

ABSTRACT
The ADA3 (Alteration/Deficiency in Activation 3) protein is an essential adaptor component of several Lysine Acetyltransferase (KAT) complexes involved in chromatin modifications. Previously, we and others have demonstrated a crucial role of ADA3 in cell cycle progression and in maintenance of genomic stability. Recently, we have shown that acetylation of ADA3 is key to its role in cell cycle progression. Here, we demonstrate that AKT activation downstream of Epidermal Growth Factor Receptor (EGFR) family proteins stimulation leads to phosphorylation of p300, which in turn promotes the acetylation of ADA3. Inhibition of upstream receptor tyrosine kinases (RTKs), HER1 (EGFR)/HER2 by lapatinib and the accompanying reduction of phospho-AKT levels led to a decrease in p300 phosphorylation and ADA3 protein levels. The p300/PCAF inhibitor garcinol also destabilized the ADA3 protein in a proteasome-dependent manner and an ADA3 mutant with K→R mutations exhibited a marked increase in half-life, consistent with opposite role of acetylation and ubiquitination of ADA3 on shared lysine residues. ADA3 knockdown led to cell cycle inhibitory effects, as well as apoptosis similar to those induced by lapatinib treatment of HER2+ breast cancer cells, as seen by accumulation of CDK inhibitor p27, reduction in mitotic marker pH3(S10), and a decrease in the S-phase marker PCNA, as well as the appearance of cleaved PARP. Taken together our results reveal a novel RTK-AKT-p300-ADA3 signaling pathway involved in growth factor-induced cell cycle progression.
KEYWORDS: ADA3, HER2, AKT, p300, acetylation, lapatinib, phosphorylation, ubiquitination, HAT complex, SAGA complex
Introduction
The Alteration/Deficiency in Activation-3 (ADA3) is an evolutionary conserved component of several lysine acetyltransferase (KAT) complexes such as STAGA (SPT3/TAFII31/GCN5 Acetyltransferase), ATAC (ADA2a containing complex), and TFTC (TATA binding protein free-TAF containing complex).1 Studies from our laboratory have shown that ADA3 is required for normal cell cycle progression, as demonstrated by the impact of conditional Ada3 deletion in mouse embryonic fibroblasts (MEFs) and ADA3 knockdown in normal human mammary epithelial cells (hMEC).2,3 We showed that ADA3, as a component of the STAGA and ATAC complexes, negatively regulates the CDK inhibitor p27 by promoting the c-Myc gene transcription.2,3 Additionally, ADA3 regulates global histone acetylation, maintains genomic stability and plays a pivotal role in mitosis by helping maintain optimal levels of the centromeric protein CENP-B at centromeres, which is required for normal chromosomal segregation.2,4,5
Aside from its function as an integral component of the classical multi-subunit KAT complexes, ADA3 also interacts with p300, that functions as a key mammalian KAT independent of the STAGA/ATAC complexes.6,7 We have also shown that ADA3 itself is acetylated by its interacting KATs.7 In the present study, we demonstrate that ADA3 acetylation is regulated by growth factor receptor activation through a novel signaling pathway that involves AKT and p300 phosphorylation.
Activation of epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases by their ligands, such as EGF, is a well-established mechanism that promotes cell proliferation under physiological conditions and in cancer.8,9 Ligand binding leads to activation of numerous downstream signaling cascades, including the phosphatidylinositol 3-kinase (PI3K) target AKT, a key regulator of physiological processes that control cell proliferation and survival.10,11 Among its wide range of targets, AKT has been shown to phosphorylate the KAT protein p300 at the Ser-1834 residue within an AKT consensus sequence RXRXXpS/T, and this phosphorylation promotes the KAT activity of p300 to regulate histone acetylation.12 How p300 Ser-1834 phosphorylation by AKT contributes to AKT-mediated regulation of cell proliferation downstream of growth factor receptor signals has not been elucidated.
In this study, we assessed the role of ADA3 in cell proliferation downstream of the EGFR family of cell surface receptors. Using EGF stimulation of normal and tumor cell line proliferation as a model, we present evidence that activation of AKT downstream of activated growth factor receptors induces p300 phosphorylation which in turn promotes ADA3 acetylation. We show that p300-mediated acetylation occurs on sites that are also the sites of ADA3 ubiquitination, suggesting a role of acetylation in stabilizing ADA3 protein by negating its ubiquitination. Indeed, treatment with the clinically used EGFR/HER2 inhibitor lapatinib, which downregulated AKT phosphorylation, led to a marked decrease in p300 phosphorylation and ADA3 protein levels. Notably, ADA3 knockdown mimicked the cell cycle and proliferation block induced by lapatinib with elevation of the levels of CDK inhibitor p27, increased apoptosis, low levels of proliferating cell nuclear antigen (PCNA) and reduced entry into mitosis. Taken together, our results establish a novel link between growth factor receptor regulation of cell proliferation and a novel downstream signaling pathway involving the AKT-p300 mediated ADA3 acetylation and stabilization.
Results
EGF induces ADA3 acetylation by activating AKT-p300 axis
We have recently shown that p300 acetylates ADA3 and that ADA3 acetylation is required for its role in promoting cell proliferation.7 To explore the upstream mechanisms that might control ADA3 acetylation during cell proliferation, we used a TERT immortalized human mammary epithelial cell line 76N-TERT, which is completely dependent on EGFR-mediated signaling for proliferation.13 Cells were deprived of EGF and serum-derived growth factors for 72 hours, and then stimulated with EGF for 15 or 30 min followed by western blotting, to assess the levels of phosphorylation of relevant downstream effectors. Treatment of cells with EGF led to an expected induction of AKT phosphorylation as well as the phosphorylation of the AKT target p300 on Ser-1834 (Fig. 1A). Due to the unavailability of antibodies to directly detect acetylated ADA3 in western blotting, we first immunoprecipitated the endogenous ADA3 from lysates of control or EGF-stimulated cells using anti-ADA3 antibodies, followed by western blotting with an anti-acetyl lysine antibody to assess the acetylation of ADA3. Notably, a significant increase in ADA3 acetylation was observed after 15 or 30 min of EGF stimulation (Fig. 1B). These results demonstrate that ADA3 acetylation is a novel downstream event in EGF-induced cell signaling. As with the 76N-TERT cells, EGF stimulation of breast cancer cell lines SKBR-3 and UACC812, and a lung cancer cell line A549, also revealed the EGF-induced acetylation of ADA3 as well as phosphorylation of AKT and p300 (Fig. 1C, D and S1A).
