

ABSTRACT
Accumulation of profibrotic myofibroblasts is involved in the process of fibrosis development during idiopathic pulmonary fibrosis (IPF) pathogenesis. TGFB (transforming growth factor β) is one of the major profibrotic cytokines for myofibroblast differentiation and NOX4 (NADPH oxidase 4) has an essential role in TGFB-mediated cell signaling. Azithromycin (AZM), a second-generation antibacterial macrolide, has a pleiotropic effect on cellular processes including proteostasis. Hence, we hypothesized that AZM may regulate NOX4 levels by modulating proteostasis machineries, resulting in inhibition of TGFB-associated lung fibrosis development. Human lung fibroblasts (LF) were used to evaluate TGFB-induced myofibroblast differentiation. With respect to NOX4 regulation via proteostasis, assays for macroautophagy/autophagy, the unfolded protein response (UPR), and proteasome activity were performed. The potential anti-fibrotic property of AZM was examined by using bleomycin (BLM)-induced lung fibrosis mouse models. TGFB-induced NOX4 and myofibroblast differentiation were clearly inhibited by AZM treatment in LF. AZM-mediated NOX4 reduction was restored by treatment with MG132, a proteasome inhibitor. AZM inhibited autophagy and enhanced the UPR. Autophagy inhibition by AZM was linked to ubiquitination of NOX4 via increased protein levels of STUB1 (STIP1 homology and U-box containing protein 1), an E3 ubiquitin ligase. An increased UPR by AZM was associated with enhanced proteasome activity. AZM suppressed lung fibrosis development induced by BLM with concomitantly reduced NOX4 protein levels and enhanced proteasome activation. These results suggest that AZM suppresses NOX4 by promoting proteasomal degradation, resulting in inhibition of TGFB-induced myofibroblast differentiation and lung fibrosis development. AZM may be a candidate for the treatment of the fibrotic lung disease IPF.
KEYWORDS: azithromycin, IPF, myofibroblast, NOX4, TGFB
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive and devastating lung parenchymal fibrosis with unknown etiology.1 Recent advances uncovered that perturbation of integrated proteostasis machineries, comprised of crosstalk between the unfolded protein response (UPR), the proteasome, and autophagy, can be involved in the pathogenesis of IPF.2 Increased endoplasmic reticulum (ER) stress accompanied by the UPR and insufficient autophagy have been demonstrated in IPF lungs, which can be associated with increased apoptosis and accelerated cellular senescence in epithelial cells, and with myofibroblast differentiation in fibroblasts.3-5 A recent paper also elucidated that enhanced proteasome activation is responsible for myofibroblast differentiation in fibroblastic foci (FF), a leading edge of fibrosis development in IPF.6 Accordingly, malfunction or aberrant crosstalk between the UPR, autophagy, and the proteasome appears to be a critical determinant in myofibroblast differentiation during IPF pathogenesis. However, precise mechanisms for myofibroblast regulation and effective medical intervention targeting myofibroblast differentiation remain to be determined.
Azithromycin (AZM), a second-generation antibacterial macrolide, has a pleiotropic effect on regulating cellular processes, including immunomodulation, antioxidant activity, and lysosome function.7 Intriguingly, AZM has been demonstrated, through immunomodulation, to have an anti-fibrotic property in the bleomycin mouse model of lung fibrosis.8 AZM accumulates particularly in phagocytes, but also in a variety of cell types, including epithelial cells, lymphocytes, hepatocytes, and fibroblasts.7 Among those cell types, fibroblasts have been proposed to be a potential reservoir, slowly releasing AZM for appropriate redistribution by phagocytes into areas of infection, indicating that azithromycin may also have a role in regulating phenotypic alteration of lung fibroblasts (LF).7 In relation to proteostasis, AZM prevents autophagosome maturation via inhibiting lysosomal acidification, suggesting that AZM may modulate crosstalk between proteostasis machineries in LF.9
Among a wide array of profibrotic cytokines, TGFB (transforming growth factor β) has been strongly implicated in IPF pathogenesis via inducing myofibroblast differentiation in LF.1 Involvement of an increased UPR, enhanced proteasome activity, and insufficient autophagy has been reported in the mechanisms for TGFB-induced myofibroblast differentiation, indicating that proteostasis machineries may play a regulatory role in the TGFB-mediated cell signaling pathway.5,6,10 NOX4 (NADPH oxidase 4)-induced reactive oxygen species (ROS) have an essential role in TGFB-mediated cell signaling and myofibroblast differentiation.11 Recent studies demonstrated that NOX4 has a crucial role in IPF pathogenesis and that NOX4 can be a potential therapeutic target for suppressing lung fibrosis development.12-14 Intriguingly, other macrolides, including erythromycin (EM) and roxithromycin (RXM) have been shown to reduce NOX4 expression levels and inhibit myofibroblast differentiation during TGFB treatment in nasal polyp-derived fibroblasts.15 NOX4 is constitutively active and its protein levels are tightly regulated via ubiquitination with subsequent proteasome degradation in LF.16,17 We hypothesized that AZM may attenuate lung fibrosis development via regulating NOX4 protein levels in LF. Accordingly, in the present study, we examined the ability of AZM to regulate NOX4 protein levels by modulating proteostasis machineries and thus inhibit myofibroblast differentiation as well as bleomycin (BLM)-induced lung fibrosis.
Results
Azithromycin inhibits TGFB1-induced NOX4 and myofibroblast differentiation in LF
Several lines of evidence, including our recent report, suggest that NOX4 is involved in the regulation of TGFB-mediated cell signaling and myofibroblast differentiation.11,18 TGFB1 treatment clearly increased NOX4 protein levels with concomitantly induced extra domain A (EDA)-containing cellular FN1 (fibronectin; EDA-FN1), COL1A1 (collagen type I α 1 chain)/COL1A2, and ACTA2 (actin, α 2, smooth muscle, aorta), all indicative of myofibroblast differentiation (Fig. 1A).19 AZM treatment clearly suppressed both NOX4 protein levels and myofibroblast differentiation (Fig. 1A). TGFB1-induced myofibroblast differentiation and the inhibitory role of AZM were further confirmed by detection of the secreted form of EDA-FN1 and COL1/type I collagen (Fig. S1). Consistent with our recent findings, siRNA-mediated NOX4 knockdown efficiently suppressed TGFB1-induced myofibroblast differentiation (Fig. 1B), suggesting that NOX4 reduction can be involved in the mechanisms for AZM-mediated inhibition of TGFB action.18
Figure 1.

Azithromycin suppresses TGFB-induced NOX4 and myofibroblast differentiation in LF. (A) Western blotting (WB) using anti-EDA-FN1, anti-COL1A1/2 (type I collagen), anti-ACTA2 (actin, α 2, smooth muscle, aorta), anti-NOX4, and anti-ACTB of cell lysates from control (lane 1, 2) and azithromycin (AZM; 10 μg/ml)-treated (lane 3, 4) LF. AZM treatment was started 1 h before TGFB1 (2 ng/ml) stimulation and protein samples were collected after 24 h treatment with TGFB1. In the lower panels are the average ( ± SEM) taken from 7 independent experiments shown as relative expressions. *p < 0.05. (B) WB of cell lysates from control siRNA (lane 1, 2) and NOX4 siRNA- (lane 3, 4) transfected LF. AZM (10 μg/ml) treatment was started 48 h post transfection and 1 h before TGFB1 (2 ng/ml) stimulation. Protein samples were collected after 24-h treatment with TGFB1. The lower panels show the average ( ± SEM) of relative expressions, which were taken from 5 independent experiments. *p < 0.05 and **p < 0.001. (C) WB using anti-phospho-SMAD2, anti-SMAD2, anti-phospho-SMAD3, anti-SMAD3, and anti-ACTB of cell lysates from control (lane 1, 2) and AZM- (10 μg/ml) treated (lane 3, 4) LF. In the lower panels are the average ( ± SEM) taken from 5 independent experiments shown as relative expressions. *p < 0.05. (D) LF were treated with TGFB1 in the presence or absence of AZM (10 μg/ml) and mRNA samples were collected after 24 h treatment with TGFB1 (n = 4). Real time-PCR was performed using primers to NOX4 or ACTB, as a control. NOX4 mRNA expression was normalized to ACTB. Shown is the fold increase ( ± SEM) relative to control-treated cells. *p < 0.05.
TGFB-induced NOX4 expression is regulated via activation of the SMAD pathway, a canonical TGFB signaling pathway.11 To elucidate the mechanisms for AZM-mediated NOX4 regulation, SMAD2/3 activation and changes of NOX4 mRNA expression levels were examined. TGFB1 significantly increased phosphorylated forms of SMAD2/3 (1 h after TGFB1 treatment) and no apparent reduction was demonstrated by AZM pretreatment (Fig. 1C). TGFB1 significantly induced NOX4 mRNA expression (increased to 15.90-fold average) (Fig. 1D). Intriguingly, AZM enhanced NOX4 mRNA expression in response to TGFB1 treatment at 24 h (increased to 27.04-fold average) (Fig. 1D), suggesting that AZM may regulate NOX4 at the protein level.
