

ABSTRACT
Small cell lung cancer (SCLC) is one of the most deadly cancers and currently lacks effective targeted treatment options. Recent advances in the molecular characterization of SCLC has provided novel insight into the biology of this disease and raises hope for a paradigm shift in the treatment of SCLC. We and others have identified activation of MYC as a driver of susceptibility to Aurora kinase inhibition in SCLC cells and tumors that translates into a therapeutic option for the targeted treatment of MYC-driven SCLC. While MYC shares major features with its paralogs MYCN and MYCL, the sensitivity to Aurora kinase inhibitors is unique for MYC-driven SCLC. In this review, we will compare the distinct molecular features of the 3 MYC family members and address the potential implications for targeted therapy of SCLC.
KEYWORDS: MYC, MYCL, MYCN, small cell lung cancer, SCLC, GEMM, mouse models
MYC family members in small cell lung cancer
Lung cancer is the leading cause of cancer-associated deaths worldwide,1 and accounts for more than 220,000 new cases annually in the US alone.2 About 15% of lung cancer cases are histologically defined as small cell lung cancer (SCLC), which represents a highly aggressive manifestation that almost exclusively arises in smokers and is characterized by rapid growth and frequent metastasis.3,4 Unfortunately, the major treatment options for SCLC – primarily platinum-based chemotherapy and radiation – have remained virtually unchanged for decades. While these treatment regimens achieve tumor regression in the majority of cases,5 this initial response is followed by rapid relapse and chemoresistance in most patients, leading to a dismal 5-year survival rate of about 6%.4,5
Several recurrent genetic aberrations have been identified in SCLC, among which MYC family genes, including MYC, MYCL and MYCN, stand out as oncogenic drivers that may constitute novel therapeutically tractable targets.6-8 The 3 MYC family proto-oncogenes are paralogs with regions of structural homology, but also functional differences. We were able to demonstrate that amplification of individual MYC family members is associated with phenotypic differences in SCLC. More specifically, we showed that the identity of the MYC family member determines the susceptibility toward the Aurora kinase inhibitor alisertib, where MYC-amplified SCLC cells are particularly sensitive.9,10 Furthermore, recent studies suggest that MYC-amplified SCLC may be more sensitive to CHK1 inhibition as well.11 This is in line with previous studies in myeloid 32D cells, where overexpression of MYC sensitizes cells to the chemotherapeutic agents adriamycin and camptothecin, while MYCL and MYCN-overexpressing cells are resistant.12 This difference in drug susceptibility based on MYC family members also seems to occur in other tumor types such as neuroblastoma.13 Thus, to successfully use MYC family members as biomarkers to predict treatment susceptibility in SCLC, it will be crucial to further dissect the molecular basis underlying the different phenotypes observed.
MYC family transcription factors
MYC family members are basic helix-loop-helix (bHLH) leucine zipper transcription factors that bind to the canonical E-box DNA element CACGTG and activate target gene expression as heterodimers with the small bHLH protein MAX.14 As paralogs, MYC family members share highly conserved domains such as a transactivation domain that recruits the transcriptional machinery, a basic region for E-box-specific DNA binding, MYC homology boxes (MB) involved in protein turnover and functional regulation, and the C-terminal HLH-leucine zipper domain responsible for MAX dimerization (Fig. 1).3,4,15-19 In the case of MYCL, 2 substantially differing transcripts have been found to be expressed in SCLC cell lines, and interestingly, the short isoform lacks the HLH domain.20 All 3 MYC paralogs can complement an activated Ha-ras gene in transforming primary rat embryo fibroblasts and are able to transform pre-neoplastic precursors of SCLC.15-18 In addition, both MYC and MYCN induce proliferation and cell cycle progression in quiescent fibroblasts.16,18,21 Taken together, these findings indicate a degree of redundancy between the family members. Indeed, during murine development, Mycn can partially complement for loss of Myc.22 Moreover, a double knockout of Myc and Mycn in haematopoietic cells has a stronger phenotype than either knockout alone,23 and both Myc and Mycn are essential genes, while Mycl is not (Fig. 1).24-27 Albeit subtle, differential binding affinities of recombinant MYC, MYCN, and MYCL with MAX to different DNA consensus motifs have been reported in vitro,28 and the transforming capacity of MYCL is lower than that of MYC in rat embryonic fibroblasts.29 Furthermore, CRISPR-mediated inactivation of Mycn or Mycl in mouse tumor-derived SCLC cell lines reduces colony formation, while that of Myc does not.30 This indicates that mechanistic differences between the family members likely exist. Interestingly, differential effects on transcriptional repression were discovered for heterotrimeric complexes of MYC family members with MAX and MIZ1.31-34 Even though MYCN/MIZ1 complex formation was described in cell line models,35,36 MYC/MIZ1 complexes were shown to be more readily detectable than MYCN/MIZ1 complexes and are considered to be relevant components for medulloblastoma subtype differentiation.32 Even though this points to biological differences between MYC family members, most data are based on overexpression of exogenous MYC, MYCN and/or MIZ1, future studies are warranted to clarify complex formation at endogenous, physiological levels. Currently no data exists regarding complexes of MYCL with MIZ1 and the role of these complexes in SCLC is yet to be determined.
Figure 1.
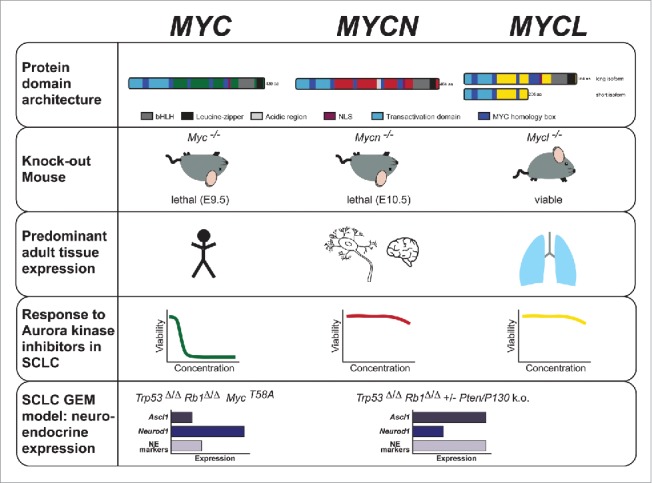
Schematic overview of major characteristics of the 3 MYC family paralogs with a focus on SCLC. Rows 1–3: Biochemical properties and physiological functions of MYC family members- protein domain architecture, phenotype of knockout mouse models and predominant distribution of MYC family member expression across tissues (broad expression of MYC, expression of MYCN primarily in neural and neuroendocrine tissues, and MYCL expression mainly in the lung). Row 4: Response of SCLC models to Aurora kinase inhibition with high sensitivity of MYC amplified/overexpressing cells. Row 5: Schematic differences of SCLC mouse models regarding genetic background, MYC family member activity/alteration and expression of neuroendocrine markers including the transcription factors Neurod1 and Ascl1. (bHLH = basic Helix-Loop-Helix, NLS = nuclear localization sequence, GEM = genetically engineered mouse, NE = neuroendocrine).
