

Abstract
The etiology underlying chronic widespread pain (CWP) remains largely unknown. An integrative biopsychosocial model seems to yield the most promising explanations for the pathogenesis of the condition, with genetic factors also contributing to disease development and maintenance. Here, we conducted a search of studies investigating the genetic and epigenetic epidemiology of CWP through electronic databases including Web of Science, Medline, PubMed, EMBASE, and Google Scholar. Combinations of keywords including CWP, chronic pain, musculoskeletal pain, genetics, epigenetics, gene, twins, single-nucleotide polymorphism, genotype, and alleles were used. In the end, a total of 15 publications were considered relevant to be included in this review: eight were twin studies on CWP, six were molecular genetic studies on CWP, and one was an epigenetic study on CWP. The findings suggest genetic and unique environmental factors to contribute to CWP. Various candidates such as serotonin-related pathway genes were found to be associated with CWP and somatoform symptoms. However, studies show some limitations and need replication. The presented results for CWP could serve as a template for genetic studies on other chronic pain conditions. Ultimately, a more in-depth understanding of disease mechanisms will help with the development of more effective treatment, inform nosology, and reduce the stigma still lingering on this diagnosis.
Keywords: chronic widespread pain, CWP, epigenetics, genetics, twin studies, environment, aetiology
Introduction
Chronic widespread pain (CWP) is defined as “pain on the left side of the body, pain on the right side of the body, pain above the waist, pain below the waist and axial skeletal pain (cervical spine or anterior chest or thoracic spine or low back) for at least 3 months.”1 Alternatively to being regarded as a distinct clinical entity, CWP is often also considered the core feature of fibromyalgia (FM) or even as being part of a spectrum, with FM representing the most extreme manifestation of CWP,2 because, in addition to widespread pain, FM is often characterized by extreme fatigue, disordered sleep, and other symptoms such as cold intolerance, multiple sensitivities, and dizziness.2 CWP is a complex and controversial disorder, with reported prevalence rates of up to 15% in the global general population.3 Because of the high population prevalence and the consequently high socioeconomic costs, as well as the high levels of distress and individual suffering that the condition causes, CWP is of considerable research interest having generated hundreds of publications related to the disease. However, only very little attention has been paid to the genetic base of the condition, even though the epidemiologic studies conducted have repeatedly shown how CWP aggregates in families, strongly indicating the influence of genetic factors.4–7 Besides a genetic predisposition to CWP, physical and psychological stress,8 affective disorders,8,9 and alterations in the central nervous system8 have shown to be potential risk factors for the condition. CWP is almost always associated with a set of psychosocial correlates10 and shows strong comorbidity with depressive and anxiety disorders.11,12
For the prevention and effective treatment of CWP and related conditions such as FM, it is crucial to understand how pain develops and how it is maintained. Based on evidence from previous studies suggesting a genetic contribution, this literature review addresses the current state of genetic and epigenetic research on CWP. Genes shape our neural structures,13 immunological14,15 and endocrinological16 processes, and subsequently our behavior and experiences. However, this is not just a one-way effect; the environment can also have an impact on our genes and alter gene expression (ie, epigenetics).17,18 There is increasing evidence that social experiences and contextual factors (eg, stress and abuse) can have long-lasting effects on an organism by impacting on gene expression and consequently manifesting themselves in behavioral changes.19 These insights further support the inseparability of biological, psychological, and social factors. Overall, knowledge about specific genes and how genes and the environment interact can thus help us to further shed light on the etiological mechanisms underlying CWP.
The main objective of this review was to determine the state of genetic and epigenetic research on CWP. This should further help answering the questions: 1) whether heritability varies from one population to another population; 2) whether there are molecular markers for the condition; 3) whether insights provided by genetic research on CWP can be applied to other chronic pain conditions; and 4) whether and how results of genetic and epigenetic studies can be translated into clinical practice.
