubMed] [Google Scholar]
- Nelson A.J., Gunewardene M.S., Hess S.T. Vol. 9169. 2014. High speed fluorescence photoactivation localization microscopy imaging. (Proc. SPIE 9169, Nanoimaging and Nanospectroscopy II). 91690p. [Google Scholar]
- Nicolson G.L. The Fluid–Mosaic model of membrane structure: still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochimica et Biophysica Acta–Biomembranes. 2014;1838(6):1451–1466. doi: 10.1016/j.bbamem.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Okabe S. Molecular dynamics of the excitatory synapse. Advances in Experimental Medicine and Biology. 2012;970:131–152. doi: 10.1007/978-3-7091-0932-8_6. [DOI] [PubMed] [Google Scholar]
- Orange J.S., Harris K.E., Andzelm M.M., Valter M.M., Geha R.S., Strominger J.L. The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proceedings of the National Academy of Sciences USA. 2003;100(24):14151–14156. doi: 10.1073/pnas.1835830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikane Y., Kataoka T., Okuda M., Hara S., Inoue H., Nakano M. Observation of nanostructure by scanning near-field optical microscope with small sphere probe. Science and Technology of Advanced Materials. 2007;8:181–185. [Google Scholar]
- Otera H., Mihara K. Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases. 2011;2(3):167–172. doi: 10.4161/sgtp.2.3.16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H., Wang C., Cleland M.M., Setoguchi K., Yokota S., Youle R.J. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. The Journal of Cell Biology. 2010;191(6):1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D.M., Rentero C., Rossy J., Magenau A., Williamson D., Rodriguez M. PALM imaging and cluster analysis of protein heterogeneity at the cell surface. Journal of Biophotonics. 2010;3(7):446–454. doi: 10.1002/jbio.200900089. [DOI] [PubMed] [Google Scholar]
- Palomo G.M., Manfredi G. Exploring new pathways of neurodegeneration in ALS: the role of mitochondria quality control. Brain Research. 2014;6(14):01338–01339. doi: 10.1016/j.brainres.2014.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavani S.R.P., Thompson M.A., Biteen J.S., Lord S.J., Liu N., Twieg R.J. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proceedings of the National Academy of Sciences USA. 2009;106(9):2995–2999. doi: 10.1073/pnas.0900245106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertsinidis A., Zhang Y., Chu S. Subnanometre single-molecule localization, registration and distance measurements. Nature. 2010;466(7306):647–651. doi: 10.1038/nature09163. [DOI] [PubMed] [Google Scholar]
- Philimonenko A.A., Janacek J., Hozak P. Statistical evaluation of colocalization patterns in immunogold labeling experiments. Journal of Structural Biology. 2000;132(3):201–210. doi: 10.1006/jsbi.2000.4326. [DOI] [PubMed] [Google Scholar]
- Pichler H., Riezman H. Where sterols are required for endocytosis. Biochimica et Biophysica Acta–Biomembranes. 2004;1666(1–2):51–61. doi: 10.1016/j.bbamem.2004.05.011. (Review) [DOI] [PubMed] [Google Scholar]
- Plowman S.J., Muncke C., Parton R.G., Hancock J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proceedings of the National Academy of Sciences USA. 2005;102(43):15500–15505. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl D.W., Denk W., Lanz M. Optical stethoscopy: image recording with resolution λ/20. Applied Physics Letters. 1984;44(7):651–653. [Google Scholar]
- Polozov I.V., Bezrukov L., Gawrisch K., Zimmerberg J. Progressive ordering with decreasing temperature of the phospholipids of influenza virus. Nature Chemical Biology. 2008;4(4):248–255. doi: 10.1038/nchembio.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pore D., Parameswaran N., Matsui K., Stone M.B., Saotome I., McClatchey A.I. Ezrin tunes the magnitude of humoral immunity. The Journal of Immunology. 2013;191(8):4048–4058. doi: 10.4049/jimmunol.1301315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl R., Soubannier V., Scholz R., Vogel F., Mendl N., Vasiljev-Neumeyer A. Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. The Journal of Cell Biology. 2009;185(6):1047–1063. doi: 10.1083/jcb.200811099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke K., Dohner K., Sodeik B. Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cellular Microbiology. 2006;8(3):387–400. doi: 10.1111/j.1462-5822.2005.00679.x. [DOI] [PubMed] [Google Scholar]
