

ABSTRACT
Dopamine agonists such as bromocriptine and cabergoline have been successfully used in the treatment of pituitary prolactinomas and other neuroendocrine tumors. However, their therapeutic mechanisms are not fully understood. In this study we demonstrated that DRD5 (dopamine receptor D5) agonists were potent inhibitors of pituitary tumor growth. We further found that DRD5 activation increased production of reactive oxygen species (ROS), inhibited the MTOR pathway, induced macroautophagy/autophagy, and led to autophagic cell death (ACD) in vitro and in vivo. In addition, DRD5 protein was highly expressed in the majority of human pituitary adenomas, and treatment of different human pituitary tumor cell cultures with the DRD5 agonist SKF83959 resulted in growth suppression, and the efficacy was correlated with the expression levels of DRD5 in the tumors. Furthermore, we found that DRD5 was expressed in other human cancer cells such as glioblastomas, colon cancer, and gastric cancer. DRD5 activation in these cell lines suppressed their growth, inhibited MTOR activity, and induced autophagy. Finally, in vivo SKF83959 also inhibited human gastric cancer cell growth in nude mice. Our studies revealed novel mechanisms for the tumor suppressive effects of DRD5 agonists, and suggested a potential use of DRD5 agonists as a novel therapeutic approach in the treatment of different human tumors and cancers.
KEYWORDS: autophagic cell death, cabergoline, dopamine agonist, dopamine receptor D5, MTOR, prolactinoma, reactive oxygen species
Introduction
Dopaminergic pathways are involved in many neurological and other physiological processes, and dopamine agonists (DAs) have been widely used clinically in the treatment of prolatinomas, Parkinson disease and other neuropsychonic disorders.1 The functions of DAs are mediated by dopamine receptors (DRs). There are 5 different DR subtypes, named DRD1 to DRD5, which can be divided into 2 groups, D1-like receptors (including DRD1 and DRD5) and D2-like receptors (DRD2, DRD3, and DRD4).2 Although both groups are G protein-coupled receptors and activated by dopamine, they have different signaling pathways: D1-like receptors are coupled with GNAS/Gsα, which activate ADCY (adenylate cyclase) and lead to the increase of intracellular levels of cyclic adenosine monophosphate (cAMP);3 whereas D2-like receptors are coupled with GNAI/Giα, which inhibit ADCY activity and cAMP production.2
DAs, such as bromocriptine (BRC) and cabergoline (CAB), have been used successfully in the treatment of prolactinomas, by suppressing prolactin secretion as well as shrinking the tumors.4-6 Previous studies on the tumor shrinkage by DAs have mainly been focused on DRD2 activation. For example, it has been shown that BRC and CAB can activate the short form of DRD2 and induce apoptosis.7,8 In the rat pituitary somatolactotroph cell line GH3, induction of DRD2 expression by epigenetic modification can result in CASP3/caspase-3 activation and apoptosis.9
Previously, we have found that CAB-induced cell death in rat pituitary cells was only partially blocked by DRD2 antagonist, suggesting other DRs may also be involved in mediating the growth suppressive function by DAs.10 It is less clear as to the involvement of D1-like receptors in the antitumor effects of DAs, especially for DRD5. Although it is well known that DRD5 is involved in diversified physiological and pathological processes, its tumor suppressive function has not been reported. In this study, we report that DRD5 mediates tumor growth suppression by activation of autophagy, leading to autophagic cell death (ACD) in pituitary tumors as well as in other human cancer cells.
Results
DRD5 is involved in CAB-mediated suppression of pituitary tumor cell growth
We have previously shown that in mouse pituitary tumor cell lines MMQ and GH3, CAB treatment decreases cell viability in a dose-dependent manner.10 To determine which specific DR is involved in the suppression of tumor cell growth, we used quinpirole, which mainly activates DRD2, and SKF83959, which activates DRD5, to investigate their effects on tumor cell growth. As shown in Fig. 1A, 48-h treatment with CAB at 50 μM significantly reduced MMQ cell viability by approximately 50%; and 100 μM of CAB treatment further reduced the viable cell count by 80%. When MMQ cells were treated with the DRD2-specific agonist quinpirole for 48 h, we observed a slight decrease in cell viability by approximately 20% to 25%; and this decrease was not affected by increasing quinpirole concentration from 1 μM to 100 μM (Fig. 1B). Treatment with the DRD5 agonist SKF83959 at 5 μM and 25 μM for 48 h decreased MMQ cell viability by approximately 35% and 56%, respectively (Fig. 1C). In another mouse pituitary tumor cell line, GH3, 48-h treatment with CAB at 100 μM significantly reduced cell viability by approximately 58% (Fig. 1D). However, treatment of GH3 cells with DRD2 agonist quinpirole had no effect on GH3 cell viability (Fig. 1E). In contrast, treatment of GH3 cells with the DRD5 agonist SKF83959 resulted in a dose-dependent decrease in cell viability (Fig. 1F).
Figure 1.

Dopamine 5 receptor is involved in CAB-mediated suppression of pituitary tumor cell growth. (A) Cell viability upon CAB treatment in MMQ cells. At 50 μM and 100 μM, 48-h CAB treatment reduced the viable cell count by 41.3% (p < 0.01) and 81.9% (p < 0.001), respectively. (B) DRD2-specific agonist quinpirole treatment at 48 h slightly reduced MMQ cell viability by 20% to 25% (p < 0.001). (C) DRD5 agonist SKF83959 treatment at 5, 25, and 50 μM reduced MMQ cell viability by 35%, 44%, and 56%, respectively (p < 0.001). (D) Cell viability upon CAB treatment in GH3 cells. At 50 μM and 100 μM, 48-h CAB treatment reduced the viable cell count by 35% (p < 0.05) and 58% (p < 0.01), respectively. (E) DRD2 agonist quinpirole treatment at 48 h had no effect on GH3 cell viability. (F) DRD5 agonist SKF83959 treatment at 5, 25, and 50 μM reduced GH3 cell viability by 19%, 26%, and 58%, respectively (p < 0.001). (G) DRD2 and DRD5 protein expression in MMQ and GH3 cells by western blotting. (H) Drd2 and Drd5 mRNA expression in MMQ and GH3 cells by qRT-PCR. (I) Western blotting showing DRD5 knockdown (KD) in GH3 cells by RNAi. (J) Drd5 KD in GH3 cells abolished CAB-induced growth suppression. At 12 h and 24 h of CAB treatment, Drd5 KD completely blocked growth suppression by CAB. At 48 h of CAB treatment, Drd5 KD reversed CAB-induced growth suppression by doubling the cell viability count. (K) Nonspecific RNAi had no effect on CAB-induced growth suppression in GH3 cells. NC, nonspecific negative control siRNA; Ctrl, control. **, p < 0.01; ***, p < 0.001.
Western blotting revealed the variant expression levels of DRs in these cell lines (Fig. S1A-B). We found that DRD2 was highly expressed in MMQ cells; but there was almost no expression in GH3 cells (Fig. 1G, 1H). In contrast, the expression level of DRD5 was much higher in GH3 cells than that in MMQ cells (Fig. 1G, 1H). Next, we used RNAi to decrease DRD5 expression in GH3 cells and tested its suppressive effect on cell growth by CAB. As shown in Fig. 1I, RNAi resulted in a decrease in DRD5 protein expression by approximately 43%. Consequently, DRD5 knockdown significantly reversed the suppression of GH3 cell growth by CAB (Fig. 1J), but nonspecific negative control siRNA had no effect on CAB-mediated growth suppression of GH3 cells (Fig. 1K).
Taken together, these data indicate that DRD5 is involved in CAB-mediated cell growth suppression.
Suppression of pituitary tumor cell growth by DRD5 activation in vitro and in vivo
Next we investigated whether DRD5 directly contributes to the suppression of tumor cell growth. We chose the GH3 cell line for this and the following parts of our study because it expresses a high level of DRD5 but virtually no DRD2 and very low DRD1 (Fig. S1A-B); and knockdown of DRD1 in GH3 cells did not affect cell viability upon SKF83959 treatment (Fig. S1C). Therefore, the growth-suppressive function of SKF83959 in GH3 cells was mainly attributed to specific activation of DRD5. Flow cytometry revealed that treatment with the DRD5 agonist SKF83959 induced apoptosis in GH3 cells in a dose-dependent manner (Fig. 2A, left). In addition, the increased CASP3 and CASP7 activities as well as cleaved (c-)CASP3 and CASP8 were observed in GH3 cells upon SKF83959 treatment (Fig. 2A, middle and right). In colony formation assays, treatment with the CAB or DRD5 agonist SKF83959 dramatically decreased the numbers of colonies in GH3 cells by approximately 60% (Fig. 2B). Moreover, we documented that the SKF83959-induced decrease in cell viability could be reversed by knockdown of DRD5 expression in GH3 cells (Fig. 2C). Knockdown of DRD5 could also reduce the SKF83959-induced expression of c-CASP3 and c-CASP8 protein (Fig. 2D).
Figure 2.