The tissue specific expression of MYCN and MYCL, in contrast to the more broad expression pattern of MYC (Fig. 1), also argues for functional differences between MYC family members. Myc is expressed in most developing tissues and sustained in many tissues in the adult mouse, while Mycn expression is restricted to early developmental stages, with elevated levels in forebrain, kidney, hindbrain and intestine of newborn mice.37-39 Mycl is also developmentally regulated and expressed in embryonic brain, kidney and lung tissue.40 Overall, this is largely consistent with the frequency of alterations in each paralog in human cancer types,41-43 with MYC being widely affected across many blood-borne and solid tumors, MYCN being frequently altered in solid tumors of neuroendocrine and neuronal origin, and MYCL predominantly in SCLC. Specifically, MYCN deregulation is frequently found in neuroblastoma,16 retinoblastoma,44 medulloblastoma,45 Wilm's tumors46 and in prostate cancer with neuroendocrine differentiation,47 while MYCL amplifications occur in SCLC,41 Merkel cell carcinoma48 and ovarian carcinoma.49 In contrast to other known oncogenes such as RAS or EGFR, MYC (with the exception of Burkitt's lymphoma) is typically not mutated in cancer, but rather amplified or deregulated resulting in increased expression. Interestingly, in SCLC MYC, MYCN and MYCL are all found to be affected in a mutually exclusive manner.3,5-8
MYC function and regulation
MYC activates gene transcription in conjunction with MAX by several mechanisms, including the recruitment of basal transcription factors, histone acetylases, chromatin remodelling enzymes, and RNA polymerases.50-55 MYC, the most frequently deregulated and best studied family member, mediates a transcriptional response that promotes cell growth and proliferation.56,57 In numerous genome-wide studies in Drosophila and mammalian cells, MYC binding sites and regulated genes have been found to cover more than 15% of genomic loci,58,59 while even regulating up to 1/3 of all transcribed genes in embryonic stem cells.51 How exactly this transcriptional response is orchestrated remains a matter of ongoing debate, likely due to the complex feedback mechanisms involved. Two opposing models have been put forward, each substantiated by numerous lines of evidence: MYC as a global amplifier of existing transcriptional programs51,60,61 or MYC as a regulator of specific target genes.33,62,63 Support for the first model comes from the direct interaction between MYC and the CDK9/Cyclin T1 complex, which has been shown to mediate pause-release of RNA Polymerase II (Pol II) and thereby enhance transcription of all expressed genes, rather than recruiting Pol II to specific target genes.51,64 Further studies in cell lines, including SCLC, demonstrate that MYC primarily acts by globally amplifying existing gene expression patterns, rather than specifically inducing a distinct set of target genes,60 a finding that could explain some of the variation in reported MYC effects between different cell types. Providing evidence for the latter of the 2 models are studies combining global chromatin immunoprecipitation (ChIP) and gene expression analysis, which derived gene-specific MYC effects and defined dedicated MYC target gene sets, including genes involved in ribosome biogenesis, translation, cell cycle regulation and energy metabolism.33,62,65,66 These 2 contrasting models may be reconciled by the recently postulated hypothesis that individual gene promoters exhibit varying affinities for binding and activation by MYC.67 According to this model, transcription of high affinity genes, such as ribosomal constituents, whose promoters are bound by MYC with high affinity, occurs at low cellular MYC levels. In contrast, the expression of low-affinity genes, e.g. genes involved in TGF-β signaling, is induced only upon strong MYC overexpression. At extreme MYC levels, DNA-binding has been reported to become increasingly unspecific and to occur sequence-independently,68 a phenomenon referred to as promoter invasion.60,62 This is in line with evidence suggesting threshold-specific tumorigenic effects of MYC depending on its expression levels.69
In addition to its transcriptional role, MYC also controls S-phase entry and replication initiation in a non-transcriptional manner by interacting with the replication initiation complex and promoting recruitment of CDC45.70-72 MYC overexpression induces activation of the DNA damage response (DDR) and results in increased genomic instability, likely a result of the well documented MYC-induced replicative stress caused by pre-mature origin firing and aberrant fork progression.70,73,74 Similarly, MYCN has also been reported to induce DDR signaling in neuroblastoma cells,13,75 which could be due to increased replicative stress.76 No reports of similar effects of MYCL overexpression are available to date.
Intriguingly, despite its indisputable central role in promoting proliferation, high levels of MYC can also induce apoptosis,77,78 and overexpression of all 3 MYC family members was found to induce apoptosis upon IL-3 withdrawal in 32D myeloid cells.12 Moreover, an “apoptosis-primed” state has been described for MYCN-overexpressing neuroblastoma,79 but also for physiological MYC levels during tissue development in young mice.80 In part, gene repression by MYC/MIZ1 is important for induction of apoptosis,81 as is MYC phosphorylation at T58,82 and downstream activation of the p53 pathway.83,84 Nonetheless, several examples of p53-independent MYC-driven apoptosis have been reported, such as during mitosis.85,86
Consistent with its central role in mediating proliferation and differentiation, MYC expression is tightly regulated in normal cells. Transcription of MYC is controlled by numerous transcription factors including CNBP, FuBP1, and TCF, as well as by structural DNA elements such as G4 quadruplexes.87-90 MYC mRNA transport, stability and translation is in turn affected by multiple factors including miRNAs,91-94 while MYC protein is post-translationally modified with ubiquitin, resulting in a short half-life of 15–30 min.95-97 Phosphorylation controls the stability of both MYC and MYCN by affecting polyubiquitination and hence proteasomal degradation.98-100 Serine 62 is phosphorylated by CDK1/CyclinA and CDK1/CyclinB1, leading to a transient stabilization of the protein, but also serving as the prerequisite for threonine 58 phosphorylation by glycogen synthase kinase 3 β (GSK3b).98,101 This phosphorylation event triggers ubiquitination and subsequent degradation by the 26S proteasome. No such regulatory mechanism has been reported for MYCL to date. The relevance of these regulatory processes for the signaling of the 3 MYC family members in the context of SCLC remains to be studied.
Targeting MYC in small cell lung cancer
Due the fact that deregulation of MYC family members is one of the most frequent oncogenic events in cancer,102 and the observation that MYC withdrawal in mouse models can lead to tumor regression,103-106 MYC family members have been considered compelling therapeutic targets for decades. Compounds directly targeting MYC or the MYC/MAX interaction have been developed,107-109 but overall this approach has proven challenging.110 This is at least partly due to the lack of intrinsic enzymatic activity and their activation by overexpression, rather than by oncogenic mutations that could be directly exploited therapeutically. This precludes the application of strategies developed for compound discovery in the context of kinase inhibition, such as inhibitory substrate-analogs and targeting mutated proteins only.
In recent years, exploiting synthetic lethality has emerged as a promising approach to overcome such limitations, and several examples demonstrate that this may be a viable option for treatment of MYC-driven tumors. In MYC-driven SCLC, we and others identified Aurora kinases (AURK) as promising synthetic lethal targets,9,10,111,112 which also emerged as potential candidate targets in other MYC-driven tumors.99,113,114 An elegant explanation for the activity of Aurora kinase inhibitors in MYCN-amplified neuroblastoma is the observation that Aurora kinase A (AURKA) binds to the MYCN/FBXW7 complex, reduces K48-linked ubiquitination of MYCN, and thus increases MYCN protein half-life.99,114,115 This MYCN-stabilizing function of AURKA is independent of its catalytic activity and compounds such as alisertib or CD532 induce a perturbation of the AURKA/MYCN complex that results in a reduction of MYCN protein levels.115 A similar stabilizing role of AURKA for MYC has also recently been proposed in NRAS-driven, MYC-expressing hepatocellular carcinoma.113 This is in contrast to what we find in SCLC, where no strong decrease of MYC stability upon alisertib treatment was observed.9,10 This indicates that additional mechanisms may sensitize MYC-overexpressing cells toward Aurora kinase inhibition independently of MYC protein abundance. Moreover, in contrast to neuroblastoma, MYCN-amplified SCLC cell lines were not particularly sensitive to AURK inhibition,9,10 suggesting that lineage-dependent factors and/or the genomic background contribute to the specific sensitivity of MYC-driven SCLC to AURK inhibition.
In another study, CDK7, a cyclin-dependent kinase that phosphorylates Pol II, was proposed as a novel therapeutic target in SCLC.116 Using the covalent CDK7 inhibitor THZ1, Christensen and colleagues demonstrated efficacy in in vitro and in vivo SCLC models. The increased THZ1-sensitivity in SCLC compared with NSCLC was in part explained by the impact of CDK7 inhibition on SCLC-specific super enhancers including those regulating MYC family members, leading to decreased MYC and MYCN levels.116 Of note, THZ1 had previously been explored in the context of neuroblastoma, where it showed selective activity against MYCN-amplified, but not against non-amplified cell lines.117 Similarly, MYC-dependent effects of CDK inhibition have been observed in other contexts. For example, MYC-addicted tumors are selectively responsive to CDK9 inhibition in hepatocellular carcinoma,118 breast cancer,119 and B-cell lymphoma.120 In cell line models overexpressing different oncogenes, inhibition of CDK1 was demonstrated to induce apoptosis only in cells overexpressing MYC.121 Whereas associations of MYC status and activity of CDK inhibition were shown in these contexts, it remains to be determined whether MYC status correlates with sensitivity to THZ1 or other CDK inhibitors in SCLC.
