

SUMMARY
AMPA receptors (AMPARs) mediate fast excitatory synaptic transmission and are crucial for synaptic plasticity, learning and memory. However, the molecular control of AMPAR stability and its neurophysiological significance remain unclear. Here we report that AMPARs are subject to lysine acetylation at their C-termini. Acetylation reduces AMPAR internalization and degradation, leading to increased cell-surface localization and prolonged receptor half-life. Through competition for the same lysine residues, acetylation intensity is inversely related to the levels of AMPAR ubiquitination. We find that sirtuin 2 (SIRT2) acts as an AMPAR deacetylase regulating AMPAR trafficking and proteostasis. SIRT2 knockout mice (Sirt2−/−) show marked up-regulation in AMPAR acetylation and protein accumulation. Both Sirt2−/− mice and mice expressing acetylation mimetic GluA1 show aberrant synaptic plasticity, accompanied with impaired learning and memory. These findings establish SIRT2-regulated lysine acetylation as a form of AMPAR post-translational modification that regulates its turnover, as well as synaptic plasticity and cognitive function.
Keywords: AMPA receptors (AMPARs), Receptor trafficking, Sirtuin 2, Deacetylase, Acetylation, Ubiquitination, Synaptic plasticity, Learning and memory, Cognitive function
eTOC Blurb
Stability of glutamatergic AMPA receptors in neurons is of vital importance for synaptic transmission and brain function. Wang et al. report that AMPA receptors are subject to acetylation, which is regulated by sirtuin 2 and controls receptor trafficking, turnover, synaptic plasticity and memory.
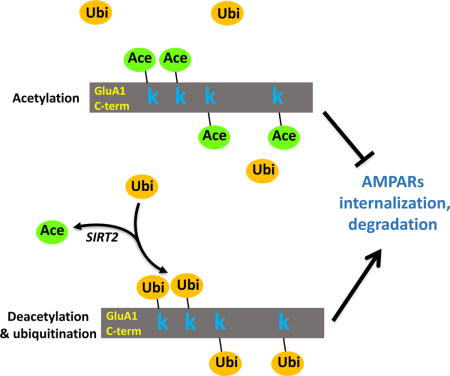
INTRODUCTION
AMPA receptors (AMPARs) are the primary mediator for the neuronal communication that underlies basal and higher brain functions. A large amount of literature has established that the primary cellular mechanism employed in synaptic plasticity, the cellular substrate for learning and memory, is the alteration of AMPAR abundance in the postsynaptic domain via dynamic receptor trafficking. An elevation in the membrane insertion and internalization of AMPARs has been shown to be the major processes underlying the expression of long-term potentiation (LTP) and long-term depression (LTD), respectively (Malinow and Malenka, 2002; Sheng and Hyoung Lee, 2003; Collingridge et al., 2004; Park et al., 2004; Palmer et al., 2005; Shepherd and Huganir, 2007). Aberrant regulation of AMPAR functions or dynamics in the brain are directly associated with cognitive impairments such as Alzheimer’s Disease (AD) and dementia (Struhl, 1998; Chang et al., 2006; Hsieh et al., 2006). In addition to trafficking-mediated rapid intracellular translocation, total protein homeostasis of AMPARs is also subject to regulation. Our recent work and studies from other groups have shown that AMPARs can be targeted for protein ubiquitination. E3 ligases such as Nedd4 and RNF167 conjugate ubiquitin units to the lysine residues at the intracellular terminal of AMPAR subunits (Schwarz et al., 2010; Lin et al., 2011; Lussier et al., 2011), leading to facilitated receptor internalization and proteasomal degradation. However, a molecular mechanism that antagonizes receptor ubiquitination and thus promotes AMPAR stabilization remains unknown.
Sirtuins are a family of highly conserved lysine deacetylases that play crucial roles in a wide range of cellular processes such as energy metabolism, DNA repair, gene transcription (Longo and Kennedy, 2006; Michan and Sinclair, 2007) and they are involved in a variety of biological and pathological conditions including development, aging, obesity and carcinogenesis (Finkel et al., 2009; Haigis and Sinclair, 2010). Sirtuins have traditionally been considered histone deacetylases, targeting the lysine residues on histones that lead to alterations in the efficacy of gene transcription (Chuang et al., 2009). However, in recent years, it has been recognized that non-histone proteins could also be targets for sirtuin-dependent modification (North et al., 2003; Brunet et al., 2004; Yeung et al., 2004; Min et al., 2010).
Among the seven members of the sirtuin family (SIRT1-7), SIRT3, 4, 5 are exclusively expressed in mitochondria, while SIRT1, 6, 7 are predominantly expressed in the nucleus. In contrast, SIRT2 is predominantly distributed and functions in the cytoplasm (Michan and Sinclair, 2007). SIRT1 and SIRT2 have been shown to be largely associated with several nervous system disorders such as Parkinson disease (PD), dementia and amyotrophic lateral sclerosis (ALS) (Kim et al., 2007; Michan and Sinclair, 2007; Outeiro et al., 2007; Harting and Knoll, 2010; Min et al., 2010). SIRT1 and SIRT2 were also identified in the molecular pathways of memory formation and cocaine addiction (Renthal et al., 2009; Gao J, 2010; Michan et al., 2010), suggesting their potential regulatory roles in synaptic plasticity and brain function.
To date, the molecular details underpinning the neural effects of sirtuins remain largely elusive. We therefore sought to investigate whether sirtuins are involved in the regulation of AMPAR expression and trafficking. In this study, we found that AMPARs are subject to lysine acetylation which is regulated by SIRT2. SIRT2 physically interacts with and deacetylates the GluA1 subunit of AMPARs. Inhibition of SIRT2 activity results in enhanced AMPAR acetylation and an elevated level of AMPAR protein amount via competitive suppression of receptor ubiquitination. Consistent with this, in SIRT2 knock-out mice (Sirt2−/−), acetylation and protein levels of AMPARs are both higher than the wild-type, without affecting other synaptic proteins including PSD-95 and GluN1. In addition, both Sirt2−/− mice and mice with intracranial viral infection of an acetylation mimetic GluA1 show aberrant synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD), accompanied with significant impairments in contextual and spatial memory. Together, these findings establish lysine acetylation as a novel post-translational modification (PTM) for AMPARs. SIRT2-dependent regulation of receptor acetylation plays a crucial role in AMPAR proteostasis, trafficking, synaptic plasticity and cognitive function.
RESULTS
SIRT2 interacts with AMPARs via lysine residues on the GluA1 C-terminus
To investigate the role of sirtuins in AMPAR expression, we over-expressed SIRT1, 2 or 3 with enhanced green fluorescent protein (EGFP) in DIV (day in vitro) 11 cultured hippocampal neurons. We found that expression of SIRT2, but not SIRT1 or SIRT3, significantly decreased endogenous AMPAR levels of transfected neurons (Figure 1A, B). Similarly, when GluA1-GFP was transfected in HEK cells together with SIRT1, 2 or 3, respectively, a decrease in GluA1 was detected only in SIRT2-expressing cells (Figure S1A, n = 4, p < 0.05). Conversely, inhibition of endogenous SIRT2 with a potent chemical inhibitor, B2 (20 μM, 24 hrs) (Bodner et al., 2006), led to an enhanced surface expression of AMPARs in cultured cortical neurons (Figure 1C, n = 4, p < 0.05). Additionally, the effect of SIRT2 showed a preference for AMPARs because no changes were found in NMDA receptors (NMDARs) (Figure S1B, n = 12 cells/group, P > 0.05) or AMPAR-associated protein GRIP1 (Figure S1C, n = 11 cells/group, P > 0.05). The SIRT2-induced reduction in AMPAR expression was dependent on the lysine residues on the C-terminus of GluA1 subunit. When neurons were transfected with a mutant GluA1 that has all four intracellular lysine residues replaced with arginine (GluA1-4KR), the SIRT2 effects were eliminated (Figure 1D, E, n = 3, p < 0.01).