Suppression of pituitary tumor cell growth by DRD5 activation in vitro and in vivo. (A) DRD5 agonist SKF83959 treatment in GH3 cells induced apoptosis. Left: After 48-h treatment at 12.5, 25, and 50 μM, the apoptotic cell population was 8.4% (p < 0.05), 30.6% (p < 0.001), and 49.5% (p < 0.001), respectively. Middle: CASP3 and CASP7 activity fold was 2.1 (p < 0.05), 4.3 (p < 0.001), and 8.5 (p < 0.001), respectively. Right: Treatment with SKF83959 increased cleaved CASP3 and CASP8. (B) In colony formation assays, SKF83959 and CAB treatment resulted in inhibition of cell colonies by 59% and 63%, respectively (p < 0.001). (C) Drd5 KD in GH3 cells partially abolished SKF83959-induced growth suppression. At 48 h and 72 h, Drd5 KD increased viable cell count from 62% to 92% and from 28% to 68%, respectively (p < 0.001). (D) Drd5 KD in GH3 cells reduced the expression of c-CASP3 and c-CASP8 induced by SKF83959. (E) DRD5 agonist SKF83959 suppressed GH3 tumor growth in nude mice. (F) Comparison of GH3 tumors from mice treated with control vehicle and with SKF83959 at d 11 of drug administration. (G) Tumor weights from mice treated with control vehicle and with SKF83959 at day 11 of drug administration. The average ratio of tumor:nude mice weight from the control group and from the SKF83959-treated group was 46 ± 13.1mg/g and 8 ± 2.5mg/g, respectively. NC, nonspecific negative control siRNA; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
More significantly, we also examined the in vivo tumor suppression by the DRD5 agonist SKF83959. GH3 cells were injected subcutaneously into nude mice, and 5 d after the tumor appearance, the mice were injected with saline for control or SKF83959. A dramatic difference in tumor size was observed between the control group and SKF83959-treatment group after drug administration for 3 d (Fig. 2E). At d 11 after drug administration, the animals were killed and tumors isolated and weighed, showing that the tumors from the SKF83959-treated group were significantly smaller in size (87 ± 33.3 mm3 vs. 669.4 ± 191.9 mm3, p = 0.017) and in tumor/nude mice weight (8 ± 2.5mg/g vs. 46 ± 13.1mg/g, p < 0.05) than those from the control group (Fig. 2F, 2G, Table S1). Therefore, our data indicate that activation of DRD5 may play a major role in the suppression of tumor cell growth in vitro and in vivo.
Inhibition of the MTOR pathway by DRD5
The AKT-MTOR pathway participates in many cellular and physiological functions, including the pathogenesis of human pituitary tumors.11-13 We have previously reported that CAB suppressed MTOR functions in MMQ and GH3 cells.10 Here we determined whether activation of DRD5 could affect the MTOR pathway. Western blotting showed that in GH3 cells, treatment with the DRD5 agonist SKF83959 reduced the protein levels of phosphorylated AKT (p-AKT), phosphorylated MTOR (p-MTOR) and one of its downstream targets, p-EIF4EBP1 (phosphorylated eukaryotic translation initiation factor 4E binding protein 1), indicating that activation of DRD5 inhibited the MTOR pathway (Fig. 3A, 3B). In contrast, we did not observe the decreases in p-MTOR and p-EIF4EBP1 in GH3 cell treated with the DRD2 agonist quinpirole (Fig. 3C). Furthermore, knockdown of DRD5 by RNAi in GH3 cells partially reversed the decrease in p-MTOR and p-EIF4EBP1 protein levels (Fig. 3D), and overexpression of DRD5 further enhanced SKF83959-induced p-MTOR and p-EIF4EBP1 reduction (Fig. 3E), suggesting that DRD5 is specifically involved in suppression of MTOR functions.
Figure 3.

Inhibition of the MTOR pathway by DRD5 activation. (A) Expression of AKT, p-AKT, MTOR, p-MTOR, EIF4EBP1, and p-EIF4EBP1 in GH3 cells after DRD5 agonist SKF83959 treatment at 25 μM by western blotting. (B) Histograms showing decreased p-EIF4EBP1, p-MTOR and p-AKT upon SKF83959 treatment. (C) DRD2 agonist quinpirole failed to reduce the expression of p-MTOR and p-EIF4EBP1 in GH3 cells by western blotting. (D) Drd5 KD abolished SKF83959-induced suppression of p-MTOR and p-EIF4EBP1 in GH3 cells by western blotting. (E) Overexpression of DRD5 in GH3 cells further enhanced SKF83959-induced p-MTOR and p-EIF4EBP1 reduction. **, p < 0.01; ***, p < 0.001.
DRD5 inhibits SOD1 activity and induces ROS production
Next, we investigated the molecular mechanisms underlying how SKF83959 inhibited the AKT-MTOR pathway. It has been reported that reactive oxygen species (ROS) increase can inhibit AKT-MTOR signaling pathways and may play important roles in cell death.14,15,16 ROS are chemically reactive molecules containing oxygen, such as peroxides, superoxide, hydroxyl radicals, and single oxygen, among which O2- is the major ROS signal for autophagy and apoptosis.17,18 SOD1 (superoxide dismutase 1) converts O2- into H2O2, which is further detoxified into H2O and O2 by CAT (catalase) or peroxidases.17,19
We found that SKF83959 inhibited SOD1 but not SOD2 expression, and decreased the SOD activity in GH3 cells in vitro (Fig. 4A). Furthermore, in the xenograft tumors of GH3 cells, we documented that SKF83959 also inhibited SOD1 expression and SOD activity in vivo (Fig. 4B). Accordingly, we observed that treatment of GH3 cells with CAB or the DRD5 agonist SKF83959 increased the ROS content in cells, and this ROS increase was inhibited by N-acetylcysteine (NAC), a ROS inhibitor (Fig. 4C, 4D, and Fig. S2A, S2B). Western blotting revealed that CAB or SKF83959 inhibited the expression of p-AKT and p-MTOR in GH3 cells in vitro. NAC abolished the decrease in the protein levels of p-AKT, p-MTOR and p-EIF4EBP1 induced by CAB or SKF83959 (Fig. 4E, 4F). Similarly, xenograft tumors from the SKF83959-treated group expressed lower levels of p-AKT and p-MTOR compared with tumors from the control group (Fig. 4G). To explore the potential involvement of ROS in DRD5-mediated growth suppression, we used NAC in the cell viability assays. As shown in Fig. 4H and 4I, NAC significantly increased cell viability in GH3 cells treated with CAB or SKF83959. For example, at 48 and 72 h after treatment, NAC reversed the SKF83959 effect by 47% and 57%, respectively (Fig. 4I). In addition, we used ROSup, a ROS-positive inducer, in GH3 cells and found that ROSup significantly inhibited the p-AKT and p-MTOR level in a time-dependent manner, increased the level of c-CASP3 and c-CASP8 and cell death (Fig. S2C-E). In summary, DRD5 activation inhibits MTOR signaling pathways by increasing the ROS level as an upstream signal.
Figure 4.

The role of ROS in CAB- and SKF83959-induced cell death. (A, B) Western blotting showing expression of SOD1, but not SOD2, was decreased after SKF83959 treatment in vitro or in vivo. SOD activity was also reduced upon SKF83959 treatment. (C, D) N-acetylcysteine (NAC), a ROS inhibitor, reversed the CAB- or SKF83959-induced increase of ROS level, from 2.7-fold to 1.6-fold (p < 0.001), and 2.5-fold to 1.8-fold (p < 0.001), respectively. (E, F) Western blotting showing the decreasing expression of p-AKT, p-MTOR and p-EIF4EBP1 by CAB or SKF83959 was reversed by NAC. (G) Western blotting showing the expression of p-AKT, p-MTOR and p-EIF4EBP1 was decreased by SKF83959 in vivo in tumors from nude mice. (H) NAC reversed CAB-induced cell death. At 48 h and 72 h, NAC increased the viable cell count from 30% to 65% and from 40% to 70%, respectively (p < 0.05). (I) NAC reversed SKF83959-induced cell death. At 48 h and 72 h, NAC increased the viable cell count from 46% to 65% and from 31% to 68%, respectively (p < 0.01).
Activation of DRD5 induces autophagy in pituitary tumor cells
The MTOR pathway participates in the regulation of autophagy, a cellular process responsible for protein degradation and turnover of the destroyed cell organelles for new cell formation.11 Autophagy can also lead to cell death under specific circumstances, known as autophagic cell death (ACD).20-23 We previously reported that treatment of MMQ and GH3 cells with CAB induced autophagy and caused cell death.10 As our data presented in Fig. 1 and Fig. 2 indicated the involvement of DRD5 in CAB-mediated cell growth suppression, we investigated whether DRD5 mediated the CAB effect on autophagy activation in these cells. A classical hallmark of autophagy activation is the conversion of MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3)-I to lipidated LC3-II. MMQ cells were treated with the DRD2 agonist quinpirole, the DRD5 agonist SKF83959, or the DRD5 antagonist SKF83566, followed by western blotting to examine LC3-I and LC3-II levels. As shown in Fig. 5A, treatment with the DRD2 agonist quinpirole and the DRD5 antagonist SKF83566 did not alter the LC3-II level in MMQ cells. In contract, treatment with the DRD5 agonist SKF83959 increased the LC3-II level, indicating that SKF83959 treatment resulted in LC3-I to LC3-II conversion. Furthermore, the increase in LC3-II level resulting from the DRD5 agonist SKF83959 treatment was partially diminished by the DRD5 antagonist SKF83566 (Fig. 5B), confirming the involvement of DRD5 in LC3-I to LC3-II conversion.
