

ABSTRACT
We previously reported that facilitating the clearance of damaged mitochondria through macroautophagy/autophagy protects against acute myocardial infarction. Here we characterize the impact of exercise, a safe strategy against cardiovascular disease, on cardiac autophagy and its contribution to mitochondrial quality control, bioenergetics and oxidative damage in a post-myocardial infarction-induced heart failure animal model. We found that failing hearts displayed reduced autophagic flux depicted by accumulation of autophagy-related markers and loss of responsiveness to chloroquine treatment at 4 and 12 wk after myocardial infarction. These changes were accompanied by accumulation of fragmented mitochondria with reduced O2 consumption, elevated H2O2 release and increased Ca2+-induced mitochondrial permeability transition pore opening. Of interest, disruption of autophagic flux was sufficient to decrease cardiac mitochondrial function in sham-treated animals and increase cardiomyocyte toxicity upon mitochondrial stress. Importantly, 8 wk of exercise training, starting 4 wk after myocardial infarction at a time when autophagy and mitochondrial oxidative capacity were already impaired, improved cardiac autophagic flux. These changes were followed by reduced mitochondrial number:size ratio, increased mitochondrial bioenergetics and better cardiac function. Moreover, exercise training increased cardiac mitochondrial number, size and oxidative capacity without affecting autophagic flux in sham-treated animals. Further supporting an autophagy mechanism for exercise-induced improvements of mitochondrial bioenergetics in heart failure, acute in vivo inhibition of autophagic flux was sufficient to mitigate the increased mitochondrial oxidative capacity triggered by exercise in failing hearts. Collectively, our findings uncover the potential contribution of exercise in restoring cardiac autophagy flux in heart failure, which is associated with better mitochondrial quality control, bioenergetics and cardiac function.
KEYWORDS: autophagy, bioenergetics, cardiovascular disease, exercise, mitochondrial fission-fusion machinery, mitochondrial quality control
Introduction
Heart failure is a major public health problem worldwide. The development of pharmacological and nonpharmacological tools has dramatically improved the clinical outcome of patients with heart failure.1 However, these interventions generally provide symptomatic relief, and do not necessarily prevent or reverse molecular changes that occur in cardiac cells.2 Therefore, the development and optimization of therapeutic strategies capable of tackling fundamental mechanisms involving the pathophysiology of heart failure are critical for better survival and quality of life.
Despite the fact that mechanisms underlying both pathogenesis and progression of heart failure are multiple and complex, recent research provides convincing evidence that defective mitochondria are a hallmark of cardiomyocyte dysfunction.3,4 The maintenance of mitochondrial bioenergetic efficiency through a dynamically integrated quality control axis seems to be crucial to keep cardiomyocytes functionally viable.5-7 Mitochondrial fusion-fission and mitophagy are key players of mitochondrial quality control in cardiac cells, which allows functionally impaired mitochondria to be rescued or eliminated upon metabolic stress.6,8,9 Indeed, disruption of critical mitochondrial quality control mechanisms such as sequestration, sorting and elimination of dysfunctional mitochondria leads to heart failure.10-12
Exercise is a well-known nonpharmacological intervention capable of improving cardiovascular fitness in both healthy and diseased individuals.13,14 Long-term exercise training was demonstrated to confer sustained improvement in quality of life and reduction in both hospitalization and cardiac death in heart failure patients.15 Overall, exercise benefits are triggered by increased energy expenditure having an impact on mitochondrial metabolism, with subsequent effects on a wide range of intracellular signaling processes.16-18 However, the impact of exercise on mitochondrial quality control mechanisms during either physiological or pathological conditions is still unknown. In general, it is expected that mitochondrial quality control has evolved within the same mechanisms that regulate bioenergetic efficiency, given that disruption of mitochondrial quality control mitigates metabolic benefits of increased energy expenditure.19,20
The question arises as to whether there is any synergy between cardiac mitochondrial quality control and bioenergetic efficiency in heart failure and whether this connectivity drives the improved cardiovascular fitness induced by exercise. In this study, we explored the impact of exercise on cardiac mitochondrial quality control in healthy and failing hearts. The results indicate that failing hearts present pro-fission mitochondrial dynamics imbalance along with impaired autophagic flux. As expected, mitochondrial quality control disruption was associated with accumulation of dysfunctional mitochondria, but over a period of 8 wk of exercise this scenario was counterbalanced by improved cardiac autophagic flux and the re-establishment of a pool of healthy mitochondria, which contributed to improved heart failure prognosis.
Results
Exercise rescues mitochondrial health in failing hearts
To investigate whether heart failure pathophysiology is associated with changes in cardiac mitochondrial health, we first evaluated mitochondrial metabolism, mitochondrial permeability transition and redox balance in a rat model of myocardial infarction (Fig. 1A). All measurements were done in the noninfarcted (remote) cardiac area 12 wk after coronary artery ligation. At this stage, post-myocardial infarction animals presented signs of heart failure such as contractile dysfunction, enlarged left ventricle, wall thinning and fetal gene reprogramming (Fig. 1B-C and Table S1). These changes were accompanied by a dramatic reduction in cardiac mitochondrial bioenergetic efficiency, decreased ATP levels, increased sensitivity to Ca2+-induced mitochondrial permeability transition, excessive release of mitochondrial reactive oxygen species, exacerbated lipid peroxidation and consequent accumulation of both 4-hydroxynonenal (4-HNE)-protein adducts and protein carbonyls in heart failure (Fig. 1D-I).
Figure 1.
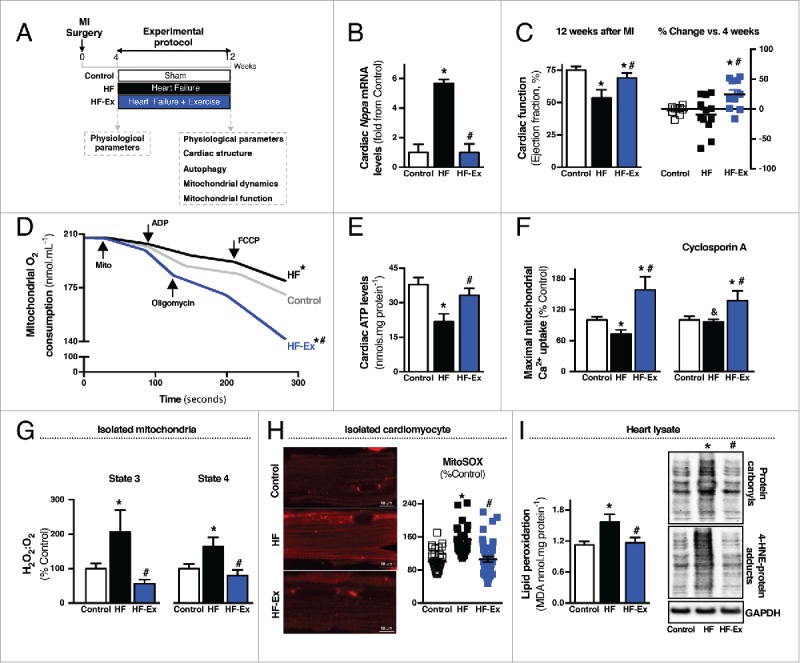
Exercise rescues mitochondrial health and improves cardiac function in heart failure. (A) Schematic panel illustrating the study design: male Wistar rats were submitted to myocardial infarction (MI) or sham surgery. Four wk later physiological parameters were evaluated and rats were randomly assigned into sedentary sham-treated (control), sedentary heart failure (HF) and exercised heart failure (HF-Ex) groups. HF-Ex rats were trained on a treadmill over 8 wk. At the end of the protocol, 12 wk after surgery, physiological parameters, cardiac structure, autophagy, mitochondrial dynamics and mitochondrial function were assessed. (B) Cardiac Nppa mRNA levels and (C) cardiac function, assessed by M-mode echocardiography in control, HF and HF-Ex rats. (D) O2 consumption in cardiac-isolated mitochondria, (E) cardiac ATP levels in heart lysate and (F) maximal Ca2+ uptake in cardiac-isolated mitochondria from control, HF and HF-Ex rats. (G) H2O2 release per O2 consumption (H2O2:O2) in state 3 and state 4 respiratory rates in cardiac-isolated mitochondria, (H) mitochondrial O2− production and representative confocal images in isolated cardiomyocytes stained with MitoSOX Red and (I) lipid peroxidation, protein carbonyl and 4-HNE-protein adducts in heart lysate from control, HF and HF-Ex rats. Data are presented as mean ± SEM.*, p < 0.05 vs. control; #, p < 0.05 vs. HF rats, and, p < 0.05 vs. HF rats without cyclosporin A.