Figure 1. SIRT2 interacts with AMPARs via the lysine residues on GluA1 C-terminus.
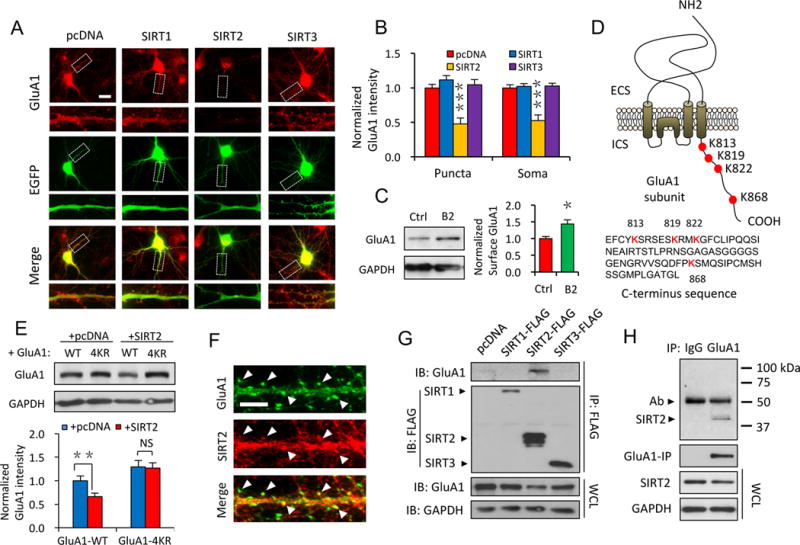
(A, B) Overexpression of SIRT2, but not SIRT1 or SIRT3, dramatically decreased the cellular GluA1 levels in cultured neurons (n ≥ 15 cells/group). Scale bar, 20 μm. (C) Inhibition of SIRT2 by B2 (20 μM, 24 hrs) led to enhanced surface expression of GluA1 in cultured neurons (n = 4). (D) A schematic illustration of the lysine residues localized on the GluA1 C-terminus (Upper panel, ECS: extracellular space; ICS: intracellular space) and the amino acids sequence of GluA1 C-terminus. (E) In HEK 293T cells, expression of SIRT2 caused a reduction in the amount of GluA1, but not GluA1-4KR (n = 4). (F) Double staining showed co-localization (arrow heads) of SIRT2 with the AMPAR GluA1 subunit in hippocampal neurons. Scale bar, 5 μm. (G, H) Co-IP assay in HEK 293T cells (G) or whole brain lysate (H) showed SIRT2, but not SIRT1 or 3, physically interacts with the GluA1 subunit of AMPARs. IP: immunoprecipitation. IB: immunoblotting. WCL: whole-cell lysates. Ab: antibody. Bar graphs represent mean ± S.E., *P < 0.05, **P < 0.01, ***P < 0.001, NS, not significant. One-way ANOVA with Tukey post-hoc test (A, B) and Student’s two-tailed t test (C, E).
To examine the subcellular distribution of SIRT2, double-staining demonstrated a high level of co-localization of SIRT2 with AMPARs (Figure 1F). Consistent with this, immunostaining and synaptosome purification revealed an enrichment of SIRT2 at synapses (Figure S2A, B). To explore the physical association between SIRT2 with AMPARs, we performed co-immunoprecipitation (co-IP) assays using transfected HEK 293T cells (Figure 1G) or rat brain lysates (Figure 1H). Among SIRT1-3, only SIRT2 showed an interaction with the GluA1 subunit of AMPARs (Figure 1G). The SIRT2-GluA1 association was further confirmed by in vitro GST pull-down assays. When incubated with rat brain lysates, purified GST-GluA1 C-terminal protein co-precipitated with endogenous SIRT2 protein (Figure S3A). More importantly, when cells were transfected with GluA1-4KR, we found that the interaction of SIRT2 with the 4KR mutant was significantly reduced (Figure S3B, C), indicating that the SIRT2-AMPAR association was dependent on the lysine residues in the GluA1 C-terminus.
AMPARs are subject to protein acetylation modifications
We wanted to know the molecular mechanisms underlying the effect of SIRT2 on AMPAR protein levels. It is known that protein stability and thus the total protein amount can be regulated by acetylation (Caron et al., 2005; Min et al., 2010). As a lysine deacetylase, SIRT2 may regulate AMPARs by direct deacetylation. However, acetylation is best known as a histone modification in the nucleus (Grunstein, 1997; Struhl, 1998), whether AMPARs are subject to this type of modification has never been investigated. To this end, we isolated the GluA1 subunit of AMPARs from rat brain lysates by immunoprecipitation and probed with antibodies specifically against acetylated lysines in western blot analysis (Inuzuka et al., 2012). Surprisingly, we observed strong acetylation signals in precipitated GluA1 from different brain regions (prefrontal cortex, hippocampus and cerebellum) (Figure 2A, B, n = 3), demonstrating for the first time that AMPARs can be modified by acetylation.
Figure 2. AMPARs are subject to protein acetylation at the intracellular lysine sites.
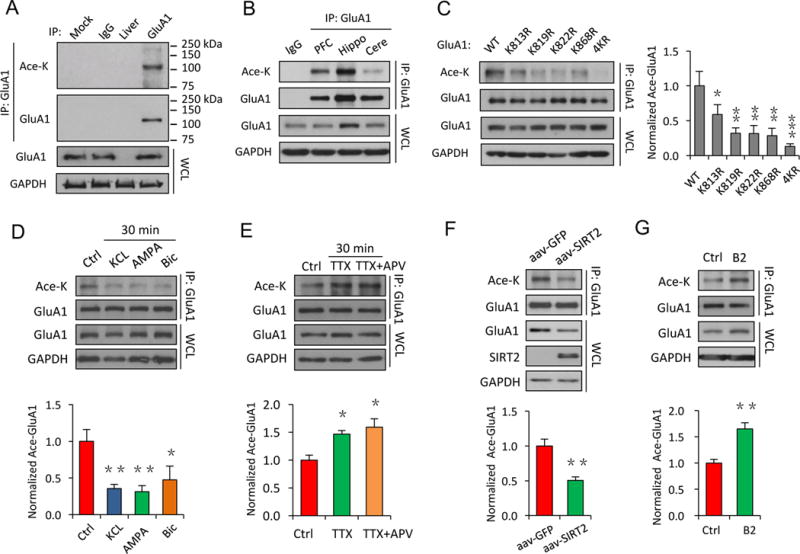
(A) In vivo acetylation assays using rat hippocampal tissue showed a strong acetylation signal on the GluA1 subunit of AMPARs. (B) AMPAR acetylation levels varied in different brain regions (PFC: pre-frontal cortex; Hippo: hippocampus; Cere: cerebellum). (C) In HEK cells, mutation of individual or all of the lysine sites to arginine on the GluA1 C-terminus (K813R, K819R, K822R, K868R, 4KR), led to reduced GluA1 acetylation (n = 3). (D, E) Neural activity-dependent regulation of AMPAR acetylation. GluA1 acetylation was reduced after increased neuronal activity with application of KCl (20 mM), AMPA (40 μM) or bicuculline (Bic, 40 μM) (n = 4) (D), while inhibition of neuronal activity for 30 min by TTX (2 μM) alone or together with APV (50 μM) significantly enhanced GluA1 acetylation (n = 3) (E). (F, G) SIRT2 is a deacetylase for AMPARs. Viral infection of SIRT2 in cultured neurons led to a reduction in GluA1 acetylation compared to a control GFP virus (n = 3) (F), while inhibition of SIRT2 activity by B2 (20 μM, 24 hrs) increased GluA1 acetylation in cultured neurons (n = 4). IP: immunoprecipitation. WCL: whole-cell lysates. Bar graphs represent mean ± S.E., *P < 0.05, **P < 0.01, ***P < 0.001. One-way ANOVA with Tukey post-hoc test (C–E) and Student’s two-tailed t test (F, G).