To further confirm the involvement of NOX4 in TGFB-induced myofibroblast differentiation, GKT137831, a NOX4 inhibitor was used.20 Efficient inhibition of myofibroblast differentiation was observed by treatment with GKT137831 (Fig. S2).
NOX4-mediated ROS production is involved in TGFB1-induced myofibroblast differentiation in LF
NOX4-mediated hydrogen peroxide (H2O2) production has been implicated in regulating TGFB-mediated cell signaling and myofibroblast differentiation.13 To confirm the involvement of NOX4-mediated ROS production in TGFB-induced myofibroblast differentiation, we examined intracellular ROS production by means of the CM-H2DCFDA assay during TGFB1 treatment. A significant increase in ROS production was observed at 24 h after TGFB1 treatment in LF (increased to 1.17-fold average) (P = 0.005, Fig. 2A). Knockdown experiments confirmed that NOX4 is mainly responsible for TGFB1-induced ROS production in LF (Fig. 2B). Consistent with NOX4 knockdown, AZM efficiently suppressed TGFB1-induced ROS production at 24 h in LF (Fig. 2C). N-acetylcysteine (NAC), a representative antioxidant, significantly suppressed TGFB1-induced myofibroblast differentiation of EDA-FN1, COL1/type I collagen, and ACTA2 at the concentration of 10 μM (Fig. 2D), supporting the notion that AZM-mediated suppression of NOX4-induced ROS is at least partly responsible for inhibiting TGFB-induced myofibroblast differentiation.
Figure 2.

NOX4-mediated ROS production is involved in TGFB-induced myofibroblast differentiation in LF. (A) Fluorescence intensity of CM-H2DCFDA staining for intracellular ROS production. After the indicated time treatment with TGFB1, incubation with CM-H2DCFDA (10 µM) was performed for 30 min, fluorescence of DCF was measured using a fluorescence microplate reader. The fluorescence level in the control without TGFB1 was designated as 1.0. Shown panels are the average ( ± SEM) taken from 7 independent experiments. *p < 0.05. (B) Fluorescence intensity of CM-H2DCFDA staining. TGFB1 (2 ng/ml for 24 h) treatment was started 48 h post-siRNA transfection. The fluorescence level in the control siRNA-transfected cells without TGFB1 treatment was designated as 1.0. Shown panels are the average ( ± SEM) taken from 5 independent experiments. *p < 0.05. (C) Fluorescence intensity of CM-H2DCFDA staining. After 24 h treatment with TGFB1in the presence or absence of AZM (10 μg/ml), incubation with CM-H2DCFDA (10 µM) was performed for 30 min. Shown panels are the average ( ± SEM) taken from 4 independent experiments. *p < 0.05. (D) WB using anti-EDA-FN1, anti-COL1A1/2, anti-ACTA2, and anti-ACTB of cell lysates from control (lane 1, 2) and NAC (10 mM) treated (lane 3, 4) LF. NAC treatment was started 1 h before TGFB1 (2 ng/ml) stimulation and protein samples were collected after 24-h treatment. Shown panels are the average ( ± SEM) taken from 3 independent experiments shown as relative expressions. *p < 0.05.
AZM enhances ubiquitination and degradation of NOX4 through STUB1 regulation and proteasome activation in LF
With respect to NOX4 activity, its protein level is a major point of regulation and the proteasome is responsible for its degradation.17,21 MG132, a proteasome inhibitor, was used. AZM-induced NOX4 reduction was efficiently abrogated by treatment with MG132 in both the presence and absence of TGFB1, suggesting the involvement of accelerated proteasomal degradation in AZM-mediated NOX4 regulation (Fig. 3A, B). To elucidate the mechanisms for enhanced proteasomal degradation of NOX4, proteasome activity was analyzed during AZM treatment. Compared to control LF, AZM-treated LF demonstrated significantly increased proteasome activity at 120 min (increased to 1.65-fold average) (P = 0.0156, Fig. 3C). K48-ubiquitinated proteins are targeted for proteasomal degradation, and AZM clearly induced accumulation of K48-ubiquitinated proteins (Fig. 3D). Immunofluorescence staining demonstrated that AZM increased colocalization of NOX4 and ubiquitin, and TGFB1 treatment clearly induced NOX4 protein expression but only slightly enhanced colocalization of NOX4 and ubiquitin (Fig. 3E). Concomitant treatment with AZM and TGFB1 showed markedly increased colocalization of NOX4 and ubiquitin, further indicating an increase in NOX4-specific ubiquitination attributable to AZM during TGFB1 treatment (Fig. 3E).
Figure 3.

AZM induces proteasomal degradation of NOX4. (A) WB using anti-NOX4 and anti-ACTB of cell lysates from control (lane 1, 3) and AZM- (10 μg/ml) treated (lane 2, 4) LF in the presence (lane 1, 2) or absence of MG132 (lane 3, 4). Protein samples were collected after 3 h treatment with AZM and MG132. Shown in the lower panel is the average ( ± SEM) taken from 6 independent experiments shown as relative expressions. *p < 0.05. (B) WB of cell lysates from TGFB1- (2 ng/ml) treated (lane 1, 2, 3, 4) and AZM (10 μg/ml) treated (lane 2, 4) LF in the presence (lane 1, 2) or absence of MG132 (lane 3, 4). MG132 and AZM treatment was started 1 h before TGFB1 stimulation. Shown in the lower panel is the average ( ± SEM) taken from 5 independent experiments shown as relative expressions. *p < 0.05. (C) Changes of 20S proteasome activity in response to AZM (10 μg/ml) treatment. After 24-h treatment with MG132 and AZM, cell lysates for measuring 20S proteasome activity were collected. Line plots show the measured relative value of fluorescence units (RFU) of average ( ± SD). The fluorescence level in the control treated cells was designated as 1.0 and shown in the lower panel is the average ( ± SEM) taken from 6 independent experiments. *p < 0.05. (D) WB using anti-K48 ubiquitin and anti-ACTB of cell lysates from control (lane 1), TGFB1- (2 ng/ml) treated (lane 2), and AZM- (10 μg/ml) treated (lane 3) LF. Shown in the lower panel is the average ( ± SEM) taken from 8 independent experiments shown as relative expressions. *p < 0.05. (E) Colocalization analysis of confocal laser scanning microscopy images of NOX4 and ubiquitin staining in LF. LF were treated with AZM (10 μg/ml) and TGFB1 (2 ng/ml) for 4 h in the presence of MG132. Staining was performed using an anti-NOX4 antibody and anti-ubiquitin antibody. Bar: 10 µm. (F) WB using anti-STUB1 and anti-ACTB of cell lysates from TGFB1- (2 ng/ml) treated (lane 3, 4) and AZM- (10 μg/ml) treated (lane 2, 4) LF. Shown in the lower panel is the average ( ± SEM) taken from 5 independent experiments shown as relative expressions. *p < 0.05. (G) Colocalization analysis of confocal laser scanning microscopy images of NOX4 and ubiquitin staining in LF. LF were transfected with control siRNA and STUB1 siRNA, and treatment with AZM (10 μg/ml) and TGFB1 (2 ng/ml) was started 48 h post-siRNA transfection. Bar: 10 µm. (H) WB using cell lysates from control and STUB1 siRNA-transfected LF. TGFB1 (2 ng/ml) and AZM (10 μg/ml) treatment were started 48 h post-siRNA transfection and protein samples were collected after 24 h treatment. Shown in the lower panel is the average ( ± SEM) taken from 7 independent experiments shown as relative expressions. *p < 0.05.
To clarify the mechanisms for enhanced NOX4-specific ubiquitination by AZM, we focused on the role of STUB1 (STIP1 homology and U-box containing protein 1), a known E3 ubiquitin ligase for NOX4.21 Intriguingly, AZM treatment significantly increased STUB1 protein levels, but a slight reduction was also observed in the presence of TGFB1 (Fig. 3F). To further elucidate the involvement of STUB1 in AZM-mediated NOX4 degradation, STUB1 siRNA was used and efficient knockdown was observed (data not shown). STUB1 knockdown attenuated AZM-induced colocalization of NOX4 and ubiquitin (Fig. 3G), and AZM-induced NOX4 reduction was efficiently restored by STUB1 knockdown (Fig. 3H), indicating that AZM accelerates NOX4 degradation via not only inducing proteasome activation but also enhancing NOX4-specific ubiquitination by STUB1.