Figure 1.

EGF induces ADA3 acetylation by activating AKT-p300 axis. (A & B) 76N-TERT cells were growth factor deprived in DFCI-3 medium for 72 h followed by stimulation with EGF for various time points. Whole cell extracts from various time points were subjected to immunoblotting with indicated antibodies (A) or immunoprecipitated with normal IgG or anti-ADA3 antibodies. Immunoprecipitates were then immunoblotted with indicated antibodies (B). (C, & D) SKBR-3 (C), and UACC812 (D) cells were serum starved for 48 h and then stimulated with EGF as indicated. Whole cell extracts from various time points were subjected to immunoprecipitation with normal IgG or anti-ADA3 antibodies. Immunoprecipitates and input fractions were then immunoblotted with indicated antibodies. Note: For immunoprecipitation (IP) with normal IgG, asynchronously (Asyn) growing cells were used. (E) HEK-293T cells were co-transfected with FLAG-ADA3 and HA-tagged wild-type AKT or kinase dead (K179M) mutant. 48 h after transfection whole cell extracts were immunoprecipitated with M2 agarose and immunoprecipitates were then eluted by 3x FLAG peptide, followed by immunoblotting-with indicated antibodies. The numbers underneath anti-acetyl lysine blot represent band intensity of FLAG-ADA3 acetylation normalized over immunoprecipitated FLAG-ADA3 as computed from ImageJ software.
Next, to assess if ADA3 acetylation by p300 requires AKT-mediated phosphorylation of p300, we used a kinase-dead dominant-negative mutant of AKT (K179M).14 For these analyses, HEK-293T cells were transfected with FLAG-ADA3 along with HA-tagged wild-type or kinase dead AKT. Anti-FLAG antibody immunoprecipitations of cell lysates were immunoblotted with a pan-anti-acetyl lysine antibody to assess ADA3 acetylation. While co-transfection with wild-type AKT led to substantial increase in ADA3 acetylation, a markedly lower level of ADA3 acetylation (about 70% as compared with wild type AKT) was observed upon co-transfection with the kinase dead AKT mutant (Fig. 1E). These results support the conclusion that ADA3 acetylation by p300 is dependent on activated AKT.
To further link the activation of AKT downstream of surface receptors to ADA3 acetylation in a different growth factor/receptor system, we stimulated A459 lung cancer cells with TNFα for 30 and 60 minutes and assessed ADA3 acetylation. As in EGF-simulated mammary epithelial and breast cancer cells, TNFα stimulation of A549 cells indeed induced the phosphorylation of AKT and p300 (Fig. S1B). Immunoblotting of anti-ADA3 immunoprecipitations of the same lysates with a pan-anti-acetyl lysine antibody revealed that ADA3 acetylation was induced after TNFα stimulation (Fig. S1B). Taken together, our results identify a novel-signaling pathway downstream of cell surface receptors that involves activation of AKT and phosphorylation of p300 to promote ADA3 acetylation.
Acetylation of ADA3 requires the p300 phosphorylation
Next, we assessed if the phosphorylation of p300 at Ser-1834, which regulates its KAT activity12 is required for the acetylation of ADA3. For this purpose, we compared the abilities of phosphorylation-defective (S1834A) and phospho-mimic (S1834E) mutants of p300 vs. its wild-type form to acetylate ADA3, using in vitro and in vivo cell-based assays. For the in vitro acetyltransferase assay, we first transfected HA-tagged wild-type or mutant p300 constructs in HEK-293T cells, performed anti-HA immunoprecipitations, and eluted p300 proteins using an excess of the HA peptide (see Materials and Methods for details) (Fig. 2A). The eluted p300 proteins were incubated with recombinant GST-ADA3 as a substrate in an in vitro acetyltransferase assay. As expected, the wild type p300 protein efficiently acetylated the recombinant ADA3, whereas the level of acetylation with the phospho-defective p300 mutant S1834A was reduced by about 60% (Fig. 2B). In contrast, the phospho-mimic mutant p300 S1834E was as efficient as the wild type for ADA3 acetylation (Fig. 2B). Recombinant histone H3 was used as a positive control, where we observed that both the wild type p300 and the phospho-mimetic S1834E mutant efficiently acetylated recombinant histone H3, whereas the phospho-defective mutant S1834A showed a reduced ability to acetylate histone H3 (Fig. 2C). Next, we co-transfected HA-tagged wild-type p300 or S1834A mutant and FLAG-tagged ADA3 in HEK-293T cells, immunoprecipitated ADA3 with anti-FLAG antibodies and assessed its acetylation by immunoblotting with a pan-anti-acetylated lysine antibody. Consistent with the results of the in vitro acetyltransferase assay, more ADA3 acetylation was seen upon co-transfection with wild type p300, while acetylation was markedly lower upon co-transfection of the phospho-defective mutant of p300 (Fig. 2D). Taken together our in vitro and cell-based assays demonstrate that phosphorylation of p300 at Ser-1834 is required for its ability to acetylate ADA3.
Figure 2.