Acetylation commonly occurs at the lysine residues on a substrate protein. On the GluA1 subunit, there are a total of four lysine residues as potential acetylation sites in its C-terminal cytoplasmic tail (Figure 1D). To verify the requirement of the lysine residues for acetylation, we substituted each of the four lysines (K) with arginines (R) to generate mutants including GluA1-K813R, K819R, K822R, K868R and GluA1-4KR with all four lysines mutated. We found that compared to GluA1 wildtype (WT), the acetylation signals were reduced on each of the individual KR GluA1 mutants, with a complete abolishment in the 4KR mutant, indicating the involvement of all lysine residues (Figure 2C, n = 3, p < 0.05, 0.01 or 0.001). These findings strongly support the existence of AMPAR acetylation at the lysine sites.
To determine whether AMPAR acetylation, like other PTMs such as phosphorylation or ubiquitination (Blackstone et al., 1994; Lussier et al., 2011), was activity dependent, we found that up-regulating neuronal activity with potassium chloride (KCl, 20 mM), AMPA (40 μM) or the GABAA receptor inhibitor bicuculline (40 μM) resulted in a reduction in AMPAR acetylation (Figure 2D, n = 4, p < 0.01 or 0.05), whereas an opposite effect was observed when neuronal activity was suppressed by tetrodotoxin (TTX, 2 μM) or TTX (2 μM) + APV (50 μM) (Figure 2E, n = 3, p < 0.05), indicating that AMPAR acetylation was dynamically regulated.
SIRT2 regulates AMPAR acetylation in neurons as a deacetylase
To examine the role of SIRT2 in AMPAR acetylation, we generated adeno-associated viruses (AAV) of SIRT2 and GFP and applied the viruses to cultured cortical neurons. We found that compared to the GFP control, overexpression of SIRT2 significantly reduced GluA1 acetylation and receptor protein levels in neurons (Figure 2F, n = 3, p < 0.01). Conversely, when we treated cultured cortical neurons with the SIRT2 specific inhibitor B2 (20 μM, 24 hrs), immunoprecipitated GluA1 showed a significant up-regulation in AMPAR acetylation (Figure 2G, n = 4, p < 0.01). Similar changes were also detected with another SIRT2 inhibitor AGK2 (20 μM, 8 hrs) in cultured neurons (Figure S3D, n = 3, p < 0.01) (Outeiro et al., 2007), or with siRNA in HEK 293T cells (Figure S4A, n = 4, p < 0.01). Collectively, these results indicate that SIRT2 functions as AMPAR deacetylase in neurons.
Acetylation regulates AMPAR protein stability and accumulation via competition against ubiquitination
Protein acetylation and ubiquitination are competing PTMs that target on the same lysine residues of a protein. Therefore, acetylated proteins, with suppressed ubiquitination, are expected to become stabilized with a reduced turnover rate and elevated protein accumulation (Caron et al., 2005). Indeed, AMPARs are known to be subject to ubiquitination which facilitates receptor internalization and subsequent proteasomal degradation (Schwarz et al., 2010; Lin et al., 2011; Widagdo et al., 2015). We therefore examined the competition between AMPAR acetylation and ubiquitination. We found that in cultured hippocampal neurons, maturation from DIV 5 to DIV 19 was accompanied with a gradual reduction in GluA1 acetylation coupled with an up-regulation in SIRT2 expression and GluA1 ubiquitination (Figure 3A), demonstrating an inverse relationship between GluA1 acetylation and ubiquitination. To further examine these two competing modifications on AMPARs, we found that SIRT2 inhibition by B2 (20 μM, 24 hrs) or knock down by siRNA led to an increase in AMPAR acetylation accompanied with a decrease in receptor ubiquitination (Figure 3B, n = 4, p < 0.001 and Figure S4A, B n = 3, p < 0.05), while the opposite was observed in HEK cells overexpressing SIRT2 (Figure 3C, n = 4, p < 0.01). In line with these findings, we also observed that an increase in AMPAR ubiquitination by the proteasomal inhibitor MG132 was coupled with a reduction in receptor acetylation (Figure S4C, n = 4, p < 0.01 or 0.05).
Figure 3. Acetylation regulates AMPAR protein stability and accumulation via competition against ubiquitination.
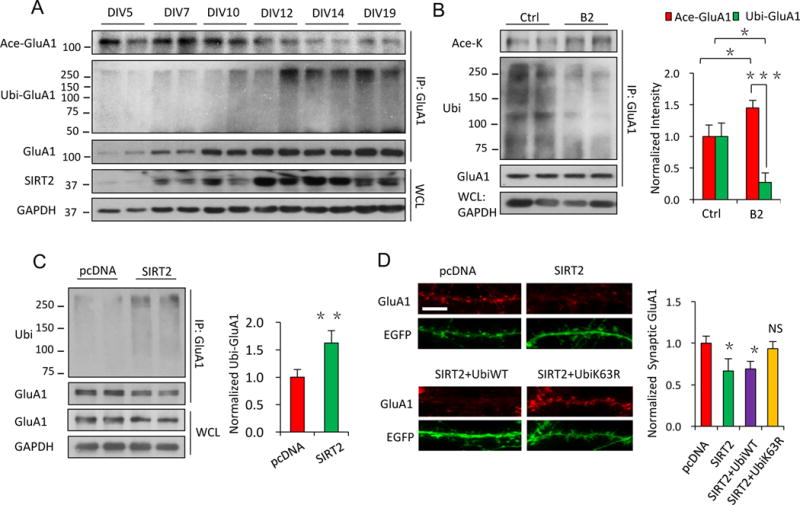
(A) Along the developmental time course of cultured neurons, an increase in SIRT2 expression was closely coupled with a decrease in GluA1 acetylation, and an increase in GluA1 ubiquitination. (B) In cultured neurons, SIRT2 inhibition by B2 resulted in an increase in acetylation but a decrease in ubiquitination of GluA1 (n = 4). (C) Overexpression of SIRT2 up-regulated GluA1 ubiquitination in HEK cells (n = 4). (D) Blockade of GluA1 ubiquitination (see Figure S4D) by mutant ubiquitin (UbiK63R) rescued the SIRT2-induced AMPAR reduction in neurons (n = 10 – 12 cells/group). Scale bar, 5 μm. IP: immunoprecipitation. WCL: whole-cell lysates. DIV: day in vitro. Bar graphs represent mean ± S.E., *P < 0.05, **P < 0.01, ***P < 0.001, NS, not significant. Student’s two-tailed t test (B, C) and one-way ANOVA with Tukey post-hoc test (D).
Because AMPAR acetylation is negatively coupled to the levels of ubiquitination, it is likely that increased receptor levels by acetylation results from a suppression in receptor ubiquitination. To confirm the role of ubiquitination in mediating the SIRT2-mediated effect on AMPAR accumulation, we utilized a mutant ubiquitin, Ubi-K63R, which can occlude GluA1 ubiquitination (Figure S4D) (Huo et al., 2015). Compared to wild-type ubiquitin (Ubi-WT), overexpression of Ubi-K63R in cultured neurons blocked the SIRT2-mediated increase in AMPAR ubiquitination (Figure S4D, n = 3, p < 0.01 or >0.05) as well as protein reduction (Figure 3D, n = 10 – 12 cells/group, p < 0.05), indicating that SIRT2 decreased AMPAR levels via competitively enhancing receptor ubiquitination.