An alternative strategy to MYC inhibition is the targeted inhibition of epigenetic regulators such as BET proteins that may reduce MYC expression levels. The most extensively characterized bromodomain inhibitor is JQ-1,122 which has been shown to reduce MYC family member expression and exhibits activity in MYC-driven acute leukemia,123 Merkel cell carcinoma,124 BRD4-NUT midline carcinoma125 and MYC-amplified medulloblastoma.126 Similarly, in a murine SCLC model, Jahchan and colleagues showed that Mycl activity is crucial for tumor-propagating SCLC cells and that their tumorigenic potential was significantly reduced after abrogation of Mycl activity by JQ-1-induced transcriptional repression or following Mycl knock-down.127 JQ-1 was moreover shown to have anti-proliferative effects in SCLC cell lines.128,129 Interestingly, key targets with reduced expression upon JQ-1 treatment in SCLC cell lines were MYC family members129 and ASCL1,130 but currently biomarkers predicting JQ-1 sensitivity in SCLC are lacking. More recently, CHK1 inhibition has been identified as an additional drug target that elicits efficacy specifically in MYC-driven SCLC,11 suggesting that MYC status is an important determinant of therapeutic response in SCLC.
Taken together, MYC family transcription factors are central signaling hubs, in one way or another affecting virtually all (proliferative) processes in a cell, which –together with their frequent deregulation- makes them attractive therapeutic targets in SCLC. However, to further define and eventually treat MYC-dependent SCLC, in vivo models that faithfully recapitulate the complexity of the human disease are crucial.
Reconstructing the role of MYC signaling in SCLC GEMM models
Multiple genetically engineered mouse models (GEMMs) for SCLC have been developed in the past decades. The first SCLC GEMMs were based on conditional loss of Rb1 and Trp53 as the key genetic alterations, leading to SCLC with high resemblance to the human disease both histologically and molecularly, albeit with long latency.131 Importantly, tumors in this model frequently exhibit focal amplifications and/or high expression of Mycl.132,133 Subsequent GEMMs incorporated the additional loss of Rb1 family member Rbl2 (p130) or Pten, both of which are lost in a subset of SCLCs.6,134-136 Loss of either tumor suppressor accelerates tumorigenesis, and has made these models more tractable for experimental use. A recent comprehensive histopathological review found that these GEMMs develop a spectrum of histopathologies including classic SCLC, NSCLC with neuroendocrine (NE) features and large cell neuroendocrine carcinoma (LCNEC).137 Notably, tumors from these GEMMs that exhibit classic SCLC morphology are characterized by high expression of neuroendocrine markers and Ascl1, a transcription factor involved in neuroendocrine differentiation. In a mouse model, Ascl1 has been shown to be required for tumorigenesis of classic SCLC,138 and was demonstrated to be essential for the survival of neuroendocrine lung cancer cell lines including NE-NSCLCs.139 Although Mycl amplifications observed in these original models occur stochastically, Mycl has been shown to play a significant functional role in SCLC tumorigenesis. Overexpression of Mycl in combination with Trp53 and Rb1 loss significantly decreases tumor latency,140 and deletion of Mycl dramatically suppresses tumor formation and leads to more mixed and NSCLC morphologies, even when targeting neuroendocrine cells.30 This suggests that beyond just driving proliferation, MYCL may play a role in SCLC differentiation.
The Rb1/Trp53/MycT58A (RPM) mouse is the first known SCLC GEMM driven by Myc, and expresses a non-phosphorylatable point mutant of MYC (T58A) causing substantially increased MYC protein levels.9 Interestingly, tumors in this model exhibit reduced neuroendocrine gene expression, including Ascl1, but in contrast display high Neurod1 expression. They thereby resemble a subset of human SCLC marked by low neuroendocrine markers, which is known as “variant” SCLC morphology.141-143 Although it is still unknown whether variant SCLC depends on Neurod1 in a similar manner as classic SCLC depends on Ascl1, it is conceivable that tumors may be addicted to their respective driver transcription factors. In addition to the variant morphology, some tumors in the RPM mice were negative for both ASCL1 and NEUROD1, a phenotype also observed in human tumors and human SCLC cell lines.9,138 The significance and the molecular mechanisms underlying these “double negative” SCLC cells are currently unclear. It is tempting to speculate that the “double negative” histological phenotype associated with high MYC expression may represent the most de-differentiated state of tumors and that MYC may be causally involved. However, whether/how MYC may control the differentiation of SCLC into the variant or “double negative” morphologies is yet to be fully understood.
Consistent with the human disease, MYC-driven murine SCLC is initially highly sensitive to the standard-of-care chemotherapy. In RPM mice, chemotherapy induces significant cell death, reduces tumor burden and increases survival. However, relapse is so rapid that animals only gain ∼10 d in survival benefit compared with untreated animals.9 We initially found that the AURK inhibitor alisertib is effective in MYC-driven human SCLC cell lines10 and notably, mice with MYC-driven tumors had a significant increase in survival when alisertib was coupled with chemotherapy.9 In line with these findings in GEMMs are reports from clinical trials for second-line SCLC treatment, which were designed to exploit such vulnerabilities discovered in vitro: 1) a phase II study investigating alisertib in pre-treated SCLC144 and 2) a recent phase II trial of relapsed SCLC patients given paclitaxel with or without alisertib (NCT02038647). In the unselected cohort of SCLC patients, the addition of alisertib to taxane treatment did not significantly prolong survival.145 However, evaluation of MYC expression by IHC revealed that in patients with high MYC levels, survival was significantly increased with the alisertib/paclitaxel combination compared with patients treated with paclitaxel only. This illustrates that MYC status may be a predictor of both SCLC subtype and therapeutic vulnerability in patients, and therefore, preclinical studies using these different GEMMs may be able to predict the outcome of clinical trials.
As mentioned above, MYCN is amplified in a small subset of SCLC patients and cell lines, but has not yet been clearly linked with variant or classic histopathology or to therapeutic response. Currently no SCLC mouse models driven by overexpression of Mycn have been published. However, David MacPherson has reported the generation of Rb1/Trp53/Mycn conditional mice (personal communication) and comparisons to existing GEMMs are awaited. It will be important to determine whether Mycn-driven SCLC GEMMs have similar or distinguishing molecular and phenotypic characteristics compared with Mycl/Myc-driven tumors.
Conclusions
Currently SCLC is treated as a homogeneous disease based on the stage at diagnosis.146 However, growing evidence of heterogeneity among SCLC patient tumors, cell lines and GEMMs based on histology147-149 and genomics9,138,150 indicates that a more differentiated treatment stratification would likely prove beneficial. Deregulated MYC signaling may play a central role in the molecular and histological heterogeneity observed in SCLC. Apart from their association with histological sub-groups, the central role of MYC family members in processes governing tumor maintenance offers new opportunities for targeted therapy, for example by exploitation of paralog-specific synthetic lethal interactions. As discussed above, the majority of functional characterization and identification of synthetic lethality has been performed for MYC and -to a lesser extent- for MYCN. In contrast, despite being the most frequently altered MYC family member in SCLC and the finding that Mycl inactivation leads to tumor suppression in mice,30 very limited knowledge is available regarding MYCL, its basic biology, its role in cancer and potential synthetic lethal partners, and this deserves intensified investigation.
In the past, in-depth molecular analyses of human SCLCs were limited by the lack of tumor tissue due to the infrequent availability of biopsy samples. The improved development of patient-derived circulating tumor cell and tissue xenograft models (CDX and PDX, respectively) may be a means to broaden the scope of human tumor models. Nevertheless, GEM models are indispensable in complementing CDX/PDX studies. Because they provide autochthonous tumors in an immune-competent background, GEMMs allow the investigation of treatments in relation to an intact tumor microenvironment and offer the possibility to study immune-checkpoint modulation in conjunction with other treatment approaches.