The phospho defective mutant p300 S1834A has reduced ability to acetylate ADA3. (A) HEK-293T cells were transfected with HA-tagged wild-type or S1834A/E mutants of p300. 48 h after transfection, whole cell extracts were subjected to immunoprecipitation by agarose conjugated anti-HA beads. Immunoprecipitates were then eluted with HA peptide and visualized on SDS gels by CBB staining. (B & C) In vitro KAT assay using 20 ng HA-tagged wild type or S1834A/E p300 mutants as enzymes obtained from A, and 1 μg of GST-ADA3 (B) or recombinant histone H3 (C) as substrates, followed by immunoblotting with anti-acetyl lysine antibody. The numbers underneath anti-Ac lysine blot in (B) represents the band intensity of GST-ADA3 acetylation normalized over total GST-ADA3 in ponceau stain as computed from ImageJ software. (D) HEK-293T cells were co-transfected with FLAG-tagged ADA3 and HA-tagged wild- type p300 or S1834A mutant. 48 h after transfection whole cell extracts were immunoprecipitated with M2 agarose and then immunoprecipitates were eluted by 3x FLAG peptide, followed by immunoblotting with indicated antibodies.
p300-mediated acetylation of ADA3 counteracts its ubiquitination and stabilizes the protein
Given our results that p300 acetylates ADA3 and that ADA3 acetylation is induced by growth factor receptor activation, we reasoned that one role of ADA3 acetylation may be to promote its stability if the sites of acetylation and ubiquitination, both of which modify the ε-amino group of lysine residues, are the same.15 Many key cellular proteins, such as SMAD7, p53, FOXP3, SREBPs, ER-α, RelA and ATP-citrate lyase are known to be stabilized, upon acetylation through competition with ubiquitination.16-22
To examine this possibility, we treated 76N-TERT hMECs with various concentrations of garcinol, a known inhibitor of p30023 and then analyzed ADA3 protein levels by immunoblotting. A dose-dependent decrease in ADA3 protein levels was observed upon garcinol treatment, suggesting that p300-mediated acetylation of ADA3 confers stability to ADA3 protein (Fig. 3A). Indeed, when cells were treated with proteasomal inhibitor MG132 along with garcinol, the garcinol-induced reduction in ADA3 protein levels was abrogated, supporting the conclusion that inhibition of p300-mediated ADA3 acetylation leads to ubiquitin-proteasome dependent degradation of ADA3 (Fig. 3A).
Figure 3.

p300 mediated acetylation of ADA3 counteracts its ubiquitination and stabilizes the protein. (A) 76N-TERT cells were treated with various doses of garcinol ± MG132 (a proteasome inhibitor) for 12 h and then whole cell lysates were subjected to immunoblotting with indicated antibodies. (B & C) HEK-293T cells were transfected with FLAG-ADA3 wild-type (B) or 5KR mutant (C). 36 hours after transfection cells were treated with 50 μg/ml of cycloheximide ± 20 μM MG132 and whole cell lysates were immunoblotted at various time points with indicated antibodies. (D) The band intensities of FLAG and HSC70 were quantified by ImageJ software. The log2 (band intensities of FLAG normalized over HSC70) were plotted against the time of cycloheximide treatment. The lines were generated by linear regression formula and each decrease of 1 unit at Y-axis is equivalent to one half-life. (E) HEK-293T cells were co-transfected with wild-type or 5KR FLAG-ADA3 mutant with or without HA-ubiquitin. 40 hours after transfection, cells were treated with 20 μM MG132 for 5 h. Equal amounts of lysates were immunoprecipitated using anti-FLAG agarose beads and subjected to SDS-PAGE. Immunoblotting was performed using indicated antibodies. Ponceau indicates the total immunoprecipitated FLAG-ADA3.
To further examine this question, we assessed the half-life of wild-type vs. a mutant ADA3 with mutations in known sites of acetylation. We have recently identified K109, −122, −124, −194 and −222 on ADA3 as sites of in vivo acetylation by p300.7 HEK-293T cells were transfected with wild-type ADA3 or an ADA3 mutant, in which all 5 of these lysine residues were mutated to arginine, referred to as 5KR mutant. These cells were then treated with cycloheximide to block new protein synthesis, and the levels of ADA3 proteins in cell lysates harvested at various time points were assessed by western blotting to determine the relative half-lives of the wild-type and mutant ADA3. Compared to a half-life of the transfected wild type ADA3 of about 4 hours (Fig. 3B and D), the 5KR mutant was highly stable and exhibited virtually no degradation during the course of cycloheximide treatment (Fig. 3C and D). These results reinforced the idea of competition between acetylation and ubiquitination of ADA3, with known sites of acetylation on ADA3 also serving as sites of ubiquitination such that the failure of the mutant ADA3 to undergo ubiquitination results in a highly stabile protein. To confirm this possibility, we performed ubiquitination analyses using HEK-293T cells co-transfected with HA-ubiquitin together with either wild type or 5KR mutant of FLAG-tagged ADA3. 40 hours after transfection, cells were treated with MG132 for 5 hours and lysates were prepared. Immunoprecipitation of cell lysates with an anti-FLAG antibody followed by immunoblotting with an anti-HA antibody showed that while the wild-type ADA3 was prominently ubiquitinated the ADA3–5KR mutant showed a marked decrease in the level of ubiquitination (Fig. 3E). These findings provide conclusive evidence that the same lysine residues on ADA3 protein are modified by acetylation or ubiquitination. Thus, we conclude that p300-mediated acetylation of ADA3 competes with its ubiquitination, thereby stabilizing ADA3 protein to promote its function.
Inhibition of upstream RTK signaling destabilizes ADA3
The analyses above in HEK-293T cells suggested that AKT-mediated p300 phosphorylation and activation could serve as a mechanism to regulate ADA3 stability downstream of RTK activation through p300-mediated acetylation of ADA3. If this was the case, we would expect that inhibition of the upstream receptor tyrosine kinases that induce AKT activation will downregulate ADA3 levels. To test this notion, two HER2 positive cell lines SKBR-3 and UACC812 were treated with increasing concentrations of a dual EGFR/HER2 tyrosine kinase inhibitor lapatinib that is known to abrogate downstream signaling through the PI3K/AKT and MAPK pathways.24 As expected, lapatinib treatment led to a marked decrease in the levels of phospho-HER2, as well phospho-AKT and phospho-p300 (Fig. 4A and B). Notably, treatment with lapatinib resulted in dose-dependent and a substantial decrease in ADA3 protein levels (Fig. 4A and B). A similar decrease in ADA3 levels was also observed upon lapatinib treatment of an EGFR+ breast cancer cell line MDA-MB-468 together with a marked decrease in p-EGFR levels (Fig. S2A). Lapatinib had essentially no impact on ADA3 mRNA levels, measured using quantitative RT-PCR, excluding the possibility that lapatinib-induced downregulation of ADA3 protein levels is due to reduction in mRNA (Fig. S2B). Consistent with this conclusion, no changes in ADA3 mRNA levels are seen when published microarray profiles of lapatinib-treated cells were examined.25,26
Figure 4.