Next, we determined whether exercise could improve mitochondrial health in heart failure. Overall, 8 wk of exercise improved oxidative phosphorylation efficiency, re-established cardiac ATP levels, increased tolerance to Ca2+-induced mitochondrial permeability transition, reduced mitochondrial reactive oxygen species release and re-established redox homeostasis (Fig. 1D-I and S1). It is important to mention that exercise started 4 wk after coronary artery ligation at a time when heart failure and mitochondrial dysfunction were already present (Fig. S2A-C and Table S1). Exercise also improved cardiac mitochondrial metabolism, reduced mitochondrial reactive oxygen species release and increased tolerance to Ca2+-induced mitochondrial permeability transition in sham-treated animals (Fig. S3A-F). Exercise had no impact on mitochondrial membrane potential in both sham-treated and heart failure groups (Fig. S3J and S4A-B).
Together, these findings reinforce our previous data showing that better mitochondrial health due to exercise is an important outcome for improving cardiovascular diseases.16 However, how exercise improves the overall mitochondrial bioenergetic efficiency and resistance to stress in failing hearts remains an open question. One possible explanation for the benefits of exercise in cardiac cells could be related to the maintenance of a healthy mitochondrial population through quality control mechanisms. Therefore, we next characterized the impact of exercise on mitochondrial quality control in heart failure.
Exercise re-establishes mitochondrial fission-fusion balance and reduces the accumulation of fragmented mitochondria in heart failure
First, we assessed mitochondrial morphology due to previous reports showing that mitochondrial architecture is directly affected by the disruption of quality control mechanisms in cardiac diseases.6 We found that failing hearts display a dramatic increase in mitochondrial number:size ratio, resulting in accumulation of smaller and spherical (fragmented) mitochondria (Fig. 2A). Despite changes in number and size, mitochondrial electron density and membrane potential were unaltered in failing hearts compared with healthy animals (Fig. 2A and S4A-B).
Figure 2.
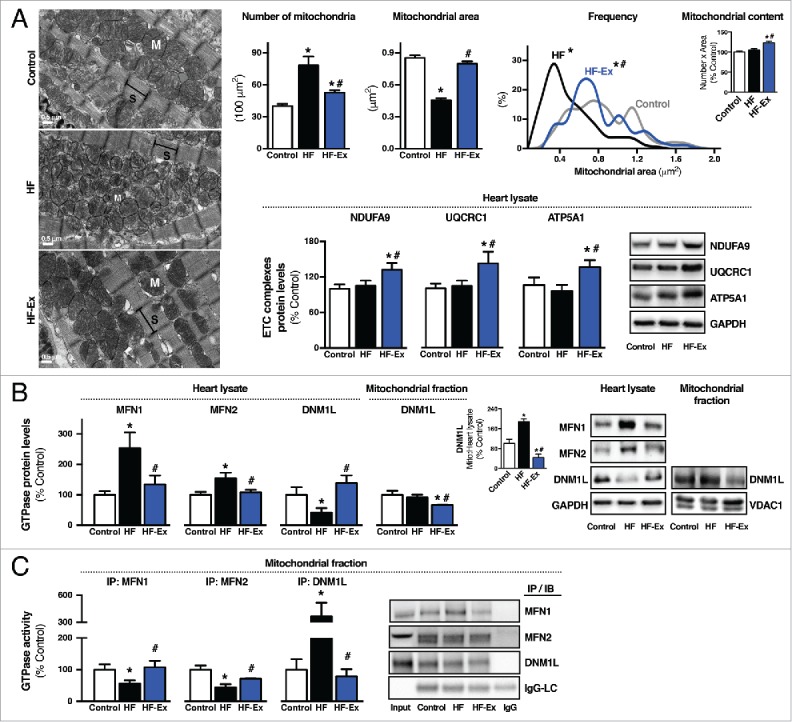
Exercise re-establishes mitochondrial fission-fusion balance in failing hearts. (A) Representative images of cardiac mitochondrial morphology (S, sarcomere; M, mitochondria), quantification of mitochondrial number and area, and frequency distribution of mitochondrial area evaluated by transmission electron microscopy, and protein levels of mitochondrial electron transport chain (ETC) complex I (NDUFA9), complex III (UQCRC1) and complex V (ATP5A1) in heart lysate from sedentary sham-treated (control), sedentary heart failure (HF) and exercised heart failure (HF-Ex) rat hearts. (B) MFN1, MFN2 and DNM1L protein levels in heart lysate and DNM1L protein levels in cardiac-isolated mitochondria from control, HF and HF-Ex rats. (C) MFN1, MFN2 and DNM1L GTPase activity in cardiac-isolated mitochondria from control, HF and HF-Ex rats. Data are presented as mean ± SEM. *, p < 0.05 vs. control; #, p < 0.05 vs. HF rats. LC, light chain.
Next, we determined whether exercise affects mitochondrial morphology in failing hearts. Cardiac mitochondria from exercised heart failure animals decreased in number and increased in size compared with nonexercised heart failure animals (Fig. 2A). As expected, exercise increased cardiac mitochondrial number in exercised sham-treated animals compared with the sedentary group (Fig. S3G). It is important to highlight that exercise increased cardiac mitochondrial electron density and content in both heart failure and sham-treated animals compared with nonexercised groups (Fig. 2A and S3G). These findings along with improved mitochondrial oxidative capacity (Fig. 1D-E) demonstrate that exercise is sufficient to re-establish the pool of healthy mitochondria in failing hearts.
Because mitochondrial number and size are tightly regulated by mitochondrial dynamics,21 we next measured the expression and activity profile of the main GTPases involved in mitochondrial fusion and fission in the heart (MFN1 [mitofusin 1], MFN2, and DNM1L [dynamin 1-like]).5 Both MFN1 and MFN2, large GTPases that mediate mitochondrial fusion and mitophagy, accumulated in failing hearts (Fig. 2B). Mitofusin accumulation usually occurs upon proteasomal inactivation.22 Therefore, elevated MFN1 and MFN2 in failing hearts might be a consequence of impaired proteasomal activity, as we previously demonstrated using an animal model of heart failure and failing human hearts.16,23 Cardiac Mfn1, Mfn2 and Dnm1l mRNA levels were significantly reduced in heart failure compared with sham-treated animals (Fig. S4C).
Despite increased cardiac MFN1 and MFN2 protein levels in heart failure, their GTPase activities were significantly reduced compared with the sham-treated group (Fig. 2C), suggesting an accumulation of dysfunctional mitofusins. A critical role of mitofusins for cardiac physiology was recently reported, where genetic disruption of Mfn1 and Mfn2 results in accumulation of fragmented mitochondria and leads to lethal heart failure in mice.21,24 In contrast, DNM1L GTPase activity was increased in failing hearts, as well as its translocation to the mitochondrial fraction (Fig. 2B-C). Overall, our findings suggest a mitochondrial fission-fusion imbalance in heart failure, favoring a pro-fission scenario accompanied by impaired mitochondrial oxidative capacity and excessive oxidative stress.
Next, we determined whether exercise affects mitochondrial dynamics in failing hearts. Eight weeks of exercise decreased accumulation of MFN1 and MFN2 in failing hearts. These results are explained, at least in part, by the positive effect of exercise in maintaining proteasomal activity in failing hearts.16 Moreover, exercise restored MFN1, MFN2 and DNM1L GTPase activities in failing hearts (Fig. 2B-C) and reversed the decreased DNM1L translocation to the mitochondria. These changes occurred in parallel to the re-establishment of the mitochondrial number:size ratio, bioenergetics efficiency and redox balance (Fig. 1D-I and Fig. 2A). In sham-treated animals, exercise increased both DNM1L translocation to the mitochondria and activity, as well as MFN2 GTPase activity (Fig. S3H-I).
Autophagic flux is impaired in failing hearts
To better understand the overall role of exercise in regulating mitochondrial quality control in heart failure, we next characterized cardiac autophagy. Basal autophagy is essential for a proper clearance of damaged/dysfunctional mitochondria.25
First, we measured at the end of the experimental protocol (Fig. 1A) the expression profile of MAP1LC3A/B (microtubule-associated protein 1 light chain 3 α/β), a key marker of autophagy. MAP1LC3A/B-II (lipidated form) levels were significantly increased in failing hearts compared with sham-treated animals (Fig. 3A). No changes in MAP1LC3A/B-I protein and Map1lc3a/b mRNA levels were detected among different groups (Fig. 3A and S5A). Moreover, failing hearts accumulated other proteins associated with autophagy such as BECN1 (Beclin 1, autophagy related), SQSTM1 (sequestosome 1) and PARK2 (Parkinson disease [autosomal recessive, juvenile] 2, parkin) (Fig. 3A and S5A). We did not find accumulation of either PARK2 or SQSTM1 in the mitochondrial fraction of failing hearts (Fig. 3A). Considering that mitochondrial depolarization is critical for PARK2-dependent mitophagy,26 these results can be explained, at least in part, by the fact that mitochondrial membrane potential was unaltered during heart failure compared with sham-treated rats (Fig. S4A-B). Of interest, accumulation of autophagy-related markers in failing hearts was accompanied by a significant increase in the proteolytic activity of CTSL (cathepsin L) (Fig. 3B), a lysosomal enzyme expressed in the heart. Using transmission electron microscopy, we also observed an accumulation of lysosome-like structures in failing hearts compared with sham-treated hearts (Fig. S5B).