Figure 5.

Activation of DRD5 induces autophagy and blocks autophagic flux in pituitary tumor cells. (A) MMQ cells were treated with the DRD2 agonist quinpirole at 100 μM (left panel), the DRD5 agonist SKF83959 at 25 μM (right panel), or the DRD5 antagonist SKF83566 at 50 μM (middle panel). Western blotting showed that only treatment with the DRD5 agonist SKF83959 resulted in an increase in the LC3-II level. (B) The DRD5 agonist SKF83959 (12.5 μM)-induced LC3-II increase was partially abolished by the DRD5 antagonist SKF83566. (C) TEM showed the formation of autophagosomes in MMQ cells treated with the DRD5 agonist SKF83959 (SKF83959 vs. Ctrl: 7.2 ± 1.3 vs. 0.9 ± 0.2, n = 40, p < 0.01) but not with the DRD2 agonist quinpirole or the DRD5 antagonist SKF83566. (D) The DRD5 agonist SKF83959 increased LC3 puncta in GH3 cells, indicating the activation of autophagy. ***, p < 0.001. (E-F) Western blotting showed increased SQSTM1 protein expression upon SKF83959 treatment (E), but there was no major change at the Sqstm1 mRNA level by qRT-PCR (F). (G) Flow analysis showing the curve of pH value shifting to the right, indicating that intracellular pH was decreased by SKF83959. (H) Fluorescence microscopy showed the lysosomal proteolytic cleavage. DQ-Red-BSA exhibited bright red fluorescence at 24 h, but not at 48 h and 72 h upon SKF83959 treatment in GH3 cells. (I) Western blotting showing the expression of CTSD and CTSB after SKF83959 treatment of GH3 cells. The expression of mature CTSD and mature CTSB were decreased following the increase of SKF83959 concentration or by SKF83959 (25μM) over time.
Another characteristic of autophagy activation is the formation of autophagosomes by sequestration of cytoplasm within double-layered membrane vesicles (autophagosomes), which fuse with lysosomes, resulting in the degradation of their contents.27 Our TEM (transmission electron microscopy) measurement revealed that treatment with the DRD5 agonist SKF83959 induced the formation of autophagosomes in MMQ cells (SKF83959 vs. Ctrl: 7.2 ± 1.3 vs. 0.9 ± 0.2, n = 40, p < 0.01) (Fig. 5C); in contrast, there was almost no autophagosome formation when MMQ cells were treated with the DRD2 agonist quinpirole or the DRD5 antagonist SKF83566 (Fig. 5C). Similarly, when GH3 cells were treated with the DRD5 agonist SKF83959, there was an increase in LC3-associated green fluorescence, indicating the formation of LC3 puncta (Fig. 5D).
To understand the mechanism of DRD5-induced LC3-II accumulation, we conducted several important experiments. First, we examined the autophagic flux upon SKF83959 treatment. We observed a time-dependent increase in the SQSTM1 protein, a marker of autophagic flux,24-26 after DRD5 agonist SKF83959 treatment in GH3 cells (Fig. 5E), although the mRNA level of Sqstm1 was not dramatically changed (Fig. 5F). These findings indicate that degradation of SQSTM1 was blocked. Confocal microscopy revealed the intact fusion between autophagosomes and lysosomes under SKF83959 exposure (Fig. S3). Flow cytometry demonstrated that SKF83959 augmented lysosome acidification, as demonstrated by staining cells with LysoSensor Green DND-189 (Fig. 5G). Next, we measured the lysosomal degradation capacity using the DQ-BSA assay. As shown in Fig. 5H, SKF83959 treatment of GH3 cells resulted in an initial increase in lysosome proteolytic activity at 24 h, followed by a loss of proteolytic activity at 48 and 72 h, indicating that proteolytic activity was inhibited in the prolonged presence of SKF83959. In addition, CTSD (cathepsin D) and CTSB (cathepsin B), the major enzymes of lysosomal degradation, were decreased in a dose- and time-dependent manner in GH3 cells following SKF83959 treatment (Fig. 5I). Taken together, these data indicate that DRD5 is involved in autophagy activation induced by DAs; however, instead of the completion of the normal autophagy cycles, DRD5 activation blocks autophagic flux by impairing lysosomal degradation.
Critical role of autophagy in DRD5-mediated suppression of pituitary tumor cell growth
Several proteins have been identified to be critical for autophagy, including ATG5, ATG7, and BECN1/Beclin 1.28,29 To confirm autophagy followed by ACD as a mechanism for DRD5-mediated growth suppression, we examined the involvement of these proteins in DRD5-mediated growth suppression. RNAi was used to knockdown the protein expression of the Atg5, Atg7, and Becn1 genes in GH3 cells. As shown in Fig. 6A and 6B, RNAi knockdown reduced the protein level of ATG5, ATG7, and BECN1 by approximately 40%, 70%, and 50%, respectively (Fig. 6A, 6B). In cell viability assays, suppression of cell growth by the DRD5 agonist SKF83959 was severely impaired in GH3 cells with the knockdown of these proteins (Fig. 6C). Knockdown of Atg7 decreased the expression of c-CASP3 and PARP protein (Fig. 6D). Next, we tested the effect of autophagy flux on cell growth suppression induced by SKF83959. Treatment of GH 3 cells with 3-methyladenine (3-MA; 5 μM), an early stage inhibitor of autophagy, increased cell viability from 60% to 77% in the presence of SKF83959 (Fig. 6E). Chloroquine (CQ), a late stage inhibitor of autophagy, inhibited the autophagic flux and decreased the GH3 cell viability in a dose-dependent manner (Fig. S4A-C). CQ at 20 μM began to affect the cell viability and inhibit autophagic flux. When CQ at 20 μM was combined with SKF83959 at 25 μM, GH3 cell viability was further decreased from 60% to 42% (Fig. 6E). The cell death was further confirmed by trypan blue staining (Fig. S5). Western blotting revealed that 3-MA treatment partially blocked SKF83959-induced LC3-II and SQSTM1 accumulation; CQ at 20 μM further increased SQSTM1 and LC3-II (Fig. 6F), as well as c-PARP protein and CASP3-CASP7 activity (Fig. S4D-F). BafA1 had a similar effect with 3-MA and partially rescued the GH3 cell death under SKF83959 exposure (Fig. S6). These findings indicate the critical role of autophagy in DRD5-mediated suppression of pituitary tumor cell growth.
Figure 6.
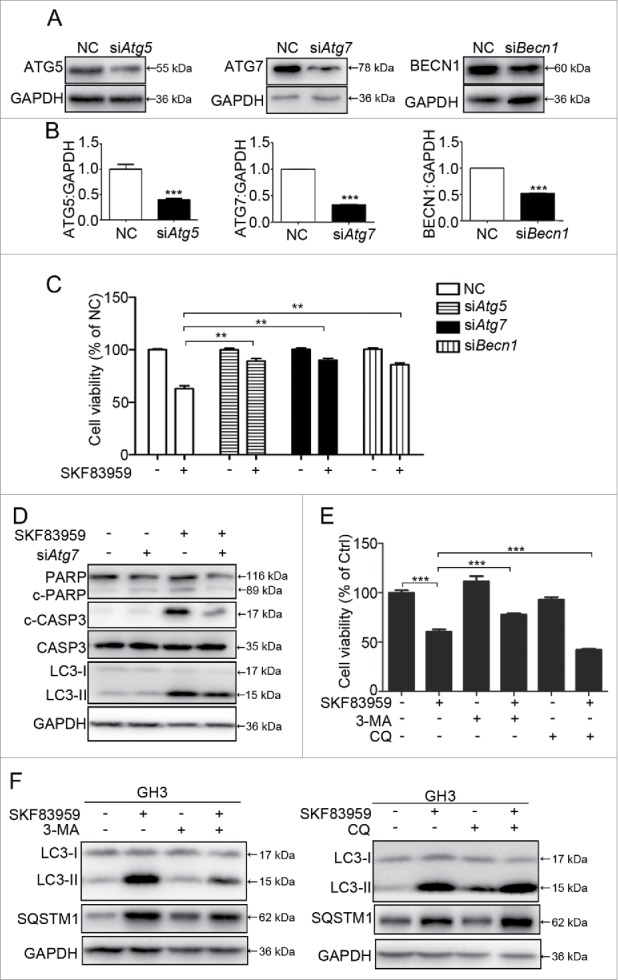
Critical role of autophagy in DRD5-mediated suppression of pituitary tumor cell growth. (A) Knockdown (KD) of ATG5, ATG7, and BECN1 protein by RNAi in GH3 cells. (B) Quantification of ATG5, ATG7, and BECN1 protein KD in GH3 cells. (C) Viable cell counts in GH3 cells with Atg5, Atg7, or Becn1 KD treated with the DRD5 agonist SKF83959, showing the abolishment of SKF83959-induced growth suppression by Atg5, Atg7, or Becn1 KD. (D) Immunoblots analysis of c-CASP3, PARP and LC3 in GH3 cells after Atg7 KD. (E) 3-MA (5 mM) partially abolished and chloroquine (20 μM) increased DRD5 agonist SKF83959-induced growth suppression in GH3 cells. (F) 3-MA (5 mM) partially abolished and chloroquine (20 μM) increased DRD5 agonist SKF83959-induced LC3-II and SQSTM1 accumulation. NC, nonspecific negative control siRNA; **, p < 0.01; ***, p < 0.001.