Inhibition of HER2 and AKT phosphorylation downregulates ADA3 protein levels. (A & B) SKBR-3 (A) and UACC-812 (B) cells were treated with increasing concentration of lapatinib for 4 h and then whole cell extracts were immunoblotted with indicated antibodies. (C & D) SKBR-3 (C) and UACC812 (D) cells were treated with 8 μg/ml cycloheximide in the presence or absence of 1 μM lapatinib. Whole cell extracts from various time points were immunoblotted with indicated antibodies. The band intensities were computed using ImageJ software and the graphs show the log2(band intensity of ADA3 normalized over β-actin) vs. time after treatment in SKBR-3 (C) and UCAA812 (D) cells. Each decrease of 1 unit at Y-axis is equivalent to one half-life. (E & F) SKBR-3 (E) and UACC-812 (F) cells were treated with 25 μM LY294002 for 24 h and then whole cell extracts were immunoblotted with indicated antibodies.
To directly assess if lapatinib-induced reduction in ADA3 protein levels was post-translational, we assessed the half-life of ADA3 protein by treating HER2+ breast cancer cell lines with cycloheximide in the presence or absence of lapatinib. Notably, ADA3 was highly stable in HER2+ cell lines, consistent with our previous finding that HER2+ tumors express higher levels of ADA3 protein.27 Importantly, a decrease in ADA3 levels was observed over time in lapatinib-treated cells (Fig. 4C and D), supporting the role of receptor tyrosine kinase signaling to stabilize ADA3 protein.
To further assess that activation of AKT downstream of receptor tyrosine kinases was indeed responsible for ADA3 stabilization, we treated SKBR-3, UACC812 and A549 cells with an AKT inhibitor LY294002. Analysis of cell lysates demonstrated that LY294002 treatment indeed led to a decrease in ADA3 protein levels (Fig. 4E, F and Fig. S2C), together with the expected reduction in p-AKT levels (Fig. 4E, F and Fig. S2C),further substantiating our conclusion that ADA3 is a downstream component of RTK signaling that is regulated at the level of protein through the AKT pathway.
ADA3 knockdown mimics lapatinib-induced inhibition of cell proliferation
Previous studies have revealed that treatment of cells dependent on HER2/EGFR signaling with lapatinib leads to inhibition of cancer cell proliferation through induction of G1 cell cycle arrest and also induces cellular apoptosis.28,29 We have previously shown that loss of ADA3 induces a proliferative block due to accumulation of the CDK inhibitor p27.2 Since lapatinib treatment led to downregulation of ADA3 levels, we hypothesized that loss of ADA3 in HER2+ cells would have cell proliferation inhibitory effects similar to those of lapatinib. To test this idea, we knocked down ADA3 in SKBR-3 cells or treated them with lapatinib, and examined the levels of p27 and phospho-H3(S10), a known mitotic marker.30 Consistent with our hypothesis, similar increase in p27 levels and reduction in phospho-H3(S10) signals were seen in SKBR-3 cells upon ADA3 knockdown or lapatinib treatment (Fig. 5A and B). We further extended our analyses to know whether ADA3 knockdown and lapatinib treatment has any effect on apoptosis that causes decreased cell growth. To test this, we examined the levels of apoptotic marker cleaved Poly(ADP-ribose) Polymerase (PARP)31 and S-phase marker proliferating cell nuclear antigen (PCNA).32 Notably, both ADA3 knockdown and lapatinib treatment resulted in the appearance of cleaved PARP (about 5-fold increase after ADA3 knockdown and a 7-fold increase after lapatinib treatment) and a significant decrease in the S-phase marker PCNA (Fig. 5C). Taken together these results strongly support a key role of ADA3 in cancer cell proliferation downstream of activated RTKs.
Figure 5.
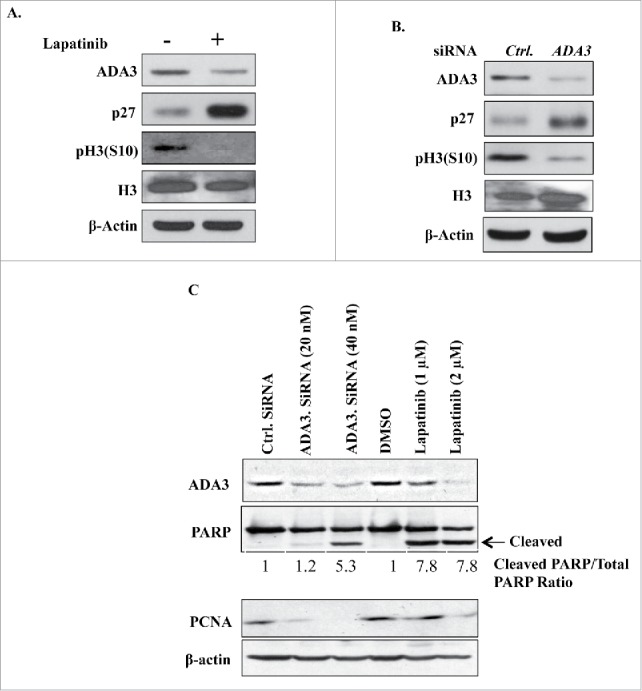
ADA3 knockdown mimics cell cycle inhibitory effect of lapatinib. (A, B & C) SKBR-3 cells were treated with either DMSO or 1 μM of lapatinib for 24 h or transiently transfected with Ctrl or ADA3 siRNA for 48 h and then whole cell extracts were immunoblotted with indicated antibodies.