- Rak G.D., Mace E.M., Banerjee P.P., Svitkina T., Orange J.S. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biology. 2011;9(9):13. doi: 10.1371/journal.pbio.1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recktenwald C.V., Lichtenfels R., Wulfaenger J., Müller A., Dressler S.P., Seliger B. Impact of the mitogen-activated protein kinase pathway on the subproteome of detergent-resistant microdomains of colon carcinoma cells. Proteomics. 2015;15(1):77–88. doi: 10.1002/pmic.201300321. [DOI] [PubMed] [Google Scholar]
- van Rheenen J., Song X., van Roosmalen W., Cammer M., Chen X., Desmarais V. EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. The Journal of Cell Biology. 2007;179(6):1247–1259. doi: 10.1083/jcb.200706206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley B.D. Modeling spatial patterns. Journal of Royal Statistical Society: Series B–Methodological. 1977;39(2):172–212. [Google Scholar]
- Rocha-Perugini V., Gordon-Alonso M., Sanchez-Madrid F. PIP2: choreographer of actin-adaptor proteins in the HIV-1 dance. Trends in Microbiology. 2014;22(7):379–388. doi: 10.1016/j.tim.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha-Perugini V., Zamai M., Gonzalez-Granado J.M., Barreiro O., Tejera E., Yanez-Mo M. CD81 controls sustained T cell activation signaling and defines the maturation stages of cognate immunological synapses. Molecular and Cellular Biology. 2013;33(18):3644–3658. doi: 10.1128/MCB.00302-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers W., Farris D., Mishra S. Merging complexes: properties of membrane raft assembly during lymphocyte signaling. Trends in Immunology. 2005;26(2):97–103. doi: 10.1016/j.it.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Rojo L., Sjoberg M.K., Hernandez P., Zambrano C., Maccioni R.B. Roles of cholesterol and lipids in the etiopathogenesis of Alzheimer's disease. Journal of Biomedicine and Biotechnology. 2006;2006(73976):1–17. doi: 10.1155/JBB/2006/73976. (Review) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollason R., Korolchuk V., Hamilton C., Jepson M., Banting G. A CD317/tetherin-RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. The Journal of Cell Biology. 2009;184(5):721–736. doi: 10.1083/jcb.200804154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom A.B., Lee S.H., To M., Lee A., Shin J.Y., Bustamante C. Optimized two-color super resolution imaging of Drp1 during mitochondrial fission with a slow-switching Dronpa variant. Proceedings of the National Academy of Sciences USA. 2014;111(36):13093–13098. doi: 10.1073/pnas.1320044111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossier O., Octeau V., Sibarita J.B., Leduc C., Tessier B., Nair D. Integrins beta1 and beta3 exhibit distinct dynamic nanoscale organizations inside focal adhesions. Nature Cell Biology. 2012;14(10):1057–1067. doi: 10.1038/ncb2588. [DOI] [PubMed] [Google Scholar]
- Rossy J., Pageon S.V., Davis D.M., Gaus K. Super-resolution microscopy of the immunological synapse. Current Opinion in Immunology. 2013;25(3):307–312. doi: 10.1016/j.coi.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Roy N.H., Chan J., Lambele M., Thali M. Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. Journal of Virology. 2013;87(13):7516–7525. doi: 10.1128/JVI.00790-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok R.W., Krijgsman P.C., de Ronde-Verloop F.M., de Jong J.C. Natural heterogeneity of shape, infectivity and protein composition in an influenza A (H3N2) virus preparation. Virus Research. 1985;3:69–76. doi: 10.1016/0168-1702(85)90042-5. [DOI] [PubMed] [Google Scholar]
- Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nature Methods. 2006;3(10):793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarikangas J., Zhao H., Lappalainen P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiological Reviews. 2010;90(1):259–289. doi: 10.1152/physrev.00036.2009. [DOI] [PubMed] [Google Scholar]
- Saini H.K., Arneja A.S., Dhalla N.S. Role of cholesterol in cardiovascular dysfunction. Canadian Journal of Cardiology. 2004;20(3):333–346. (Review) [PubMed] [Google Scholar]