Figure 3.
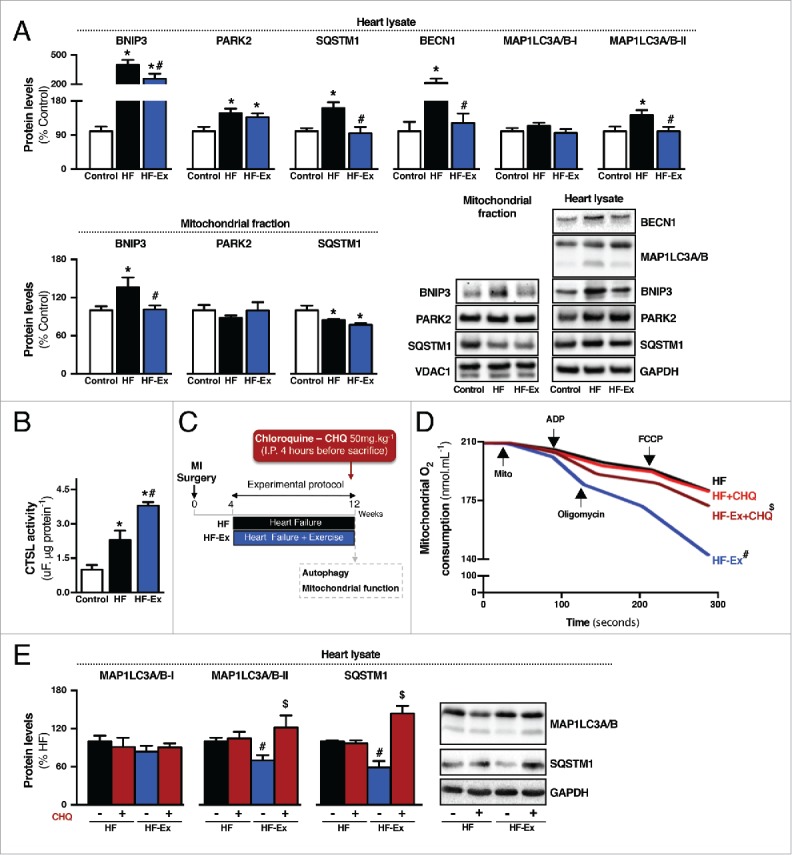
Exercise re-establishes autophagic flux in failing hearts. (A) Protein levels of BNIP3, PARK2, SQSTM1, BECN1 and MAP1LC3A/B in heart lysate and BNIP3, PARK2 and SQSTM1 in isolated mitochondria from sedentary sham-treated (control), sedentary heart failure (HF) and exercised heart failure (HF-Ex) rat hearts. (B) Cardiac CTSL activity in control, HF and HF-Ex rats. Data are presented as mean ± SEM.*, p < 0.05 vs. control; #, p < 0.05 vs. HF rats. (C) Schematic panel illustrating the study design: male Wistar rats were submitted to myocardial infarction (MI) surgery. 4 wk later rats were randomly assigned into sedentary heart failure (HF) and exercised heart failure (HF-Ex) groups. Exercised heart failure rats were trained on a treadmill over 8 wk. At the end of the protocol, 12 wk after surgery, rats were treated with a single intraperitoneal injection containing CHQ (50 mg.kg−1) to inhibit autophagic flux. Four h after injection animals were killed and autophagy-related markers and mitochondrial function were assessed. (D) O2 consumption in cardiac-isolated mitochondria and (E) protein levels of autophagy-related markers (MAP1LC3A/B and SQSTM1) in heart lysate from HF and HF-Ex rats treated with saline (−) or CHQ (+). Data are presented as mean ± SEM. #, p < 0.05 vs. HF (−) rats; $, p < 0.05 vs. HF-Ex (−) rats.
To better understand the meaning of increased levels of autophagy-related markers in heart failure, we performed an autophagic flux assay27 and treated the rats at the end of the experimental protocol with chloroquine (CHQ; 50mg.kg−1) for 4 h to block lysosomal function (Fig. 3C and Fig. 4D). As expected, CHQ treatment increased cardiac autophagy-related markers (MAP1LC3A/B-II and SQSTM1) in sham-treated animals (Fig. 4F), but not in those experiencing heart failure (Fig. 3E). These findings provide evidence that autophagic flux is impaired in failing hearts.
Figure 4.

Exercise does not change autophagic flux in healthy hearts. (A) Schematic panel illustrating the study design: male Wistar rats were submitted to physiological parameters evaluation and were randomly assigned into sedentary sham-treated (control) and exercised sham-treated (Ex) groups. Exercised rats were trained on a treadmill over 8 wk. At the end of the protocol, physiological parameters, cardiac structure, autophagy, mitochondrial dynamics and mitochondrial function were assessed. (B) Protein levels of BNIP3, PARK2, SQSTM1, BECN1 and MAP1LC3A/B in heart lysate and BNIP3, PARK2 and SQSTM1 in isolated mitochondria from sedentary (control) and exercised (Ex) rat hearts. (C) Cardiac CTSL activity in control and Ex rats. Data are presented as mean ± SEM.*, p < 0.05 vs. control. (D) Schematic panel illustrating the study design: male Wistar rats were randomly assigned into control and Ex groups. Exercised rats were trained on a treadmill over 8 wk. At the end of the protocol, rats were treated with a single intraperitoneal injection containing CHQ (50 mg.kg−1) to inhibit autophagic flux. Four h after injection animals were killed and autophagy-related markers and mitochondrial function were assessed. (E) O2 consumption in cardiac-isolated mitochondria and (F) protein levels of autophagy-related markers (MAP1LC3A/B and SQSTM1) in heart lysate from control and Ex rats treated with saline (−) or CHQ (+). Data are presented as mean ± SEM.*, p < 0.05 vs. control (−) rats. #, p < 0.05 vs. Ex (−) rats.
Next, we evaluated whether reduced autophagic flux is associated with accumulation of damaged mitochondria in heart failure. First, pharmacological disruption of autophagic flux was enough to decrease cardiac mitochondrial bioenergetic efficiency in sham-treated animals (Fig. 4E). In contrast, 4 h of CHQ treatment did not worsen mitochondrial dysfunction during heart failure (Fig. 3D). Of interest, transmission electron microscopy images provided evidence of excessive accumulation of fragmented mitochondria associated with lysosome-like structures in failing hearts (Fig. S5B). Overall, these findings suggest that reduced autophagic flux is associated with impaired cardiac bioenergetics through accumulation of dysfunctional mitochondria in both healthy and failing hearts.
Exercise re-establishes autophagic flux in failing hearts
Next, we determined whether exercise affects autophagy in failing hearts. Eight wk of exercise, starting 4 wk after coronary artery ligation at a time when impaired autophagic flux and mitochondrial dysfunction were already present (Fig. S2), did not affect mRNA transcript levels of autophagy related-genes in failing hearts (Fig. S5A). However, exercise significantly reduced cardiac MAP1LC3A/B-II, BECN1 and SQSTM1 protein levels during heart failure (Fig. 3A), suggesting an improvement in the cardiac autophagic flux. Exercise also decreased cardiac protein levels of SQSTM1, but not other autophagy-related markers, in sham-treated animals (Fig. 4B). Of interest, exercise training significantly increased CTSL activity in both sham-treated and heart failure animals (Fig. 3B and Fig. 4C).
To test whether exercise training affects cardiac autophagic flux we treated at the end of the experimental protocol (Fig. 3C and Fig. 4D) both exercised heart failure and exercised sham-treated rats with CHQ for 4 h and measured cardiac levels of autophagy-related markers. Exercise re-established cardiac responsiveness to CHQ treatment in heart failure animals demonstrated by a significant CHQ-induced accumulation of MAP1LC3A/B-II and SQSTM1 in exercised heart failure animals compared with non-exercised heart failure animals (Fig. 3E). Of interest, exercise did not change autophagic flux in sham-treated animals (Fig. 4F). These findings provide evidence that 8 wk of exercise is sufficient to re-establish cardiac autophagic flux in a rat model of heart failure.
Next, we evaluated whether increased cardiac mitochondrial oxidative capacity seen in exercised heart failure animals was associated with the re-establishment of autophagic flux. Supporting an autophagy mechanism for exercise-induced improvements in mitochondrial bioenergetics, 4 h of CHQ treatment was enough to mitigate the increased mitochondrial oxidative capacity triggered by exercise training in heart-failure animals (Fig. 3D). Of interest, CHQ also reduced the benefits of exercise with regard to mitochondrial bioenergetics in sham-treated animals (Fig. 4E). Finally, the re-establishment of cardiac mitochondrial quality control in exercised heart failure was associated with fewer fragmented mitochondria associated with lysosome-like structures in failing hearts (Fig. S5B).