Overall, several central questions remain open and need to be addressed: Are synthetic lethal interactions with MYC overexpression also relevant to MYCN and/or MYCL? What is the best strategy to discover novel synthetic lethal interactions in MYCL and MYCN-driven SCLC? How well do findings in in vitro and in vivo models translate into the clinic? To answer these questions, the investigation of human SCLC samples will certainly remain a cornerstone. However, integrative analysis of orthogonal tumor models including cell lines, GEMMs and CDX/PDXs will be an essential factor to gain biological insights and develop novel treatment strategies. Given the important phenotypic differences associated with different MYC family members in GEM models, it will be important to characterize MYC status in patients and model systems. A more complete understanding of the role of MYC family members in tumor phenotype and drug response ultimately holds great promise for improved outcome for patients with SCLC.
Disclosure of potential conflicts of interest
Martin L. Sos receives a research grant from Novartis.
Funding
This work was supported by the German Ministry of Science and Education (BMBF) as part of the e:Med program (MILES, grant no. 01ZX1406 to M.L.S), and a V Scholar award from The V Foundation for Cancer Research, the American Cancer Society (Research Scholar Award no. RSG-13–300–01-TBG), and the NIH (no. R01CA187457) to T.G.O. T.G.O. is a Damon Runyon-Rachleff Innovation Awardee and this work is supported by the Damon Runyon Cancer Research Foundation (no. DRR-26–13).
References
- [1].Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65:87-108; PMID:25651787; https://doi.org/ 10.3322/caac.21262 [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66:7-30; PMID:26742998; https://doi.org/ 10.3322/caac.21332 [DOI] [PubMed] [Google Scholar]
- [3].Bunn PA, Minna JD, Augustyn A, Gazdar AF, Ouadah Y, Krasnow MA, Berns A, Brambilla E, Rekhtman N, Massion PP, et al.. Small cell lung cancer: Can recent advances in biology and molecular biology be translated into improved outcomes? J Thorac Oncol 2016; 11:453-74; PMID:26829312; https://doi.org/ 10.1016/j.jtho.2016.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pietanza MC, Byers LA, Minna JD, Rudin CM. Small cell lung cancer: Will recent progress lead to improved outcomes? Clin Cancer Res 2015; 21:2244-55; PMID:25979931; https://doi.org/ 10.1158/1078-0432.CCR-14-2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sharp A, Bhosle J, Abdelraouf F, Popat S, O'Brien M, Yap TA. Development of molecularly targeted agents and immunotherapies in small cell lung cancer. Eur J Cancer 2016; 60:26-39; PMID:27060747; https://doi.org/ 10.1016/j.ejca.2016.03.004 [DOI] [PubMed] [Google Scholar]
- [6].George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al.. Comprehensive genomic profiles of small cell lung cancer. Nature 2015; 524:47-53; PMID:26168399; https://doi.org/ 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al.. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 2012; 44:1111-6; PMID:22941189; https://doi.org/ 10.1038/ng.2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al.. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012; 44:1104-10; PMID:22941188; https://doi.org/ 10.1038/ng.2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, et al.. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 2017; 31:270-85; PMID:28089889; https://doi.org/ 10.1016/j.ccell.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sos ML, Dietlein F, Peifer M, Schöttle J, Balke-Want H, Müller C, Koker M, Richters A, Heynck S, Malchers F, et al.. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A 2012; 109:17034-9; PMID:23035247; https://doi.org/ 10.1073/pnas.1207310109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, Redwood A, Fan Y, Li L, Glisson BS, et al.. CHK1 inhibition in small cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res 2017; 77(14):1-15; PMID:28490518; https://doi.org/ 10.1158/0008-5472.CAN-16-3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nesbit CE, Grove LE, Yin X, Prochownik EV. Differential apoptotic behaviors of c-myc, N-myc, and L-myc oncoproteins. Cell Growth Differ 1998; 9:731-41; PMID:9751117 [PubMed] [Google Scholar]
- [13].Petroni M, Veschi V, Prodosmo A, Rinaldo C, Massimi I, Carbonari M, Dominici C, McDowell HP, Rinaldi C, Screpanti I, et al.. MYCN sensitizes human neuroblastoma to apoptosis by HIPK2 activation through a DNA damage response. Mol Cancer Res 2011; 9:67-77; PMID:21173028; https://doi.org/ 10.1158/1541-7786.MCR-10-0227 [DOI] [PubMed] [Google Scholar]
- [14].Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993; 72:233-45; PMID:8425220; https://doi.org/ 10.1016/0092-8674(93)90663-B [DOI] [PubMed] [Google Scholar]
- [15].DePinho RA, Hatton KS, Tesfaye A, Yancopoulos GD, Alt FW. The human myc gene family: Structure and activity of L-myc and an L-myc pseudogene. Genes Dev 1987; 1:1311-26; PMID:3322939; https://doi.org/ 10.1101/gad.1.10.1311 [DOI] [PubMed] [Google Scholar]
- [16].Schwab M, Varmus HE, Bishop JM. Human N-myc gene contributes to neoplastic transformation of mammalian cells in culture. Nature 1985; 316:160-2; PMID:4040214; https://doi.org/ 10.1038/316160a0 [DOI] [PubMed] [Google Scholar]
- [17].Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983; 304:596-602; PMID:6308472; https://doi.org/ 10.1038/304596a0 [DOI] [PubMed] [Google Scholar]
- [18].Yancopoulos GD, Nisen PD, Tesfaye A, Kohl NE, Goldfarb MP, Alt FW. N-myc can cooperate with ras to transform normal cells in culture. Proc Natl Acad Sci USA 1985; 82:5455-9; PMID:3860871; https://doi.org/ 10.1073/pnas.82.16.5455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li LH, Nerlov C, Prendergast G, MacGregor D. c-Myc represses transcription in vivo by a novel mechanism dependent on the initiator element and Myc box II. EMBO J 1994; 13:4070-9; PMID:8076602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kaye F, Battey J, Nau M, Brooks B. Structure and expression of the human L-myc gene reveal a complex pattern of alternative mRNA processing. Mol Cell Biol 1988; 8:186-95; PMID:2827002; https://doi.org/ 10.1128/MCB.8.1.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cavalieri F, Goldfarb M. N-myc proto-oncogene expression can induce DNA replication in Balb/c 3T3 fibroblasts. Oncogene 1988; 2:289-91; PMID:3281096 [PubMed] [Google Scholar]
- [22].Malynn BA, de Alboran IM, O'Hagan RC, Bronson R, Davidson L, DePinho RA, Alt FW. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev 2000; 14:1390-9; PMID:10837031 [PMC free article] [PubMed] [Google Scholar]
- [23].Zimmerman KA, Yancopoulos GD, Collum RG, Smith RK, Kohl NE, Denis KA, Nau MM, Witte ON, Toran-Allerand D, Gee CE, et al.. Differential expression of myc family genes during murine development. Nature 1986; 319:780-3; PMID:2419762; https://doi.org/ 10.1038/319780a0 [DOI] [PubMed] [Google Scholar]
- [24].Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev 1993; 7:671-82; PMID:8458579; https://doi.org/ 10.1101/gad.7.4.671 [DOI] [PubMed] [Google Scholar]
- [25].Hatton KS, Mahon K, Chin L, Chiu FC, Lee HW, Peng D, Morgenbesser SD, Horner J, DePinho RA. Expression and activity of L-Myc in normal mouse development. Mol Cell Biol 1996; 16:1794-804; PMID:8657155; https://doi.org/ 10.1128/MCB.16.4.1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sawai S, Shimono A, Wakamatsu Y, Palmes C, Hanaoka K, Kondoh H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development 1993; 117:1445-55; PMID:8404543 [DOI] [PubMed] [Google Scholar]