AZM-mediated autophagy inhibition and the UPR are involved in STUB1 regulation and proteasome activation in LF
AZM blocks autophagy flux by preventing lysosomal acidification, and autophagy has an essential role in removing disorganized ER, which is linked to ER-associated protein degradation by the proteasome.4,9 Hence, to elucidate the mechanisms for proteasome activation, we first focused on AZM-mediated autophagy inhibition. Autophagy was examined by detecting the conversion of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3) from LC3B-I (free form) to LC3B-II (phosphatidylethanolamine-conjugated form) in the presence of protease inhibitors (E64d and pepstatin A). LC3B conversion experiments clarified that AZM may enhance autophagosome formation but inhibit further degradation because AZM increased the accumulation of LC3B-II, but the concomitant addition of protease inhibitors demonstrated no additional increase in LC3B-II accumulation (Fig. 4A). Inhibition of lysosomal acidification by AZM in association with inappropriate autolysosome maturation was examined by using LF expressing mRFP-GFP tandem fluorescent-tagged LC3 (Fig. 4B). During maturation of the autolysosome GFP is quenched/degraded and only the mRFP signal remains. Consistent with bafilomycin A1 (Baf A1), a representative lysosomal acidification inhibitor, AZM treatment showed accumulation of yellow dots, which indicates the coexistence of GFP and mRFP reflecting insufficient lysosome acidification (and a block in autophagosome-lysosome fusion) or autolysosomal acidification (resulting in a block in degradation). Autophagy inhibition by AZM was further confirmed by accumulation of SQSTM1/p62, a receptor protein of selective autophagy (Fig. 4C).
Figure 4.

Autophagy inhibition and enhanced unfolded protein responses (UPR) by AZM in association with STUB1 regulation and proteasome activation in LF. (A) WB using anti-LC3B and anti-ACTB of cell lysates from control (lane 1, 2) and AZM (lane 3, 4) treated LF in the presence or absence of protease inhibitors (E64d, pepstatin A). Protein samples were collected after 24-h treatment with AZM (10 μg/ml). In the lower panel is the average ( ± SEM) taken from 3 independent experiments shown as relative expression. *p < 0.05. (B) Colocalization analysis of mRFP and EGFP by using LF expressing mRFP-GFP tandem fluorescent-tagged LC3B. After 24 h treatment with bafilomycin A1 (Baf A1; 10 nM) and AZM (10 μg/ml), fixed LF were evaluated by confocal laser scanning microscopy. Bar: 20 μm. (C) WB using anti-SQSTM1 and anti-ACTB of cell lysates from control (lane 1) and AZM (10 μg/ml) treated (lane 2) LF. Protein samples were collected after 24 h treatment. In the lower panel is the average ( ± SEM) taken from 3 independent experiments shown as relative expression. *p < 0.05. (D) WB using anti-ATG5, anti-HSPA5, anti-p-ERN1, anti-ATF4, anti-ATF6, anti-STUB1, and anti-ACTB of cell lysates from control siRNA (lane 1) and ATG5 siRNA- (lane 2) transfected LF. Protein samples were collected after 72 h transfection. In the right panels are the average ( ± SEM) taken from 4 independent experiments shown as relative expression. *p < 0.05. (E) Changes of 20S proteasome activity by ATG5 knockdown. Cell lysates for measuring 20S proteasome activity were collected after 72-h transfection with control and ATG5 siRNA. Line plots show the measured relative value of fluorescence units (RFU) of the averages ( ± SD). Shown in the lower panel is the average ( ± SEM) taken from 3 independent experiments. *p < 0.05. (F) WB of cell lysates from control (lane 1) and AZM (10 μg/ml) treated (lane 2) LF. In the right panels are the average ( ± SEM) taken from 3 independent experiments shown as relative expression. *p < 0.05. (G) WB of cell lysates from control (lane 1) and tunicamycin (TM; 0.1 μg/ml) treated LF (lane 2). Protein samples were collected after 24 h treatment with TM. In the right panels are the average ( ± SEM) taken from 4 independent experiments shown as relative expression. *p < 0.05. (H) Changes of 20S proteasome activity by TM treatment. Cell lysates for measuring 20S proteasome activity were collected after 24 h treatment with control and TM. Line plots show the measured relative value of fluorescence units (RFU) of the averages ( ± SD). Shown in the right panel is the average ( ± SEM) taken from 3 independent experiments. *p < 0.05. (I) 20S proteasome activity of concomitant AZM and TM treatment, and concomitant ATG5 knockdown and TM treatment. AZM and TM treatments were started 48 h post-siRNA transfection and cell lysates for measuring 20S proteasome activity were collected after 24 h treatment with AZM and TM. Shown in the panel is the average ( ± SEM) taken from 3 independent experiments. *p < 0.05.
To clarify whether autophagy regulates proteasome activity in LF, autophagy was inhibited by siRNA-mediated knockdown of ATG5, one of the key regulators of autophagosome formation.22 ATG5 siRNA demonstrated efficient knockdown (Fig. 4D) and ATG5 knockdown clearly reduced autophagy as measured by LC3B conversion (data not shown). ATG5 knockdown significantly enhanced proteasome activity in LF (Fig. 4E). ATG5 knockdown also induced the UPR to overexpress HSPA5 (heat shock protein family A [Hsp70] member 5), phosphorylated (p)-ERN1 (endoplasmic reticulum to nucleus signaling 1), ATF4 (activating transcription factor 4), ATF6 (activating transcription factor 6), and STUB1 (Fig. 4D). Accordingly, we next examined the involvement of the UPR in AZM-mediated proteasome activation. The AZM-induced UPR was confirmed by an increase in HSPA5, p-ERN1, ATF4, and ATF6 (Fig. 4F). To see the effect of the UPR on proteasome activity, tunicamycin (TM), a glycosylation inhibitor inducing ER stress, was used. TM induced the UPR and significantly enhanced proteasome activity, but no alteration of STUB1 protein level was observed (Fig. 4G, H). To further elucidate the functional crosstalk between autophagy and UPR in terms of regulating proteasome activity by AZM, AZM-treated LF and ATG5 knockdown LF were simultaneously incubated with TM, respectively. Interestingly, when compared with TM-treated LF, no additive effect on increasing proteasome activity was demonstrated by AZM treatment and ATG5 knockdown (Fig. 4I). These data suggest that the UPR induced by AZM, which can be attributed to autophagy inhibition, is mainly responsible for enhanced proteasome activity and that autophagy inhibition is involved in STUB1 regulation.
TGFBI induces autophagy, the UPR, and proteasome activation in LF
We have previously reported autophagy activation by TGFB in LF.4 Consistently, TGFB1-induced autophagy activation was demonstrated by means of LC3B-II conversion and reduced SQSTM1 protein levels (Fig. 5A, B). TGFB1-induced autophagy activation was attenuated in the presence of AZM (Fig. 5A, B). TGFB1 markedly activated the UPR, causing overexpression of HSPA5, p-ERN1, ATF4, and ATF6. A semi-additive effect on the UPR was also observed by combination treatment with AZM and TGFB1, showing increased HSPA5, ATF4, and p-ERN1 but not ATF6 (Fig. 5C). Consistent with recently published results,6 TGFB1 treatment induces proteasome activation, which was comparable to that of AZM, but concomitant treatment with TGFB1 and AZM demonstrated no further increase in proteasome activation (Fig. 5D), suggesting that the AZM-mediated UPR alone is sufficient for proteasome activation.
Figure 5.