Because heart failure and exercise affected both mitochondrial dynamics and autophagy, we next used differentiated H9c2 cells (a cell line established from rat embryonic ventricular tissue) (Fig. 5A) to better understand the isolated impact of each system on mitochondrial health and cellular viability. Differentiation of H9c2 myoblasts into myotubes was validated by a significant increase of cardiomyocyte markers such as cardiac sarcomeric proteins (TNNI3 [troponin I, cardiac 3] and ACTC1 [actin, α cardiac muscle 1]), calcium transporters (CACNA1A [calcium channel, voltage-dependent, P/Q type, α 1A subunit]) and mitochondrial components (COX4I1 [cytochrome c oxidase subunit IV isoform 1] and PARK2) (Fig. 5B).
Figure 5.
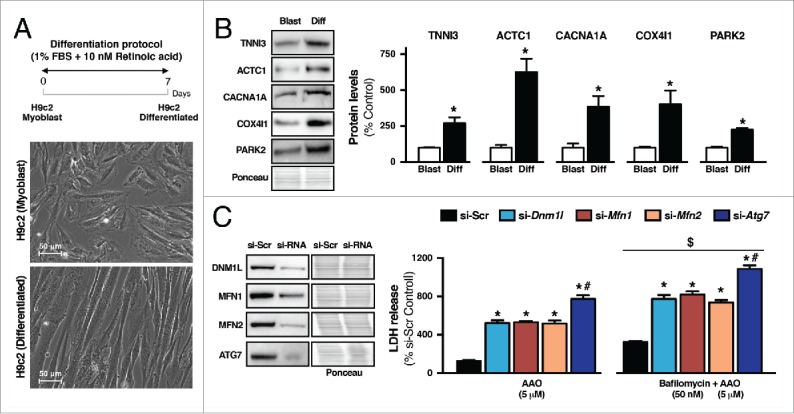
Downregulation of mitochondrial dynamics- or autophagy-related genes exacerbates mitochondrial dysfunction-induced cytotoxicity in H9c2 cells. (A) Schematic panel illustrating the study design and representative images of morphological changes after the differentiation protocol in H9c2 cells. H9c2 cells were differentiated from myoblasts by reducing FBS from 10% to 1% for 7 d. To improve cardiac myocyte yield, the media was daily supplemented in the dark with 10 nM retinoic acid. (B) Protein levels of specific markers of cardiac differentiation TNNI3, ACTC1, CACNA1A, COX4I1 and PARK2 in H9c2 myoblasts (Blast) and H9c2 differentiated (Diff) cells. Data are presented as mean ± SEM. *, p < 0.05 vs. Blast. (C) H9c2 differentiated cells were transfected with siRNA targeting Dnm1l (si-Dnm1l), Mfn1 (si-Mfn1), Mfn2 (si-Mfn2), Atg7 (si-Atg7) or with a nonspecific siRNA control (si-Scr). LDH release was measured after 6-h treatment with antimycin A + oligomycin (AAO; 5 μM each) or 2-h treatment with bafilomycin A1 (50 nM) followed by 6 h with AAO. Data are presented as mean ± SEM. *, p < 0.05 vs. si-Scr; #, p < 0.05 vs. si-Dnm1l, si-Mfn1 and si-Mfn2; $, p < 0.05 vs. AAO.
Next, we determined whether disruption of either mitochondrial fission-fusion or autophagy affects cell viability upon mitochondrial stress (AAO; antimycin A + oligomycin for 6 h), mimicking our heart failure model. For that, differentiated H9c2 cells were transfected with small interfering RNA (siRNA) targeting mitochondrial fission (si-Dnm1l), mitochondrial fusion (si-Mfn1 and si-Mfn2) or autophagy-related (si-Atg7) genes (Fig. 5C). The knockdown of mitochondrial dynamics-related genes significantly increased cytotoxicity upon AAO-induced mitochondrial stress compared with control (nonspecific siRNA [siScr]) cells (Fig. 5C). Of interest, reduction of autophagic flux by knocking down ATG7 had a prominent increase in H9c2 cytotoxicity compared with other groups (Fig. 5C). Supporting an autophagy mechanism for impaired mitochondrial bioenergetics-induced cytotoxicity, pharmacological inhibition of autophagy using bafilomycin A1 (50 nM for 2 h) exacerbated H9c2 cytotoxicity in all groups (Fig. 5C), including those treated with siRNA for mitochondrial fission-fusion-related genes. These findings reinforce our in vivo model showing that autophagy is critical for the maintenance of cardiac myocyte viability upon stress. These findings highlight the benefits of exercise as a strategy to improve autophagy and restore mitochondrial bioenergetics to mitigate the effects of heart failure (Fig. 3D).
Discussion
Heart failure is a multifactorial syndrome and represents a major and growing public health problem. Despite advances in pharmacological and nonpharmacological interventions, the incidence and prevalence of heart failure continue to increase.1 Consequently, critical mechanisms related to the establishment and progression of heart failure must be extensively studied.
Considering their critical role in ATP generation, redox balance, Ca2+ homeostasis and cell death, mitochondria have become an attractive target for novel therapies against heart failure. The maintenance of mitochondrial bioenergetics relies on a dynamically integrated quality control axis that allows segregation of damaged mitochondria, which are further sequestered and removed by autophagy. Disruption of genes that regulate mitochondrial quality control mechanisms such as mitochondrial fusion, fission or mitophagy reduces both myocardial functionality and viability.21,24,25,28-30 We have recently reported that either inhibition of excessive mitochondrial fragmentation or improvement of mitochondrial clearance protects against acute cardiac ischemia-reperfusion injury.6,8 These findings demonstrate the pivotal role of mitochondrial quality control mechanisms in the maintenance of myocardial homeostasis and protection against acute stress.
Here, using an in vivo model of postmyocardial infarction-induced heart failure, we provided evidence that failing hearts exhibit disrupted mitochondrial quality control, characterized by loss of mitochondrial fusion/fission balance and reduced autophagic flux. These changes were followed by increased mitochondrial fragmentation with reduced bioenergetic efficiency and excessive oxidative stress. Moreover, we demonstrated that 8 wk of exercise training re-established both autophagic flux and the pool of healthy mitochondria in failing hearts, characterized by the increased efficiency of mitochondrial oxidative phosphorylation. Further supporting an autophagy mechanism for exercise-induced improvements in mitochondrial bioenergetics, acute inhibition of autophagic flux using CHQ was sufficient to abrogate the increased mitochondrial oxidative capacity triggered by exercise training in a heart failure model. In sham-treated animals, exercise improved both mitochondrial content and oxidative capacity without affecting autophagic flux. These findings suggest that improved mitochondrial oxidative capacity as well as mitochondrial biogenesis observed in exercised sham-treated animals appears to occur by a mechanism independent of changes in autophagic function. However, acute CHQ treatment was sufficient to abolish the increased mitochondrial oxidative capacity triggered by exercise. Future studies exploring the contribution of autophagy to exercise-induced mitochondrial changes under physiological conditions are required.
Autophagy is a dynamic process essential for the maintenance of cardiac function. Changes in autophagic activity can exacerbate or mitigate cardiac pathophysiology. Activation of autophagy protects the heart against acute ischemia-reperfusion injury.8,31 In contrast, impaired autophagy is associated with ventricular dysfunction and cardiomyocyte death upon ischemia-reperfusion injury.32,33 During chronic conditions (i.e., heart failure), impaired autophagy seems to contribute to myocardial degeneration. Evidence supporting the detrimental role of autophagy in heart failure emerged from ultrastructural analysis of hearts explanted from end-stage heart failure patients. These studies demonstrated that failing hearts accumulate autophagic vacuoles, which are associated with increased levels of polyubiquitinated proteins, vacuole-associated mitochondria and reduced integrity of cardiac myofibers.34,35 However, those findings do not necessarily provide evidence of increased autophagy during heart failure. Accumulation of autophagic vacuoles could instead be a consequence of impaired autophagic flux.27
Our study provides evidence that increased protein levels of autophagy-related markers along with the accumulation of lysosome-like structures in heart failure are related to impaired autophagic flux instead of excessive autophagy. In fact, blocking autophagic flux using CHQ increased cardiac levels of MAP1LC3A/B-II, BECN1 and SQSTM1 in sham-treated, but not in heart failure animals. The loss of responsiveness to CHQ treatment was previously demonstrated in hearts exposed to ischemia-reperfusion injury.32 A similar phenotype of accumulation of cardiac autophagic vacuoles has been found in mice lacking LAMP2 (lysosomal-associated membrane protein 2), which is an important constituent of the lysosomal membrane30 or CTSL, a lysosomal cysteine protease within the myocardium. Either LAMP2 or CTSL deficiency causes impaired autophagic flux and cardiomyopathy in rodents and humans.29,30,36,37 Of interest, we found an increased cardiac CTSL activity in heart failure animals compared with sham-treated, suggesting a compensatory mechanism due to impaired autophagic flux. However, future studies are required to validate this hypothesis.