- Saka S.K., Honigmann A., Eggeling C., Hell S.W., Lang T., Rizzoli S.O. Multi-protein assemblies underlie the mesoscale organization of the plasma membrane. Nature Communications. 2014;5:4509. doi: 10.1038/ncomms5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sako Y., Kusumi A. Compartmentalized structure of the plasma membrane for receptor movements as revealed by a nanometer-level motion analysis. The Journal of Cell Biology. 1994;125(6):1251–1264. doi: 10.1083/jcb.125.6.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salbreux G., Charras G., Paluch E. Actin cortex mechanics and cellular morphogenesis. Trends in Cell Biology. 2012;22(10):536–545. doi: 10.1016/j.tcb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Saltel F., Mortier E., Hytonen V.P., Jacquier M.C., Zimmermann P., Vogel V. New PI(4,5)P2- and membrane proximal integrin-binding motifs in the talin head control beta3-integrin clustering. The Journal of Cell Biology. 2009;187(5):715–731. doi: 10.1083/jcb.200908134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P., Rietveld A., Wilk T., Simons K. Influenza viruses select ordered lipid domains during budding from the plasma membrane. The Journal of Biological Chemistry. 1999;274(4):2038–2044. doi: 10.1074/jbc.274.4.2038. [DOI] [PubMed] [Google Scholar]
- Scheiffele P., Roth M.G., Simons K. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO Journal. 1997;16(18):5501–5508. doi: 10.1093/emboj/16.18.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh L., Heintzmann R., Leonhardt H. A guide to super-resolution fluorescence microscopy. The Journal of Cell Biology. 2010;190(2):165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P., Korlach J., Webb W.W. Anomalous subdiffusion of proteins and lipids in membranes observed by fluorescence correlation spectroscopy. Biophysical Journal. 1999;76(1):A391. [Google Scholar]
- Semrau S., Schmidt T. Particle image correlation spectroscopy (PICS): retrieving nanometer-scale correlations from high-density single-molecule position data. Biophysical Journal. 2007;92(2):613–621. doi: 10.1529/biophysj.106.092577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P., Jovanovic-Talisman T., Skoko D., Renz M., Veatch S.L., Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nature Methods. 2011;8(11):969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Varma R., Sarasij R.C., Ira, Gousset K., Krishnamoorthy G. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell. 2004;116(4):577–589. doi: 10.1016/s0092-8674(04)00167-9. [DOI] [PubMed] [Google Scholar]
- Sharonov A., Hochstrasser R.M. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proceedings of the National Academy of Sciences USA. 2006;103(50):18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M.L., Stone K.L., Colangelo C.M., Gulcicek E.E., Palese P. Cellular proteins in influenza virus particles. Plos Pathogens. 2008;4(6):e1000085. doi: 10.1371/journal.ppat.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz M.P. Membrane skeletal dynamics: role in modulation of red cell deformability, mobility of transmembrane proteins, and shape. Seminars in Hematology. 1983;20(3):175–188. [PubMed] [Google Scholar]
- Shelby S.A., Holowka D., Baird B., Veatch S.L. Distinct stages of stimulated FcepsilonRI receptor clustering and immobilization are identified through superresolution imaging. Biophysical Journal. 2013;105(10):2343–2354. doi: 10.1016/j.bpj.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman E., Barr V., Manley S., Patterson G., Balagopalan L., Akpan I. Functional nanoscale organization of signaling molecules downstream of the T cell antigen receptor. Immunity. 2011;35(5):705–720. doi: 10.1016/j.immuni.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman E., Barr V., Samelson L.E. Super-resolution characterization of TCR-dependent signaling clusters. Immunological Reviews. 2013;251(1):21–35. doi: 10.1111/imr.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S.H., Xia C., Zhong G., Babcock H.P., Vaughan J.C., Huang B. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proceedings of the National Academy of Sciences USA. 2012;109(35):13978–13983. doi: 10.1073/pnas.1201882109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff H., Galbraith C.G., Galbraith J.A., Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nature Methods. 2008;5(5):417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff H., Galbraith C.G., Galbraith J.A., White H., Gillette J., Olenych S. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proceedings of the National Academy of Sciences USA. 2007;104(51):20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtengel G., Galbraith J.A., Galbraith C.G., Lippincott-Schwartz J., Gillette J.M., Manley S. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proceedings of the National Academy of Sciences USA. 2009;106(9):3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist S.J., Sabatini B.L. Optical super-resolution microscopy in neurobiology. Current Opinion in Neurobiology. 2012;22(1):86–93. doi: 10.1016/j.conb.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Simons K., Gerl M.J. Revitalizing membrane rafts: new tools and insights. Nature Reviews Molecular Cell Biology. 2010;11(10):688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387(6633):569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons K., van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27(17):6197–6202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- Simons K., Wandinger-Ness A. Polarized sorting in epithelia. Cell. 1990;62(2):207–210. doi: 10.1016/0092-8674(90)90357-k. [DOI] [PubMed] [Google Scholar]