- [27].Stanton BR, Reid SW, Parada LF. Germ line transmission of an inactive N-myc allele generated by homologous recombination in mouse embryonic stem cells. Mol Cell Biol 1990; 10:6755-8; PMID:1701023; https://doi.org/ 10.1128/MCB.10.12.6755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Prochownik EV, VanAntwerp ME. Differential patterns of DNA binding by myc and max proteins. Proc Natl Acad Sci USA 1993; 90:960-4; PMID:8430110; https://doi.org/ 10.1073/pnas.90.3.960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Barrett J, Birrer MJ, Kato GJ, Dosaka-Akita H, Dang CV. Activation domains of L-Myc and c-Myc determine their transforming potencies in rat embryo cells. Mol Cell Biol 1992; 12:3130-7; PMID:1620120; https://doi.org/ 10.1128/MCB.12.7.3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kim D-W, Wu N, Kim Y-C, Cheng PF, Basom R, Kim D, Dunn CT, Lee AY, Kim K, Lee CS, et al.. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev 2016; 30:1289-99; PMID:27298335; https://doi.org/ 10.1101/gad.279307.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Peukert K. An alternative pathway for gene regulation by Myc. EMBO J 1997; 16:5672-86; PMID:9312026; https://doi.org/ 10.1093/emboj/16.18.5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vo BT, Wolf E, Kawauchi D, Gebhardt A, Rehg JE, Finkelstein D, Walz S, Murphy BL, Youn YH, Han Y-G, et al.. The interaction of Myc with Miz1 defines medulloblastoma subgroup identity. Cancer Cell 2016; 29:5-16; PMID:26766587; https://doi.org/ 10.1016/j.ccell.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al.. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014; 511:483-7; PMID:25043018; https://doi.org/ 10.1038/nature13473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wiese KE, Haikala HM, von Eyss B, Wolf E, Esnault C, Rosenwald A, Treisman R, Klefstrom J, Eilers M. Repression of SRF target genes is critical for Myc-dependent apoptosis of epithelial cells. EMBO J 2015; 34:1554-71; PMID:25896507; https://doi.org/ 10.15252/embj.201490467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Akter J, Takatori A, Hossain MS, Ozaki T, Nakazawa A, Ohira M, Suenaga Y, Nakagawara A. Expression of NLRR3 orphan receptor gene is negatively regulated by MYCN and Miz-1, and its downregulation is associated with unfavorable outcome in neuroblastoma. Clin Cancer Res 2011; 17:6681-92; PMID:21908575; https://doi.org/ 10.1158/1078-0432.CCR-11-0313 [DOI] [PubMed] [Google Scholar]
- [36].Iraci N, Diolaiti D, Papa A, Porro A, Valli E, Gherardi S, Herold S, Eilers M, Bernardoni R, Valle GD, et al.. A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TRKA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer Res 2011; 71:404-12; PMID:21123453; https://doi.org/ 10.1158/0008-5472.CAN-10-2627 [DOI] [PubMed] [Google Scholar]
- [37].Downs KM, Martin GR, Bishop JM. Contrasting patterns of myc and N-myc expression during gastrulation of the mouse embryo. Genes Dev 1989; 3:860-9; PMID:2663644; https://doi.org/ 10.1101/gad.3.6.860 [DOI] [PubMed] [Google Scholar]
- [38].Hirvonen H, Makela TP, Sandberg M, Kalimo H, Vuorio E, Alitalo K. Expression of the myc proto-oncogenes in developing human fetal brain. Oncogene 1990; 5:1787-97; PMID:2284098 [PubMed] [Google Scholar]
- [39].Mugrauer G, Alt FW, Ekblom P. N-myc proto-oncogene expression during organogenesis in the developing mouse as revealed by in situ hybridization. J Cell Biol 1988; 107:1325-35; PMID:3049618; https://doi.org/ 10.1083/jcb.107.4.1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cole MD. The myc oncogene: Its role in transformation and differentiation. Annu Rev Genet 1986; 20:361-84; PMID:3028245; https://doi.org/ 10.1146/annurev.ge.20.120186.002045 [DOI] [PubMed] [Google Scholar]
- [41].Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, McBride OW, Bertness V, Hollis GF, Minna JD. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 1985; 318:69-73; PMID:2997622; https://doi.org/ 10.1038/318069a0 [DOI] [PubMed] [Google Scholar]
- [42].Wasylishen AR, Stojanova A, Oliveri S, Rust AC, Schimmer AD, Penn LZ. New model systems provide insights into Myc-induced transformation. Oncogene 2011; 30:3727-34; PMID:21441954; https://doi.org/ 10.1038/onc.2011.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J 1997; 16:2985-95; PMID:9214616; https://doi.org/ 10.1093/emboj/16.11.2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol 2013; 14:327-34; PMID:23498719; https://doi.org/ 10.1016/S1470-2045(13)70045-7 [DOI] [PubMed] [Google Scholar]
- [45].Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, Lau J, Masic S, Nguyen K, Yakovenko S, et al.. Pleiotropic role for MYCN in medulloblastoma. Genes Dev 2010; 24:1059-72; PMID:20478998; https://doi.org/ 10.1101/gad.1907510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Williams RD, Chagtai T, Alcaide-German M, Apps J, Wegert J, Popov S, Vujanic G, van Tinteren H, van den Heuvel-Eibrink MM, Kool M, et al.. Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget 2015; 6:7232-43; PMID:25749049; https://doi.org/ 10.18632/oncotarget.3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, Baertsch R, Sokolov A, Meyerowitz JG, Mathis C, et al.. N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell 2016; 29:536-47; PMID:27050099; https://doi.org/ 10.1016/j.ccell.2016.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Paulson KG, Lemos BD, Feng B, Jaimes N, Penas PF, Bi X, Maher E, Cohen L, Leonard JH, Granter SR, et al.. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol 2009; 129:1547-55; PMID:19020549; https://doi.org/ 10.1038/jid.2008.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wu R, Lin L, Beer DG, Ellenson LH, Lamb BJ, Rouillard JM, Kuick R, Hanash S, Schwartz DR, Fearon ER, et al.. Amplification and overexpression of the L-MYC proto-oncogene in ovarian carcinomas. Am J Pathol 2003; 162:1603-10; PMID:12707044; https://doi.org/ 10.1016/S0002-9440(10)64294-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 1998; 94:363-74; PMID:9708738; https://doi.org/ 10.1016/S0092-8674(00)81479-8 [DOI] [PubMed] [Google Scholar]
- [51].Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell 2010; 141:432-45; PMID:20434984; https://doi.org/ 10.1016/j.cell.2010.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Vervoorts J, Lüscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Lüscher B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 2003; 4:484-90; PMID:12776737; https://doi.org/ 10.1038/sj.embor.embor821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet 1999; 22:102-5; PMID:10319872; https://doi.org/ 10.1038/8811 [DOI] [PubMed] [Google Scholar]
- [54].Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan H-M, Livingston DM, Amati B. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep 2003; 4:575-80; PMID:12776177; https://doi.org/ 10.1038/sj.embor.embor861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S, et al.. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell 2015; 58:440-52; PMID:25818646; https://doi.org/ 10.1016/j.molcel.2015.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Armelin HA, Armelin MC, Kelly K, Stewart T, Leder P, Cochran BH, Stiles CD. Functional role for c-myc in mitogenic response to platelet-derived growth factor. Nature 1984; 310:655-60; PMID:6088986; https://doi.org/ 10.1038/310655a0 [DOI] [PubMed] [Google Scholar]
- [57].Kelly K, Cochran BH, Stiles CD, Leder P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 1983; 35:603-10; PMID:6606489; https://doi.org/ 10.1016/0092-8674(83)90092-2 [DOI] [PubMed] [Google Scholar]