Growth suppression by DRD5 activation is functional in multiple human cancer cell types
So far we have shown that in the pituitary tumor cell lines MMQ and GH3, representing prolactinomas and GH- and PRL-secreting tumors, activation of DRD5 by DAs suppressed tumor cell growth in vitro and in vivo, through a mechanism of inhibition of the MTOR pathway and activation of autophagy followed by induction of ACD. Next we investigated whether this growth suppressive function of DRD5 was specifically limited in such pituitary tumor cells, or it was a general function working in different types of human pituitary tumors and other cancers. Western blotting demonstrated that DRD5 protein was expressed in the majority of human pituitary adenomas, including PRL-secreting, GH-secreting, and ACTH-secreting adenomas as well as clinically non-functioning adenomas (Fig. 7A). Next, we tested the effect of the DRD5 agonist SKF83959 on in vitro growth of the primary culture of several human pituitary adenomas, including 3 clinically nonfunctioning tumors, 1 GH-secreting tumor, and 1 PRL-secreting tumor (Table S2). As shown in Fig. 7B, SKF83959 treatment at 25 μM resulted in a decrease of cell viability in 4 of the cases; only one clinically nonfunctioning pituitary tumor primary culture did not respond to SKF83959 treatment (Fig. 7B). Immunohistochemical staining showed that the 4 primary pituitary adenomas that were responsive to SKF83959 all expressed DRD5 protein; while in the one tumor that did not respond to SKF83959 (NFPA-3), DRD5 expression was extremely low (Fig. 7C).
Figure 7.
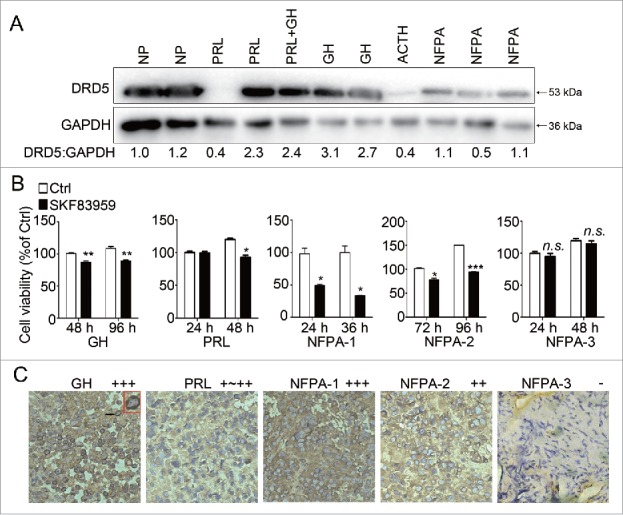
Growth suppression by DRD5 activation in human pituitary tumor primary cultures. (A) Western blotting showing DRD5 expression in human pituitary adenomas of different types. NP, normal human pituitary; PRL, PRL-secreting pituitary adenoma; PRL+GH, human pituitary adenoma secreting both PRL and GH; GH, GH-secreting pituitary adenoma; ACTH, ACTH-secreting pituitary adenoma; NFPA, clinically non-functioning pituitary adenoma. (B) Primary cultures of 5 human pituitary adenomas were treated with DRD5 agonist SKF83959 at 25 μM. The viable cell counts were measured at the indicated time points. SKF83959 decreased viable cells significantly in 4 of 5 pituitary tumor primary cultures. (C) Immunostaining showed high levels of DRD5 in 4 pituitary adenomas responsive to SKF83959 but a very low level of DRD5 in one nonresponsive pituitary tumor.
We also retrieved DRD5 mRNA expression data in cancer cells from The Cancer Genome Atlas (TCGA) database with cBioportal; the results showed that the majority of cancers had high expression of DRD5 mRNA (Fig. S7).30,31 Western blotting confirmed DRD5 protein expression in several different human cancer cell lines, including glioblastoma U87, U251, SHG66; colon cancer SW480; and gastric cancer SCG7901 (Fig. S8). Confocal microscopy also revealed the presence of DRD5 in the membrane and cytoplasm of GH3, U87 and SCG7901 cells (Fig. S9). Therefore, we investigated the effect of SKF83959 in these human cancer cells. As shown in Fig. 8A, SKF83959 suppressed cell growth in U87, U251, SW480, SCG7901, and SHG66 by 41%, 20%, 38%, 42%, and 21%, respectively. SKF83959 increased the expression of c-CASP3 and c-PARP protein (Fig. 8B). Furthermore, in these human cancer cells, SKF83959 treatment increased LC3-II protein and decreased p-MTOR and p-EIF4EBP1, similar to what we observed in GH3 cells (Fig. 8B). LC3-associated green fluorescence (Fig. 8C) and TEM (Fig. 8D and Fig. S10) revealed the formation of LC3 puncta and autophagosomes in these human cancer cells upon SKF83959 treatment. In addition, SKF83959 suppressed cell growth in mouse neuroblastoma N2a cells, and increased LC3-II level and decreased p-EIF4EBP1 level (Fig. S11). These data suggest that activation of DRD5 suppresses in vitro cell growth in multiple human tumors and cancers by inhibiting MTOR functions and inducing autophagy followed by cell death.
Figure 8.

Growth suppression and autophagy activation by DRD5 activation in human cancer cells. (A) SKF83959 (25 μM) treatment at 48 h significantly reduced the viable cell count in human glioblastoma cell lines U87, U251, SHG66, colon cancer cell line SW480, and gastric cancer SCG7901, as measured by MTS assays. (B) Western blotting showing that in these human cancer cell lines, SKF83959 treatment (25 μM) for 48 h resulted in a decrease of p-MTOR and p-EIF4EBP1 and an increase in LC3-II, c-CASP3 and c-PARP, indicating inhibition of MTOR signaling and activation of autophagy. (C) SKF83959 increased LC3-associated green fluorescence in these human cancer cell lines, indicating the activation of autophagy. (D) TEM showed the formation of autolysosomes in SCG7901 and SHG66 cells. (E) DRD5 agonist SKF83959 suppressed tumor growth from human gastric cancer SCG7901 in nude mice. (F) Comparison of SCG7901 tumors from mice treated with control vehicle and with SKF83959 (1 mg/kg) at d 11 of drug administration. (G) Comparison of the ratio of tumor:mouse weights from mice injected with SCG7901 treated with control vehicle and with SKF83959 at d 11 of drug administration. (H) Western blotting showed that compared with the tumors from the control mice the tumors from SKF83959-treated mice expressed more LC3-II, c-CASP3, c-PARP, and less p-MTOR and p-EIF4EBP1. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Finally, using the human gastric cancer cell line SCG7901 as a model, we examined whether DRD5 activation was able to suppress in vivo tumor growth from this human cancer cell line. SCG7901 cells were injected subcutaneously into nude mice to form tumors; and the animals received one injection of saline (for control) or SKF83959 (1 mg/kg). A significant difference in tumor sizes between the control and the SKF83959 group was observed at d 5 after drug administration, and the difference kept increasing over time (Fig. 8E). The animals were killed at d 11 and the tumors isolated and weighed. As shown in Fig. 8F and 8G, the tumors from the SKF83959-treated group were significantly smaller in size (401.5 ± 76 mm3 vs. 916.7 ± 260.3 mm3, p < 0.001) and weight (15 ± 1.3 mg/g vs. 27 ± 1.7mg/g, p < 0.001) than those from the control group, indicating the in vivo tumor suppression ability of DRD5 activation in this human gastric cancer cell line. Western blotting showed that tumors from the SKF83959-treated group expressed more LC3-II, c-CASP3, c-PARP, and less p-MTOR and p-EIF4EBP1 compared with tumors from the control group (Fig. 8H), similar to what we observed in in vitro studies.
Taken together, our data clearly demonstrated the in vitro and in vivo tumor suppressive function by DRD5 activation in different human tumor and cancer cells, presumably using the mechanisms of inhibiting MTOR functions and inducing autophagy followed by cell death.
Discussion
Most of the previous studies on DRD5 have shown its involvement in diversified physiological and pathological processes such as memory and learning,32 hypertension,33,34 immunological modulation,35-37 paranoid schizophrenia,38 and attention-deficit hyperactivity disorder.39 Our study demonstrates the first time that activation of DRD5 suppresses tumor growth in vitro and in vivo in different cell types, by inducing ROS production, inhibiting the MTOR pathway, inducing autophagy, and causing ACD.