- Singer S.J. The molecuar organization of biological membranes. In: Rothfield L.I., editor. Structure and function of biological membranes. Academic Press; New York: 1971. pp. 145–222. [Google Scholar]
- Singer S.J., Nicolson G.L. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Singleton P.A., Dudek S.M., Chiang E.T., Garcia J.G. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB Journal. 2005;19(12):1646–1656. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- Siraganian R.P. Mast cell signal transduction from the high-affinity IgE receptor. Current Opinion in Immunology. 2003;15(6):639–646. doi: 10.1016/j.coi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annual Review of Biochemistry. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. (Review) [DOI] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Influenza haemagglutinin. Vaccine. 2002;20:S51–S54. doi: 10.1016/s0264-410x(02)00131-7. [DOI] [PubMed] [Google Scholar]
- Sneath P.H. The application of computers to taxonomy. Journal of General Microbiology. 1957;17(1):201–226. doi: 10.1099/00221287-17-1-201. [DOI] [PubMed] [Google Scholar]
- Soares H., Henriques R., Sachse M., Ventimiglia L., Alonso M.A., Zimmer C. Regulated vesicle fusion generates signaling nanoterritories that control T cell activation at the immunological synapse. The Journal of Experimental Medicine. 2013;210(11):2415–2433. doi: 10.1084/jem.20130150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., Kenworthy A.K., Sanders C.R. Cholesterol as a co-solvent and a ligand for membrane proteins. Protein Science. 2014;23(1):1–22. doi: 10.1002/pro.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spendier K., Lidke K.A., Lidke D.S., Thomas J.L. Single-particle tracking of immunoglobulin E receptors (FcepsilonRI) in micron-sized clusters and receptor patches. FEBS Letters. 2012;586(4):416–421. doi: 10.1016/j.febslet.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahelin R.V. Could the Ebola virus matrix protein VP40 be a drug target? Expert Opinion on Therapeutic Targets. 2014;18(2):115–120. doi: 10.1517/14728222.2014.863877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottrup B.L., Veatch S.L., Keller S.L. Nonequilibrium behavior in supported lipid membranes containing cholesterol. Biophysical Journal. 2004;86(5):2942–2950. doi: 10.1016/S0006-3495(04)74345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradal T.E., Rottner K., Disanza A., Confalonieri S., Innocenti M., Scita G. Regulation of actin dynamics by WASP and WAVE family proteins. Trends in Cell Biology. 2004;14(6):303–311. doi: 10.1016/j.tcb.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Stryer L. Fluorescence energy-transfer as a spectroscopic ruler. Annual Review of Biochemistry. 1978;47:819–846. doi: 10.1146/annurev.bi.47.070178.004131. (Review) [DOI] [PubMed] [Google Scholar]
- Stump R.F., Pfeiffer J.R., Seagrave J., Oliver J.M. Mapping gold-labeled IgE receptors on mast cells by scanning electron microscopy: receptor distributions revealed by silver enhancement, backscattered electron imaging, and digital image analysis. Journal of Histochemistry and Cytochemistry. 1988;36(5):493–502. doi: 10.1177/36.5.2965720. [DOI] [PubMed] [Google Scholar]