- [58].Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev 2003; 17:1115-29; PMID:12695333; https://doi.org/ 10.1101/gad.1067003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zeller KI, Zhao X, Lee CWH, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, et al.. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci USA 2006; 103:17834-9; PMID:17093053; https://doi.org/ 10.1073/pnas.0604129103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012; 151:56-67; PMID:23021215; https://doi.org/ 10.1016/j.cell.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al.. c-Myc is a universal amplifierof expressed genes in lymphocytes and embryonic stem cells. Cell 2012; 151:68-79; PMID:23021216; https://doi.org/ 10.1016/j.cell.2012.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al.. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014; 511:488-92; PMID:25043028; https://doi.org/ 10.1038/nature13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Schuhmacher M, Kohlhuber F, Hölzel M, Kaiser C, Burtscher H, Jarsch M, Bornkamm GW, Laux G, Polack A, Weidle UH, et al.. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res 2001; 29:397-406; PMID:11139609; https://doi.org/ 10.1093/nar/29.2.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gargano B, Amente S, Majello B, Lania L. P-TEFb is a crucial co-factor for Myc transactivation. Cell Cycle 2007; 6:2031-7; PMID:17700062; https://doi.org/ 10.4161/cc.6.16.4554 [DOI] [PubMed] [Google Scholar]
- [65].Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, et al.. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 2011; 6:e26057; PMID:22039435; https://doi.org/ 10.1371/journal.pone.0026057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Perna D, Fagà G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung W-K, Sanges R, et al.. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene 2012; 31:1695-709; PMID:21860422; https://doi.org/ 10.1038/onc.2011.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, Kisker C, Wolf J, Eilers M, Wolf E. Different promoter affinities account for specificity in MYC-dependent gene regulation. Elife 2016; 5:e15161; PMID:27460974; https://doi.org/ 10.7554/eLife.15161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Guo J, Li T, Schipper J, Nilson KA, Fordjour FK, Cooper JJ, Gordân R, Price DH. Sequence specificity incompletely defines the genome-wide occupancy of Myc. Genome Biol 2014; 15:482; PMID:25287278; https://doi.org/ 10.1186/s13059-014-0482-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. Distinct thresholds govern Myc's biological output In Vivo. Cancer Cell 2008; 14:447-57; PMID:19061836; https://doi.org/ 10.1016/j.ccr.2008.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J, Dalla-Favera R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007; 448:445-51; PMID:17597761; https://doi.org/ 10.1038/nature05953 [DOI] [PubMed] [Google Scholar]
- [71].Srinivasan SV, Dominguez-Sola D, Wang LC, Hyrien O, Gautier J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep 2013; 3:1629-39; PMID:23643534; https://doi.org/ 10.1016/j.celrep.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sankar N, Kadeppagari RK, Thimmapaya B. c-Myc-induced aberrant DNA synthesis and activation of DNA damage response in p300 knockdown cells. J Biol Chem 2009; 284:15193-205; PMID:19332536; https://doi.org/ 10.1074/jbc.M900776200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Karlsson A, Deb-Basu D, Cherry A, Turner S, Ford J, Felsher DW. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc Natl Acad Sci USA 2003; 100:9974-9; PMID:12909717; https://doi.org/ 10.1073/pnas.1732638100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Menssen A, Epanchintsev A, Lodygin D, Rezaei N, Jung P, Verdoodt B, Diebold J, Hermeking H. c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: Identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle 2007; 6:339-52; PMID:17297307; https://doi.org/ 10.4161/cc.6.3.3808 [DOI] [PubMed] [Google Scholar]
- [75].Carr-Wilkinson J, Griffiths R, Elston R, Gamble LD, Goranov B, Redfern CPF, Lunec J, Tweddle DA. Outcome of the p53-mediated DNA damage response in neuroblastoma is determined by morphological subtype and MYCN expression. Cell Cycle 2011; 10:3778-87; PMID:22052359; https://doi.org/ 10.4161/cc.10.21.17973 [DOI] [PubMed] [Google Scholar]
- [76].Petroni M, Sardina F, Heil C, Sahun-Roncero M, Colicchia V, Veschi V, Albini S, Fruci D, Ricci B, Soriani A, et al.. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ 2016; 23:197-206; PMID:26068589; https://doi.org/ 10.1038/cdd.2015.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 1991; 6:1915-22; PMID:1923514 [PubMed] [Google Scholar]
- [78].Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992; 69:119-28; PMID:1555236; https://doi.org/ 10.1016/0092-8674(92)90123-T [DOI] [PubMed] [Google Scholar]
- [79].Ham J, Costa C, Sano R, Lochmann TL, Sennott EM, Patel NU, Dastur A, Gomez-Caraballo M, Krytska K, Hata AN, et al.. Exploitation of the apoptosis-primed state of MYCN- amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell 2016; 29:159-72; PMID:26859456; https://doi.org/ 10.1016/j.ccell.2016.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, Cantlon A, Fisch S, Golomb-Mello G, Ryan JA, et al.. Developmental regulation of mitochondrial apoptosis by c-Myc governs age- and tissue- specific sensitivity to cancer therapeutics. Cancer Cell 2017; 31:142-56; PMID:28017613; https://doi.org/ 10.1016/j.ccell.2016.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Seoane J, Le H-V, Massagué J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 2002; 419:729-34; PMID:12384701; https://doi.org/ 10.1038/nature01119 [DOI] [PubMed] [Google Scholar]
- [82].Chang DW, Claassen GF, Hann SR, Cole MD. The c-Myc transactivation domain is a direct modulator of apoptotic versus proliferative signals. Mol Cell Biol 2000; 20:4309-19; PMID:10825194; https://doi.org/ 10.1128/MCB.20.12.4309-4319.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev 1998; 12:2424-33; PMID:9694806; https://doi.org/ 10.1101/gad.12.15.2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science 1994; 265:2091-3; PMID:8091232; https://doi.org/ 10.1126/science.8091232 [DOI] [PubMed] [Google Scholar]
- [85].Amanullah A, Liebermann DA, Hoffman B. p53-independent apoptosis associated with c-Myc-mediated block in myeloid cell differentiation. Oncogene 2000; 19:2967-77; PMID:10871848; https://doi.org/ 10.1038/sj.onc.1203638 [DOI] [PubMed] [Google Scholar]
- [86].Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L, Ridgway RA, Huels D, Littler S, Schandl C, et al.. MYC is a major determinant of mitotic cell fate. Cancer Cell 2015; 28:129-40; PMID:26175417; https://doi.org/ 10.1016/j.ccell.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Yochum GS, Sherrick CM, Macpartlin M, Goodman RH. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5“ and 3” Wnt responsive enhancers. Proc Natl Acad Sci U S A 2010; 107:145-50; PMID:19966299; https://doi.org/ 10.1073/pnas.0912294107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Jang M, Park BC, Kang S, Chi S-W, Cho S, Chung SJ, Lee SC, Bae K-H, Park SG. Far upstream element-binding protein-1, a novel caspase substrate, acts as a cross-talker between apoptosis and the c-myc oncogene. Oncogene 2009; 28:1529-36; PMID:19219071; https://doi.org/ 10.1038/onc.2009.11 [DOI] [PubMed] [Google Scholar]
- [89].Hsiao H-H, Nath A, Lin C-Y, Folta-Stogniew EJ, Rhoades E, Braddock DT. Quantitative characterization of the interactions among c-myc transcriptional regulators FUSE, FBP, and FIR. Biochemistry 2010; 49:4620-34; PMID:20420426; https://doi.org/ 10.1021/bi9021445 [DOI] [PubMed] [Google Scholar]
- [90].Borgognone M, Armas P, Calcaterra NB. Cellular nucleic-acid-binding protein, a transcriptional enhancer of c-Myc, promotes the formation of parallel G-quadruplexes. Biochem J 2010; 428:491-8; PMID:20394585; https://doi.org/ 10.1042/BJ20100038 [DOI] [PubMed] [Google Scholar]
- [91].Weidensdorfer D, Stohr N, Baude A, Lederer M, Kohn M, Schierhorn A, Buchmeier S, Wahle E, Huttelmaier S. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA 2008; 15:104-15; PMID:19029303; https://doi.org/ 10.1261/rna.1175909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Marderosian M, Sharma A, Funk AP, Vartanian R, Masri J, Jo OD, Gera JF. Tristetraprolin regulates Cyclin D1 and c-Myc mRNA stability in response to rapamycin in an Akt-dependent manner via p38 MAPK signaling. Oncogene 2006; 25:6277-90; PMID:16702957; https://doi.org/ 10.1038/sj.onc.1209645 [DOI] [PubMed] [Google Scholar]