We also reported that failing hearts accumulated autophagic vacuole-associated fragmented mitochondria, suggesting a defect in mitochondria recruitment and uptake by lysosomes. This scenario was accompanied by accumulation of fragmented mitochondria with reduced bioenergetic efficiency and oxidative stress. Of interest, CHQ-induced disruption of autophagic flux did not exacerbate the already poor mitochondrial function and oxidative stress seen during heart failure. In contrast, 4 h of CHQ treatment was sufficient to reduce mitochondrial bioenergetic efficiency in sham-treated animals. Our findings suggest that autophagy is critical for the maintenance of cardiac mitochondrial bioenergetics. We speculate that disrupted autophagic flux along with accumulation of dysfunctional mitochondria is linked to impaired mitophagy—an essential catabolic process involving elimination of mitochondria by lysosomes.
Together, these data indicate that loss of autophagic flux is detrimental to the maintenance of a healthy mitochondrial population, which contributes to the pathophysiology of heart failure. In fact, recent evidence demonstrates that activation of autophagy is critical to remove damaged mitochondria and protect the heart against acute and chronic stresses such as ischemia-reperfusion injury5 and cardiac proteinopathy,38 respectively. Moreover, knocking out individual genes involved in the formation of autophagosomes (i.e., Becn1, Atg5 or Pik3c3/Vps34), fusion with lysosomes (i.e., Lamp2) or lysosomal proteolysis (i.e., Ctsl) results in the accumulation of damaged mitochondria and development of cardiac dysfunction in mice.25,28-30
Finally, we found that 8 wk of exercise, starting 4 wk after myocardial infarction at a time when impaired autophagic flux and mitochondrial dysfunction were already present, improved mitochondrial quality control by re-establishing both mitochondrial fission-fusion balance and autophagic flux. Overall, these changes contributed to the maintenance of a healthy mitochondrial population, which positively affected cardiac metabolism, redox balance and contractility properties of failing hearts. Moreover, exercise improved cardiac CTSL activity in both sham-treated and heart failure groups; increased cathepsin activity is associated with protection against pathological cardiac remodeling.39
Different groups recently reported the positive impact of exercise in activating cardiac autophagy.38,40 Voluntary running was shown to be sufficient to improve autophagy and increase survival in mice with cardiac proteinopathy.38 Moreover, impaired exercise-induced autophagy reduced running performance in transgenic mice.40 Our results using a moderate intensity running exercise provide new evidence that regular (5 d/wk) exercise re-establishes both autophagic flux and mitochondrial dynamics in a heart failure model, which contributes to the maintenance of cardiac bioenergetic efficiency and heart failure outcome. We speculate that maintenance of autophagic efficiency is critical for the exercise-induced metabolic adjustments, mainly triggered by increased energy expenditure having an impact on mitochondrial health.
Overall, our study provides insights into the benefits of exercise in regulating mitochondrial quality control to mitigate the effects of heart failure. We find an excessive accumulation of fragmented/dysfunctional mitochondria in failing hearts, most likely resulting from impaired mitochondrial fusion and defective autophagy. In addition, exercise re-establishes mitochondrial quality control and affects the cardiac pool of healthy mitochondria. We propose that exercise improves the synergy between cardiac mitochondrial quality control and bioenergetic efficiency in heart failure, therefore contributing to better disease prognosis.
Materials and methods
Study design
1) A cohort of male Wistar rats was submitted to myocardial infarction surgery or an equal procedure without ligation of the left anterior descending coronary artery (sham surgery). Four wk after surgery, physiological parameters (cardiac function, blood pressure and maximal aerobic capacity) were evaluated and rats were randomly assigned into sedentary sham-treated (control), sedentary heart failure (HF) and exercised heart failure (HF-Ex) groups. HF-Ex rats were trained on a treadmill over 8 wl, 5 d/wk, 60 min/d at 60% of maximal aerobic capacity, as described elsewhere.15 At the end of the protocol, 12 wk after surgery, measurements of physiological parameters were repeated, the rats were killed and cardiac structure, autophagy, mitochondrial dynamics and mitochondrial function were assessed. Another set of animals exposed to the same experimental protocol was treated, at the end of the protocol, 12 wk after surgery, with a single intraperitoneal injection containing CHQ (50 mg.kg−1) to inhibit autophagic flux. Four h after injection rats were killed and autophagy-related markers and mitochondrial function were assessed.
2) A cohort of male Wistar rats was submitted to physiological parameters evaluation (cardiac function, blood pressure and maximal aerobic capacity) and rats were randomly assigned into sedentary sham-treated (control) and exercised sham-treated (Ex) groups. Exercised rats were trained using the same conditions described above. At the end of the protocol, measurements of physiological parameters were repeated, the rats were killed and cardiac structure, autophagy, mitochondrial dynamics and mitochondrial function were assessed. Another set of animals exposed to the same experimental protocol was treated, at the end of the protocol, with a single intraperitoneal injection containing CHQ (50 mg.kg−1) to inhibit autophagic flux. Four h after injection rats were killed and autophagy-related markers and mitochondrial function were assessed.
Animals
A cohort of male Wistar rats (250–300 g) was selected for the study. Rats were maintained in a 12:12-h light-dark cycle and temperature-controlled environment (22°C) with free access to standard laboratory chow (Nuvital Nutrientes, Nuvilab CR-1) and tap water. This study was conducted in accordance with the ethical principles in animal research adopted by the Brazilian College of Animal Experimentation (www.cobea.org.br). The animal care and protocols in this study were reviewed and approved by the Ethics Committee of the School of Physical Education and Sport of the University of Sao Paulo (2009/56).
Myocardial infarction-induced heart failure model
We have chosen this model because myocardial infarction is the underlying etiology of heart failure in nearly 70% of patients.41 Male Wistar rats (12th wk of age), were anesthetized intraperitoneally with ketamine (50 mg.kg−1) and xylazine (10 mg.kg−1), endotracheally intubated, and mechanically ventilated with room air (respiratory rate of 60–70 breaths/min and tidal volume of 2.5 mL). Left thoracotomy between the fourth and fifth ribs was performed and the left anterior descending coronary artery was permanently ligated.42 After the surgery, animals were monitored daily and, as previously published,16 heart failure was observed 4 wk after coronary artery ligation and was defined when an animal presented pathological cardiac remodeling accompanied by left ventricle dysfunction and cardiac dilation (Fig. 1 and Table S1), according to the Guidelines of American Heart Association.43 Left thoracotomy with equal procedure duration to that of the HF group, but without left anterior descending coronary artery ligation, was undertaken in the sham-treated group (control, n = 21). After 4 wk, HF animals were randomly assigned into sedentary (HF, n = 14) and exercise groups (HF-Ex, n = 13) (Fig. 1A).
Cardiovascular measurements
Four and 12 wk after surgery, noninvasive cardiac function evaluation was performed by M-mode echocardiography in anesthetized (isoflurane 3%) rats. Briefly, rats were positioned in the supine position with front paws wide open and ultrasound transmission gel was applied to the precordium. Transthoracic echocardiography was performed using an Acuson Sequoia model 512 echocardiographer (SIEMENS) equipped with a 14-MHz linear transducer. Left ventricle systolic function was estimated by ejection fraction (EF) and fractional shortening (FS) as follows: EF (%) = [(LVEDD3 – LVESD3)/LVEDD3] x 100 and FS (%) = [(LVEDD – LVESD)/LVEDD] x 100, where LVEDD is the left ventricular end-diastolic diameter, and LVESD is the left ventricular end-systolic diameter.
Graded treadmill exercise test and running training protocol
All animals were submitted to graded exercise testing on a motor treadmill adapted to experimental models before and after the experimental period. After being adapted to treadmill exercises and the test environment for over 1 wk (10 min each session), rats were placed in the treadmill and allowed to acclimatize for at least 30 min. Treadmill speed started at 6 m.min−1 and was increased by 3 m.min−1 every 3 min at 0% grade until exhaustion, where rats could no longer maintain a running speed over 3 min. This test provided the total distance run, and peak workload was measured at the termination of the test.44
Exercised sham-treated (Ex) and exercised heart failure (HF-Ex) rats performed moderate-intensity running training on a motor treadmill over 8 wk (from 12th–20th wk of age), 5 d/w, 60 min/d, as described elsewhere.15 Running speed and duration of exercise were progressively increased to elicit 60% of maximal speed (corresponding to the maximal lactate steady-state workload) at the second wk of training.45 At the fourth wk of training, run capacity was evaluated to readjust exercise training intensity. Treadmill running skills were maintained in sedentary sham-treated (control) and sedentary heart failure rats (HF) by treadmill running for 5 min, twice a wk. This procedure was performed to avoid any interference of treadmill stress on the variables studied. This latter activity did not seem to alter maximal exercise capacity.
ATP content
ATP content in the ventricular remote area was determined by light emission at 560 nm on a spectrofluorometer (Molecular Devices SPECTRAmax) using a commercial luciferin-luciferase kit (ATP Determination Kit; Invitrogen, A22066).