Acetylation regulates AMPAR trafficking, turnover and synaptic accumulation
Our previous work has shown that ubiquitination regulates AMPAR dynamics by facilitating EPS15-mediated receptor internalization (Lin and Man, 2014). To test whether acetylation affects AMPAR trafficking, we performed AMPAR internalization and insertion assays in cultured hippocampal neurons. We found that SIRT2 inhibition by B2 (20 μM, 24hrs) resulted in a suppression of agonist-induced (AMPA, 100 μM) AMPAR internalization (Beattie et al., 2000) (Figure 4A, n ≥ 15 cells/group, p < 0.05) without affecting glycine (200 μM)-induced receptor insertion (Lu et al., 2001) (Figure 4B, n ≥ 15 cells/group, p > 0.05). Overnight inhibition of SIRT2 by B2 (20 μM) also resulted in an increase in cell-surface (Figure 4A, B, n ≥ 15 cells/group, p < 0.05) and synaptic AMPARs (Figure 4C, n ≥ 15 cells/group, p < 0.001). Consistent with this, patch-clamp recordings showed significantly larger mEPSC amplitudes in neurons treated with B2 (20 μM, 24 hrs) (Figure 4D, n = 8 cells/group, p < 0.05). Similar effects on AMPAR expression (Figure S3E, n = 4, p < 0.05) and synaptic strength (Figure S3F, n = 8 cells/group, p < 0.05) were also observed in neurons treated with a different SIRT2 inhibitor, AGK2 (20 μM, 8 hrs).
Figure 4. Acetylation regulates AMPAR trafficking, turnover and accumulation in neuron.
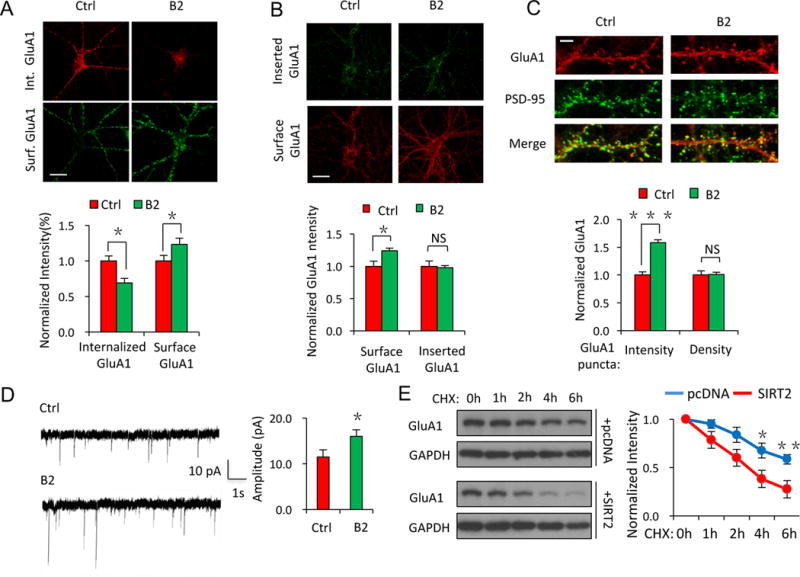
(A–D) In cultured hippocampal neurons, inhibition of SIRT2 activity by B2 (20 μM, 24 hrs) resulted in a suppression in internalization (A, n ≥ 15 cells/group), an increase in surface expression (A, B, n ≥ 15 cells/group) and an elevated cellular accumulation of AMPARs (C, n ≥ 15 cells/group), as well as an increase in AMPAR-mediated synaptic transmission (D, n = 8 cells/group), without affecting the insertion of AMPARs (B, n ≥ 15 cells/group) or the density of AMPAR puncta (C, n ≥ 15 cells/group). Scale bar, 20 μm (A, B), 5 μm (C). (E) In HEK cells expressing GluA1 with pcDNA control, or GluA1 with SIRT2, cycloheximide (CHX) tracing showed faster degradation of GluA1 in cells co-expressing SIRT2 (lower panel) (n = 3). Bar graphs represent mean ± S.E., *P < 0.05, **P < 0.01, ***P < 0.001, NS, not significant. Student’s two-tailed t test.
To directly determine the role of AMPAR acetylation in receptor protein stability, we measured the turnover rate of AMPARs in transfected HEK cells in the presence of the protein synthesis inhibitor cycloheximide (CHX, 100 μM) (Figure 4E). Indeed, in cells co-expressing GluA1 and SIRT2, GluA1 showed a more rapid decrease compared with cells transfected with GluA1 alone (Figure 4E, n = 3, p < 0.05 or 0.01), indicating an increased turnover rate for AMPARs in the presence of SIRT2.
SIRT2 knock-out mice show enhanced AMPAR acetylation and impaired synaptic plasticity
To further investigate the role of SIRT2 in AMPAR acetylation and related neural function in vivo, we employed SIRT2 knock-out mice (Sirt2−/−). The transgenic mice showed normal body weight and gross brain anatomy (Figure S5A, B), with no measurable differences in hippocampal neuron spine density (Figure 5A, n = 8 cells/group, p > 0.05) or gross hippocampal structure (Figure 5B). Importantly, in line with our findings in cultured neurons, Sirt2−/− mice showed a marked increase in AMPAR acetylation and receptor protein amounts (Figure 5C, n = 8 mice/group, p < 0.001). In comparison, no significant changes were detected in other synaptic proteins including GluN1 or PSD-95 in the Sirt2−/− mice (Figure 5C, n = 8 mice/group, p > 0.05).
Figure 5. SIRT2 knock-out mice show enhanced AMPAR acetylation and impaired synaptic plasticity.
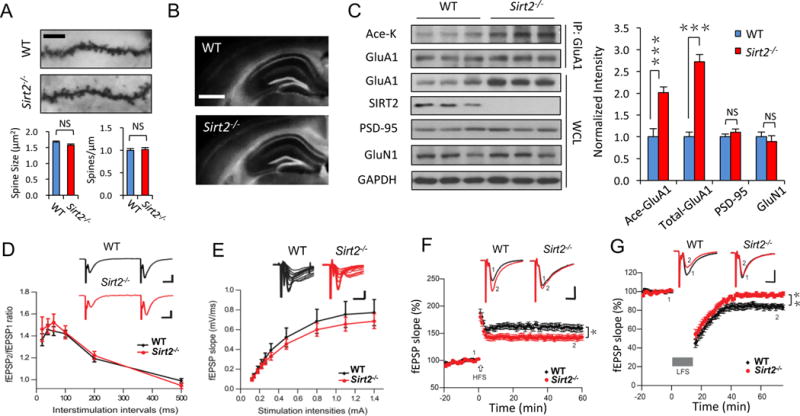
(A, B) Golgi staining showed no change in spine morphology and density in hippocampal neurons (n = 8 cells/group, scale bar, 5 μm) (A), and Hoechst staining showed normal gross morphology of the hippocampus (B) in Sirt2−/− mice, scale bar, 500 μm. (C) GluA1 acetylation and protein expression were significantly elevated in SIRT2 knock-out mice (Sirt2−/−), compared to C57BL/6 wild-type (WT) control mice (n = 8 mice/group). No significant differences were observed in other synaptic proteins like PSD-95 or GluN1 (n = 8 mice/group). (D, E) There is no significant difference between WT and Sirt2−/− mice in synaptic paired pulse facilitation (D) and input/output curve (E) (n = 12 slice/group). (F, G) Recordings of field potentials in hippocampal CA1 regions of acute brain slices showed impairments in LTP (F) and LTD (G) in Sirt2−/− mice (n = 12 slice/group). Scale bars: 10 ms, 0.5 mV. Bar graphs represent mean ± S.E., *P < 0.05, **P < 0.01, ***P < 0.001, NS, not significant. Student’s two-tailed t test (A–C) and one-way ANOVA (D–G).
Because AMPAR expression and trafficking are crucial for synaptic plasticity, we next examined LTP and LTD in hippocampal slices from the Sirt2−/− mice. Paired pulse facilitation and the input/output relationship indicated that loss of SIRT2 did not cause changes in basal synaptic properties compared to WT control mice (Figure 5D, E, n = 12 slices/group, p > 0.05). Consistent with a specific effect on AMPARs (Figure 5C), Sirt2−/− hippocampal CA1 neurons showed an elevated amplitude of AMPAR-, but not NMDAR-mediated evoked currents, and an increased ratio of AMPAR/NMDAR currents (Figure S5C, n = 9 cells/group, p < 0.001). Importantly, compared to the WT control, Sirt2−/− mice showed a reduction in LTP and almost abolished LTD in hippocampal slices (Figure 5F, G, n = 12 slices/group, p < 0.05 or 0.01), demonstrating impaired synaptic plasticity in the absence of SIRT2.
