

ABSTRACT
RavZ, an effector protein of pathogenic Legionella pneumophila, inhibits host macroautophagy/autophagy by deconjugation of lipidated LC3 proteins from phosphatidylethanolamine (PE) on the autophagosome membrane. The mechanism for how RavZ specifically recognizes and deconjugates the lipidated LC3s is not clear. To understand the structure-function relationship of LC3-deconjugation by RavZ, we prepared semisynthetic LC3 proteins modified with different fragments of PE or 1-hexadecanol (C16). We find that RavZ activity is strictly dependent on the conjugated PE structure and RavZ extracts LC3–PE from the membrane before deconjugation. Structural and biophysical analysis of RavZ-LC3 interactions suggest that RavZ initially recognizes LC3–PE on the membrane via its N-terminal LC3-interacting region (LIR) motif. RavZ specifically targets to autophagosome membranes by interaction with phosphatidylinositol 3-phosphate (PtdIns3P) via its C-terminal domain and association with membranes via the hydrophobic α3 helix. The α3 helix is involved in extraction of the PE moiety and docking of the fatty acid chains into the lipid-binding site of RavZ, which is related in structure to that of the phospholipid transfer protein Sec14. The LIR interaction and lipid binding facilitate subsequent proteolytic cleavage of LC3–PE. The findings reveal a novel mode of host-pathogen interaction.
KEYWORDS: autophagy, crystal structure, host-pathogen interaction, LC3, Legionella pneumophila, lipidated proteins, native chemical ligation, RavZ, Sec14
Autophagy serves as a defense mechanism against invading pathogens such as bacteria, viruses and parasites (termed xenophagy). The pathogens are sequestered, engulfed by the phagophore, the precursor to the autophagosome, and subsequently eliminated through autophagosome-lysosome fusion. Antibacterial xenophagy recognizes invasive bacteria through autophagy receptors, which bind to ubiquitinated substrates through the ubiquitin-binding domain (UBD) and connect them to the phagophore membrane through the interaction with LC3 proteins via the LC3-interacting region (LIR) motif.
Some bacteria have evolved mechanisms to combat autophagy for their survival. Infection by Legionella pneumophila is a common cause of community and hospital-acquired pneumonia. This bacterium inhibits autophagy by injecting the effector protein RavZ into the host cell. Previous studies showed that RavZ displays an activity of cysteine protease to irreversibly deconjugate lipidated Atg8-family proteins, which could be the key mechanism to inhibit autophagy. RavZ represents an intriguing pathogenic effector for the mechanistic study of autophagosome biogenesis. However, how RavZ recognizes and deconjugates LC3–phosphatidylethanolamine (PE) remains elusive.
To address the structure-function relationship, we produced semisynthetic LC3–PE proteins with different fragments of the PE moiety using chemical approaches. Unlike endogenous ATG4B, RavZ only cleaves the PE-modified LC3, which is independent of membrane binding but strictly dependent on the conjugated lipid structure. RavZ cleaves neither LC3 modified with soluble fragments of PE nor LC3 with 1-hexadecanol. Moreover, RavZ displays a 12-fold reduction of catalytic efficiency toward LC3–PE (6:0) with shorter fatty acid chains. Because the fatty acid chains are buried in the membrane, we speculated that RavZ should contain a lipid-binding site and extract LC3–PE from the membrane before cleavage.
The extraction activity of RavZ was confirmed by in vitro and in vivo assays. Indeed, protease-deficient RavZC258A extracts lipidated LC3 from the cellular membrane in a dose-dependent manner. Overexpression of RavZC258A leads to reduction of GFP-LC3 puncta in the cell and a decrease of SQSTM1/p62 degradation. However, overexpression does not affect the total amount of lipidated LC3 in the cell. These results suggest that RavZC258A inhibits autophagy by extraction of lipidated LC3 from membranes without interfering with the autophagy machinery upstream of LC3. This is in contrast to protease-deficient ATG4BC74A, which inhibits LC3 lipidation by sequestration of unlipidated LC3 through formation of stable complexes in the cytosol. These results also indicate that lipidated Atg8-family proteins are essential for autophagy.
The crystal structure of RavZ exhibits 2 major domains, the N-terminal catalytic domain (residues 49–320) and C-terminal domain (residues 329–432). RavZ1–331 shows similar LC3-binding affinity and catalytic efficiency to the wild type, but its extraction and cleavage activities are reduced in cells. The C-terminal domain has been found to specifically associate with phosphatidylinositol 3-phosphate (PtdIns3P). Therefore, the membrane binding of RavZ is required for extraction and deconjuation of LC3–PE in vivo.