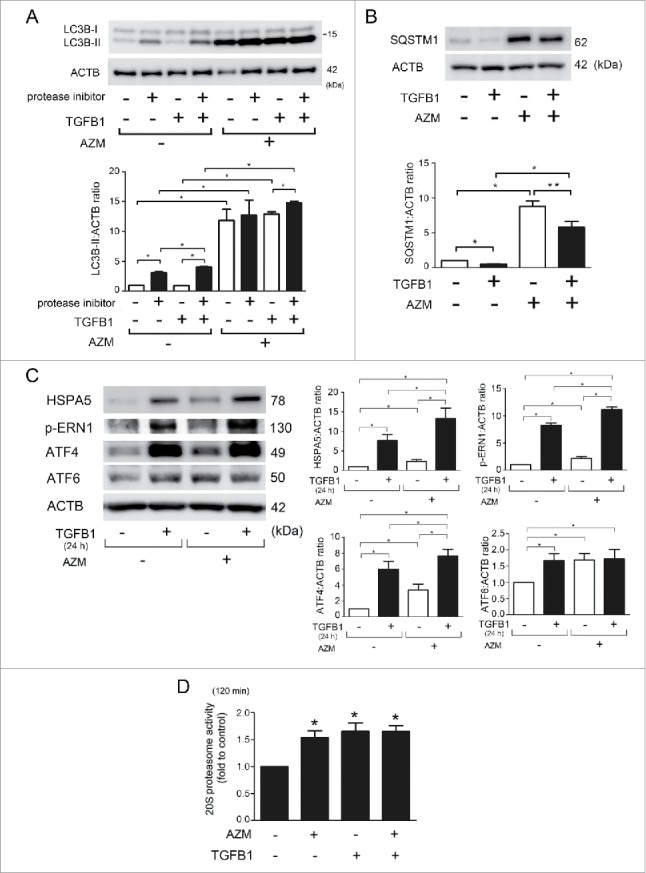
Effect of AZM on TGFB-induced autophagy and TGFB-mediated UPR and proteasome activation in LF. (A) WB using anti-LC3 and anti-ACTB of cell lysates from control (lane 1, 2), TGFB1-treated (lane 3, 4, 7, 8) and AZM-treated (lane 5, 6, 7, 8) LF in the presence or absence of protease inhibitors (E64d, pepstatin A). Protein samples were collected after 24 h treatment with AZM (10 μg/ml) and TGFB1 (2 ng/ml). In the lower panel is the average ( ± SEM) taken from 3 independent experiments shown as relative expression. *p < 0.05. (B) WB using anti-SQSTM1 and anti-ACTB of cell lysates from control (lane 1), TGFB1- (2 ng/ml) treated (lane 2, 4), and AZM (10 μg/ml) treated (lane 3, 4) LF. Protein samples were collected after 24 h treatment. In the lower panel is the average ( ± SEM) taken from 3 independent experiments shown as relative expression. *p < 0.05. (C) WB using anti-HSPA5, anti-p-ERN1, anti-ATF4, anti-ATF6, and anti-ACTB of cell lysates from control (lane 1), TGFB1- (2 ng/ml) treated (lane 2, 4), and AZM (10 μg/ml) treated (lane 3, 4) LF. Protein samples were collected after 24 h treatment. In the right panels are the average ( ± SEM) taken from 3 independent experiments shown as relative expression. *p < 0.05. (D) Changes of 20S proteasome activity by TGFB1 and AZM treatment. Cell lysates for measuring 20S proteasome activity were collected after 24 h treatment with control, TGFB1 (2 ng/ml), and concomitant TGFB1 and AZM (10 μg/ml). Shown in the right panel is the average ( ± SEM) taken from 3 independent experiments. *p < 0.05.
AZM-mediated autophagy inhibition does not induce mitochondrial ROS production and cellular dysfunction in LF
In line with our previous findings, ATG5 knockdown clearly induced ACTA2 in LF (Fig. 6A), a hallmark of myofibroblast differentiation.4 We have recently reported that enhanced mitochondrial ROS is responsible for myofibroblast differentiation in the setting of insufficient autophagy/mitophagy through the activation of the PDGFR (platelet-derived growth factor receptor) signaling pathway.23 Although AZM inhibits autophagy,9 AZM does not induce myofibroblast differentiation but rather inhibits TGFB-induced myofibroblast differentiation, suggesting the existence of a substantial difference in autophagy inhibition between ATG5 knockdown and AZM, especially with respect to mitochondrial ROS regulation. Therefore, mitochondrial integrity was examined during AZM treatment.
Figure 6.

AZM is not associated with increase in mitochondrial damage or mitochondrial ROS production in LF. (A) WB using anti-ACTA2 and anti-ACTB of cell lysates from control siRNA (lane 1) and ATG5 siRNA- (lane 2) transfected LF. Protein samples were collected after 72 h transfection. In the right panels are the average ( ± SEM) taken from 3 independent experiments shown as relative expression. *p < 0.05. (B) Photographs of fluorescence staining of control siRNA, ATG5 siRNA, and AZM-treated LF with MitoSOX Red (upper panels) and with Hoechst 33258 (lower panels). LF were transfected with nonsilencing control siRNA and ATG5 siRNA, and AZM treatment (10 μg/ml) was started 48 h post-siRNA transfection. LF were treated with AZM for 24 h. Bar: 100 µm (C) WB using anti-SDHA, anti-TOMM20, and anti-ACTB of cell lysates from control siRNA (lane 1), ATG5 siRNA (lane 2), and AZM (10 μg/ml) (lane 3) treated LF. AZM treatment was started 48 h post-siRNA transfection and protein samples were collected after 24 h treatment with AZM. Right panels are the average ( ± SEM) taken from 4 independent experiments shown as relative expressions. * p <0.05. (D) Intracellular ATP levels were determined in LF after control (open bar) or AZM treatment (10 μg/ml for 24 h; black bar), respectively. ATP levels in the control cells were designated as 1.0. (E) Cell proliferation was evaluated by MTT assay. LF were treated with control and AZM, respectively. Cell proliferation was assessed after 24 h treatment. Control treated cells were designated as 1.0. (F) Cell death was evaluated by LDH cytotoxicity assay. LF were treated with control and AZM, respectively. Cell death was assessed after 24 h treatment.
In contrast to increased mitochondrial ROS production seen with ATG5 knockdown, no obvious enhancement of mitochondrial ROS was demonstrated by AZM treatment as measured by Mitosox Red staining (Fig. 6B). Although increased expression levels of the mitochondrial proteins SDHA and TOMM20 was observed in ATG5 knockdown (reflecting increased mitochondrial mass), no such changes were observed during AZM treatment (Fig. 6C). Mitochondrial structural integrity was examined by electron microscopy, which showed no difference in the percentage of normal structured mitochondria between control and AZM-treated LF (Fig. S3). Mitochondrial functional integrity (as measured by ATP production), was not hampered by AZM treatment (Fig. 6D). Furthermore, no alteration in cell function and cell damage as measured by proliferation (MTT assay) and death (LDH cytotoxicity assay) was present (Fig. 6E, F). Taken together, mitochondrial ROS production and mitochondrial functional integrity were appropriately preserved without cellular damage during AZM treatment in LF.
Azithromycin attenuates bleomycin-induced NOX4 and lung fibrosis development with enhanced STUB1 and proteasome activity
To further confirm the anti-fibrotic role of AZM in more physiological conditions, we used a bleomycin (BLM)-induced lung fibrosis model. Generally, d 7 after BLM treatment is recognized to be the beginning of the fibrotic phase. To elucidate a potential clinical relevance of AZM treatment of IPF with progressive fibrosis, intraperitoneal AZM injection was initiated on d 7 following BLM treatment. Compared to control-treated mice, BLM-treated mice showed significant body weight loss, which was markedly recovered during AZM treatment (Fig. 7A). AZM treatment significantly attenuated lung fibrosis development at d 21 as shown by Sircol collagen assay and Masson trichrome staining (Fig. 7B, C). Reduced NOX4 protein levels following AZM treatment was also demonstrated by means of immunohistochemistry and WB of lung homogenates (Fig 7D, E). To elucidate the participation of proteasome-mediated NOX4 degradation in the anti-fibrotic property of AZM, K48-ubiquitin and STUB1 protein levels and proteasome activity were examined in mouse lungs. Consistent with in vitro experiments, AZM significantly increased both K48-ubiquitinated proteins and STUB1 protein levels in lung homogenates (Fig. 7F). Significantly enhanced proteasome activity in AZM-treated lungs was also demonstrated (Fig. 7G).
Figure 7.