Sirt2−/− mice show impairments in learning and memory
Given the crucial role of hippocampal synaptic plasticity in learning and memory (Shepherd and Huganir, 2007; Collingridge et al., 2010), we next performed different tests on memory behavior in Sirt2−/− mice (Figure 6). In fear conditioning tests (Figure S6A), the Sirt2−/− mice showed significantly impaired contextual fear memory (Figure 6A, B, n = 15 mice/group, p < 0.05 or 0.01), a function mainly dependent on the hippocampus (McEchron et al., 1998). However, the tone-associated fear memory, which is amygdala-dependent, remained intact (Figure S6B, n = 15 mice/group, p > 0.05), suggesting a preferential impact on hippocampal learning. Between Sirt2−/− and wild-type mice, no significant differences in basal locomotive behavior and sensitivity to electrical shocks (ES) were observed (Figure 6C, n = 15 mice/group, p > 0.05). In novel object discrimination tests, we found that the Sirt2−/− mice showed significantly slower learning during the training sessions compared to their respective WT controls (Figure 6D, n = 15 mice/group, p < 0.01 or 0.001) whereas their basal exploration of the objects showed no difference (Figure S6C, n = 15 mice/group, p > 0.05). In probe sessions, Sirt2−/− mice showed memory impairments to the old objects (Figure 6E, n = 15 mice/group, p < 0.001). Finally, we employed the Barnes maze test to assess spatial memory. Sirt2−/− mice seemed to be normal in locating the target escape hole across training sessions when compared to wild-type animals (Figure S6E, F, n = 15 mice/group, p > 0.05). However, in Sirt2−/− mice, we observed significantly reduced accuracy and a prolonged latency in finding the target hole (Figure 6F, G, n = 15 mice/group, p < 0.01 or 0.05), as well as reduced exploration time in the target quadrant (Figure 6H, I, n = 15 mice/group, p < 0.01). These findings strongly indicate impairments in long-term memory in Sirt2−/− mice.
Figure 6. Sirt2−/− mice show impairments in learning and memory.
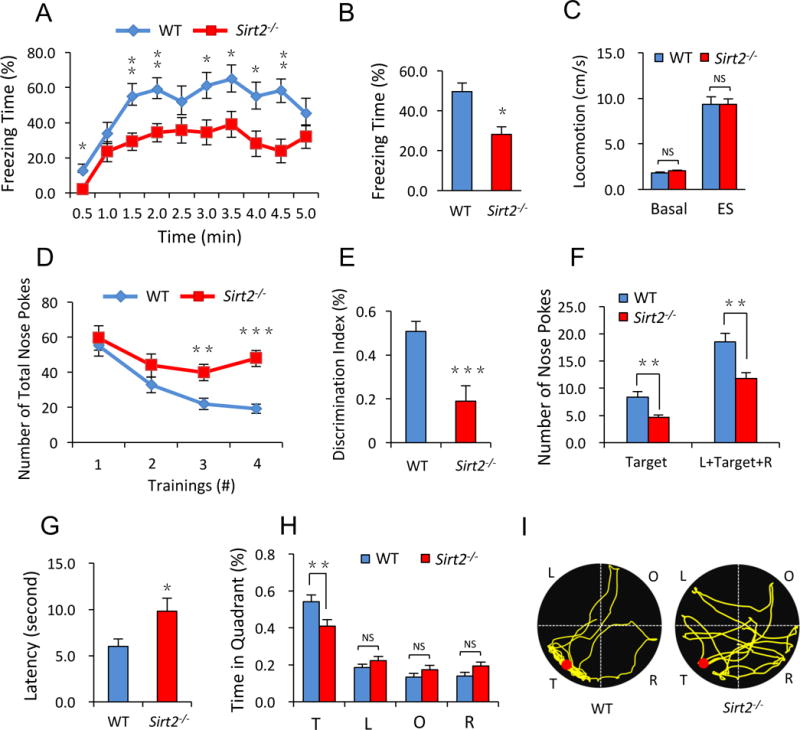
(A–C) Sirt2−/− mice showed significant defects in hippocampus-dependent contextual fear conditioning memory (A, B), but not in basal locomotive activity and responses to electrical shock (ES) (C) (n = 15 mice/group). (D, E) Novel object discrimination assays showed impaired memory in Sirt2−/− mice. Sirt2−/− mice were significantly slower than WT mice in recognizing the old objects during the training session (D) and showed a reduced preference toward the novel object (E) (n = 15 mice/group). (F–I) The Barnes maze test revealed a spatial memory impairment in Sirt2−/− mice. The numbers of nose pokes to the target hole, or the adjacent holes (L+Target+R) were reduced significantly in Sirt2−/− mice (F). Sirt2−/− mice took longer time to reach the target hole (G), and spent less time in the target quandrant (T) (H), as was shown in the representative path tracing (I) (n = 15 mice/group). T, target; L, left; O, opposite; R, right. Bar graphs represent mean ± S.E., *P < 0.05, **P < 0.01, ***P < 0.001, NS, not significant. Student’s two-tailed t test.
Mice with overexpression of acetylation mimetic AMPARs show defects in synaptic plasticity and memory similar to Sirt2−/− mice
In order to verify a direct involvement of AMPAR acetylation in synaptic plasticity, we generated an acetylation mimetic of GluA1 by replacing the four lysines (K) to glutamines (Q) (GluA1-4KQ) (Kemper et al., 2009; Inuzuka et al., 2012) (Figure S7A). Immunostainings showed that both GluA1-WT and 4KQ accumulated at the synapse and co-localized with the postsynaptic scaffolding protein PSD-95 (Figure S7B). Compared to GluA1-WT, GluA1-4KQ showed a slower turnover rate (Figure 7A, n = 3, p < 0.05 or 0.01), consistent with the expected biochemical property for an acetylated protein. We injected GluA1-WT and GluA1-4KQ viruses into the lateral ventricles of C57BL/6 wild-type mouse pups on postnatal day 2 (P2). At P60, biochemical analysis showed that both GluA1-WT and GluA1-4KQ were well expressed in extended brain regions including the hippocampus and the prefrontal cortex (Figure 7B). Strikingly, the GluA1-4KQ infected mice showed a reduction in LTP and an abolishment of LTD (Figure 7C, D, n = 8 slices/group, p < 0.05), similar to changes observed in the Sirt2−/− mice (Figure 5F, G). Furthermore, behavioral tests showed that the GluA1-4KQ viral infected mice had impairments in learning and memory similar to the defects observed in the Sirt2−/− mice (Figure 7E–G). In fear conditioning tests, GluA1-4KQ viral infected mice showed dramatically impaired contextual memory (Figure 7E, n = 12 WT or 15 4KQ, p < 0.05). Compared to the GluA1-WT viral infected control mice, the 4KQ mice also showed slower learning curve to old objects (Figure 7F, n = 12 WT or 15 4KQ, p < 0.05) and a drastically impaired ability in novel object recognition (Figure 7G, n = 12 WT or 15 4KQ, p < 0.001). These findings strongly indicate that the impairments in synaptic plasticity and memory in Sirt2−/− mice more likely result from enhanced acetylation of AMPARs in the brain.
Figure 7. Mice with overexpression of acetylation mimetic AMPARs show defects in synaptic plasticity and memory.