Mitochondrial isolation
Heart mitochondria were isolated as described by Cancherini et al.46 Briefly, cardiac samples from a remote area were minced and homogenized in isolation buffer (300 mM sucrose [Amresco, 0335], 10 mM HEPES, 2 mM EGTA, pH 7.2, 4°C) containing 0.1 mg/mL of type I protease (bovine pancreas; Sigma, P4630) to release mitochondria from within muscle fibers and later washed in the same buffer in the presence of 1 mg/mL bovine serum albumin (Amresco, 0332). The suspension was homogenized in a 40 mL tissue grinder and centrifuged at 950 g for 5 min. The resulting supernatant was centrifuged at 9500 g for 10 min. The mitochondrial pellet was washed, resuspended in isolation buffer and submitted to a new centrifugation (9500 g for 10 min). The mitochondrial pellet was washed and the final pellet was resuspended in a minimal volume of isolation buffer.
Mitochondrial function
Mitochondrial function was assessed in isolated cardiac mitochondria from a remote area as described elsewhere.47 All experiments with isolated mitochondria (0.125 mg mitochondrial protein/mL) were monitored in experimental buffer containing 125 mM sucrose, 65 mM KCl, 10 mM HEPES, 2 mM inorganic phosphate, 2 mM MgCl2, 100 µM EGTA, 0.01% BSA, pH 7.2 and were made in the presence of succinate (Sigma, S3674), malate (Sigma, M1000) and glutamate (Sigma, G1251) substrates (2 mM of each) with continuous stirring at 37°C.
Mitochondrial O2 consumption was monitored using a computer-interfaced Clark-type electrode (OROBOROS, Oxygraph-2k). ADP (1 mM; Amresco, 0160) was added to induce state 3 respiratory rates. A subsequent addition of oligomycin (1 µg.mL−1; Sigma, 4876) was used to determine state 4 rates. Respiratory control ratios were calculated by the ratio state 3:state 4. Additionally, 0.1 mM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; Enzo, BML-CM120) was added to evaluate O2 consumption during the mitochondrial uncoupling state.47
Mitochondrial Ca2+ uptake was measured by monitoring Calcium Green (100 mM; Molecular Probes, C3010MP) fluorescence with consecutive additions of 50 μM CaCl2 until the mitochondria failed to reduce extramitochondrial Ca2+. The fluorescence was monitored using a fluorescence spectrophotometer (F-2500, Hitachi) operating at excitation and emission wavelengths of 506 and 532 nm, respectively, and slit widths of 5.0 nm.48 We therefore plotted a calibration curve that correlates fluorescence and Ca2+ concentration.
Mitochondrial H2O2 release was measured using an Amplex Red (25 μM; Molecular Probes, A12222)-horseradish peroxidase (0.5 U.mL−1; Sigma, P8125) system.47 Amplex Red is oxidized in the presence of extramitochondrial horseradish peroxidase bound to H2O2, generating resorufin, which can be detected using a fluorescence spectrophotometer (ʎex = 563/ʎem = 587 nm) (F-2500, Hitachi). In an attempt to estimate H2O2 release during state 3 and state 4 respiratory rates, and during the uncoupling state, we added ADP (1 mM), oligomycin (1 µg.mL−1) and FCCP (0.1 mM), respectively. Calibration was conducted by adding H2O2 at known concentrations (A240 = 43.6 M−1.cm−1) to the experimental buffer.
Mitochondrial inner membrane potential was estimated through fluorescence changes of 5 μM safranin O (Amresco, 0574) (ʎex = 485/ʎem = 586 nm) in the presence of oligomycin using a fluorescence spectrophotometer (F-2500, Hitachi), as described previously.49 Data obtained were calibrated using a potassium gradient.
Adult cardiomyocyte isolation
Adult ventricular myocytes were freshly isolated as described previously.23 Briefly, hearts were removed and perfused via the Langendorff method with Ca2+-free modified Tyrode solution (130 mM NaCl, 5.4 mM KCl, 25 mM HEPES, 0.33 mM NaH2PO4, 1 mM MgCl2, 1 mM lactic acid [Sigma, L6402], 3 mM sodium pyruvate, 22 mM glucose, pH 7.4) supplemented with 10 U.L−1 insulin (Sigma, I4011) until the blood was washed out. Hearts were then perfused with modified Tyrode solution containing type 2 collagenase (Worthington, LS004177), removed from the perfusion apparatus, minced into 1-mm chunks and stirred. Cells were then filtered through a 200-μm mesh, and extracellular Ca2+ concentration was raised to 1.8 mM through 3 centrifuge cycles. Cells were stored in Dulbecco's modified Eagle's medium; Sigma, D6429) until they were used for experiments.
Mitochondrial superoxide anion production
Mitochondrial superoxide anion (O2−) production was quantified in isolated cardiomyocytes. Isolated cardiomyocytes were incubated at 37°C for 30 min with 3 µM MitoSOX Red (Molecular Probes, M7513). The cells were then washed, mounted and viewed with a laser scanning confocal microscope and mitochondrial superoxide production was estimated through fluorescence changes (ʎex = 510/ʎem = 580 nm) (n = 3 per group, 15 cells per rat). Experiments were performed at room temperature (22–24°C). Images were obtained using the ZEISS Meta confocal microscope (Zeiss 510 Meta) from CAPI (Centro de Aquisição e Processamento de Imagens ICB/UFMG).
Lipid hydroperoxidation
Lipid hydroperoxides were evaluated in heart lysate from a remote area using the thiobarbituric acid reactive substances assay, as described in the Oxi-Tek TBARS Assay Kit (Zeptometrix, 22–156–701). Thiobarbituric acid reacts with malondialdehyde, an end product of lipid peroxidation, generating a fluorescent product.
Immunoblotting
Protein levels were measured by immunoblotting in heart lysate and mitochondrial fraction from the ventricular remote area, and in the whole lysate of H9c2 cells. Briefly, samples were subjected to SDS-PAGE in polyacrylamide gels (6–15%) depending upon protein molecular weight. After electrophoresis, proteins were electrotransferred onto nitrocellulose membranes. Equal gel loading and transfer efficiency were monitored using 0.5% Ponceau S staining of blot membrane. Blotted membrane was then blocked (5% nonfat dry milk, 10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Tween 20 [Amresco, M147]) for 2 h at room temperature and then incubated overnight at 4°C with specific antibodies against 4-HNE (4-hydroxy-2-nonenal; Calbiochem 393207), ACTC1 (Santa Cruz Biotechnology, sc-58670), ATP5A1 (ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit 1; Abcam, ab14748), ATG7 (Novus Biologicals, MAB6608), BECN1 (Cell Signaling Technology, 3738S), BNIP3 (Cell Signaling Technology, 3769S), CACNA1A (Boster Biotechnology, PA2292), COX4I1 (Santa Cruz Biotechnology, sc-58348), DNM1L (Biosciences, BD-611113), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Advanced Immunochemical, RGM2), MAP1LC3A/B (Cell Signaling Technology, 4108S), MFN1 (Abnova, H00055669-M04), MFN2 (Abnova, H00009927-M01), NDUFA9 (NADH dehydrogenase [ubiquinone] 1 α subcomplex, 9; Mitosciences, MS111), PARK2 (Santa Cruz Biotechnology, sc-32282), SQSTM1 (Cell Signaling Technology, 5114S), TNNI3 (Santa Cruz Biotechnology, sc-365446), UQCRC1 (ubiquinol-cytochrome c reductase core protein 1; Mitosciences, MS303) and VDAC1 (voltage-dependent anion channel 1; Santa Cruz Biotechnology, sc-32063). Binding of the primary antibody was detected with the use of peroxidase-conjugated secondary antibodies (rabbit, mouse or goat, depending on the protein, for 2 h at room temperature; Thermo Fisher Scientific, 31460, 31430 and A27014) and developed using enhanced chemiluminescence detected by autoradiography. Oxidized proteins levels were assessed using an OxyBlot Protein Oxidation Kit (Millipore, S7150). Quantification analysis of blots was performed with the use of NIH ImageJ software. Samples were normalized to relative changes in GAPDH (heart lysate), VDAC1 (mitochondrial fraction) or Ponceau S (H9c2 cells) and expressed as percentage of the control group.
Transmission electron microscopy
Rat hearts were fixed, embedded and stained as described previously.50 Small blocks (5 blocks per animal) from a cardiac remote area were cut and fixed with 2% glutaraldehyde and 4% paraformaldehyde in sodium cacodylate buffer, pH 7.3 for 1 h at room temperature and cut into ∼1-mm3 blocks. After several buffer washes, samples were post-fixed in 2% osmium tetroxide and 1% uranyl acetate for 2 h, rinsed in water, dehydrated through ascending concentrations of ethanol followed by 100% acetone, and then infiltrated and embedded in Eponate 12 (Ted Pella, NC0541169). Final blocks were used to evaluate mitochondrial and lysosomal number and morphology. Images were acquired using a JEOL1230 Gatan 967 CCD transmission electron microscope at 80kV and a Gatan Orius 4k X 4k digital camera (Gatan). We performed qualitative analysis in at least 10 fields per rat (3 rats per group).