- [93].Challagundla KB, Sun XX, Zhang X, DeVine T, Zhang Q, Sears RC, Dai MS. Ribosomal protein L11 recruits miR-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol Cell Biol 2011; 31:4007-21; PMID:21807902; https://doi.org/ 10.1128/MCB.05810-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Li Y, Challagundla KB, Sun X-X, Zhang Q, Dai M-S. MicroRNA-130a associates with ribosomal protein L11 to suppress c-Myc expression in response to UV irradiation. Oncotarget 2015; 6:1101-14; PMID:25544755; https://doi.org/ 10.18632/oncotarget.2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Tworkowski KA, Salghetti SE, Tansey WP. Stable and unstable pools of Myc protein exist in human cells. Oncogene 2002; 21:8515-20; PMID:12466972; https://doi.org/ 10.1038/sj.onc.1205976 [DOI] [PubMed] [Google Scholar]
- [96].Jaenicke LA, von Eyss B, Carstensen A, Wolf E, Xu W, Greifenberg AK, Geyer M, Eilers M, Popov N. Ubiquitin-dependent turnover of MYC antagonizes MYC/PAF1C complex accumulation to drive transcriptional elongation. Mol Cell 2016; 61:54-67; PMID:26687678; https://doi.org/ 10.1016/j.molcel.2015.11.007 [DOI] [PubMed] [Google Scholar]
- [97].Welcker M, Orian A, Jin J, Grim JE, Grim JA, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA 2004; 101:9085-90; PMID:15150404; https://doi.org/ 10.1073/pnas.0402770101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 complex plays a prime role in regulating N-Myc phosphorylation and turnover in neural precursors. Dev Cell 2005; 9:327-38; PMID:16139224; https://doi.org/ 10.1016/j.devcel.2005.07.014 [DOI] [PubMed] [Google Scholar]
- [99].Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, Jamin Y, Thway K, Robinson SP, Roels F, et al.. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013; 24:75-89; PMID:23792191; https://doi.org/ 10.1016/j.ccr.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Malempati S, Tibbitts D, Cunningham M, Akkari Y, Olson S, Fan G, Sears RC. Aberrant stabilization of c-Myc protein in some lymphoblastic leukemias. Leukemia 2006; 20:1572-81; PMID:16855632; https://doi.org/ 10.1038/sj.leu.2404317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lutterbach B, Hann SR. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol Cell Biol 1994; 14:5510-22; PMID:8035827; https://doi.org/ 10.1128/MCB.14.8.5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al.. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463:899-905; PMID:20164920; https://doi.org/ 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Annibali D, Whitfield JR, Favuzzi E, Jauset T, Serrano E, Cuartas I, Redondo-Campos S, Folch G, Gonzàlez-Juncà A, Sodir NM, et al.. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat Commun 2014; 5:4632; PMID:25130259; https://doi.org/ 10.1038/ncomms5632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999; 4:199-207; PMID:10488335; https://doi.org/ 10.1016/S1097-2765(00)80367-6 [DOI] [PubMed] [Google Scholar]
- [105].Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, Swigart LB, Evan GI. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev 2013; 27:504-13; PMID:23475959; https://doi.org/ 10.1101/gad.205542.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature 2008; 455:679-83; PMID:18716624; https://doi.org/ 10.1038/nature07260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, Vogt PK. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci USA 2002; 99:3830-5; PMID:11891322; https://doi.org/ 10.1073/pnas.062036999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. In Vitro Cytotoxicity and In Vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J Pharmacol Exp Ther 2010; 335:715-27; PMID:20801893; https://doi.org/ 10.1124/jpet.110.170555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zirath H, Frenzel A, Oliynyk G, Segerström L, Westermark UK, Larsson K, Munksgaard Persson M, Hultenby K, Lehtiö J, Einvik C, et al.. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc Natl Acad Sci U S A 2013; 110:10258-63; PMID:23733953; https://doi.org/ 10.1073/pnas.1222404110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Fletcher S, Prochownik EV. Small-molecule inhibitors of the Myc oncoprotein. Biochim Biophys Acta 2015; 1849:525-43; PMID:24657798; https://doi.org/ 10.1016/j.bbagrm.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Hook KE, Garza SJ, Lira ME, Ching KA, Lee NV, Cao J, Yuan J, Ye J, Ozeck M, Shi ST, et al.. An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Mol Cancer Ther 2012; 11:710-9; PMID:22222631; https://doi.org/ 10.1158/1535-7163.MCT-11-0184 [DOI] [PubMed] [Google Scholar]
- [112].Helfrich BA, Kim J, Gao D, Chan DC, Zhang Z, Tan AC, Bunn PA. Barasertib (AZD1152), a small molecule aurora B inhibitor, inhibits the growth of SCLC cell lines In Vitro and In Vivo. Mol Cancer Ther 2016; 15:2314-22; PMID:27496133; https://doi.org/ 10.1158/1535-7163.MCT-16-0298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Dauch D, Rudalska R, Cossa G, Nault JC, Kang TW, Wuestefeld T, Hohmeyer A, Imbeaud S, Yevsa T, Hoenicke L, et al.. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat Med 2016; 22:744-53; PMID:27213815; https://doi.org/ 10.1038/nm.4107 [DOI] [PubMed] [Google Scholar]
- [114].Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, et al.. Stabilization of N-Myc is a critical function of aurora A in human neuroblastoma. Cancer Cell 2009; 15:67-78; PMID:19111882; https://doi.org/ 10.1016/j.ccr.2008.12.005 [DOI] [PubMed] [Google Scholar]
- [115].Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, Simonds EF, Seeger R, Matthay KK, Hertz NT, et al.. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 2014; 26:414-27; PMID:25175806; https://doi.org/ 10.1016/j.ccr.2014.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al.. Targeting transcriptional addictions in small cell lung cancerwith a covalent CDK7 inhibitor. Cancer Cell 2014; 26:909-22; PMID:25490451; https://doi.org/ 10.1016/j.ccell.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al.. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014; 159:1126-39; PMID:25416950; https://doi.org/ 10.1016/j.cell.2014.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Huang C-H, Lujambio A, Zuber J, Tschaharganeh DF, Doran MG, Evans MJ, Kitzing T, Zhu N, de Stanchina E, Sawyers CL, et al.. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev 2014; 28:1800-14; PMID:25128497; https://doi.org/ 10.1101/gad.244368.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Sengupta S, Biarnes MC, Jordan VC. Cyclin dependent kinase-9 mediated transcriptional de-regulation of cMYC as a critical determinant of endocrine-therapy resistance in breast cancers. Breast Cancer Res Treat 2013; 143:113-24; PMID:24309997; https://doi.org/ 10.1007/s10549-013-2789-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O, Lefebure M, Martin BP, Dawson MA, Johnstone RW, et al.. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 2015; 29:1437-41; PMID:25578475; https://doi.org/ 10.1038/leu.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med 2007; 13:820-7; PMID:17589519; https://doi.org/ 10.1038/nm1606 [DOI] [PubMed] [Google Scholar]