Discussion
Cell cycle involves a tightly-regulated sequence of events to accomplish cell proliferation while maintaining genomic stability, and deviations from normal cell cycle control represent important contributors to diseases including cancer.33 The physiological or cancer-associated cell cycle progression commonly involves the inducible or constitutive activation of cell surface receptor tyrosine kinases, but how this activation leads to the full spectrum of events that culminate in cell proliferation still remains incomplete. ADA3 has recently emerged as a key positive regulator of cell cycle progression and studies from our laboratory and by others have shown that ADA3 is required for G1 to S phase transition, as well as to maintain genomic stability and for mitosis.2,4,5,34 We have shown that ADA3 protein is mislocalized from nucleus to cytoplasm, and is overexpressed in breast cancer patients, in particular those with EGFR/HER2 overexpression, and the mislocalization/overexpression of ADA3 correlates with poor prognosis and short survival of patients.3,27 To begin to explore the mechanistic links between ADA3 and cell proliferation, we recently demonstrated that ADA3 is post-translationally modified by acetylation, and that acetylation is critical for the role of ADA3 to promote cell cycle progression.7 In this study, we reveal new mechanistic links that control ADA3 acetylation during cell cycle progression downstream of prototype receptor tyrosine kinases of the EGFR family.
Using a series of normal mammary epithelial and tumor cell lines, we present evidence that ADA3 acetylation is a downstream event of ligand-induced EGFR signaling. Using a kinase dead AKT mutant, we demonstrate that AKT phosphorylation is upstream of ADA3 acetylation. Furthermore, using the phospho-defective or phospho-mimetic mutants of histone acetyltransferase p300, we demonstrate that AKT-mediated phosphorylation is a necessary event for p300 activation, which in turn governs ADA3 acetylation. In this manner, we defined a novel RTK-AKT-p300 pathway that enhances ADA3 acetylation as part of the biochemical events required to promote cell proliferation (Fig. 6).
Figure 6.

Model depicting RTK signaling by EGF to acetylation of ADA3 to regulate cell cycle progression. Activation of RTK signaling by EGF leads to phosphorylation of AKT, which then phosphorylates p300 to acetylate ADA3, leading to stabilization of ADA3 by preventing its ubiquitination, and thereby promoting cell cycle progression. Treatment of cells by RTK inhibitors such as lapatinib inhibits phospho-AKT and consequently p300 phosphorylation, which in turn prevents ADA3 acetylation leading to its poly-ubiquitination (Ubn) and destabilization and causes cell cycle block.
Our study also revealed that competition between acetylation and ubiquitination of ADA3 maintains ADA3 levels in cells, providing a novel mechanism by which acetylation of ADA3 downstream of RTK signaling promotes ADA3 function during cell cycle progression. We demonstrate that lysine residues we have previously established as sites of ADA3 are also critical for its ubiquitination and instability (Fig. 3). Importantly, ADA3 becomes highly unstable when either p300 or upstream kinases HER2/EGFR or AKT are chemically inhibited in HER2-ovexpressing breast cancer cell lines (Figs. 3 and 4). The stabilization of ADA3 by HER2 (and potentially other RTK) signaling is consistent with the increased ADA3 levels seen in breast cancer specimens, especially those from HER2+ patients.27 How the relative levels of ADA3 acetylation vs. ubiquitination are regulated during cell cycle and potentially other cellular activities regulated by ADA3 remain to be fully defined. As ADA3 protein stability is governed by p300 mediated acetylation and ubiquitination of same lysine residues, it is possible that aside from activation of p300, the level and/or activity of deubiquitinating enzymes may also critically control the balance of acetylation vs. ubiquitination and ADA3 stability and its availability to drive cell cycle related events. It is intriguing that the STAGA complex, of which ADA3 is a component within the HAT module, harbors a deubiquitinase USP22; importantly, USP22 is part of an 11 gene signature that correlates with poor prognosis in multiple cancers.35 Whether USP22 is the deubiquitinase for ADA3 that may contribute to its acetylation-dependent stabilization will be of considerable interest in future studies.
We used lapatinib, an FDA-approved dual HER2/EGFR kinase inhibitor24 to examine the involvement of RTK signaling in regulating ADA3. Lapatinib is approved for treatment of patients with advanced HER2-positive breast cancer in combination with capecitabine.36,37 Previous studies have revealed that lapatinib arrests tumor cells in G0/G1 phase, thus inhibiting cancer cell proliferation.28,38 However, the molecular basis of lapatinib induced cell cycle arrest remains poorly understood. Our finding that lapatinib destabilizes ADA3, a critical positive regulator of G1-S cell cycle progression, suggests the idea that the mechanism of function of lapatinib and potentially other HER2/EGFR targeted agents may involve destabilization of ADA3.
In further support of this scenario, both EGFR/HER2 inhibition and ADA3 knockdown lead to an increase in the levels of CDK inhibitor p27 (Fig. 5). We have previously established that increase in p27 levels after loss of ADA3 involves reduced expression of its ubiquitin ligase SKP2.2,3 However, p27 and SKP2 are also direct substrates of AKT, with their phosphorylation promoting degradation and activation, respectively.39-42 SKP2 also activates AKT via a positive feedback loop involving non-proteolytic ubiquitination.43 Thus, further studies are warranted to dissect the relative role of ADA3-mediated vs. other signaling pathways downstream of RTKs and their relevance to targeted therapies.
Since ADA3 is a core component of multiple HAT complexes, our studies suggest the potential of targeting ADA3-regulated downstream events to accentuate the effectiveness of and counter resistance to RTK directed therapies since de novo or acquired resistance to these agents is a major clinical issue. Resistance to RTK inhibitors commonly arises due to upregulation of parallel RTK pathways that lead to activation of key downstream signaling cascades such as MAP kinase or PI3K/AKT and efforts to inhibit these mediators to overcome resistance are currently being tested.44,45 Thus, future studies to assess if ADA3 represents a nodal point downstream of AKT and potentially other signaling pathways activated by RTKs could help develop a rationale to target ADA3-dependent pathways.
Materials and methods
Cell lines and cell culture
76N-TERT, immortalized human mammary epithelial cell line was grown in DFCI-1 medium and where necessary, was growth factor deprived in DFCI-3 medium, as described earlier.3,13 HEK-293T and A549, and UACC812 and MDA-MB-468, and SKBR-3 were cultured in DMEM, α-MEM and RPMI 1640, respectively. All media were supplemented with 10% fetal bovine serum (FBS), 10 μg/ml gentamycin, 1 mM sodium pyruvate, 10 mM HEPES, 2 mM L-glutamine, 1x minimal non-essential amino acids. For serum starvation experiments, cells were first starved in media without glutamine, minimal non-essential amino acids, and fetal bovine serum followed by stimulation with 25 ng/ml EGF for the time points indicated in the results section.