Mitochondrial morphology
Mitochondrial distribution was quantified in adult isolated cardiomyocytes. Cells were incubated at 37°C for 30 min with 200 nM MitoTracker Green (Molecular Probes, M7514), washed, mounted and viewed with a laser scanning confocal microscope. Cardiac mitochondria were visualized through fluorescence changes (ʎex = 490/ʎem = 516 nm) (n = 3 per group, 15 cells per rat). Experiments were performed at room temperature (22–24°C). Images were obtained using the ZEISS Meta confocal microscope (Zeiss, 510 Meta) from CAPI (Centro de Aquisição e Processamento de Imagens ICB/UFMG).
GTPase activity
MFN1, MFN2 and DNM1L were immunoprecipitated from a mitochondrial fraction of rat hearts. GTPase activity was assessed using a GTPase Assay Kit (Innova Biosciences, 602). Briefly, the mitochondrial fraction of rat heart (250 μg protein) was incubated in 1 mL immunoprecipitation (IP) lysis buffer (150 mM NaCl, 5 mM EDTA, 10 mM Tris-HCl, 0.1% Triton X-100 [Sigma, X-100], pH 7.4) with antibodies against MFN1, MFN2 and DNM1L for 3 h at 4°C, followed by incubation with protein G PLUS-agarose beads (Santa Cruz Biotechnology, sc-2002) for 1 h at 4°C. After low speed centrifugation, 2300 g for 3 min at room temperature, the pellet was washed 3 times in 1 mL IP lysis buffer with low speed centrifugation after each wash. The immunoprecipitate was kept in 10 µL of IP lysis buffer. GTPase activity was normalized to the immunoprecipitate, measured by immunoblotting.
CTSL activity
CTSL activity was determined in 25 µg of a mitochondria- and lysosomes-enriched cardiac fraction by fluorescence excitation at 400 nm and emission at 505 nm on a Molecular Devices SPECTRAmax spectrofluorometer using a commercially available kit (Cathepsin L Activity Assay Kit [Fluorometric]; Abcam, ab65306) following the manufacturer's instructions. Data are presented as fold change from the control group.
Chloroquine treatment
To confirm impaired autophagy during heart failure and to test whether autophagy contributes to mitochondrial dysfunction we decided to inhibit the autophagic process in vivo. Four and 12 wk after surgery, 4 h before sacrifice, rats were treated with a single intraperitoneal injection containing CHQ 50 mg.kg−1 (Sigma, C6628). CHQ inhibits lysosomal activity and allows the measurement of autophagic flux in vivo.51
Cell culture
H9c2 cells were maintained in Dulbecco's modified Eagle's medium, supplemented with 10% (v/v) fetal bovine serum (FBS; Life Technologies, 26140–079) and 1% (v/v) penicillin/streptomycin (Sigma, P4333) at 37°C in 5% CO2 in 95% air. H9c2 myoblasts have been constantly used as an alternative for cardiomyocytes; however, their metabolism relies on glycolysis. Considering that adult heart maintains a predominantly mitochondrial oxidative metabolism,52 and H9c2 cells have the ability to differentiate toward a cardiac phenotype,53 we differentiate H9c2 cells by culturing them in low serum medium (1% FBS) in the presence of retinoic acid (10 nM; Sigma, R2625) for 7 d. The differentiation of H9c2 cells induced cell fusion and the formation of larger cells compared with the common spindle-to-stellate shape exhibited for myoblasts (Fig. 5A), a morphological indication of H9c2 differentiated cells.
siRNA transfection
Differentiated H9c2 cells were transfected with small interfering RNA (siRNA) for Atg7 (Thermo Fisher Scientific, s161900), Dnm1l (Thermo Fisher Scientific, s220305), Mfn1 (Thermo Fisher Scientific, s140577) and Mfn2 (Thermo Fisher Scientific, s134143), or with a nonspecific siRNA control (Silencer Select Negative Control; Thermo Fisher Scientific, 4390843) using Effectene Transfection Reagent (Qiagen, B00118) following the manufacturer's instructions. The media was replaced after 24 h incubation with fresh medium, and the cells were maintained for another 24 h. Subsequently, LDH (lactate dehydrogenase) release was measured after 6 h treatment with antimycin A (Sigma, A8674) + oligomycin (Sigma, 04876) (5 μM each) or 2-h treatment with bafilomycin A1 (50 nM; EMD Millipore, 19–148) followed by 6 h with AAO. LDH release was determined by absorbance at 450 nm on a spectrofluorometer (Molecular Devices SPECTRAmax) using a commercial LDH Activity Assay Kit (Sigma, MAK066).
Statistical analysis
Data are presented as means ± standard error of the mean (SEM). Shapiro-Wilk normality test was used to verify data normal distribution. One-way analysis of variance (ANOVA) with a post-hoc testing by Duncan was used to analyze data from Figs. 1, 2, 3A-B and 5C. Two-way ANOVA with a post-hoc testing by Duncan was used to analyze data from Figs. 3D-E and 4E-F. An unpaired Student t test was used to analyze data from Figs. 4B-C and 5B. Statistical significance was considered achieved when the value of P was < 0.05.
Supplementary Material
Abbreviations
- 4-HNE
4-hydroxy-2-nonenal
- AAO
antimycin A + oligomycin
- ACTC1
actin, α cardiac muscle 1
- ATG7
autophagy-related 7
- ATP5A1
ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit 1
- BECN1
Beclin 1, autophagy related
- BNIP3
BCL2/adenovirus EiB interacting protein 3
- CACNA1A
calcium channel, voltage-dependent, P/Q type, α 1A subunit
- CHQ
chloroquine
- COX4I1
cytochrome c oxidase subunit IV isoform 1
- CTSL
cathepsin L; DNM1L, dynamin 1-like
- Ex
exercised rats
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HF
heart failure
- IP
immunoprecipitation
- LAMP2
lysosomal-associated membrane protein 2
- LDH
lactate dehydrogenase
- MAP1LC3A
microtubule-associated protein 1 light chain 3 α
- MFN1
mitofusin 1
- MFN2
mitofusin 2
- NDUFA9
NADH dehydrogenase (ubiquinone) 1 α subcomplex, 9
- PARK2
Parkinson disease (autosomal recessive, juvenile) 2, parkin
- SOD
superoxide dismutase
- SQSTM1/p62
sequestosome 1
- TNNI3
troponin I, cardiac 3
- UQCRC1
ubiquinol-cytochrome c reductase core protein 1
- VDAC1
voltage-dependent anion channel 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Katt C. Mattos, Marcele C. Coelho, and Camille C. C. da Silva for technical assistance.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo, São Paulo - SP (FAPESP #2009/12349–2, #2010/00028–4, #2010/51906–1, #2012/05765–2, #2015/20783–5 and #2016/01633–5), National Institute of Health Grant HL52141 to DM-R, Conselho Nacional de Pesquisa e Desenvolvimento – Brasil (CNPq), Instituto Nacional de Ciência e Tecnologia, and Centro de Pesquisa e Desenvolvimento de Processos Redox em Biomedicina.