- [122].Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al.. Selective inhibition of BET bromodomains. Nature 2010; 468:1067-73; PMID:20871596; https://doi.org/ 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Coudé M-M, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, Raffoux E, Itzykson R, Delord M, Riveiro ME, et al.. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015; 6:17698-712; PMID:25989842; https://doi.org/ 10.18632/oncotarget.4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Shao Q, Kannan A, Lin Z, Stack BC, Suen JY, Gao L. BET protein inhibitor JQ1 attenuates Myc-Amplified MCC tumor growth In Vivo. Cancer Res 2014; 74:7090-102; PMID:25277525; https://doi.org/ 10.1158/0008-5472.CAN-14-0305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Grayson AR, Walsh EM, Cameron MJ, Godec J, Ashworth T, Ambrose JM, Aserlind AB, Wang H, Evan GI, Kluk MJ, et al.. MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene 2014; 33:1736-42; PMID:23604113; https://doi.org/ 10.1038/onc.2013.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, et al.. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res 2014; 20:912-25; PMID:24297863; https://doi.org/ 10.1158/1078-0432.CCR-13-2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Jahchan NS, Lim JS, Bola B, Morris K, Seitz G, Tran KQ, Xu L, Trapani F, Morrow CJ, Cristea S, et al.. Identification and targeting of long-term tumor- propagating cells in small cell lung cancer. Cell Rep 2016; 16:644-56; PMID:27373157; https://doi.org/ 10.1016/j.celrep.2016.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lam LT, Lin X, Faivre EJ, Yang Z, Huang X, Wilcox DM, Bellin RJ, Jin S, Tahir SK, Mitten M, et al.. Vulnerability of small cell lung cancer to apoptosis induced by the combination of BET bromodomain proteins and BCL2 inhibitors. Mol Cancer Ther 2017; [Epub ahead of print]; PMID:28468776; https://doi.org/ 10.1158/1535-7163.MCT-16-0459 [DOI] [PubMed] [Google Scholar]
- [129].Kato F, Fiorentino FP, Alibés A, Perucho M, Sánchez-Céspedes M, Kohno T, Yokota J. MYCL is a target of a BET bromodomain inhibitor, JQ1, on growth suppression efficacy in small cell lung cancer cells. Oncotarget 2016; 7:77378-88; PMID:27764802; https://doi.org/ 10.18632/oncotarget.12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Lenhart R, Kirov S, Desilva H, Cao J, Lei M, Johnston K, Peterson R, Schweizer L, Purandare A, Ross-Macdonald P, et al.. Sensitivity of small cell lung cancer to BET inhibition is mediated by regulation of ASCL1 gene expression. Mol Cancer Ther 2015; 14:2167-74; PMID:26253517; https://doi.org/ 10.1158/1535-7163.MCT-15-0037 [DOI] [PubMed] [Google Scholar]
- [131].Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003; 4:181-9; PMID:14522252; https://doi.org/ 10.1016/S1535-6108(03)00220-4 [DOI] [PubMed] [Google Scholar]
- [132].Dooley AL, Winslow MM, Chiang DY, Banerji S, Stransky N, Dayton TL, Snyder EL, Senna S, Whittaker CA, Bronson RT, et al.. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev 2011; 25:1470-5; PMID:21764851; https://doi.org/ 10.1101/gad.2046711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Huijbers IJ, Bin Ali R, Pritchard C, Cozijnsen M, Kwon M-C, Proost N, Song J-Y, de Vries H, Badhai J, Sutherland K, et al.. Rapid target gene validation in complex cancer mouse models using re-derived embryonic stem cells. EMBO Mol Med 2014; 6:212-25; PMID:24401838; https://doi.org/ 10.1002/emmm.201303297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Schaffer BE, Park KS, Yiu G, Conklin JF, Lin C, Burkhart DL, Karnezis AN, Sweet-Cordero EA, Sage J. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res 2010; 70:3877-83; PMID:20406986; https://doi.org/ 10.1158/0008-5472.CAN-09-4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Cui M, Augert A, Rongione M, Conkrite K, Parazzoli S, Nikitin AY, Ingolia N, MacPherson D. PTEN is a potent suppressor of small cell lung cancer. Mol Cancer Res 2014; 12:654-9; PMID:24482365; https://doi.org/ 10.1158/1541-7786.MCR-13-0554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, Bhutkar A, McKenna A, Dooley A, Vernon A, et al.. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014; 156:1298-311; PMID:24630729; https://doi.org/ 10.1016/j.cell.2014.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Gazdar AF, Savage TK, Johnson JE, Berns A, Sage J, Linnoila RI, MacPherson D, McFadden DG, Farago A, Jacks T, et al.. The comparative pathology of genetically engineered mouse models for neuroendocrine carcinomas of the lung. J Thorac Oncol 2015; 10:553-64; PMID:25675280; https://doi.org/ 10.1097/JTO.0000000000000459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, Girard L, Minna JD, Gazdar AF, Cobb MH, et al.. ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep 2016; 16:1259-72; PMID:27452466; https://doi.org/ 10.1016/j.celrep.2016.06.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Augustyn A, Borromeo M, Wang T, Fujimoto J, Shao C, Dospoy PD, Lee V, Tan C, Sullivan JP, Larsen JE, et al.. ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc Natl Acad Sci U S A 2014; 111:14788-93; PMID:25267614; https://doi.org/ 10.1073/pnas.1410419111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Semenova EA, Kwon M-C, Monkhorst K, Song J-Y, Bhaskaran R, Krijgsman O, Kuilman T, Peters D, Buikhuisen WA, Smit EF, et al.. Transcription Factor NFIB Is a driver of small cell lung cancer progression in mice and marks metastatic disease in patients. Cell Rep 2016; 16:631-43; PMID:27373156; https://doi.org/ 10.1016/j.celrep.2016.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Carney DN, Gazdar AF, Bepler G, Guccion JG, Marangos PJ, Moody TW, Zweig MH, Minna JD. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res 1985; 45:2913-23; PMID:2985257 [PubMed] [Google Scholar]
- [142].Gazdar AF, Carney DN, Nau MM, Minna JD. Characterization of variant subclasses of cell lines derived from small cell lung cancer having distinctive biochemical, morphological, and growth properties. Cancer Res 1985; 45:2924-30; PMID:2985258 [PubMed] [Google Scholar]
- [143].Poirier JT, Dobromilskaya I, Moriarty WF, Peacock CD, Hann CL, Rudin CM. Selective tropism of seneca valley virus for variant subtype small cell lung cancer. J Natl Cancer Inst 2013; 105:1059-65; PMID:23739064; https://doi.org/ 10.1093/jnci/djt130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Melichar B, Adenis A, Lockhart AC, Bennouna J. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: A five-arm phase 2 study. Lancet Oncol 2015; 16:395-405; PMID:25728526; https://doi.org/ 10.1016/S1470-2045(15)70051-3 [DOI] [PubMed] [Google Scholar]
- [145].Owonikoko TK, Nackaerts K, Csoszi T, Ostoros G, Baik C, Mark Z, Sheldon-Waniga E, Huebner D, Leonard EJ, Spigel DR. Randomized phase 2 study of investigational aurora A kinase (AAK) inhibitor alisertib (MLN8237) + paclitaxel (P) vs placebo + P as second line therapy for small-cell lung cancer (SCLC). Ann Oncol 2016; 27:14230; https://doi.org/ 10.1093/annonc/mdw389.01 [DOI] [Google Scholar]
- [146].Rudin CM, Ismaila N, Hann CL, Malhotra N, Movsas B, Norris K, Pietanza MC, Ramalingam SS, Turrisi AT III, Giaccone G. Treatment of small-cell lung cancer: American society of clinical oncology endorsement of the american college of chest physicians guideline. J Clin Oncol 2015; 33:4106-11; PMID:26351333; https://doi.org/ 10.1200/JCO.2015.63.7918 [DOI] [PubMed] [Google Scholar]
- [147].Hamanaka W, Motoi N, Ishikawa S, Ushijima M, Inamura K, Hatano S, Uehara H, Okumura S, Nakagawa K, Nishio M, et al.. A subset of small cell lung cancer with low neuroendocrine expression and good prognosis: A comparison study of surgical and inoperable cases with biopsy. Hum Pathol 2014; 45:1045-56; PMID:24746210; https://doi.org/ 10.1016/j.humpath.2014.01.001 [DOI] [PubMed] [Google Scholar]
- [148].Rekhtman N. Neuroendocrine tumors of the lung: An update. Arch Pathol Lab Med 2010; 134:1628-38; PMID:21043816; https://doi.org/ 10.1043/2009-0583-RAR.1 [DOI] [PubMed] [Google Scholar]
- [149].Travis WD. Lung tumours with neuroendocrine differentiation. Eur J Cancer 2009; 45:251-66; PMID:19775623; https://doi.org/ 10.1016/S0959-8049(09)70040-1 [DOI] [PubMed] [Google Scholar]
- [150].Poirier JT, Gardner EE, Connis N, Moreira AL, de Stanchina E, Hann CL, Rudin CM. DNA methylation in small cell lung cancer de. 2015; 34:5869-78; PMID:25746006; https://doi.org/ 10.1038/onc.2015.38 [DOI] [PMC free article] [PubMed] [Google Scholar]