Kou and colleagues reported that activation of D1-like receptors, including DRD1 and DRD5, is able to suppress IGF1 (insulin like growth factor 1)-induced vascular smooth muscle cell proliferation via blocking IGF1 expression and phosphorylation.40 Interestingly, this IGF1-induced vascular smooth muscle cell proliferation was also blocked by MTOR inhibitors, suggesting a crosstalk between dopamine signaling and MTOR pathways. Our results here showed that in pituitary tumor cells as well as other human cancer cells, activation of DRD5 inhibited phosphorylation of both MTOR and its downstream targets, and induced ACD, indicating that the suppression of MTOR functions resulted from DRD5 activation.
It has been reported that ROSs are involved in the regulation of autophagy through inhibiting the AKT-MTOR pathway17,19,41 In HeLa cells, α-eleostearic acid induced ROS generation and inhibited the MTOR pathway by AKT suppression, therefore activating autophagy and resulting in ACD.16 Meanwhile, activation of DRD1 has been reported to promote oxidative stress in human neuroblastoma SK-N-MC cells,,42,43 and dopamine induced autophagy-associated cell death in human neuroblastoma SK-SY5Y cells.44 Consistent with these studies, our data showed that activation of DRD5 in GH3 cells inhibited SOD1 activity and expression, and increased ROS content, but blocking of ROS generation by NAC diminished DRD5-induced autophagy and MTOR suppression, and thus protected cells from ACD.
We and others recently showed that DAs induce autophagy.10,45,46 Wang et al. reported that activation of DRD2 and DRD3 induce autophagy in several cell lines, including rat adrenal medulla cell PC12, rat/mouse hybrid mesencephalon cell MES23.5, and human neuroblastoma SH-SY5Y.45 However, according to their report, activation by the D2-like agonist pramipexole is MTOR-independent. Also, the effect of D2-like receptor activation on cell proliferation was not explored in their study. Interestingly, DRD2 antagonists also induce autophagy in cardiac myocytes and SH-SY5Y neuroblastoma cells via an MTOR-independent pathway.47,48 In addition, the effect of DR agonists/antagonists on tumor growth was reported through different mechanisms.49,50 It has also been reported that DR antagonists selectively target cancer stem cells and DR is a potential target of tumor cells.51 Obviously, the mechanisms and biological consequences for D1-like and D2-like receptor activation are quite different.52 We showed here that DRD5 activation was MTOR-dependent, which could result in tumor cell growth suppression.
It has been reported that activation of DRD5 can result in PLC activation, leading to phosphatidylinositol-4,5-bisphosphate hydrolysis and, subsequently, inhibition of PDK1 and AKT, and suppression of the MTOR pathway.3 However, little is known about the biological pathways downstream to MTOR mediated by DRD5. We previously reported that CAB induced autophagy by inhibiting the MTOR pathway.10 Typically autophagy is a process of clearance of unwanted intracellular components. CAB treatment can enhance the formation of autophagosomes; however, CAB can also block autophagic flux and cause the autophagy process to be incomplete, thus leading to autolysosome accumulation within the cells, and ACD.10 Our data in Fig. 5C and Fig. S3 clearly demonstrated the formation of autolysosomes in cells treated with the DRD5 agonist SKF83959, and highlighted the importance of autophagy in DRD5-mediated growth suppression. Conversely, SKF83959 can augment lysosome acidification, while decreasing the lysosomal degradation ability, leading to the blockage of autophagic flux with accumulation of SQSTM1 and LC3. The autophagy inhibitor 3-MA blocked the early initial steps of autophagy, and thus impaired DRD5-mediated growth suppression. In contrast, CQ as a late stage inhibitor of autophagy can further block the autophagic flux.53 Consistently, we have found that CQ was capable of enhancing DRD5-mediated growth suppression, as revealed in Fig. 6E and 6F.
DAs such as bromocriptine and cabergoline have been used to treat prolactinoma successfully, and it has been considered that their pharmacological functions were mainly mediated by DRD2.4,54 However, our study here demonstrated that the tumor suppression effect of DAs is mainly mediated by DRD5, through the inhibition of MTOR signaling and the activation of ACD. Our study has also revealed that DRD5 is widely expressed in different types of human pituitary tumors, including PRL-secreting, GH-secreting, and ACTH-secreting adenomas, as well as clinically non-functioning pituitary adenomas, for which there is currently no approved medical therapy. In addition, DRD5 is also expressed in many malignant human cancer cells such as brain, colon, and gastric cancers. Our data have shown for the first time that treatment with DRD5 agonist can result in growth suppression of human pituitary tumor cells. Furthermore, this DRD5-mediated tumor suppression is not limited in the pituitary. Rather, in several other human cancer cells, DRD5 activation can result in growth suppression and tumor formation. Therefore, our study suggests a potential use of DRD5 agonists as a novel therapeutic approach for the treatment of different human tumors and cancers.
Materials and methods
Cell culture, pituitary tumor tissues, and other reagents
DMEM (11995065), F12 medium (11765062), fetal bovine serum (16000044), horse serum (16050122) and penicillin/streptomycin (15240062) were purchased from Gibco/Life Technologies.
Rat pituitary cell lines GH3 and MMQ cells were purchased from the American Type Culture Collection (ATCC, CCL−82.1, CCL−10609) and were cultured in DMEM (Gibco, c11995500BT) and F12 medium (Gibco, 11765062), respectively, supplemented with 2.5% fetal bovine serum (Gibco, 26140079), 15% horse serum (Gibco, 26050088). Primary human pituitary tumor cells were obtained from pituitary tumor patients who underwent surgery for pituitary tumors and were cultured in DMEM with 10% fetal bovine serum and 100 u/ml penicillin/streptomycin (Gibco, 15240112). All other human cancer cell lines (U87, U251, SW480, SCG7901, SHG66) were gifts from Liang-Fu Zhou (Department of Neurosurgery, Huashan Hospital, Fudan University, Shanghai, China) and were cultured in DMEM medium (Gibco, c11995500BT) supplemented with 10% fetal bovine serum. All of the cells were cultured in an atmosphere of 5% CO2:95% air at 37°C.
Human pituitary tumor tissues were obtained from pituitary tumor patients, who underwent surgery between 2008 and 2014 at the Department of Neurosurgery, Ruijin Hospital of Shanghai Jiaotong University. Non-neoplastic pituitary specimens were obtained from autopsy. Nine pituitary tumor tissues (PRL [prolactin]-secreting adenomas, n = 2; GH [growth hormone]-secreting adenoma, n = 2; mixed hormone secreting tumor, n = 1; POMC/ACTH-secreting adenoma, n = 1; clinically non-functioning pituitary adenomas, n = 3) and 2 normal pituitary tissues were analyzed for DRD5 expression by immunohistochemistry and western blotting. The procedures related to human subjects were approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine. Written informed consent was obtained from all patients whose tumor tissues were used in this study.
Chemical compounds used in this study are listed in Table 1.
Table 1.
Chemical compounds used in this study.
| Chemical compounds | Catalog number | Company |
|---|---|---|
| SKF83959 | 2074 | Tocris Bioscience |
| quinpirole | 1061 | Tocris Bioscience |
| SKF83566 | 1586 | Tocris Bioscience |
| cabergoline | 2664 | Tocris Bioscience |
| DMSO | D2650 | Sigma-Aldrich Chemistry |
| N-acetyl-L-cysteine | S0077 | Beyotime Biotechnology |
| ROSup | S0033 | Beyotime Biotechnology |
| DQ-BSA | D12051 | Thermo Fisher Scientific |
| SOD Assay Kit | S0101 | Beyotime Biotechnology |
| DND-189 | L7535 | Invitrogen |
Cell viability and cell death assays
Cells were plated in 96-well tissue culture plates and then treated with appropriate doses of each drug for a certain time as indicated. Cell survival was assayed using the MTS-based CellTiter 96®AQueous One solution cell proliferation assay (Promega, G3580) and Trypan Blue Staining Cell Viability Assay Kit (Beyotime, S0033) according to the manufacturer's instructions. Upon addition of MTS solution, the reaction plate was incubated at 37°C for 3 h, and the absorbance was read at 490 nm with a plate reader (TECAN, Männedorf, Switzerland). For phosphatidylserine exposure, cells were stained with ANXA5/annexin V-PE as described by the manufacturer (BD Biosciences, San Jose, CA, USA), and assayed by flow cytometry (CyAn ADP, Beckman Coulter, Brea, CA, USA).
Measurement of CASP3-CASP7 activity
Cells were plated in 96-well tissue culture plates and treated with appropriate doses of each drug for a certain time as indicated. CASP3-CASP7 activity was assayed using the Caspase-Glo®3/7 Assay kit (Promega, G8093) according to the manufacturer's instructions with a standard chemiluminescence plate reader (TECAN, Switzerland).