Reagents
Various reagents were purchased from indicated companies. FBS (10437–028) and gentamycin (15750–060) (Gibco). 100x Sodium pyruvate (11360070), 100x HEPES (15630080), 100x L-glutamine (25030081), 100x minimal non-essential amino acids (11140050) (Thermo Fisher Scientific). LY294002 (S1105) (Selleckchem). Lapatinib (L-4899) (L.C. laboratories). Trichostatin A (TSA, T8552), Nicotinamide (NAM, N0636), cycloheximide (C7698), MG-132 (M7449), acetyl co-enzyme A sodium salt (A2056), garcinol (G5173), HA peptide (I2149), 3X FLAG peptide (F4799), TNFα (T0157) and EGF (E9644) (all from Sigma).
Plasmids and transient transfection
Generation of FLAG-tagged ADA3 wild-type or 5K/R mutant was described previously.7 HA-AKT wild-type (plasmid# 9004–903) and K179M (plasmid# 9007–904) were purchased from Addgene. HA-p300 WT, S1834A and S1834E were generous gift from Dr. Denise Galloway (Fred Hutchinson Cancer Research Center, Seattle, WA). HA-Ub was described earlier.46 For transient transfection experiments the indicated plasmids were transfected using X-tremeGene HP transfection reagent (Roche # 06366236001) according to manufacturer's protocol. For ADA3 knockdown experiments, cells were transfected with 50 nM of control (sc-37007, Santa Cruz Biotechnology) or ADA3 siRNA (sc-78466, Santa Cruz Biotechnology), using the DharmaFECT Transfection Reagent (T-2001–03, Dharmacon RNAi Technologies).
RNA extraction and quantitative real-time PCR
TRIzol reagent (ThermoFisher Scientific, Waltham, MA) was used to isolate total RNA from cells. 2 μg of total RNA was reverse transcribed using SuperScriptTM II reverse transcriptase (Invitrogen). Real-time PCR quantification was performed in triplicates using SYBR Green PCR master mix (Applied Biosystems). 18s rRNA primers were from Thermo Fisher Scientific (AM1716) whereas the primer sequence for hADA3 are as follows Forward: 5′TAGGAGGCCGCAGAGAGGAGGTAG3′ and Reverse: 5′ AGAGGTGATGCTCGTCAAGTGCCC 3′
Immunoprecipitation
For immunoprecipitation, cells were harvested in lysis buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.5% Nonidet P-40, 0.1 mm Na4VO3, 1 mm NaF, and protease inhibitor mixture, 2 μM TSA and 10 mM NAM (for acetylation experiments) and whole cell extracts were subjected to immunoprecipitation with indicated antibodies overnight at 4°C. Beads were then washed 5 times at 5000 rpm for 1 min with lysis buffer. For elution by FLAG or HA peptide, the immunoprecipitated FLAG or HA-tagged proteins were eluted with 0.25 μg/μl peptide (Sigma) into lysis buffer. The elutes were subjected to SDS-PAGE and then analyzed by immunoblotting, as indicated.
Antibodies
Generation of anti-ADA3 mouse monoclonal antiserum has been described previously.2 Purified anti-phosphotyrosine mAb 4G1047 was provided by Dr. Brian Druker (Oregon Health Science University, Portland, OR). Antibodies against HSC-70 (sc-7298), pHER2 (sc-12352), EGFR (sc-31155), HER2 (sc-284), pAKT (sc-7985), AKT (sc-5298), p300 (sc-584 and sc-585) were from Santa Cruz Biotechnology. Agarose conjugated anti-FLAG (A2220) and anti-HA (A2095); anti-ADA3 rabbit polyclonal (HPA042250), FLAG (A8592), β-actin (A5441) were from Sigma. Histone H3 (06–755) was from EMD Millipore. p27 (610241), PCNA (610665) and PARP (551025) from BD Biosciences, pH3(S10) (ab-14955) was from Abcam. pp300-S1834 (PA5–12735) was from Thermo Fisher Scientific. Anti-acetyl lysine (9681), anti-acetyl lysine-HRP (6952) and anti-HA (2999) antibodies were from Cell Signaling Technologies.
In vitro KAT assay
For in vitro acetylation reactions, HA-p300 WT, S1834A or S1834E were ectopically expressed and then immunoprecipitated from HEK293T cells. The immunoprecipitates were eluted with HA peptide (I2149, Sigma) and a fraction of eluate was analyzed on SDS-PAGE by CBB staining with known amounts of BSA used as control. 1 μg GST-ADA3 or purified histone H3 was incubated with 20 ng HA-p300 WT, S1834A or S1834E in KAT buffer (50 mM Tris HCl pH 8.0, 50 mM KCl, 5% glycerol, 0.1 mM EDTA, 1mM DTT, 2 μM TSA, 50 μM Acetyl co-enzyme, a sodium salt and 1 mM PMSF) at 30°C for 30 min. The reaction was stopped by adding 6x SDS sample buffer and the products were subjected to SDS-PAGE analysis and immunoblotted with the indicated antibodies.
ADA3 half-life experiments
HEK-293T cells were transfected with wild-type or acetylation deficient FLAG-ADA3 constructs. 24 hours post transfection, cells were trypsinized and then plated in 6 well plates. After 18 hours, cells were treated with 50 μg/ml of cycloheximide ± 20 μM MG132 for indicated time points. Equivalent amounts of cell lysates were resolved on SDS-PAGE followed by Western blotting. To examine effects of lapatinib on half-life of endogenous ADA3 in SKBR-3 or UACC812 cells, cells were treated with 8 μg/ml of cycloheximide, and then analyzed similarly as described above. Densitometry analysis was performed on scanned images using ImageJ software.