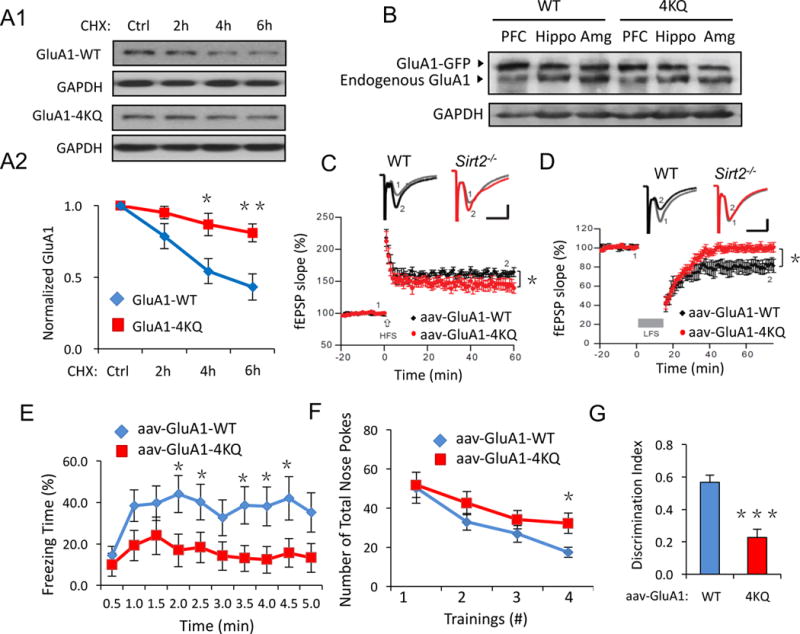
(A) GluA1-WT, 4KQ degradation assays in transfected HEK cells. Compared to GluA1-WT, the GluA1-4KQ showed a reduced degradation rate (n = 3). (B) Adenovirus of GFP-tagged GluA1-WT or GluA1-4KQ was injected into the lateral ventricles of wild-type mice at P2 and brains were collected at P60. Western blot revealed high expression levels of viral GFP-GluA1 in brain regions including prefrontal cortex (PFC), hippocampus (Hippo) and amygdala (Amg). (C, D) Hippocampal brain slice recordings of LTP and LTD. GluA1-4KQ viral infected mice showed impaired LTP and LTD compared to the GluA1-WT control (n = 12 WT or 15 4KQ, p < 0.05). (E–G) Similar to the Sirt2−/− mice, GluA1-4KQ viral infected mice showed impaired contextual fear memory (E) and learning memory (F, G). AAV, adeno-associated virus. Bar graphs represent mean ± S.E., *P < 0.05, **P < 0.01, ***P < 0.001. Student’s two-tailed t test (A, E–G) and one-way ANOVA (C, D).
DISCUSSION
This study demonstrates that AMPARs are subject to lysine acetylation as a new form of PTM. Acetylation has been considered a modification mainly for nuclear histone proteins, although recently a few other cytosolic proteins including tubulin, AMP-activated protein kinase (AMPK) and tau have also been shown to be substrates (North et al., 2003; Min et al., 2010; Lu et al., 2011). Here we find that AMPARs have high levels of acetylation during basal conditions, which can be efficiently regulated by neuronal activity. We also identify SIRT2 as the deacetylase down-regulating AMPAR acetylation. Through competition for the lysine sites at the GluA1 C-terminus, acetylation and ubiquitination of AMPARs reach a balance in order to maintain receptor proteostasis (Caron et al., 2005). Deacetylation of AMPARs by the brain-enriched SIRT2 facilitates receptor ubiquitination, leading to receptor internalization and proteasomal degradation. Surprisingly, stabilization of AMPARs via elevated acetylation levels in Sirt2−/− mice or with the expression of a GluA1 acetylation mimetic, results in aberrant synaptic plasticity and impaired memory. Therefore, regulation of AMPAR acetylation by related enzymes such as SIRT2 may be of crucial importance for AMPAR turnover, synaptic plasticity and cognitive brain function.
Acetylation is a novel PTM for AMPARs
AMPARs are known to be subject to multiple forms of protein modification. Among the three currently described major PTMs for AMPARs, phosphorylation and palmitoylation mainly influence the receptor channel conductance and intracellular trafficking, while ubiquitination serves as the signal for receptor internalization and proteasomal degradation (Lu and Roche, 2012). In this study, we establish that lysine acetylation is a novel PTM for AMPARs which represents the only modification known to prolong AMPAR half-life and to stabilize receptors on the cell membrane. Considering the crucial role of surface AMPAR stability in synaptic transmission and plasticity, AMPAR acetylation is expected to have major functional importance in brain function.
The mechanism for acetylation-dependent AMPAR stabilization seems to directly compete with ubiquitination. During ubiquitination, individual ubiquitin units or poly-ubiquitin chains are conjugated to the lysine residues of a protein, which serve as a signal for proteasome recognition and degradation. Therefore, when a lysine site is occupied by an acetyl group during acetylation, AMPAR subunits are protected from being ubiquitinated by an E3 ligase such as Need4 (Lin et al., 2011). We observed a clear competition between AMPAR acetylation and ubiquitination in cultured neurons (Figure 3). When AMPAR acetylation or ubiquitination is enhanced, the counterpart modification is down-regulated, and vice versa. However, these opposing modifications on AMPARs are not necessarily mutually exclusive. There are four lysine sites at the intracellular C-terminal of GluA1 subunits. Although all of the sites seem to be targeted for ubiquitination (Widagdo et al., 2015), and acetylation as shown in this study (Figure 2C), a specific site may have preference for one type of modification. For instance, the last lysine (K868) is a dominant site for ubiquitination compared to other sites (Lin et al., 2011). It is possible that another lysine site(s) may be preferentially targeted for acetylation. Therefore, both acetylation and ubiquitination can co-exist on AMPARs, and the functional consequence will depend on the relative weight of a modification not only between acetylation and ubiquitination, but also other forms of PTM including receptor phosphorylation.
Developmental down regulation of AMPAR acetylation
During synapse formation and maturation, AMPARs begin to be synthesized and increasingly accumulated in the cell and at the synaptic sites. We find that AMPAR acetylation is down-regulated, accompanied with increasing levels in ubiquitination, over the time course of neuronal maturation (Figure 3A). Our data reveals that receptor acetylation leads to reduced endocytosis and increased stability of surface AMPARs. Whereas the physiological function of this developmental regulation remains to be further investigated, down regulation of acetylation, and therefore higher dynamics in mature neurons may be critical for synaptic plasticity and brain function (Malinow and Malenka, 2002; Shepherd and Huganir, 2007). Thus, in the brain, neurons may need to maintain a suppression on AMPAR acetylation while maintaining active receptor ubiquitination so as to ensure an appropriate level of AMPAR dynamics.
Over-acetylation of AMPARs leads to impaired synaptic plasticity and memory
In line with the physiological down regulation of AMPAR acetylation during neuron development, our findings indicate detrimental effects with abnormal increases in AMPAR acetylation. By pharmacological manipulation or knock out of the deacetylase SIRT2, increased receptor acetylation results in an increase in AMPAR protein levels as expected. While elevated AMPAR acetylation indeed enhanced basal synaptic transmission strength in vitro and in vivo, it surprisingly causes significant defects in synaptic plasticity including hippocampal LTP and LTD. Consistent with this, SIRT2 knock-out mice or mice overexpressing an acetylation mimetic GluA1 (GluA1-4KQ) demonstrate marked impairments in learning and memory. These findings strongly indicate that cognitive functions are not likely improved simply by increasing synaptic AMPAR accumulation; rather, the newly added receptors must possess normal properties in dynamics. Therefore, aiming at optimization on AMPAR trafficking and dynamics, rather than addition to augmentation of total receptor amount, should be the ultimate goal in the pursuit of improving higher brain functions.
EXPERIMENTAL PROCEDURES
Primary Neuron Culture
Primary rat (Sprague Dawley) cortical and hippocampal neuron cultures were prepared as described previously (Wang et al., 2015). Briefly, cortices and hippocampi were dissected out from embryonic day 18 (E18) rats of either sex, then digested with papain (0.5 mg/ml in EBSS) and plated on poly-L-lysine coated coverslips or Petri dishes. Neurons were plated in plating medium [MEM containing 10% fetal bovine serum (FBS), 5% horse serum (HS), 31 mg L-cysteine, and 1% penicillin/streptomycin and L-glutamine mixture (1% P/S/G); Invitrogen] which was then replaced 24 hr later by feeding medium (Neurobasal medium supplemented with 1% HS, 2% B-27 and 1% P/S/G). Thereafter, neurons were maintained in feeding medium and fed twice a week until use around 2–3 weeks later.