Denaturing SDS-PAGE and western blotting
Cells were lysed in RIPA buffer (Beyotime, P0013C), and total protein concentration was measured using a bicinchoninic acid protein assay kit (Tiangen Biotech, PA115). Proteins from whole cell extracts were resolved using denaturing SDS-PAGE and analyzed by western blotting. Antibodies used in western blotting analyses are listed in Table 2. Immunoblots shown in the accompanying figures are derived from 3 independent experiments. Intensities of protein bands were quantified by densitometry using the software ImageJ (http://imagej.nih.gov/ij/).
Table 2.
Antibodies used in this study.
| Antibodies | Catalog number | Company |
|---|---|---|
| anti-GAPDH | ab181602 | Abcam |
| anti-ATG7 | ab133528 | Abcam |
| anti-ATG5 | ab108327 | Abcam |
| anti-BECN1/beclin1 | 3738 | Cell Signaling Technology |
| anti-ACTB/β-actin | 5057 | Cell Signaling Technology |
| anti-MTOR | 2983 | Cell Signaling Technology |
| anti-phospho-MTOR | 5536 | Cell Signaling Technology |
| anti-EIF4EBP1 | 9644 | Cell Signaling Technology |
| anti-phospho-EIF4EBP1 | 2855 | Cell Signaling Technology |
| anti-DRD2 | sc-7523 | Santa Cruz Biotechnology |
| anti-DRD5 | sc-25650 | Santa Cruz Biotechnology |
| anti-LC3 | L7543 | Sigma-Aldrich Chemistry |
| anti-AKT | 4691 | Cell Signaling Technology |
| anti-p-AKT | 4060 | Cell Signaling Technology |
| anti-SOD1 | 10269 | Proteintech Group |
| anti-SOD2 | 24147 | Proteintech Group |
| anti-DRD1 | ab20066 | Abcam |
| anti-DRD3 | ab42114 | Abcam |
| anti-CTSD | 2284 | Cell Signaling Technology |
| anti-CTSB | 31718 | Cell Signaling Technology |
Gene silencing and overexpression
Cells were transfected with 100 nM nontargeting siRNA or rat Drd5, Drd1, Atg7, Atg5 and Becn1-specific siRNAs using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, 13778–075), in accordance with the manufacturer's instructions, for 72 h. The target sequences of rat Drd5- or Drd1-specific ONTARGET plus siRNAs (Bionner, Korea) were siDrd5 (5′-CGUAUCCCUAGCUGUCUCA-3′, 5′-UGAGACAGCUAGGGAUACG-3′) and siDrd1 (5′-GCCUGUCGAAUGUUCUCAATT-3′, 5′-UUGAGAACAUUCGACAGGCTT-3′) respectively. SiAtg7, siAtg5 and siBecn1-specific siRNAs were synthesized at GenePharma (China), according to the previously published study.10
For overexpression of DRD5, the open reading frame of DRD5 (NM_012768) was cloned into the lentiviral vector GV358 (GeneChem Co., Ltd., China). Transfections were performed using polybrene and enhanced infection solution (GeneChem Co., GOSL71609), according to the manufacturer's instructions.
Real-time RT-PCR
Total RNA was isolated from cultured cells using the Trizol reagent (Invitrogen, 1596018), according to the manufacturer's instructions. PCR primers used were as follows: Drd2 (Rat-forward): 5′-CACCACGGCCTACATAGCAA-3′; Drd2 (Rat- reverse): 5′-GGCTGCCCATTCTTCTCT-3′. Drd5 (Rat-forward): 5′-CCACTGCGCTCCATCCTGAATC-3′; Drd5 (Rat- reverse): 5′-GGCTACTCGCTGGGTCATCTTG-3′. Sqstm1 (Rat-forward): 5′-GACCCCACTTGAGATTCGT-3′; Sqstm1 (Rat-reverse):5′-TGCTCCATCAGAGGATCCCA-3′; Gapdh (Rat-forward): 5′-ACCCTGTTGCTGTAGCCATATTC −3′; Gapdh (Rat- reverse): 5′-ACCCTGTTGCTGTAGCCATATTC-3′.
Transmission electron microscopy (TEM)
Samples were processed in the Electron Microscopy Core at Fudan Univeristy. Cell pellets were fixed with 2.5% glutaraldehyde overnight and post-fixed with 1% osmium tetroxide (pH 7.4) for 2 h at room temperature. The pellets were then dehydrated in a graded ethanol series, infiltrated and embedded in Spurr's resin (Polysciences, 01916–1). Samples were then polymerized for 48 h at 60°C, cut into 60-nm-thick sections on an LKB-I microtome (LKB, Sweden), positioned on 200-mesh grids (Polysciences, 22896–1), and stained with uranyl acetate and lead citrate. TEM was performed on a PHILIPS CM120 TEM at an accelerating voltage of 120 kV. Images were acquired with Gatan type UltraScan 4000SP CCD camera connected to the TEM.
Immunocytochemistry
Cells were grown to 50% confluence on glass coverslips in a 12-well tissue culture plate, and subsequently treated with the appropriate drug at the indicated concentrations for the stated durations. They were washed 3 times with sterile phosphate-buffered saline (PBS; 8.4 mM Na2HPO4, 1.5 mM KH2PO4, 136.9 mM NaCl, 2.7 mM KCl, pH 7.2), fixed and permeabilized in acetone for 10 min, and blocked with 3% BSA (Beyotime, ST023) for 1 h. Primary immunostaining with LC3B antibody (Sigma, L7543) or DRD5 antibody (Santa Cruz Biotechnology, sc-25650) was performed at room temperature overnight, followed by immunostaining with Alexa Fluor 488-conjugated secondary antibody (Invitrogen, A-11008). Cellular DNA was subsequently counterstained with DAPI (Sigma, 28718–90–3) for 15 min at room temperature. Staining was visualized and photographed using an LSM710 laser scanning confocal microscope with a × 63 oil immersion lens (Carl Zeiss, Oberkochen, Germany). Cell images were independently validated by performing cell counting on 5 different magnification fields per image: LC3B puncta per cell = number of LC3B puncta/total number of cells.
Colony formation assay
Cells (2 × 103) were plated in 6-well plates and then treated with CAB at 100 μM or SKF83959 at 25 μM for 2 weeks. Cells were washed twice in 1 × PBS and fixed with methanol for 10 min, followed by staining with 0.5% crystal violet dye (in methanol/deionized water, 1:5; Beyotime, C0121) for 10 min. Excess crystal violet dye was removed by 5 washes of deionized water on a shaker (10 min per wash), and microscopy was obtained by using an Axiovert 200 microscope (Carl Zeiss, Oberkochen, Germany) and cell colonies were counted using ImageJ.
In vivo tumor formation
Female athymic nude mice were purchased from the SLAC (Shanghai Slack Laboratory Animal Co., Ltd., Shanghai, China) and were kept under specific pathogen-free (SPF) conditions. One million GH3 cells or SCG7901 gastric cancer cells were mixed in 1 × PBS and injected subcutaneously into the flanks of 5-wk-old athymic nude mice (n = 10 mice for each cell line). The animals were assigned randomly to 2 groups with 5 animals in each group; 1 group for drug treatment and the other for control. For drug administration, the animals were injected intraperitoneally with 100μl vehicle control (0.1% DMSO in 0.9% saline) or 1 mg/kg SKF83959 daily for 11 d. The tumors were allowed to grow to ∼50 mm3 in size. Tumor ellipsoid volume was estimated as described previously.55 Weight of mice and tumor dimensions were measured every other day after drug injection. All mice were killed and tumors were harvested, followed by photography and western blotting. All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The care and use of athymic nude mice (nu/nu) was approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine.
Determination of ROS levels
Drug-treated cells were washed once with warm PBS and were incubated with 10 μM 2′,7′ -dichlorodihydrofluorescein diacetate (Beyotime, S0033) in warm PBS supplemented with 5.5 mM glucose. After 20 min at 37°C, PBS was replaced with complete culture medium, and cells were incubated for an additional 10–15 min, washed once again with warm PBS, and analyzed using a microplate reader (TECAN, Männedorf, Switzerland).
Tracking of lysosomal pH
For staining with LysoSensor Green DND-189 (Invitrogen, L7535), a pH indicator that exhibits a pH-dependent increase in fluorescence intensity upon acidification, cells were labeled with 1 μM LysoSensor Green DND-189 for 30 min, and then the probe-containing medium was replaced with fresh medium. Then cells were harvested with 0.25% trypsin, and washed twice with PBS, the pH level determined by flow cytometry.
DQ-BSA proteolytic assay
DQ-Red-BSA (Thermo Fisher Scientific, D12051) was used to evaluate lysosomal proteolytic activity as described previously.56 Briefly, GH3 cells were plated in a chamber slide and incubated with DQ-Red-BSA (10 µg/mL) for 2 h at 37°C, followed by washing with PBS. After adding fresh medium, the cells were treated with SKF83959 and control (DMSO) and examined by fluorescence microscopy at 24, 48, and 72 h. Upon lysosomal proteolytic cleavage, DQ-Red-BSA exhibits red fluorescence.
Detection of SOD activity
After incubation with SKF83959 for 48h as described above, cells were harvested with 0.25% trypsin, and washed twice with PBS. The tumor tissues harvested from nude mice were treated with an ultrasound homogenizer. Then, the contents of SOD (superoxide dismutase) were determined using the corresponding detection kits (Total Superoxide Dismutase Assay Kit with WST-8, Beyotime, S0101) according to the manufacturer's instructions.