- Sun Y., Thapa N., Hedman A.C., Anderson R.A. Phosphatidylinositol 4,5-bisphosphate: targeted production and signaling. Bioessays. 2013;35(6):513–522. doi: 10.1002/bies.201200171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sund S.E., Axelrod D. Actin dynamics at the living cell submembrane imaged by total internal reflection fluorescence photobleaching. Biophysical Journal. 2000;79(3):1655–1669. doi: 10.1016/S0006-3495(00)76415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sverdlov M., Shinin V., Place A.T., Castellon M., Minshall R.D. Filamin A regulates caveolae internalization and trafficking in endothelial cells. Molecular Biology of the Cell. 2009;20(21):4531–4540. doi: 10.1091/mbc.E08-10-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. Cholesterol in health and disease. The Journal of Clinical Investigation. 2002;110(5):583–590. doi: 10.1172/JCI16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M., Leser G.P., Russell C.J., Lamb R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proceedings of the National Academy of Sciences USA. 2003;100(25):14610–14617. doi: 10.1073/pnas.2235620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanford C. Hydrophobic effect and organization of living Matter. Science. 1978;200(4345):1012–1018. doi: 10.1126/science.653353. [DOI] [PubMed] [Google Scholar]
- Tanford C. 2nd ed. Krieger; Malabar, Fla: 1991. The hydrophobic effect: Formation of micelles and biological membranes. [Google Scholar]
- Tank D.W., Wu E.S., Webb W.W. Enhanced molecular diffusibility in muscle membrane blebs: release of lateral constraints. The Journal of Cell Biology. 1982;92(1):207–212. doi: 10.1083/jcb.92.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavano R., Contento R.L., Baranda S.J., Soligo M., Tuosto L., Manes S. CD28 interaction with filamin-A controls lipid raft accumulation at the T-cell immunological synapse. Nature Cell Biology. 2006;8(11):1270–1276. doi: 10.1038/ncb1492. [DOI] [PubMed] [Google Scholar]
- Thompson T.E., Tillack T.W. Organization of glycosphingolipids in bilayers and plasma membranes of mammalian cells. Annual Review of Biophysics and Biophysical Chemistry. 1985;14:361–386. doi: 10.1146/annurev.bb.14.060185.002045. [DOI] [PubMed] [Google Scholar]
- Treanor B., Depoil D., Bruckbauer A., Batista F.D. Dynamic cortical actin remodeling by ERM proteins controls BCR microcluster organization and integrity. The Journal of Experimental Medicine. 2011;208(5):1055–1068. doi: 10.1084/jem.20101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji A., Ohnishi S. Restriction of the lateral motion of band 3 in the erythrocyte membrane by the cytoskeletal network: dependence on spectrin association state. Biochemistry. 1986;25(20):6133–6139. doi: 10.1021/bi00368a045. [DOI] [PubMed] [Google Scholar]
- Van Engelenburg S.B., Shtengel G., Sengupta P., Waki K., Jarnik M., Ablan S.D. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science. 2014;343(6171):653–656. doi: 10.1126/science.1247786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S.L. From small fluctuations to large-scale phase separation: lateral organization in model membranes containing cholesterol. Seminars in Cell and Developmental Biology. 2007;18(5):573–582. doi: 10.1016/j.semcdb.2007.08.016. (Review) [DOI] [PubMed] [Google Scholar]
- Veatch S.L., Chiang E.N., Sengupta P., Holowka D.A., Baird B.A. Quantitative nanoscale analysis of IgE-FcepsilonRI clustering and coupling to early signaling proteins. The Journal of Physical Chemistry B. 2012;116(23):6923–6935. doi: 10.1021/jp300197p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S.L., Cicuta P., Sengupta P., Honerkamp-Smith A., Holowka D., Baird B. Critical fluctuations in plasma membrane vesicles. ACS Chemical Biology. 2008;3(5):287–293. doi: 10.1021/cb800012x. [DOI] [PubMed] [Google Scholar]
- Veatch S.L., Gawrisch K., Keller S.L. Closed-loop miscibility gap and quantitative tie-lines in ternary membranes containing diphytanoyl PC. Biophysical Journal. 2006;90(12):4428–4436. doi: 10.1529/biophysj.105.080283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S.L., Keller S.L. Organization in lipid membranes containing cholesterol. Physical Review Letters. 2002;89(26):268101. doi: 10.1103/PhysRevLett.89.268101. [DOI] [PubMed] [Google Scholar]
- Veatch S.L., Keller S.L. A closer look at the canonical “raft mixture” in model membrane studies. Biophysical Journal. 2003;84(1):725–726. doi: 10.1016/S0006-3495(03)74891-7. (Letter) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S.L., Keller S.L. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophysical Journal. 2003;85(5):3074–3083. doi: 10.1016/S0006-3495(03)74726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S.L., Keller S.L. Miscibility phase diagrams of giant vesicles containing sphingomyelin. Physical Review Letters. 2005;94(14):148101. doi: 10.1103/PhysRevLett.94.148101. [DOI] [PubMed] [Google Scholar]