RavZ contains 3 LIR motifs, LIR1 (residues 14–19) and LIR2 (27–32) at the N-terminal region, and LIR3 (433–438) at the C-terminal region. RavZ containing any of the LIR motifs or the LIR2 peptide (DIDEFDLLEGDE) per se binds LC3 in an affinity similar to the RavZ-LC3 interaction (Kd = ca. 300 nM). Therefore, the LIR motif contributes the major binding energy for RavZ-LC3 interaction. However, only LIR2 is crucial for RavZ activity. The LIR2-LC3 interaction is further elucidated by the crystal structure of the LIR2-LC3 fusion protein, which adopts the classical LIR-LC3 binding mode as shown before. However, the crystal structure of the RavZ-LC3 complex displays a different binding mode involving interaction of LIR2 with the α3 loop of its symmetric mate. We showed that such an interaction is due to the crystal packing but does not exist outside the crystal. The results indicate that there is no defined LC3-binding site on RavZ except for the LIR2 loop. It is unclear why RavZ needs multiple LIR motifs. It is possible that RavZ evolved multiple LIRs particularly 2 LIRs at the N-terminal loop to ensure initial recognition of LC3. Because LIR2 has no preference with regard to Atg8-family proteins, RavZ can deconjugate all Atg8-type proteins in the host cell, whereas ATG4 proteins show specificity to individual members of the Atg8 family; it would be interesting to investigate if the LIR motif of ATG4 plays a role in the substrate selectivity. Based on this finding, we were able to inhibit RavZ activity using the LIR2 peptide (IC50 = 43 µM), suggesting a promising avenue for developing therapeutics against Legionella.
The lipid-binding site (LBS) on RavZ was postulated by superimposition of the catalytic domain of RavZ with the yeast phospholipid transfer proteins (Sec14 family). RavZ (α2-α4, β6-β9) shows a similar fold to the core lipid binding domain of Sec14 (α7-α10, β1-β5). Indeed, all LBS mutants exhibit significant reduction of RavZ activity by in vitro cleavage assay, or in vivo extraction and cleavage assays. The structural analysis suggests a highly dynamic and hydrophobic nature of the α3 helix, which may function similarly to the lid helices (α9-α10) of Sec14 that move inward upon lipid binding to form a closed conformation. Because α3 is involved in membrane binding, α3 may play an essential role in digging the conjugated PE moiety out of the membrane. However, the LBS of RavZ is closed without showing a defined pocket for lipid, whereas a clear lipid binding cavity is observed in free Sec14 structures. There must be a conformational change to open the lipid-binding site to accommodate the fatty acid chains of PE.
Our results have provided new insights into how Legionella subverts host autophagy (Fig. 1). The Legionella effector RavZ exhibits both a lipid extraction function structurally related to the phospholipid transfer protein Sec14 family, and cysteine protease activity structurally related to the ubiquitin-like (Ubl)-specific protease family. The former determines the specificity and the latter confers the turnover. Although extraction alone is sufficient to inhibit autophagy, proteolytic cleavage would be more cost effective, as below stoichiometric amounts of RavZ molecules are required. It would be interesting to examine if the RavZ-model also exists in other bacterial effectors that target lipidated proteins, such as the Shigella effector IpaJ with N-myristoylated ARF GTPases and the Yersinia effector YopT with prenylated RHOA GTPases. Legionella bacteria have evolved a specific and cost-effective mechanism during the long-term battle for survival in host cells.
Figure 1.
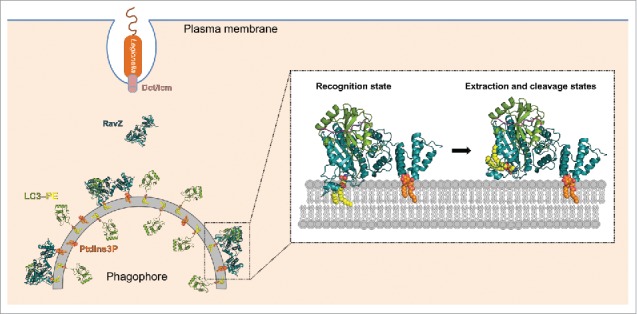
Schematic diagram of the anti-host autophagy mechanism of Legionella pneumophila. The bacterium injects its effector RavZ into the host cell through a type IV secretion system called Dot/Icm. RavZ targets to autophagosome membranes for LC3–PE deconjugation, thereby inhibiting autophagy. RavZ (deep teal) initially recognizes an LC3 molecule (green) on the membrane via its LIR2 motif (magenta). RavZ targets to the autophagosome membrane by interaction with PtdIns3P (orange) via its C-terminal domain and association with membranes via the α3 helix. The α3 facilitates extraction of the PE moiety (yellow) from the membrane and docking of the fatty acid chains into the lipid-binding site of RavZ. The interaction of the PE moiety with LBS and the LIR2-LC3 binding facilitate the binding of the C-terminal tail with the active site for proteolytic cleavage.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Daniel J. Klionsky for critical reading of this manuscript.
Funding
This work was supported by Deutsche Forschungsgemeinschaft, DFG (grant No.: SPP 1623), Behrens Weise Stiftung and European Research Council, ERC (ChemBioAP) to Y.W.W. and by the Introduction of Innovative R&D Team Program of Guangdong Province (2009010058) to A.Y.