In vivo ubiquitination assays
For in vivo ubiquitination assays, HEK-293T cells were transfected with 1 μg of wild-type or acetylation defective FLAG-tagged ADA3 construct with or without 2 μg pcDNA3.1-HA-Ubiquitin construct. 40 hours post transfection, cells were treated with 20 μM MG132 for 5 hours, lysates immunoprecipitated with anti-FLAG antibody, followed by immunoblotting with anti-HA-HRP antibody.
Supplementary Material
Disclosure of potential conflicts of interest
Authors disclose no potential conflicts of interest.
Funding
This work was supported by the NIH grants CA116552, CA87986 and CA105489 to HB, and CA96844 and CA144027 to VB; Department of Defense grants W81WH-11–1–0167 to HB, W81XWH-07–1–0351, W81XWH-11–1–0171 and W81XWH-14–1–0567 to VB. We acknowledge the support from the NCI Cancer Center Support Grant (P30CA036727) to Fred & Pamela Buffett Cancer Center and the Nebraska Research Initiative. SM was a postdoctoral fellow of the Susan G. Komen Foundation.
References
- [1].Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 2007; 26:5341-57; PMID:17694077; DOI: 1210604 [pii] [DOI] [PubMed] [Google Scholar]
- [2].Mohibi S, Gurumurthy CB, Nag A, Wang J, Mirza S, Mian Y, Quinn M, Katafiasz B, Eudy J, Pandey S, et al.. Mammalian alteration/deficiency in activation 3 (Ada3) is essential for embryonic development and cell cycle progression. J Biol Chem 2012; 287:29442-56; PMID:22736770; https://doi.org/ 10.1074/jbc.M112.378901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Griffin NI, Sharma G, Zhao X, Mirza S, Srivastava S, Dave BJ, Aleskandarany M, Rakha E, Mohibi S, Band H, et al.. ADA3 regulates normal and tumor mammary epithelial cell proliferation through c-MYC. Breast Cancer Res 2016; 18:113; PMID:27852327; https://doi.org/ 10.1186/s13058-016-0770-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mirza S, Katafiasz BJ, Kumar R, Wang J, Mohibi S, Jain S, Gurumurthy CB, Pandita TK, Dave BJ, Band H, et al.. Alteration/deficiency in activation-3 (Ada3) plays a critical role in maintaining genomic stability. Cell Cycle 2012; 11:4266-74; PMID:23095635; https://doi.org/ 10.4161/cc.22613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mohibi S, Srivastava S, Wang-France J, Mirza S, Zhao X, Band H, Band V. Alteration/deficiency in activation 3 (ADA3) protein, a cell cycle regulator, associates with centromere through CENP-B and regulates chromosome segregation. J Biol Chem 2015; PMID:26429915; DOI: jbc.M115.685511 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang T, Kobayashi T, Takimoto R, Denes AE, Snyder EL, el-Deiry WS, Brachmann RK. hADA3 is required for p53 activity. EMBO J 2001; 20:6404-13; PMID:11707411; https://doi.org/ 10.1093/emboj/20.22.6404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mohibi S, Srivastava S, Bele A, Mirza S, Band H, Band V. Acetylation of mammalian ADA3 is required for its functional roles in histone acetylation and cell proliferation. Mol Cell Biol 2016; PMID:27402865; DOI: MCB.00342-16 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res 2003; 284:2-13; PMID:12648462; DOI: S0014482702001052 [pii] [DOI] [PubMed] [Google Scholar]
- [9].Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol 2009; 218:460-6; PMID:19006176; https://doi.org/ 10.1002/jcp.21635 [DOI] [PubMed] [Google Scholar]
- [10].Song G, Ouyang G, Bao S. The activation of akt/PKB signaling pathway and cell survival. J Cell Mol Med 2005; 9:59-71; PMID:15784165; DOI: 009.001.07 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: A double-edged sword in cell proliferation and genome stability. J Oncol 2012; 2012:951724; PMID:22481935; https://doi.org/ 10.1155/2012/951724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Huang WC, Chen CC. Akt phosphorylation of p300 at ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol 2005; 25:6592-602; PMID:16024795; DOI: 25/15/6592 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Band V, Sager R. Distinctive traits of normal and tumor-derived human mammary epithelial cells expressed in a medium that supports long-term growth of both cell types. Proc Natl Acad Sci U S A 1989; 86:1249-53; PMID:2919173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK. The akt kinase: Molecular determinants of oncogenicity. Proc Natl Acad Sci U S A 1998; 95:14950-5; PMID:9843996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: Role in the control of protein stability. Bioessays 2005; 27:408-15; PMID:15770681; https://doi.org/ 10.1002/bies.20210 [DOI] [PubMed] [Google Scholar]
- [16].Giandomenico V, Simonsson M, Gronroos E, Ericsson J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol 2003; 23:2587-99; PMID:12640139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gronroos E, Hellman U, Heldin CH, Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell 2002; 10:483-93; PMID:12408818; DOI: S1097276502006391 [pii] [DOI] [PubMed] [Google Scholar]
- [18].Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, Lei QY. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell 2013; 51:506-18; PMID:23932781; https://doi.org/ 10.1016/j.molcel.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ma Y, Fan S, Hu C, Meng Q, Fuqua SA, Pestell RG, Tomita YA, Rosen EM. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Mol Endocrinol 2010; 24:76-90; PMID:19887647; https://doi.org/ 10.1210/me.2009-0218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li H, Wittwer T, Weber A, Schneider H, Moreno R, Maine GN, Kracht M, Schmitz ML, Burstein E. Regulation of NF-kappaB activity by competition between RelA acetylation and ubiquitination. Oncogene 2012; 31:611-23; PMID:21706061; https://doi.org/ 10.1038/onc.2011.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 2002; 277:50607-11; PMID:12421820; https://doi.org/ 10.1074/jbc.C200578200 [DOI] [PubMed] [Google Scholar]
- [22].van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, Brenkman AB, Hijnen DJ, Mutis T, Kalkhoven E, et al.. Regulation of treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010; 115:965-74; PMID:19996091; https://doi.org/ 10.1182/blood-2009-02-207118 [DOI] [PubMed] [Google Scholar]