Immunohistochemical staining of DRD5 in pituitary tumor tissue sections
Antigens were retrieved from formaldehyde-fixed, paraffin-embedded (FFPE) tumor tissue sections by boiling in sodium citrate buffer (pH 6.0) for 30 min using a microwave histoprocessor. The tissue sections were dehydrated and subjected to peroxidase blocking. Immunohistochemical staining was performed by incubating tissue sections using mouse DRD5 primary antibody (Santa Cruz Biotechnology, sc-376088; 1:50 in 1% BSA in TBST [Sangon Biotech, C520009]) overnight at 4°C with gentle shaking, followed by incubation with goat anti-mouse HRP secondary antibody (Abcam, ab6788; 1:200 in 1% BSA in TBST) for 1 h at room temperature. The sections were then exposed to DAB substrate (dissolved in Dako substrate buffer; Roche, 760–500), followed by Gill's Hematoxylin counterstaining (Sangon Biotech, E607317) and standard dehydration treatment. The staining images were obtained by using an Axiovert 200 microscope (Carl Zeiss, Oberkochen, Germany).
Statistics
Statistical significance in experiments was assessed using GraphPad Prism, version 5 (GraphPad Software, La Jolla, CA, USA). Student unpaired, 2-tailed t tests with a 95% confidence interval were used to analyze data involving direct comparison of an experimental group with a control group. Reported P values were adjusted to account for multiple comparisons. A P value less than 0.05 was considered statistically significant.
Abbreviations
- 3-MA
3-methyladenine
- ACD
autophagic cell death
- act-CTSB
activated cathepsin B
- ATG
autophagy related
- BafA1
bafilomycin A1
- BECN1
Beclin 1
- BRC
bromocriptine
- CAB
cabergoline
- cAMP
cyclic adenosine monophosphate
- CASP3
caspase 3
- CASP8
caspase 8
- c-CASP3
cleaved-CASP3
- c-CASP8
cleaved-CASP8
- c-PARP
cleaved-PARP
- CQ
chloroquine
- Ctrl
control
- CTSB
cathepsin B
- CTSD
cathepsin D
- DAPI 4′
6-diamidino-2-phenylindole
- DAs
dopamine agonists
- DRD1
dopamine receptor D1
- DRD2
dopamine receptor D2
- DRD3
dopamine receptor D3
- DRD4
dopamine receptor D4
- DRD5
dopamine receptor D5
- DRs
dopamine receptors
- EIF4EBP1
elongation translation initiation factor 4E binding protein 1
- GH
growth hormone
- IGF1
insulin-like growth factor 1
- im-CTSD
immature cathepsin D
- LV
lentivirus
- MAP1LC3-I/LC3-I
microtubule-associated protein 1 light chain 3-I
- MAP1LC3-II/LC3-II
microtubule-associated protein 1 light chain 3-II
- mat-CTSD
mature cathepsin D
- MTOR
mechanistic target of rapamycin
- NFPA
nonfunctioning pituitary adenoma
- PARP
poly-(ADP-ribose) polymerase
- p-EIF4EBP1
phosphorylated EIF4EBP1
- p-MTOR
phosphorylated MTOR
- POMC/ACTH
proopiomelanocortin
- PRL
prolactin
- pro-CTSB
proenzyme cathepsin B
- RNAi
RNA interference
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- SOD1
superoxide dismutase 1
- SOD2
superoxide dismutase 2
- SPF
specific pathogen-free
- SQSTM1
sequestosome 1
- TEM
transmission electron microscopy
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Natural Science Foundation of China under Grant number 81471392 and 81271523; the Zhejiang Open Foundation of the Top Key Discipline under Grant number LKYJ015; and Shanghai Municipal Education Commission Gaofeng Clinical Medicine Grant Support under Grant number 20161407.
References
- [1].Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev 2006; 27:485-534; PMID:16705142; https://doi.org/ 10.1210/er.2005-9998 [DOI] [PubMed] [Google Scholar]
- [2].Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiological Rev 1998; 78:189-225. PMID:9457173 [DOI] [PubMed] [Google Scholar]
- [3].Undieh AS. Pharmacology of signaling induced by dopamine D(1)-like receptor activation. Pharmacol Ther 2010; 128:37-60; PMID:20547182; https://doi.org/ 10.1016/j.pharmthera.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bevan JS, Webster J, Burke CW, Scanlon MF. Dopamine agonists and pituitary tumor shrinkage. Endocr Rev 1992; 13:220-40; PMID:1352243; https://doi.org/ 10.1210/edrv-13-2-220 [DOI] [PubMed] [Google Scholar]
- [5].Wu ZB, Yu CJ, Su ZP, Zhuge QC, Wu JS, Zheng WM. Bromocriptine treatment of invasive giant prolactinomas involving the cavernous sinus: results of a long-term follow up. J Neurosurg 2006; 104:54-61; PMID:16509147; https://doi.org/ 10.3171/jns.2006.104.1.54 [DOI] [PubMed] [Google Scholar]
- [6].Colao A, Savastano S. Medical treatment of prolactinomas. Nat Rev Endocrinol 2011; 7:267-78; PMID:21423245; https://doi.org/ 10.1038/nrendo.2011.37 [DOI] [PubMed] [Google Scholar]
- [7].Li Q, Su Z, Liu J, Cai L, Lu J, Lin S, Xiong Z, Li W, Zheng W, Wu J, et al.. Dopamine receptor D2S gene transfer improves the sensitivity of GH3 rat pituitary adenoma cells to bromocriptine. Mol Cell Endocr 2014; 382:377-84; PMID:24184771; https://doi.org/ 10.1016/j.mce.2013.10.021 [DOI] [PubMed] [Google Scholar]
- [8].Radl DB, Ferraris J, Boti V, Seilicovich A, Sarkar DK, Pisera D. Dopamine-induced apoptosis of lactotropes is mediated by the short isoform of D2 receptor. PLoS One 2011; 6:e18097; PMID:21464994; https://doi.org/ 10.1371/journal.pone.0018097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rowther FB, Richardson A, Clayton RN, Farrell WE. Bromocriptine and dopamine mediate independent and synergistic apoptotic pathways in pituitary cells. Neuroendocrinology 2010; 91:256-67; PMID:20110659; https://doi.org/ 10.1159/000279753 [DOI] [PubMed] [Google Scholar]
- [10].Lin SJ, Leng ZG, Guo YH, Cai L, Cai Y, Li N, Shang HB, Le WD, Zhao WG, Wu ZB. Suppression of mTOR pathway and induction of autophagy-dependent cell death by cabergoline. Oncotarget 2015; 6:39329-41; PMID:26513171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015; 125:25-32; PMID:25654547; https://doi.org/ 10.1172/JCI73939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Monsalves E, Juraschka K, Tateno T, Agnihotri S, Asa SL, Ezzat S, Zadeh G. The PI3K/AKT/mTOR pathway in the pathophysiology and treatment of pituitary adenomas. Endocrine-Related Cancer 2014; 21:R331-44; PMID:25052915; https://doi.org/ 10.1530/ERC-14-0188 [DOI] [PubMed] [Google Scholar]
- [13].Lee M, Wiedemann T, Gross C, Leinhauser I, Roncaroli F, Braren R, Pellegata NS. Targeting PI3K/mTOR Signaling Displays Potent Antitumor Efficacy against Nonfunctioning Pituitary Adenomas. Clin Cancer Res 2015; 21:3204-15; PMID:25838390; https://doi.org/ 10.1158/1078-0432.CCR-15-0288 [DOI] [PubMed] [Google Scholar]
- [14].Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A 2006; 103:4952-7; PMID:16547133; https://doi.org/ 10.1073/pnas.0511288103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, et al.. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006; 10:241-52; PMID:16959615; https://doi.org/ 10.1016/j.ccr.2006.08.009 [DOI] [PubMed] [Google Scholar]
- [16].Eom JM, Seo MJ, Baek JY, Chu H, Han SH, Min TS, Cho CS, Yun CH. Alpha-eleostearic acid induces autophagy-dependent cell death through targeting AKT/mTOR and ERK1/2 signal together with the generation of reactive oxygen species. Biochem Biophys Res Commun 2010; 391:903-8; PMID:19951696; https://doi.org/ 10.1016/j.bbrc.2009.11.161 [DOI] [PubMed] [Google Scholar]
- [17].Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36:30-8; PMID:20728362; https://doi.org/ 10.1016/j.tibs.2010.07.007 [DOI] [PubMed] [Google Scholar]
- [18].Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009; 16:1040-52; PMID:19407826; https://doi.org/ 10.1038/cdd.2009.49 [DOI] [PubMed] [Google Scholar]
- [19].Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 2010; 6:838-54; PMID:20505317; https://doi.org/ 10.4161/auto.6.7.12113 [DOI] [PubMed] [Google Scholar]
- [20].Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990-5; PMID:15528435; https://doi.org/ 10.1126/science.1099993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115:2679-88; PMID:16200202; https://doi.org/ 10.1172/JCI26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res 2014; 74:647-51; PMID:24459182; https://doi.org/ 10.1158/0008-5472.CAN-13-2966 [DOI] [PubMed] [Google Scholar]