Immunoprecipitation (IP)
Brain tissues or cultured neurons were lysed in 1X radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4; 50 mM NaCl, 1% NP-40, 1% SDoC, 0.1-1% SDS) supplemented with protease inhibitor cocktail tablets (11697498001, Roche) to minimize protein degradation. Stringent RIPA buffer (1% SDS) was used in IP experiments to ensure clean protein immunoprecipitation while mild RIPA buffer (0.1% SDS) was used in Co-IP experiments for protein-protein interaction assays. To break down the tissues and cells, 10 bursts of sonication were applied to the samples before they were head-to-toe rotated for 30 min and centrifuged for another 10 min at the speed of 13,000 rpm under 4°C. The clear supernatant part was then subjected to BCA assay to balance the protein concentration before IP while the pellets were discarded. Cell lysates were then incubated with specific antibodies for 1 hour then another 4 hours with protein A-agarose beads (sc-2001, Santa Cruz Biotechnology). Agarose beads were then rinsed in NP-40 buffer with vortexing (25 mM Tris-HCl, pH 7.4; 150 mM NaCl, 1 mM EDTA, 1% NP-40) for at least three times to ensure a clean immunoprecipitation. Thereafter, the agarose beads were boiled with Laemmli 2X sample buffer (4% SDS, 10% 8 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M Tris HCl) for 10 minutes at 95°C before being subjected to immunoblotting analysis.
In vivo/in vitro acetylation assay
To detect the acetylation signal of endogenous AMPARs in brain (in vivo) or recombinant receptor protein overexpressed in HEK 293T cells (in vitro), the GluA1 subunit was first immunoprecipitated following the IP procedure described previously, with a specific antibody by Protein A-agarose beads from the whole brain extracts or homogenized 293T cells lysates. Lysis buffer (RIPA buffer with 1% SDS) used to homogenize the brain tissue was supplemented with deacetylase inhibitors cocktail (100 μM trichostatin A, 50 mM sodium butyrate and 50 mM nicotinamide) to sufficiently block deacetylase activity. The acetylation signal was then assessed by immunoblotting with an antibody that detects general acetylated lysine residues (ab80178, Abcam).
Immunoblotting (IB)
Samples of whole tissue/cell lysis, IP or Co-IP, were prepared for immunoblotting by boiling in 2X Laemmli sample buffer (4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M Tris HCl) for 10 min at 95°C to denature all proteins then stored at −20°C. After resolving by SDS-page electrophoresis, proteins were transferred to a PVDF membrane and probed with specific primary antibodies. 8 – 15% SDS-page gels were used in electrophoresis depending on the molecular weights of the target proteins. Experiments involving immunoblotting were all repeated at least 3 times or more (n ≥ 3) to ensure the reproducibility and consistency of the results.
Immunocytochemistry
Hippocampal neurons on coverslips were washed once in 1X artificial cerebrospinal fluid (ACSF) then fixed in a 4% paraformaldehyde/4% sucrose solution for 10 minutes. To immunostain the total protein, neurons were permeabilized with 0.3% Triton-X-100 (FisherBiotech) in phosphate buffered saline (PBS) for 8 minutes, followed with three rinses with 1X PBS. Neurons were then subjected to a one-hour blocking procedure in 10% goat-serum/PBS followed with another one-hour incubation with primary antibody at room temperature. After thoroughly rinsing to remove excess primary antibody, neurons were incubated with Alexa Fluor®-conjugated fluorescent secondary antibodies (1:700, Life Technologies) for another hour. Neurons were then mounted to microscopy glass slides with ProlongGold anti-fade mounting reagent (Life Technologies) and stored at 4°C until subsequent visualization.
AMPA receptor internalization/insertion assay
After drug treatment, neurons were incubated with GluA1Nt antibody (Millipore, 1:100) at 37°C for 10 min then washed three times with feeding medium to get rid of extra antibody. Neurons were then treated with AMPA (100 μM, 1 min) to induce drastic AMPAR internalization. After rinsing off extra AMPA from the medium, neurons were further incubated with a high concentration of secondary antibody (Alexa Fluor® dye, 1:80, 10 min) to bind all the AMPARs remaining on the cell surface. Thereafter, neurons were rinsed in 1X ACSF then fixed in 4% paraformaldehyde/4% sucrose solution for 10 minutes. Neurons were permeabilized with 0.3% Triton-X-100/PBS and incubated with a lower concentration of secondary antibody with a different color (1:700, 45 min) to specifically label the internalized AMPARs. AMPAR insertion assays were performed as previously described (Lu et al., 2001). Surface AMPARs of 2 week-old hippocampal neurons were live labeled with anti-GluA1Nt antibodies at a saturating concentration (1:80) and fluorescent secondary antibodies (red). Cells were then incubated with glycine plus a receptor antagonist cocktail in a CO2 cell culture incubator for 3 min and, following replacing the treatment to normal culture medium, cells were placed in the incubator for 20 min (Lu et al., 2001). Neurons were then fixed and the newly inserted AMPARs were labeled with the same anti-GluA1Nt and a fluorescent secondary antibody (green). Quantitation of the GluA1 puncta was performed in ImageJ. A threshold was set to all the cell pictures so that all the GluA1 puncta were marked. Only the puncta near, but not in, the dendritic shaft with sizes ranging from 20 pixels (0.45 μm2) to 50 pixels (1.13 μm2) were included in the analysis.
Immunofluorescence microscopy
Mounted coverslips were stored in the dark at 4°C for no longer than a week before fluorescence microscopy. An inverted Carl Zeiss fluorescent microscope was used to collect all the images. A 63× oil-immersion objective (numerical aperture, 1.4) along with the AxioVision software (release 4.5) were used to take pictures of the neurons. The exposure duration of the fluorescence signal was established manually to ensure that it was within the full dynamic range by using a glow scale look-up table. For each experiment, images of at least 15 neurons per group (n ≥ 15/group) were randomly taken and used for quantification. NIH Image J software was used for the image quantification. When quantifying the GluA1 puncta, we included only the puncta near, but not in, the dendritic shaft with sizes ranging from 20 pixels (0.45 μm2) to 50 pixels (1.13 μm2).
Electrophysiology
Whole cell patch-clamp recording of excitatory postsynaptic potentials (EPSP) or recording of field excitatory postsynaptic potentials (fEPSP) was performed on acute hippocampal slices from C57BL/6J wild-type and SIRT2 knock-out (Sirt2−/−) mice (8–12 weeks old) of either sex. Whole cell patch-clamp recording of miniature excitatory postsynaptic potentials (mEPSP) was performed on 2–3 weeks old cultured rat hippocampal neurons. The postsynaptic potentials were amplified using an Axon Instruments 200B amplifer and digitized with a Digidata 1322A digitizer. Traces were obtained by pClamp 9.2 and analyzed using Clampfit 9.2. More technical details about electrophysiological recording could be found in the Supplemental Experimental Procedure.
Adeno-associated virus construction and purification
The adeno-associated virus vector (AAV2) used for sub-cloning was from Addgene (#50954). The open-reading frame (ORF) for ReaChR-citrine was removed by restriction enzymes and replaced by the ORFs of the target proteins (GFP, SIRT2, GluA1-WT and GluA1-4KQ). This vector contains a human synapsin 1 promoter that ensures expression of the target proteins in neurons. The expression efficiency of the constructs was tested and verified by transfection and immunostaining in cultured neurons.