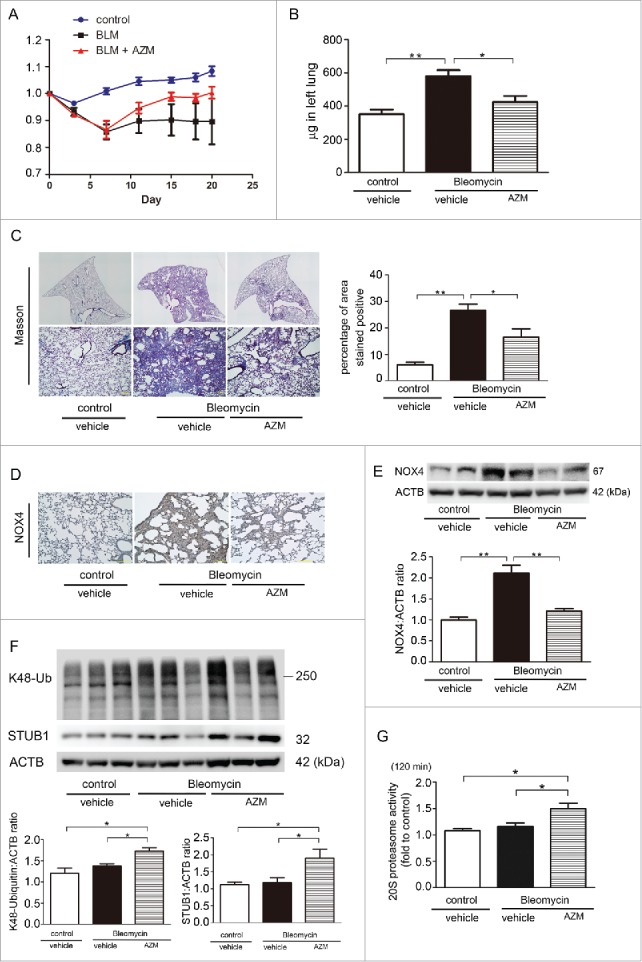
Effect of AZM on bleomycin-induced lung fibrosis development in mice. (A) Body weight (BW) changes after BLM treatment. BW at d 0 before treatment was designated as 1.0. (B) Shown in the panel is the average ( ± SEM) soluble collagen measurement from Sircol assay using control (n = 12), BLM-treated (n = 15), and BLM-treated with subsequent AZM injection mouse lungs (n = 15) at d 21. Open bar is control, filled bar is BLM-treated, and horizontal crosshatched bar is BLM-treated with subsequent AZM injection. *p < 0.05. (C) Photomicrographs of Masson trichrome staining of mouse lungs at d 21. Upper panels are low magnification view of original magnification × 40. Lower panels are high magnification view of original magnification × 100. The right panel shows the average ( ± SEM) percentage of positively stained area quantified using ImageJ. *p < 0.05. (D) Immunohistochemical staining of NOX4 in mouse lungs at day 21. Original magnification × 100. (E) WB using anti-NOX4 and anti-ACTB of lung homogenates. Lower panel is the average ( ± SEM) shown as relative expressions. Treatment groups were composed of control (n = 7), BLM-treated (n = 6), and BLM-treated with subsequent AZM injection mouse lungs (n = 6). *p < 0.05. (F) WB using anti-K48 ubiquitin, anti-STUB1, and anti-ACTB of lung homogenates. Lower panels are the average ( ± SEM) shown as relative expressions. Treatment groups were composed of control (n = 4), BLM-treated (n = 7), and BLM-treated with subsequent AZM injection mouse lungs (n = 8). *p < 0.05. (G) 20S proteasome activity of mouse lungs. Mouse lungs for measuring 20S proteasome activity were collected at d 21. The fluorescence level in the control-treated lung homogenate from one representative mouse was designated as 1.0. Shown panel is the average ( ± SEM) and treatment groups were composed of the same number of mice (n = 5). *p < 0.05.
AZM has been reported to have an anti-fibrotic role in BLM-induced lung fibrosis development via suppressing inflammatory cell accumulation.8 Hence we also performed cell counting of bronchiole-alveolar lavage fluid (BALF) at d 21. BLM treatment significantly increased total cell, macrophage, and lymphocyte counts, but only slightly enhanced neutrophil counts (Fig. S4). Although no change was observed in macrophage and neutrophil counts, AZM significantly reduced total cell and lymphocyte counts, suggesting that not only enhanced NOX4 degradation by the proteasome but also immunomodulation can be involved in the mechanisms for AZM-mediated attenuation of BLM-induced lung fibrosis.
Azithromycin inhibits TGFB1-induced NOX4 and myofibroblast differentiation in LF derived from IPF patients
We have recently reported an increase in NOX4 protein levels in LF derived from IPF patients.18 To further confirm the clinical relevance of using AZM as a treatment of IPF patients, we used LF derived from IPF patients. TGFB1 treatment increased NOX4 protein levels with concomitant induction of myofibroblast differentiation. Consistent with the experimental results from LF derived from non-IPF patients (Fig. 1), AZM efficiently suppressed TGFB1-induced NOX4 expression and myofibroblast differentiation with concomitantly increased STUB1 protein levels in LF derived from IPF patients (Fig. 8). AZM-mediated regulation of TGFB-induced myofibroblast differentiation in LF derived from IPF patients was further confirmed by detection of the secreted form of EDA-FN1 and COL1/type I collagen (Fig. S5).
Figure 8.

Azithromycin suppresses TGFB-induced NOX4 and myofibroblast differentiation in LF derived from IPF patients. WB using anti-EDA-FN1, anti-COL1A1/2, anti-ACTA2, anti-NOX4, anti-STUB1, and anti-ACTB of cell lysates from control (lane 1, 2) and azithromycin (AZM; 10 μg/ml) treated (lane 3, 4) LF. AZM treatment was started 1 h before TGFB1 (2 ng/ml) stimulation and protein samples were collected after 24 h treatment with TGFB1. In the right panels are the average ( ± SEM) taken from 5 LF derived from different IPF patients shown as relative expressions. *p < 0.05.
Discussion
In the present study, we showed that AZM-mediated proteasomal degradation of NOX4 is responsible for inhibiting TGFB-induced myofibroblast differentiation in LF and also for attenuating BLM-induced lung fibrosis. Autophagy inhibition by AZM is involved in enhanced ubiquitination of NOX4 via increased protein levels of STUB1, an E3 ubiquitin ligase. Increase in proteasome activity appeared to be mainly attributed to an increased UPR caused by AZM, which may be resulting from functional crosstalk between proteostasis machineries (Fig. 9). Inhibition of autolysosomal acidification may explain the mechanisms for AZM-mediated autophagy inhibition. Although AZM-mediated autophagy inhibition and also NOX4 targeting as treatments of IPF have been previously proposed,9,14 we clearly elucidated the direct link between modifying proteostasis machineries and NOX4 regulation as a part of the anti-fibrotic mechanisms of AZM treatment.
Figure 9.
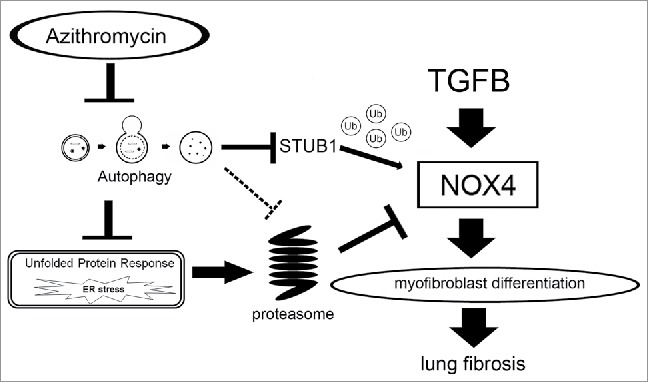
Hypothetical model of Azithromycin-mediated inhibition of myofibroblast differentiation and lung fibrosis development. AZM-mediated NOX4 degradation can result from enhanced proteasome activation and NOX4 ubiquitination by STUB1, an E3 ubiquitin ligase. Autophagy inhibition by AZM is involved in enhanced ubiquitination of NOX4 via increased protein levels of STUB1. The increase in proteasome activity appeared to be mainly attributable to an increased UPR caused by AZM, which may be resulting from functional crosstalk between proteostasis machineries. AZM can be a novel modality of IPF treatment of targeting NOX4 degradation resulting from modulation of crosstalk between proteostasis machineries.
AZM has been demonstrated to have a pleiotropic effect on modulating cellular functions,7 and we were the first to describe that AZM enhances proteasome activation. During proteostasis, enhanced proteasomal degradation in response to the UPR is well known to induce ER-associated protein degradation, and autophagy inhibition can induce the UPR. Based on the experimental results of ATG5 knockdown and TM treatment, it is likely that an increased UPR is mainly responsible for proteasome activation during AZM treatment, and UPR induction can also be attributed to autophagy inhibition by AZM (Fig. 4). However, autophagy has been demonstrated to be involved in regulating proteasome activity through degradation of the proteasome,24,25 hence it is plausible that not only the subsequent UPR but also AZM-mediated autophagy inhibition itself can also be involved in regulating proteasome levels and activity, which remains to be determined (Fig. 9). Interestingly, a recent paper proposed that increased proteasome activity is responsible for TGFB-induced myofibroblast differentiation and fibrosis development during IPF pathogenesis.6 Increased proteasome activity accompanied by the UPR was also shown by TGFB1 treatment (Fig. 5). Concomitant treatment with TGFB1 and AZM demonstrated additive effect on UPR but no increase in proteasome activity in LF (Fig. 5). We also observed no additive effect on proteasome activity by TM, a representative UPR inducer, during AZM treatment (Fig. 3C, 4I), suggesting that AZM-mediated UPR may be sufficient for proteasome activation.
Our present results and a recent paper indicate that proteasomal degradation of NOX4 is crucial for attenuating TGFB signaling and fibrosis development.17 However, increased NOX4 protein levels and proteasome activation have been shown in FF fibroblasts in IPF lungs, indicating that enhanced proteasome activation does not always efficiently target NOX4 for degradation.12 In seeming contrast to the proteasomal activation seen in TGFB treatment and in IPF lung, AZM-mediated proteasome activation induces efficient NOX4 degradation and subsequent downstream fibrosis inhibition.