- [23].Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, Kundu TK. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem 2004; 279:33716-26; PMID:15155757; https://doi.org/ 10.1074/jbc.M402839200 [DOI] [PubMed] [Google Scholar]
- [24].Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL. Anti-tumor activity of GW572016: A dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 2002; 21:6255-63; PMID:12214266; https://doi.org/ 10.1038/sj.onc.1205794 [DOI] [PubMed] [Google Scholar]
- [25].Hegde PS, Rusnak D, Bertiaux M, Alligood K, Strum J, Gagnon R, Gilmer TM. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Cancer Ther 2007; 6:1629-40; PMID:17513611; DOI: 6/5/1629 [pii] [DOI] [PubMed] [Google Scholar]
- [26].O'Neill F, Madden SF, Aherne ST, Clynes M, Crown J, Doolan P, O'Connor R. Gene expression changes as markers of early lapatinib response in a panel of breast cancer cell lines. Mol Cancer 2012; 11:41,4598-11-41; PMID:22709873; https://doi.org/ 10.1186/1476-4598-11-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mirza S, Rakha EA, Alshareeda A, Mohibi S, Zhao X, Katafiasz BJ, Wang J, Gurumurthy CB, Bele A, Ellis IO, et al.. Cytoplasmic localization of alteration/deficiency in activation 3 (ADA3) predicts poor clinical outcome in breast cancer patients. Breast Cancer Res Treat 2013; 137:721-31; PMID:23288344; https://doi.org/ 10.1007/s10549-012-2363-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tang L, Wang Y, Strom A, Gustafsson JA, Guan X. Lapatinib induces p27(Kip1)-dependent G(1) arrest through both transcriptional and post-translational mechanisms. Cell Cycle 2013; 12:2665-74; PMID:23907131; https://doi.org/ 10.4161/cc.25728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhao H, Faltermeier CM, Mendelsohn L, Porter PL, Clurman BE, Roberts JM. Mislocalization of p27 to the cytoplasm of breast cancer cells confers resistance to anti-HER2 targeted therapy. Oncotarget 2014; 5:12704-14; PMID:25587029; DOI: 2871 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sawicka A, Seiser C. Histone H3 phosphorylation - a versatile chromatin modification for different occasions. Biochimie 2012; 94:2193-201; PMID:22564826; https://doi.org/ 10.1016/j.biochi.2012.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].D'Amours D, Sallmann FR, Dixit VM, Poirier GG. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: Implications for apoptosis. J Cell Sci 2001; 114:3771-8; PMID:11707529 [DOI] [PubMed] [Google Scholar]
- [32].Essers J, Theil AF, Baldeyron C, van Cappellen WA, Houtsmuller AB, Kanaar R, Vermeulen W. Nuclear dynamics of PCNA in DNA replication and repair. Mol Cell Biol 2005; 25:9350-9; PMID:16227586; DOI: 25/21/9350 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432:316-23; PMID:15549093; DOI: nature03097 [pii] [DOI] [PubMed] [Google Scholar]
- [34].Orpinell M, Fournier M, Riss A, Nagy Z, Krebs AR, Frontini M, Tora L. The ATAC acetyl transferase complex controls mitotic progression by targeting non-histone substrates. EMBO J 2010; 29:2381-94; PMID:20562830; https://doi.org/ 10.1038/emboj.2010.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest 2005; 115:1503-21; PMID:15931389; https://doi.org/ 10.1172/JCI23412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ryan Q, Ibrahim A, Cohen MH, Johnson J, Ko CW, Sridhara R, Justice R, Pazdur R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008; 13:1114-9; PMID:18849320; https://doi.org/ 10.1634/theoncologist.2008-0816 [DOI] [PubMed] [Google Scholar]
- [37].Gomez HL, Doval DC, Chavez MA, Ang PC, Aziz Z, Nag S, Ng C, Franco SX, Chow LW, Arbushites MC, et al.. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J Clin Oncol 2008; 26:2999-3005; PMID:18458039; https://doi.org/ 10.1200/JCO.2007.14.0590 [DOI] [PubMed] [Google Scholar]
- [38].Kim JW, Kim HP, Im SA, Kang S, Hur HS, Yoon YK, Oh DY, Kim JH, Lee DS, Kim TY, et al.. The growth inhibitory effect of lapatinib, a dual inhibitor of EGFR and HER2 tyrosine kinase, in gastric cancer cell lines. Cancer Lett 2008; 272:296-306; PMIDPMID:18774637; https://doi.org/ 10.1016/j.canlet.2008.07.018 [DOI] [PubMed] [Google Scholar]
- [39].Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol 2009; 11:397-408; PMID:19270695; https://doi.org/ 10.1038/ncb1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, Yang WL, Erdjument-Bromage H, Nakayama KI, Nimer S, et al.. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by akt/PKB. Nat Cell Biol 2009; 11:420-32; PMID:19270694; https://doi.org/ 10.1038/ncb1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem 2002; 277:28706-13; PMID:12042314; https://doi.org/ 10.1074/jbc.M203668200 [DOI] [PubMed] [Google Scholar]
- [42].Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, et al.. PKB/akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 2002; 8:1153-60; PMID:12244302; https://doi.org/ 10.1038/nm761 [DOI] [PubMed] [Google Scholar]
- [43].Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, et al.. The Skp2-SCF E3 ligase regulates akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012; 149:1098-111; PMID:22632973; https://doi.org/ 10.1016/j.cell.2012.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, et al.. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 2008; 68:9221-30; PMID:19010894; https://doi.org/ 10.1158/0008-5472.CAN-08-1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, et al.. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov 2012; 2:934-47; PMID:22961667; https://doi.org/ 10.1158/2159-8290.CD-12-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gao Q, Kumar A, Singh L, Huibregtse JM, Beaudenon S, Srinivasan S, Wazer DE, Band H, Band V. Human papillomavirus E6-induced degradation of E6TP1 is mediated by E6AP ubiquitin ligase. Cancer Res 2002; 62:3315-21; PMIDPMID:12036950 [PubMed] [Google Scholar]
- [47].Druker BJ, Mamon HJ, Roberts TM. Oncogenes, growth factors, and signal transduction. N Engl J Med 1989; 321:1383-91; PMID:2682241; https://doi.org/ 10.1056/NEJM198911163212007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.