To package the adenoviruses, the target viral plasmids were co-transfected with the helper XR2 and XX6.80 plasmids (obtained from National Gene Vector Biorepository) in HEK 293T cells with polyethylenimine (PEI). The adenoviruses were purified using the PEG-it™ virus purification solution (System Bioscience) according to the manufacturer’s instructions. 72 hrs after transfection, the HEK cells were scrapped off the culture dishes with sterile 1X PBS and went through four freeze-thaw cycles to fully lyse the cells and release the viral particles. After centrifugation to remove the debris, the supernatant with viral particles was mixed with the PEG-it™ virus purification solution (1:5 dilution) and refrigerated overnight at 4°C. The precipitated viral particles were spun down thereafter and re-suspended in 500 μL sterile 1X PBS. Re-suspended adenoviruses were stored at −80°C for experiments.
Behavioral analysis of memory
Fear conditioning test, novel object recognition test and Barnes maze test have been utilized to analyze the fear memory, learning memory and spatial memory, respectively, of Sirt2−/− mice or AAV infected mice (8–15 weeks old of either sex). More details of the behavior test procedures could be found in the Supplemental Experimental Procedure.
Animal use
All the procedures involving animal use were in compliance with the policies of the Institutional Animal Care and Use Committee (IACUC) at Boston University (protocol #: 14-033). C57BL/6J wild-type (#000664) and SIRT2 knock-out mice (B6.129-Sirt2tm1.1Fwa/J, #012772) were obtained from Jackson Laboratory (Bar Harbor, ME). Mouse colonies were maintained in the Laboratory Animal Care Facility (LACF) at Boston University, Charles River Campus. The mice aged 8–15 weeks of either sex were randomly selected from different litters. Only mice with normal body size, morphology and no observable disabilities were used for the experiments.
Data Analysis and Statistics
All values are presented as mean ± S.E. Statistical analysis was performed using the student’s two-tailed t test to compare two groups, or analysis of variance (ANOVA) with Tukey post-hoc test for multiple groups. The highest and lowest values of each experimental group were excluded from the statistical analysis. P < 0.05 is considered as statistically significant. P values are presented as P > 0.05 (NS, not significant), *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Material
HIGHLIGHTS.
AMPA receptors are subject to lysine acetylation within their C-terminal domains.
Acetylation increases AMPA receptor stability and reduces receptor trafficking.
SIRT2 causes deacetylation of AMPA receptors and regulates receptor protein stability.
Elevated acetylation of AMPA receptors leads to impaired synaptic plasticity and memory.
Acknowledgments
This work was supported by NIH grant MH079407 (H.Y.M.), Alzheimer’s Association grant NIRG-12-242825 (S.L.), Pew Foundation (X.H.) and International Fulbright Sci&Tech Outstanding Student Award (G.W.). We thank all of the Man Lab members for providing insightful advice and technical assistance in this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of financial interests.
AUTHOR CONTRIBUTIONS
H.Y.M. designed, directed and coordinated the project. G.W. designed and performed biochemistry, molecular biology, behavior experiments and did data analysis. H.J.G. helped G.W. design and coordinate the behavior test experiments. S.L. and Z.W. did the hippocampus slice electrophysiology recordings and data analysis. Z.L. helped S.L. with the data analysis. J.G. did electrophysiology recordings in cultured neurons and data analysis. D.J.S. and X.H. helped design and coordinate the electrophysiology (DJS) and behavior test experiments (XH). G.W. and H.Y.M. wrote, and J. G., S.L., H.J.G. edited the manuscript. All the authors read the manuscript.
References
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Blackstone C, Murphy TH, Moss SJ, Baraban JM, Huganir RL. Cyclic AMP and synaptic activity-dependent phosphorylation of AMPA-preferring glutamate receptors. J Neurosci. 1994;14:7585–7593. doi: 10.1523/JNEUROSCI.14-12-07585.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, McLean PJ, Young AB, Housman DE, Kazantsev AG. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. Proc Natl Acad Sci U S A. 2006;103:4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays. 2005;27:408–415. doi: 10.1002/bies.20210. [DOI] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, W W, Mao YW, Gräff J, Guan JS, Pan L, Mak G, Kim D, Su SC, Tsai LH. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–1109. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harting K, Knoll B. SIRT2-mediated protein deacetylation: An emerging key regulator in brain physiology and pathology. Eur J Cell Biol. 2010;89:262–269. doi: 10.1016/j.ejcb.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Khatri N, Hou Q, Gilbert J, Wang G, Man HY. The deubiquitinating enzyme USP46 regulates AMPA receptor ubiquitination and trafficking. J Neurochem. 2015;134:1067–1080. doi: 10.1111/jnc.13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H, Gao D, Finley LW, Yang W, Wan L, Fukushima H, Chin YR, Zhai B, Shaik S, Lau AW, et al. Acetylation-dependent regulation of Skp2 function. Cell. 2012;150:179–193. doi: 10.1016/j.cell.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D, Tsang S, Wu SY, Chiang CM, Veenstra TD. FXR Acetylation Is Normally Dynamically Regulated by p300 and SIRT1 but Constitutively Elevated in Metabolic Disease States. Cell Metab. 2009;10:392–404. doi: 10.1016/j.cmet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Hou Q, Jarzylo L, Amato S, Gilbert J, Shang F, Man HY. Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J Neurochem. 2011;119:27–39. doi: 10.1111/j.1471-4159.2011.07221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Man HY. Endocytic adaptor epidermal growth factor receptor substrate 15 (Eps15) is involved in the trafficking of ubiquitinated alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. J Biol Chem. 2014;289:24652–24664. doi: 10.1074/jbc.M114.582114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Lu JY, Lin YY, Sheu JC, Wu JT, Lee FJ, Chen Y, Lin MI, Chiang FT, Tai TY, Berger SL, et al. Acetylation of yeast AMPK controls intrinsic aging independently of caloric restriction. Cell. 2011;146:969–979. doi: 10.1016/j.cell.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Lu W, Roche KW. Posttranslational regulation of AMPA receptor trafficking and function. Curr Opin Neurobiol. 2012;22:470–479. doi: 10.1016/j.conb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier MP, Nasu-Nishimura Y, Roche KW. Activity-dependent ubiquitination of the AMPA receptor subunit GluA2. J Neurosci. 2011;31:3077–3081. doi: 10.1523/JNEUROSCI.5944-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- McEchron MD, Bouwmeester H, Tseng W, Weiss C, Disterhoft JF. Hippocampectomy disrupts auditory trace fear conditioning and contextual fear conditioning in the rat. Hippocampus. 1998;8:638–646. doi: 10.1002/(SICI)1098-1063(1998)8:6<638::AID-HIPO6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Michan S, Li Y, Chou MMH, Parrella E, Ge HY, Long JM, Allard JS, Lewis K, Miller M, Xu W, et al. SIRT1 Is Essential for Normal Cognitive Function and Synaptic Plasticity. J Neurosci. 2010;30:9695–9707. doi: 10.1523/JNEUROSCI.0027-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD(+)-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- Palmer CL, Cotton L, Henley JM. The molecular pharmacology and cell biology of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol Rev. 2005;57:253–277. doi: 10.1124/pr.57.2.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, et al. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron. 2009;62:335–348. doi: 10.1016/j.neuron.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Hall BJ, Patrick GN. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci. 2010;30:16718–16729. doi: 10.1523/JNEUROSCI.3686-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hyoung Lee S. AMPA receptor trafficking and synaptic plasticity: major unanswered questions. Neurosci Res. 2003;46:127–134. doi: 10.1016/s0168-0102(03)00040-3. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- Wang G, Amato S, Gilbert J, Man HY. Resveratrol up-regulates AMPA receptor expression via AMP-activated protein kinase-mediated protein translation. Neuropharmacology. 2015;95:144–153. doi: 10.1016/j.neuropharm.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, Anggono V. Activity-Dependent Ubiquitination of GluA1 and GluA2 Regulates AMPA Receptor Intracellular Sorting and Degradation. Cell Rep. 2015;10:783–795. doi: 10.1016/j.celrep.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.