With respect to the ubiquitin-proteasome system, ubiquitination is a crucial step for NOX4 degradation.17,21 We elucidated that AZM is an efficient inducer of STUB1, a representative E3 ubiquitin ligase for NOX4.21 In contrast to the mechanisms for enhanced proteasome activity, AZM-mediated autophagy inhibition, but not the UPR, is responsible for STUB1 regulation (Fig. 4), suggesting the existence of autophagy-associated regulatory mechanisms for STUB1. Functional association between STUB1 and NOX4 ubiquitination was clarified by STUB1 knockdown experiments (Fig. 3G, H). TGFB1-induced autophagy activation and AZM-mediated STUB1 induction were significantly reduced by TGFB1, further supporting the notion that autophagy inhibition but not the UPR is responsible for STUB1 regulation (Fig. 3F). Hence, proteasomal degradation of NOX4 by AZM is largely attributable to enhanced STUB1 protein levels.
We have recently reported that autophagy/mitophagy inhibition induces myofibroblast differentiation and that mitochondrial ROS is responsible for myofibroblast differentiation through enhanced activation of the PDGFR signaling pathway.23 Corresponding to a recent report showing AZM-mediated autophagy inhibition,9 AZM induces conversion from LC3B-I to LC3B-II but inhibits further degradation, which appears to be attributable to attenuation of autolysosomal acidification (Fig. 4B). Intriguingly, in contrast to autophagy inhibition by ATG5 knockdown, no increase in mitochondrial ROS production or mitochondrial mass were demonstrated in AZM-treated LF. Furthermore, preservation of ATP production without affecting cell proliferation and cell death and only slight accumulation of structurally damaged mitochondria were demonstrated (Fig. 6, Fig. S3). Hence, it is likely that mitochondrial functional integrity is not hampered during AZM-mediated autophagy inhibition. There are several potential mechanisms for the maintenance of mitochondrial integrity during AZM treatment, including an anti-oxidative property of AZM,26 compensatory enhanced proteasomal degradation,27 increased mitochondrial turnover,28 and mitophagy selective preservation, which should be clarified in future studies.
AZM has been widely used for patients with chronic respiratory infection, including non-tuberculosis Mycobacterium (NTM), for a relatively long time with acceptable adverse events.7,29 Although we used higher concentrations of AZM (10 μg/ml) in in vitro experiments compared with clinically achievable plasma AZM concentrations,30 maximum AZM concentration in lung tissue has been reported to be approximately 10 μg/ml after an oral dose of 500 mg azithromycin.30 Attenuation of BLM-induced lung fibrosis development was demonstrated in mice treated with 150 mg/kg of AZM during the fibrotic phase, which is estimated to be comparable to an AZM dose of approximately 500 mg/d for a 60 kg human. Accordingly, the AZM dose for pulmonary infection (500 mg/day) might be sufficient to show the anti-fibrotic properties of AZM in IPF.
Taken together, it is plausible that AZM, via NOX4 degradation, is a promising anti-fibrotic modality of treatment of fibrotic lung disorders affected by TGFB. AZM-mediated NOX4 degradation is governed through enhanced proteasome activation and NOX4 ubiquitination by STUB1. Proteasome activation by AZM can be attributable mainly to an increased UPR through autophagy inhibition. Although detailed mechanisms for autophagy inhibition without apparent mitochondrial damage and proteasome activation have yet to be determined, AZM can be a novel modality for IPF treatment.
Materials and methods
Cell culture, antibodies, and reagents
Normal lung tissues were obtained from pneumonectomy and lobectomy specimens from primary lung cancer. Informed consent was obtained from all surgical participants as part of an approved ongoing research protocol by the ethical committee of Jikei University School of Medicine. Lung fibroblasts (LF) were isolated and characterized as described previously.23 Briefly, LF outgrown from lung fragments were cultured in fibroblast growth medium (DMEM with 10% fetal calf serum and penicillin-streptomycin). LF were serially passaged and used for experiments until passage 6. LF demonstrated >95% positive staining with anti-VIM/vimentin antibodies (Sigma-Aldrich, V 6630), and <5% positive staining with the anti-KRT/cytokeratin antibody (BioCare Medical, Lu-5; data not shown). Antibodies used were rabbit anti-NOX4 (Novus, NB110–58849; Proteintech, 14347–1-AP), goat anti-COL1/type I collagen (Southern Biotech, 1310–01), mouse anti-ACTA2 (Sigma-Aldrich, A2547), rabbit anti-SMAD2 (Cell Signaling Technology, 3122), rabbit anti-SMAD3 (Cell Signaling Technology, 9513), rabbit anti-phospho-SMAD2 (Cell Signaling Technology, 3101), rabbit anti-phospho-SMAD3 (Cell Signaling Technology, 8769), rabbit anti-MAP1LC3B/LC3B (Novus, 600–1384), rabbit anti-ATG5 (Cell Signaling Technology, 2630), rabbit anti-SQSTM1 (MBL International, PM045), mouse anti-poly-monoubiquitin (Enzo Life Sciences, BML-PW8810), rabbit anti-K48-linkage-specific polyubiquitin (Cell Signaling Technology, 4289), rabbit anti-STUB1 (C3B6; Cell Signaling Technology, 2080), rabbit anti-HSPA5 (C50B12; Cell Signaling Technology, 3177), rabbit anti-phosho-ERN1 (Abcam, ab48187), mouse anti-SDHA (2E3GC12FB2AE2; Abcam, ab14715), mouse anti-TOMM20 (F-10; Santa Cruz Biotechnology, sc-17764), mouse anti-FN1/ cellular fibronectin containing extra domain A (Abcam, ab6328), mouse anti-ATF6 (Abam, ab122897) and mouse anti-ACTB/β-actin (Sigma-Aldrich, A5316). The following reagents were used: Azithromycin (LKT Laboratories, Inc., A9834), recombinant human TGFB1/TGF-β1 (R&D Systems, 100-B), N-acetylcysteine (NAC; Wako, 017–05131), bafilomycin A1 (Baf A1; Sigma-Aldrich, B1793), MG-132 (Enzo Life Sciences, BML-P102), CM-H2DCFDA (Life Technologies, C6827), Hoechst 33258 (Sigma-Aldrich, B2883), MitoSox Red (Molecular Probes/Life Technologies, M36008), pepstatin A (Peptide Institute, 4397), E64d (Peptide Institute, 4321-v), GKT137831 (Cayman, 17764) and bleomycin (Nippon Kayaku Co., Tokyo, Japan).
Plasmids, small interfering RNA and transfection
An mRFP-EGFP-LC3B plasmid was obtained from Addgene (21074; deposited by Tamotsu Yoshimori Lab). Small interfering RNA (siRNA) targeting NOX4 (QIAGEN, Hs_NOX4_6 FlexiTube siRNA, SI02642507 and Applied Biosystems Life Technologies, 4392420, ID: 27013), STUB1 (Applied Biosystems Life Technologies, 4392420, ID: s195027), and negative control siRNAs (Applied Biosystems Life Technologies, AM4635 and AM4641) were purchased. Specific knockdowns of NOX4 were validated using 2 different siRNAs. Transfections of LF were performed using the Neon® Transfection System (Invitrogen Life Technologies, MPK5000), using matched optimized transfection kits (Invitrogen Life Technologies, MPK10096).
Measurement of ROS production
LF, at a density of 1 × 104 per well, were seeded in a 96-well microplate (Thermo Fisher Scientific, 237105). CM-H2DCFDA was used to measure total cellular ROS according to the manufacturer's instructions. After incubation with CM-H2DCFDA (10 µM) for 30 min at 37°C, fluorescence of DCF was measured at an excitation wavelength of 485 nm and an emission wavelength of 535 nm by a fluorescence microplate reader (Infinite F 200; Tecan Japan, Kanagawa, Japan). Mitochondrial ROS production was analyzed by MitoSOX Red staining according to the manufacturer's instructions, which was evaluated by fluorescence microscopy (Olympus, Tokyo, Japan and Keyence, BZ-X700).
Western blotting
LF grown on 6-well culture plates were lysed in RIPA buffer (Thermo Fisher Scientific, 89900) containing a protease inhibitor cocktail (Roche Diagnostics, 11697498001) and 1 mM sodium orthovanadate (Wako, 13721–39–6), or lysed with Laemmli sample buffer. Western blotting was performed as described previously.23 For each experiment, equal amounts of total protein were resolved by 7.5–10% SDS-PAGE. After SDS-PAGE, proteins were transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, ISEQ00010), and incubation with specific primary antibody was performed for 1 h at 37°C, or 24 h at 4°C. After washing several times with PBST (Wako, 161–25521), the membrane was incubated with anti-rabbit IgG, HRP-linked secondary antibody (Cell Signaling Technology, 7074), anti-mouse IgG, HRP-linked secondary antibody, 7076) or anti-goat IgG (H+I), HRP-linked secondary antibody (BETHYL, A50–100P) followed by chemiluminescence detection (Thermo scientific, 34080, and BIO-RAD, 1705061) with the ChemiDocTM Touch Imaging System (BIO-RAD, CA, USA).