- [23].Fulda S, Kogel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015; 34:5105-13; PMID:25619832; https://doi.org/ 10.1038/onc.2014.458 [DOI] [PubMed] [Google Scholar]
- [24].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; https://doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- [25].Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181-97; PMID:19200883 [DOI] [PubMed] [Google Scholar]
- [26].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; https://doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-21; PMID:11099404; https://doi.org/ 10.1126/science.290.5497.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651-62; PMID:23406030; https://doi.org/ 10.1056/NEJMra1205406 [DOI] [PubMed] [Google Scholar]
- [29].Lebovitz CB, Bortnik SB, Gorski SM. Here, there be dragons: charting autophagy-related alterations in human tumors. Clin Cancer Res 2012; 18:1214-26; PMID:22253413; https://doi.org/ 10.1158/1078-0432.CCR-11-2465 [DOI] [PubMed] [Google Scholar]
- [30].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; https://doi.org/ 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; https://doi.org/ 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].da Silva WC, Kohler CC, Radiske A, Cammarota M. D1/D5 dopamine receptors modulate spatial memory formation. Neurobiol Learn Mem 2012; 97:271-5; PMID:22266268; https://doi.org/ 10.1016/j.nlm.2012.01.005 [DOI] [PubMed] [Google Scholar]
- [33].Banday AA, Lokhandwala MF. Dopamine receptors and hypertension. Curr Hypertens Rep 2008; 10:268-75; PMID:18625155; https://doi.org/ 10.1007/s11906-008-0051-9 [DOI] [PubMed] [Google Scholar]
- [34].Li H, Armando I, Yu P, Escano C, Mueller SC, Asico L, Pascua A, Lu Q, Wang X, Villar VA, et al.. Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J Clin Invest 2008; 118:2180-9; PMID:18464932; https://doi.org/ 10.1172/JCI33637C1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Prado C, Bernales S, Pacheco R. Modulation of T-cell mediated immunity by dopamine receptor d5. Endocr Metab Immune Disord Drug Targets 2013; 13:184-94; PMID:23701196; https://doi.org/ 10.2174/1871530311313020007 [DOI] [PubMed] [Google Scholar]
- [36].Mikulak J, Bozzo L, Roberto A, Pontarini E, Tentorio P, Hudspeth K, Lugli E, Mavilio D. Dopamine inhibits the effector functions of activated NK cells via the upregulation of the D5 receptor. J Immunol 2014; 193:2792-800; PMID:25127864; https://doi.org/ 10.4049/jimmunol.1401114 [DOI] [PubMed] [Google Scholar]
- [37].Prado C, Contreras F, Gonzalez H, Diaz P, Elgueta D, Barrientos M, Herrada AA, Á Lladser, Bernales S, Pacheco R. Stimulation of dopamine receptor D5 expressed on dendritic cells potentiates Th17-mediated immunity. J Immunol 2012; 188:3062-70; PMID:22379034; https://doi.org/ 10.4049/jimmunol.1103096 [DOI] [PubMed] [Google Scholar]
- [38].Zhao Y, Ding M, Pang H, Xu XM, Wang BJ. Relationship between genetic polymorphisms in the DRD5 gene and paranoid schizophrenia in northern Han Chinese. Genet Mol Res 2014; 13:1609-18; PMID:24668635; https://doi.org/ 10.4238/2014.March.12.13 [DOI] [PubMed] [Google Scholar]
- [39].Mill J, Curran S, Richards S, Taylor E, Asherson P. Polymorphisms in the dopamine D5 receptor (DRD5) gene and ADHD. Am J Med Genet B Neuropsychiatr Genet 2004; 125B:38-42; PMID:14755441; https://doi.org/ 10.1002/ajmg.b.20127 [DOI] [PubMed] [Google Scholar]
- [40].Kou X, Han Y, Yang D, Liu Y, Fu J, Zheng S, He D, Zhou L, Zeng C. Dopamine d(1)-like receptors suppress proliferation of vascular smooth muscle cell induced by insulin-like growth factor-1. Clin Exp Hypertens 2014; 36:140-7; PMID:23713966; https://doi.org/ 10.3109/10641963.2013.789048 [DOI] [PubMed] [Google Scholar]
- [41].Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem 2008; 283:31153-62; PMID:18768473; https://doi.org/ 10.1074/jbc.M805056200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen J, Wersinger C, Sidhu A. Chronic stimulation of D1 dopamine receptors in human SK-N-MC neuroblastoma cells induces nitric-oxide synthase activation and cytotoxicity. J Biol Chem 2003; 278:28089-100; PMID:12738794; https://doi.org/ 10.1074/jbc.M303094200 [DOI] [PubMed] [Google Scholar]
- [43].Chen J, Rusnak M, Luedtke RR, Sidhu A. D1 dopamine receptor mediates dopamine-induced cytotoxicity via the ERK signal cascade. J Biol Chem 2004; 279:39317-30; PMID:15247297; https://doi.org/ 10.1074/jbc.M403891200 [DOI] [PubMed] [Google Scholar]
- [44].Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res 2003; 73:341-50; PMID:12868068; https://doi.org/ 10.1002/jnr.10663 [DOI] [PubMed] [Google Scholar]
- [45].Wang JD, Cao YL, Li Q, Yang YP, Jin M, Chen D, Wang F, Wang GH, Qin ZH, Hu LF, et al.. A pivotal role of FOS-mediated BECN1/Beclin 1 upregulation in dopamine D2 and D3 receptor agonist-induced autophagy activation. Autophagy 2015; 11:2057-73; PMID:26649942; https://doi.org/ 10.1080/15548627.2015.1100930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wei C, Gao J, Li M, Li H, Wang Y, Li H, Xu C. Dopamine D2 receptors contribute to cardioprotection of ischemic post-conditioning via activating autophagy in isolated rat hearts. Int J Cardiol 2016; 203:837-9; PMID:26599746; https://doi.org/ 10.1016/j.ijcard.2015.11.006 [DOI] [PubMed] [Google Scholar]
- [47].Yan H, Li WL, Xu JJ, Zhu SQ, Long X, Che JP. D2 dopamine receptor antagonist raclopride induces non-canonical autophagy in cardiac myocytes. J Cell Biochem 2013; 114:103-10; PMID:22886761; https://doi.org/ 10.1002/jcb.24306 [DOI] [PubMed] [Google Scholar]
- [48].Shin JH, Park SJ, Kim ES, Jo YK, Hong J, Cho DH. Sertindole, a potent antagonist at dopamine D(2) receptors, induces autophagy by increasing reactive oxygen species in SH-SY5Y neuroblastoma cells. Biol Pharm Bull 2012; 35:1069-75; PMID:22791154; https://doi.org/ 10.1248/bpb.b12-00009 [DOI] [PubMed] [Google Scholar]
- [49].Sheikhpour M, Ahangari G, Sadeghizadeh M, Deezagi A. A novel report of apoptosis in human lung carcinoma cells using selective agonist of D2-like dopamine receptors: a new approach for the treatment of human non-small cell lung cancer. Int J Immunopathol Pharmacol 2013; 26:393-402; PMID:23755754; https://doi.org/ 10.1177/039463201302600212 [DOI] [PubMed] [Google Scholar]
- [50].Yin T, He S, Shen G, Ye T, Guo F, Wang Y. Dopamine receptor antagonist thioridazine inhibits tumor growth in a murine breast cancer model. Mol Med Rep 2015; 12:4103-8; PMID:26095429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, et al.. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012; 149:1284-97; PMID:22632761; https://doi.org/ 10.1016/j.cell.2012.03.049 [DOI] [PubMed] [Google Scholar]
- [52].Borcherding DC, Tong W, Hugo ER, Barnard DF, Fox S, LaSance K, Shaughnessy E, Ben-Jonathan N. Expression and therapeutic targeting of dopamine receptor-1 (D1R) in breast cancer. Oncogene 2015; 35(24):3103-13; PMID:26477316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Poole B, Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol 1981; 90:665-9; PMID:6169733; https://doi.org/ 10.1083/jcb.90.3.665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wu ZB, Zheng WM, Su ZP, Chen Y, Wu JS, Wang CD, Lin C, Zeng YJ, Zhuge QC. Expression of D2RmRNA isoforms and ERmRNA isoforms in prolactinomas: correlation with the response to bromocriptine and with tumor biological behavior. J Neurooncol 2010; 99:25-32; PMID:20063113; https://doi.org/ 10.1007/s11060-009-0107-y [DOI] [PubMed] [Google Scholar]
- [55].Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 1989; 24:148-54; PMID:2544306; https://doi.org/ 10.1007/BF00300234 [DOI] [PubMed] [Google Scholar]
- [56].Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant-Genevier M, Southall NT, Marugan J, et al.. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci U S A 2015; 112:E1373-81; PMID:25733853; https://doi.org/ 10.1073/pnas.1419669112 [DOI] [PMC free article] [PubMed] [Google Scholar]