- Veatch S.L., Polozov I.V., Gawrisch K., Keller S.L. Liquid domains in vesicles investigated by NMR and fluorescence microscopy. Biophysical Journal. 2004;86(5):2910–2922. doi: 10.1016/S0006-3495(04)74342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M., Ma X., Adelstein R.S., Horwitz A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nature Reviews Molecular Cell Biology. 2009;10(11):778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola A., Gupta N. Tether and trap: regulation of membrane-raft dynamics by actin-binding proteins. Nature Reviews Immunology. 2007;7(11):889–896. doi: 10.1038/nri2193. (Review) [DOI] [PubMed] [Google Scholar]
- Wang J., Richards D.A. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biology Open. 2012;1(9):857–862. doi: 10.1242/bio.20122071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrezinieck L., Rigneault H., Marguet D., Lenne P.F. Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophysical Journal. 2005;89(6):4029–4042. doi: 10.1529/biophysj.105.067959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner A. Head to tail polymerization of actin. Journal of Molecular Biology. 1976;108(1):139–150. doi: 10.1016/s0022-2836(76)80100-3. [DOI] [PubMed] [Google Scholar]
- Weigel A.V., Simon B., Tamkun M.M., Krapf D. Ergodic and nonergodic processes coexist in the plasma membrane as observed by single-molecule tracking. Proceedings of the National Academy of Sciences USA. 2011;108(16):6438–6443. doi: 10.1073/pnas.1016325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal V., Rizzoli S.O., Lauterbach M.A., Kamin D., Jahn R., Hell S.W. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320(5873):246–249. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- Wiley D.C., Skehel J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza-virus. Annual Review of Biochemistry. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. (Review) [DOI] [PubMed] [Google Scholar]
- Williamson D.J., Owen D.M., Rossy J., Magenau A., Wehrmann M., Gooding J.J. Pre-existing clusters of the adaptor Lat do not participate in early T cell signaling events. Nature Immunology. 2011;12(7):655–662. doi: 10.1038/ni.2049. [DOI] [PubMed] [Google Scholar]
- Winckler P., Lartigue L., Giannone G., De Giorgi F., Ichas F., Sibarita J.B. Identification and super-resolution imaging of ligand-activated receptor dimers in live cells. Scientific Reports. 2013;3:2387. doi: 10.1038/srep02387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.Y., Lin M.W., Wu D.C., Huang Y.B., Huang H.T., Chen C.L. The role of phosphoinositide-regulated actin reorganization in chemotaxis and cell migration. British Journal Pharmacology. 2014;171(24):5541–5554. doi: 10.1111/bph.12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K., Babcock H.P., Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nature Methods. 2012;9(2):185–188. doi: 10.1038/nmeth.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeomans J.M. Oxford University Press; Oxford: 2002. Statistical mechanics of phase transitions. [Google Scholar]
- Yonemura S., Matsui T., Tsukita S. Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: an essential role for polyphosphoinositides in vivo. Journal of Cell Science. 2002;115(Pt 12):2569–2580. doi: 10.1242/jcs.115.12.2569. [DOI] [PubMed] [Google Scholar]
- Yoon Y., Pitts K.R., McNiven M.A. Mammalian dynamin-like protein DLP1 tubulates membranes. Molecular Biology of the Cell. 2001;12(9):2894–2905. doi: 10.1091/mbc.12.9.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias D.A., Violin J.D., Newton A.C., Tsien R.Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296(5569):913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- van Zanten T.S., Cambi A., Koopman M., Joosten B., Figdor C.G., Garcia-Parajo M.F. Hotspots of GPI-anchored proteins and integrin nanoclusters function as nucleation sites for cell adhesion. Proceedings of the National Academy of Sciences USA. 2009;106(44):18557–18562. doi: 10.1073/pnas.0905217106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbes R.M., van der Klei I.J., Veenhuis M., Pfanner N., van der Laan M., Bohnert M. Mitofilin complexes: conserved organizers of mitochondrial membrane architecture. Biological Chemistry. 2012;393(11):1247–1261. doi: 10.1515/hsz-2012-0239. [DOI] [PubMed] [Google Scholar]
- Zhong G., He J., Zhou R., Lorenzo D., Babcock H.P., Bennett V. Developmental mechanism of the periodic membrane skeleton in axons. eLife. 2014;23(3):04581. doi: 10.7554/eLife.04581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R., Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochimica et Biophysica Acta. 2007;6(24):6. doi: 10.1016/j.bbamem.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]