Immunofluorescence staining
LF were grown on 8-well culture slides. LF transfected with a plasmid encoding mRFP-GFP-LC3B were treated with BafA1 or AZM at 24 h posttransfection and evaluated by fluorescence microscopy (Olympus, Tokyo, Japan and Keyence, BZ-X700). LF were transfected with control siRNA or STUB1 siRNA, and treatment was started at 48 h posttransfection. LF were fixed with 4% paraformaldehyde for 15 min followed by permeabilization with 0.03% Triton X-100 (Wako, 160–24751) for 60 min. After blocking with 1% BSA (Sigma-Aldrich, A2153) for 60 min, the primary and secondary antibodies were applied according to the manufacturers' instructions. Confocal laser scanning microscopy analysis of LF was performed using mouse anti-poly-monoubiquitin (Enzo Life Sciences, BML-PW8810) and rabbit anti-NOX4 (Novus, NB110–58849) staining (Carl Zeiss Japan, Tokyo, Japan).
Proteasome activity
LF, at a density of 1 × 107 per well, were seeded in a 10-cm dish (Thermo Fisher Scientific, 172931). For measuring proteasome activity, Cyclex® Proteasome Enrichment and Activity Assay Kit (Cyclex Co., Ltd. CY-7002) was used according to the manufacturer's instructions. The 20S proteasome was isolated from LF and lung tissue lysate by using hHR23B ubiquitin-like domain resin (UbL-resin; Cyclex Co., Ltd. CY-7002), and 20S proteasome activity was measured by adding a fluorogenic proteasome substrate (Suc-LLVY-AMC; Cyclex Co., Ltd. CY-7002). The intensity of fluorescence of each reaction mixture was measured using a 380/460-nm filter set in a fluorometer.
RNA isolation, polymerase chain reaction
RNA isolation, reverse transcription and real-time PCR were performed using the SYBR green (Power SYBR® Green PCR Master Mix; Applied Biosystems™, 4367659) method as described previously.31 The primers used were NOX4 sense primer, 5′-CAGATGTTGGGGCTAGGATTG-3′; NOX4 antisense primer, 5′-GAGTGTTCGGCACATGGGTA-3′; NOX4 sense primer, 5′-TGTGCCGAACACTCTTGGC-3′; NOX4 antisense primer, 5′- ACATGCACGCCTGAGAAAATA-3′; ACTB sense primer 5′-CATGTACGTTGCTATCCAGGC-3′ ACTB antisense primer 5′-CTCCTTAATGTCACGCACGAT-3′. These primer sets yielded PCR products of 96 base pair and 250 base pair for NOX4 and ACTB, respectively. PCRs of NOX4 were validated using 2 different primers. Primer sequences were from Primer Bank (http://pga.mgh.harvard.edu/primerbank.)
Electron microscopy
Electron microscopy was performed as described previously.32 LF transfected with siRNA and treated with AZM were fixed with 2% glutaraldehyde, 0.1 M phosphate buffer (pH 7.4) after 48 h of incubation and dehydrated with a graded series of ethanol. Fixed LF were then embedded in epoxy resin (Epok812; Oken, 02–1001). Ultrathin sections were stained with uranyl acetate and lead citrate and observed with the Hitachi H-7500 transmission electron microscope (Hitachi). For quantitative evaluation of mitochondria in LF, 10 image fields (× 10,000) were selected for each sample. Mitochondria >2 µm longer than other mitochondria were counted as elongated. Mitochondrial damage was determined by abnormal swelling and crista disruption.
Measurement of cellular ATP level
LF, at a density of 1 × 104 per well, were seeded in a 96-well microplate (Thermo Fisher Scientific, 236105). ATP levels in cell lysates were measured using a CellTiter-Glo luminescent cell viability assay (Promega, G7572).
MTT assay
LF, at a density of 1 × 104 per well, were seeded in a 96-well microplate (Thermo Fisher Scientific, 167008). The MTT assay was performed according to the manufacturer's instructions (Roche, 11465007001).
LDH cytotoxicity assay
LF, at a density of 1 × 104 per well, were seeded in a 96-well microplate (Thermo Fisher Scientific, 167008). The MTT assay was performed according to the manufacturer's instructions (Roche, 04744926001).
Mouse models
C57BL/6J mice were purchased (CLEA Japan INC, Tokyo, Japan) and were maintained in the animal facility at the Jikei University School of Medicine. All experimental procedures were approved by the Jikei University School of Medicine Animal Care Committee. A dose of 3 U/kg bleomycin (Nippon Kayaku Co., 4234400D4032) was intratracheally administered in 50 μL saline using the MicroSprayerTM Aerosolizer and a high pressure syringe (PennCentury, Philadelphia, PA, USA). Intraperitoneal doses of azithromycin (150 mg/kg) were given daily from d 7 to d 20. On the 21st d the lungs were removed, fixed overnight in 10% buffered formalin, embedded in paraffin, and the sections stained with hematoxylin and eosin (HE) and Masson's trichrome according to conventional protocols for histopathological evaluation.23 Immunohistochemistry was performed on the paraffin-embedded lung tissues as described previously with minor modifications.23 Tracheotomy was performed to insert a tracheal tube for collecting bronchoalveolar lavage fluid (BALF) sample with PBS (Wako, 041–20211; total 3 ml). The cell numbers in BALF were counted using a hemocytometer. Differential cell counts in BALF were analyzed on 300 cells stained with Diff-Quick (Sysmex, 16920).
Sircol soluble collagen assay
For quantitatively measuring collagen in the left lungs of the mice, the Sircol soluble collagen assay was performed according to the manufacturer's instructions (Biocolor Life Science Assay, S100).
Statistics
Data are shown as the average ( ± SEM) taken from at least 3 independent experiments. Student t test was used for comparison of 2 data sets, analysis of variance for multiple data sets. Tukey's or Dunn's test were used for parametric and nonparametric data, respectively, to find where the difference lay. Significance was defined as p < 0.05. Statistical software used was Prism v.5 (GraphPad Software, Inc., San Diego, CA).
Abbreviations
- ACTA2 actin
α 2, smooth muscle, aorta
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- ATG5
autophagy-related 5
- AZM
azithromycin
- BALF
bronchoalveolar lavage fluid
- BLM
bleomycin
- COL1A1
collagen type I α 1 chain
- COL1A2
collagen type I α 2 chain
- EDA-FN1
EDA-containing cellular fibronectin 1
- EM
electron microscopy
- EM
erythromycin
- ER
endoplasmic reticulum
- ERN1
endoplasmic reticulum to nucleus signaling 1
- FF
fibroblastic foci
- H2O2
hydrogen peroxide
- HSPA5
heat shock protein family A (Hsp70) member 5
- IPF
idiopathic pulmonary fibrosis
- LF
lung fibroblasts
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3
- NAC
N-acetylcysteine
- NOX4
NADPH oxidase 4
- NTM
non-tuberculosis mycobacterium
- RXM
roxithromycin
- SDHA
succinate dehydrogenase complex flavoprotein subunit A
- TGFB
transforming growth factor β
- TM
tunicamycin
- TOMM20
translocase of outer mitochondrial membrane 20
- UbL-resin
ubiquitin-like domain resin
- UPR
unfolded protein response
- STUB1
STIP1 homology and U-box containing protein 1
- PDGFR
platelet derived growth factor receptor
- ROS
reactive oxygen species
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank Stephanie Cambier (University of Washington, Seattle, USA), Emi Kikuchi and Dr. Toshiaki Tachibana (Jikei University School of Medicine, Tokyo, Japan) for technical support.
Funding
This work was supported by grants from the Jikei University Graduate Research Grant to N.Saito, M.Yoshida, and Y.Kurita, JSPS KAKENHI Grant Number JP15K09231 to J.A. and JP15K09232 to K.Kuwano, Satoshi Okamoto Memorial Foundation of Pulmonary Fibrosis to J.A., the Practical Research Project for Rare Intractable Diseases from Japan Agency for Medical Research and development, AMED to K.Kuwano, and the Ministry of Health, Labor and Welfare of Japan awarded to the Study Group on Diffuse Pulmonary Disorders, Scientific Research/Research on intractable diseases K.Kuwano.