References
- [1].Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, et al.. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation 2015; 131:e29-322; PMID:25520374; https://doi.org/ 10.1161/CIR.0000000000000152 [DOI] [PubMed] [Google Scholar]
- [2].Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am College Cardiol 2013; 61:599-610; PMID:23219298; https://doi.org/ 10.1016/j.jacc.2012.08.1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang ZV, Li DL, Hill JA. Heart failure and loss of metabolic control. J Cardiovasc Pharmacol 2014; 63:302-13; PMID:24336014; https://doi.org/ 10.1097/FJC.0000000000000054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gomes KM, Campos JC, Bechara LR, Queliconi B, Lima VM, Disatnik MH, Magno P, Chen CH, Brum PC, Kowaltowski AJ, et al.. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc Res 2014; 103:498-508; PMID:24817685; https://doi.org/ 10.1093/cvr/cvu125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Disatnik MH, Hwang S, Ferreira JC, Mochly-Rosen D. New therapeutics to modulate mitochondrial dynamics and mitophagy in cardiac diseases. J Mol Med (Berlin, Germany) 2015; 93:279-87; PMID:25652199; https://doi.org/ 10.1007/s00109-015-1256-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc 2013; 2:e000461; PMID:24103571; https://doi.org/ 10.1161/JAHA.113.000461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et al.. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012; 485:251-5; PMID:22535248; https://doi.org/ 10.1038/nature10992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yogalingam G, Hwang S, Ferreira JC, Mochly-Rosen D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cdelta (PKCdelta) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem 2013; 288:18947-60; PMID:23653351; https://doi.org/ 10.1074/jbc.M113.466870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 2013; 288:915-26; PMID:23152496; https://doi.org/ 10.1074/jbc.M112.411363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW 2nd. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circulation Res 2014; 114:257-65; PMID:24192653; https://doi.org/ 10.1161/CIRCRESAHA.114.302734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson ÅB. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 2013; 27:1365-77; https://doi.org/ 10.1101/gad.215871.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Campos JC, Bozi LH, Bechara LR, Lima VM, Ferreira JC. Mitochondrial quality control in cardiac diseases. Front Physiol 2016; 7:479; PMID:27818636; https://doi.org/ 10.3389/fphys.2016.00479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Adams V, Schuler G. Heart failure: Exercise training–a magic bullet for chronic heart failure? Nat Rev 2012; 9:677-8; PMID:23149829 [DOI] [PubMed] [Google Scholar]
- [14].Levine BD. Can intensive exercise harm the heart? The benefits of competitive endurance training for cardiovascular structure and function. Circulation 2014; 130:987-91; PMID:25223769; https://doi.org/ 10.1161/CIRCULATIONAHA.114.008142 [DOI] [PubMed] [Google Scholar]
- [15].Belardinelli R, Georgiou D, Cianci G, Purcaro A. 10-year exercise training in chronic heart failure: a randomized controlled trial. J Am College Cardiol 2012; 60:1521-8; PMID:22999730; https://doi.org/ 10.1016/j.jacc.2012.06.036 [DOI] [PubMed] [Google Scholar]
- [16].Campos JC, Queliconi BB, Dourado PM, Cunha TF, Zambelli VO, Bechara LR, Kowaltowski AJ, Brum PC, Mochly-Rosen D, Ferreira JC. Exercise training restores cardiac protein quality control in heart failure. PloS One 2012; 7:e52764; PMID:23300764; https://doi.org/ 10.1371/journal.pone.0052764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Emter CA, Baines CP. Low-intensity aerobic interval training attenuates pathological left ventricular remodeling and mitochondrial dysfunction in aortic-banded miniature swine. Am J Physiol 2010; 299:H1348-56; PMID:20817828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Campos JC, Gomes KM, Ferreira JC. Impact of exercise training on redox signaling in cardiovascular diseases. Food Chem Toxicol 2013; 62:107-19; PMID:23978413; https://doi.org/ 10.1016/j.fct.2013.08.035 [DOI] [PubMed] [Google Scholar]
- [19].Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011; 13:589-98; PMID:21478857; https://doi.org/ 10.1038/ncb2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 2013; 17:491-506; PMID:23562075; https://doi.org/ 10.1016/j.cmet.2013.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 2012; 111:1012-26; PMID:22904094; https://doi.org/ 10.1161/CIRCRESAHA.112.274142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010; 191:1367-80; PMID:21173115; https://doi.org/ 10.1083/jcb.201007013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ferreira JC, Boer BN, Grinberg M, Brum PC, Mochly-Rosen D. Protein quality control disruption by PKCbetaII in heart failure; rescue by the selective PKCbetaII inhibitor, betaIIV5-3. PloS One 2012; 7:e33175; PMID:22479367; https://doi.org/ 10.1371/journal.pone.0033175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen Y, Liu Y, Dorn GW 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 2011; 109:1327-31; PMID:22052916; https://doi.org/ 10.1161/CIRCRESAHA.111.258723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shires SE, Gustafsson AB. Mitophagy and heart failure. J Mol Med (Berlin, Germany) 2015; 93:253-62; PMID:25609139; https://doi.org/ 10.1007/s00109-015-1254-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen Y, Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science (New York, NY) 2013; 340:471-5; https://doi.org/ 10.1126/science.1231031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gottlieb RA, Andres AM, Sin J, Taylor DP. Untangling autophagy measurements: all fluxed up. Circ Res 2015; 116:504-14; PMID:25634973; https://doi.org/ 10.1161/CIRCRESAHA.116.303787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al.. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007; 13:619-24; PMID:17450150; https://doi.org/ 10.1038/nm1574 [DOI] [PubMed] [Google Scholar]
- [29].Sun M, Chen M, Liu Y, Fukuoka M, Zhou K, Li G, Dawood F, Gramolini A, Liu PP. Cathepsin-L contributes to cardiac repair and remodelling post-infarction. Cardiovasc Res 2011; 89:374-83; PMID:21147810; https://doi.org/ 10.1093/cvr/cvq328 [DOI] [PubMed] [Google Scholar]
- [30].Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000; 406:902-6; PMID:10972293; https://doi.org/ 10.1038/35022595 [DOI] [PubMed] [Google Scholar]
- [31].Godar RJ, Ma X, Liu H, Murphy JT, Weinheimer CJ, Kovacs A, Crosby SD, Saftig P, Diwan A. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy 2015; 11:1537-60; PMID:26103523; https://doi.org/ 10.1080/15548627.2015.1063768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 2012; 125:3170-81; PMID:22592897; https://doi.org/ 10.1161/CIRCULATIONAHA.111.041814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Diwan A. Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy 2012; 8:1394-6; PMID:22889942; https://doi.org/ 10.4161/auto.21036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn WP, et al.. Myocytes die by multiple mechanisms in failing human hearts. Circ Res 2003; 92:715-24; PMID:12649263; https://doi.org/ 10.1161/01.RES.0000067471.95890.5C [DOI] [PubMed] [Google Scholar]
- [35].Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, Suma H. Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Japanese Circ J 2001; 65:965-8; PMID:11716248; https://doi.org/ 10.1253/jcj.65.965 [DOI] [PubMed] [Google Scholar]
- [36].Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al.. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000; 406:906-10; PMID:10972294; https://doi.org/ 10.1038/35022604 [DOI] [PubMed] [Google Scholar]
- [37].Stypmann J, Glaser K, Roth W, Tobin DJ, Petermann I, Matthias R, Mönnig G, Haverkamp W, Breithardt G, Schmahl W, et al.. Dilated cardiomyopathy in mice deficient for the lysosomal cysteine peptidase cathepsin L. Proc Natl Acad Sci U S A 2002; 99:6234-9; PMID:11972068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 2013; 123:5284-97; PMID:24177425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Beynat J, Charles A, Astruc K, Metral P, Chirpaz L, Bron AM, Creuzot-Garcher C. Screening for diabetic retinopathy in a rural French population with a mobile non-mydriatic camera. Diabetes Metab 2009; 35:49-56; PMID:19097818 [DOI] [PubMed] [Google Scholar]
- [40].He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al.. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481:511-5; PMID:22258505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gheorghiade M, Bonow RO. Chronic heart failure in the United States: a manifestation of coronary artery disease. Circulation 1998; 97:282-9; PMID:9462531 [DOI] [PubMed] [Google Scholar]
- [42].Johns TN, Olson BJ. Experimental myocardial infarction. I. A method of coronary occlusion in small animals. Ann Surgery 1954; 140:675-82; PMID:13208115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, et al.. 2009 focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391-479; PMID:19324966 [DOI] [PubMed] [Google Scholar]
- [44].Ferreira JC, Bacurau AV, Bueno CR Jr, Cunha TC, Tanaka LY, Jardim MA, Ramires PR, Brum PC. Aerobic exercise training improves Ca2+ handling and redox status of skeletal muscle in mice. Exp Biol Med (Maywood, NJ) 2010; 235:497-505 [DOI] [PubMed] [Google Scholar]
- [45].Ferreira JC, Rolim NP, Bartholomeu JB, Gobatto CA, Kokubun E, Brum PC. Maximal lactate steady state in running mice: effect of exercise training. Clin Exp Pharmacol Physiol 2007; 34:760-5; PMID:17600553 [DOI] [PubMed] [Google Scholar]
- [46].Cancherini DV, Queliconi BB, Kowaltowski AJ. Pharmacological and physiological stimuli do not promote Ca(2+)-sensitive K+ channel activity in isolated heart mitochondria. Cardiovasc Res 2007; 73:720-8; PMID:17208207; https://doi.org/ 10.1016/j.cardiores.2006.11.035 [DOI] [PubMed] [Google Scholar]
- [47].Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med 2009; 46:1283-97; PMID:19245829; https://doi.org/ 10.1016/j.freeradbiomed.2009.02.008 [DOI] [PubMed] [Google Scholar]
- [48].Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci U S A 1996; 93:9893-8; PMID:8790427; https://doi.org/ 10.1073/pnas.93.18.9893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kowaltowski AJ, Cosso RG, Campos CB, Fiskum G. Effect of Bcl-2 overexpression on mitochondrial structure and function. J Biol Chem 2002; 277:42802-7; PMID:12207028; https://doi.org/ 10.1074/jbc.M207765200 [DOI] [PubMed] [Google Scholar]
- [50].Karnovsky MJ. The localization of cholinesterase activity in rat cardiac muscle by electron microscopy. J Cell Biol 1964; 23:217-32; PMID:14222810; https://doi.org/ 10.1083/jcb.23.2.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy 2008; 4:322-9; PMID:18216495; https://doi.org/ 10.4161/auto.5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Taegtmeyer H. Energy metabolism of the heart: from basic concepts to clinical applications. Curr Probl Cardiol 1994; 19:59-113; PMID:8174388; https://doi.org/ 10.1016/0146-2806(94)90008-6 [DOI] [PubMed] [Google Scholar]
- [53].Branco AF, Pereira SP, Gonzalez S, Gusev O, Rizvanov AA, Oliveira PJ. Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype. PloS One 2015; 10:e0129303; PMID:26121149; https://doi.org/ 10.1371/journal.pone.0129303 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.