References
- [1].Araya J, Nishimura SL. Fibrogenic reactions in lung disease. Annu Rev Pathol 2010; 5:77-98; PMID:20078216; https://doi.org/ 10.1146/annurev.pathol.4.110807.092217 [DOI] [PubMed] [Google Scholar]
- [2].Mulugeta S, Nureki S, Beers MF. Lost after translation: insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol 2015; 309:L507-25; PMID:26186947; https://doi.org/ 10.1152/ajplung.00139.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, Weaver TE, Guenther A. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008; 178:838-46; PMID:18635891; https://doi.org/ 10.1164/rccm.200802-313OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Kawabata Y, Hano H, Nakayama K, Kuwano K. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2013; 304:L56-69; PMID:23087019; https://doi.org/ 10.1152/ajplung.00213.2012 [DOI] [PubMed] [Google Scholar]
- [5].Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol 2012; 46:731-9; PMID:21852685; https://doi.org/ 10.1165/rcmb.2011-0121OC [DOI] [PubMed] [Google Scholar]
- [6].Semren N, Welk V, Korfei M, Keller IE, Fernandez IE, Adler H, Gunther A, Eickelberg O, Meiners S. Regulation of 26S Proteasome Activity in Pulmonary Fibrosis. Am J Respir Crit Care Med 2015; 192:1089-101; PMID:26207697; https://doi.org/ 10.1164/rccm.201412-2270OC [DOI] [PubMed] [Google Scholar]
- [7].Parnham MJ, Erakovic Haber V, Giamarellos-Bourboulis EJ, Perletti G, Verleden GM, Vos R. Azithromycin: mechanisms of action and their relevance for clinical applications. Pharmacol Ther 2014; 143:225-45; PMID:24631273; https://doi.org/ 10.1016/j.pharmthera.2014.03.003 [DOI] [PubMed] [Google Scholar]
- [8].Wuyts WA, Willems S, Vos R, Vanaudenaerde BM, De Vleeschauwer SI, Rinaldi M, Vanhooren HM, Geudens N, Verleden SE, Demedts MG, Thomeer M, Verbeken EK, Verleden GM. Azithromycin reduces pulmonary fibrosis in a bleomycin mouse model. Exp Lung Res 2010; 36:602-14; PMID:20874225; https://doi.org/ 10.3109/01902148.2010.492895 [DOI] [PubMed] [Google Scholar]
- [9].Renna M, Schaffner C, Brown K, Shang S, Tamayo MH, Hegyi K, Grimsey NJ, Cusens D, Coulter S, Cooper J, Bowden AR, Newton SM, Kampmann B, Helm J, Jones A, Haworth CS, Basaraba RJ, DeGroote MA, Ordway DJ, Rubinsztein DC, Floto RA. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest 2011; 121:3554-63; PMID:21804191; https://doi.org/ 10.1172/JCI46095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D. Autophagy in idiopathic pulmonary fibrosis. PLoS One 2012; 7:e41394; PMID:22815997; https://doi.org/ 10.1371/journal.pone.0041394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jiang F, Liu GS, Dusting GJ, Chan EC. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol 2014; 2:267-72; PMID:24494202; https://doi.org/ 10.1016/j.redox.2014.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 2009; 15:1077-81; PMID:19701206; https://doi.org/ 10.1038/nm.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010; 65:733-8; PMID:20685750; https://doi.org/ 10.1136/thx.2009.113456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jarman ER, Khambata VS, Cope C, Jones P, Roger J, Ye LY, Duggan N, Head D, Pearce A, Press NJ, Bellenie B, Sohal B, Jarai G. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am J Respir Cell Mol Biol 2014; 50:158-69; PMID:23977848 [DOI] [PubMed] [Google Scholar]
- [15].Park HH, Park IH, Cho JS, Lee YM, Lee HM. The effect of macrolides on myofibroblast differentiation and collagen production in nasal polyp-derived fibroblasts. Am J Rhinol Allergy 2010; 24:348-53; PMID:21244734; https://doi.org/ 10.2500/ajra.2010.24.3520 [DOI] [PubMed] [Google Scholar]
- [16].Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 2006; 18:69-82; PMID:15927447; https://doi.org/ 10.1016/j.cellsig.2005.03.023 [DOI] [PubMed] [Google Scholar]
- [17].Desai LP, Zhou Y, Estrada AV, Ding Q, Cheng G, Collawn JF, Thannickal VJ. Negative regulation of NADPH oxidase 4 by hydrogen peroxide-inducible clone 5 (Hic-5) protein. J Biol Chem 2014; 289:18270-8; PMID:24831009; https://doi.org/ 10.1074/jbc.M114.562249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J, Saito N, Fujita Y, Kurita Y, Kobayashi K, Ito S, Hara H, Kadota T, Yanagisawa H, Hashimoto M, Utsumi H, Wakui H, Kojima J, Numata T, Kaneko Y, Odaka M, Morikawa T, Nakayama K, Kohrogi H, Kuwano K. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res 2016; 17:107; PMID:27576730; https://doi.org/ 10.1186/s12931-016-0420-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].White ES, Muro AF. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011; 63:538-46; PMID:21698758; https://doi.org/ 10.1002/iub.493 [DOI] [PubMed] [Google Scholar]
- [20].Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol 2012; 47:718-26; PMID:22904198; https://doi.org/ 10.1165/rcmb.2011-0418OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gil Lorenzo AF, Costantino VV, Appiolaza ML, Cacciamani V, Benardon ME, Bocanegra V, Valles PG. Heat Shock Protein 70 and CHIP Promote Nox4 Ubiquitination and Degradation within the Losartan Antioxidative Effect in Proximal Tubule Cells. Cell Physiol Biochem 2015; 36:2183-97; PMID:26279425; https://doi.org/ 10.1159/000430184 [DOI] [PubMed] [Google Scholar]
- [22].Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458-67; PMID:19491929; https://doi.org/ 10.1038/nrm2708 [DOI] [PubMed] [Google Scholar]
- [23].Kobayashi K, Araya J, Minagawa S, Hara H, Saito N, Kadota T, Sato N, Yoshida M, Tsubouchi K, Kurita Y, Ito S, Fujita Y, Takasaka N, Utsumi H, Yanagisawa H, Hashimoto M, Wakui H, Kojima J, Shimizu K, Numata T, Kawaishi M, Kaneko Y, Asano H, Yamashita M, Odaka M, Morikawa T, Nakayama K, Kuwano K. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J Immunol 2016; https://doi.org/ 10.4049/jimmunol.1600265 [DOI] [PubMed] [Google Scholar]
- [24].Wang C, Wang X. The interplay between autophagy and the ubiquitin-proteasome system in cardiac proteotoxicity. Biochim Biophys Acta 2015; 1852:188-94; PMID:25092168; https://doi.org/ 10.1016/j.bbadis.2014.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang XJ, Yu J, Wong SH, Cheng AS, Chan FK, Ng SS, Cho CH, Sung JJ, Wu WK. A novel crosstalk between two major protein degradation systems: regulation of proteasomal activity by autophagy. Autophagy 2013; 9:1500-8; PMID:23934082; https://doi.org/ 10.4161/auto.25573 [DOI] [PubMed] [Google Scholar]
- [26].Chen M, Yang T, Meng X, Sun T. Azithromycin attenuates cigarette smoke extract-induced oxidative stress injury in human alveolar epithelial cells. Mol Med Rep 2015; 11:3414-22; PMID:25607112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010; 191:1367-80; PMID:21173115; https://doi.org/ 10.1083/jcb.201007013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Okamoto K, Kondo-Okamoto N. Mitochondria and autophagy: critical interplay between the two homeostats. Biochim Biophys Acta 2012; 1820:595-600; PMID:21846491; https://doi.org/ 10.1016/j.bbagen.2011.08.001 [DOI] [PubMed] [Google Scholar]
- [29].Duran JM, Amsden GW. Azithromycin: indications for the future? Expert Opin Pharmacother 2000; 1:489-505; PMID:11249533; https://doi.org/ 10.1517/14656566.1.3.489 [DOI] [PubMed] [Google Scholar]
- [30].Foulds G, Shepard RM, Johnson RB. The pharmacokinetics of azithromycin in human serum and tissues. J Antimicrob Chemother 1990; 25 Suppl A:73-82; PMID:2154441; https://doi.org/ 10.1093/jac/25.suppl_A.73 [DOI] [PubMed] [Google Scholar]
- [31].Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus VC, Sheppard D, Barzcak A, Xiao Y, Erle DJ, Nishimura SL. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest 2007; 117:3551-62; PMID:17965775; https://doi.org/ 10.1172/JCI32526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii S, Kojima J, Shimizu K, Numata T, Kawaishi M, Odaka M, Morikawa T, Harada T, Nishimura SL, Kaneko Y, Nakayama K, Kuwano K. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015; 11:547-59; PMID:25714760; https://doi.org/ 10.1080/15548627.2015.1017190 [DOI] [PMC free article] [PubMed] [Google Scholar]