Methods
Data sources and searches
A systematic search of studies was conducted through a number of well-known online databases such as Web of Science (all Databases; http://apps.webofknowledge.com),20 Medline (http://www.nlm.nih.gov),21 PubMed (http://www.ncbi.nlm.nih.gov/pubmed),22 EMBASE (https://www.embase.com),23 and Google Scholar (https://scholar.google.com)24 using a combination of terms related to CWP (eg, “pain disorder,” “chronic widespread pain,” and “chronic pain”) as well as genetics (“genetic,” “epigenetic,” “gene,” “haplotype,” “twins,” “SNP,” and “genotype”). In addition, the “Rechercheportal” (the University of Zurich’s online library collection)25 was searched for textbooks and other specialized books on the topics of interest. Electronic searches were supplemented with reference lists and additional citations suggested by experts. Further relevant articles were identified in the reference lists of already-selected publications.
Evidence selection
Publications were considered relevant to this review if they focused exclusively on human studies (general population and clinical samples) and included: 1) univariate and multivariate quantitative genetic studies including twin and family studies estimating the relative contribution of genes and environmental factors in CWP; 2) molecular genetic studies using genome-wide and candidate gene approaches to identify specific candidate alleles (ie, single-nucleotide polymorphisms [SNPs]) involved in CWP; 3) studies investigating gene expression by means of currently available strategies, including methylation and chromatin remodeling in CWP; and 4) any other study design (eg, controlled observational studies) focusing on genetics and using molecular genetic approaches in CWP. In the end, a total of 15 publications were considered relevant to be included in this review. All the studies on these topics were considered, no matter how far back they dated. Eight were twin studies on CWP, six were molecular genetic studies on CWP, and one was an epigenetic study on CWP.
Basic principles of genetics and epigenetics and relevant study designs
Quantitative genetic modeling
Population genetics studies the distribution of genetic variation within a population and offers a set of approaches and methodologies to investigate the sources of phenotypic variation.26 Most relevant to this review is the concept of the so-called quantitative or complex traits as opposed to simple traits or Mendelian phenotypes. A complex trait is determined by an interplay of numerous genetic and environmental factors that each contribute to phenotype expression, whereas a simple trait is determined by one single gene that is responsible for disease or phenotypic expression (eg, cystic fibrosis).26 Phenotypes such as sensory processing disorder and CWP – similar to most human traits and behaviors – are assumed to be polygenic and therefore influenced by a multitude of genes and environmental factors.
A common quantitative approach used to disentangle the relative contribution of genetic and environmental factors to variation in a trait is the study of twins, where monozygotic (MZ) twins – who share ~100% of their genes – and dizygotic (DZ) twins – who only share on average 50% of their genetic material – are compared in terms of a specific phenotype. Hereby, the presence of genetic and environmental contributions is indicated through different patterns of within-pair correlations in MZ and DZ twins.36 In summary, the total phenotypic variation is made up of additive genetic influence (A), dominant genetic influence (D), and common (C) and unique (E) environmental factors.27 In this context, heritability (h2) is defined “as the proportion of phenotypic variance attributable to genetic variance,” or in other words, the “extent to which genetic individual differences contribute to individual differences in observed behavior (or phenotypic individual differences).”28 Although additive genetic influences reflect the effects of individual alleles at loci that influence a trait, dominant genetic influences refer to the interaction of alleles at the same locus. For MZ twins, the correlations of both additive and dominant gene effects are rA,D=1.0, whereas for DZ twins the correlation for additive genetic effects is rA=0.5 and for dominant genetic effects rD=0.25.29 Twin studies are based on several assumptions, the most notable of them being the equal environment assumption, which states that MZ and DZ twins share the pre- and postnatal environment to the same degree and that greater phenotypic similarity of MZ twins results from the twofold greater genetic similarity.36 Univariate twin analyses can be extended to multivariate analyses, where sophisticated modeling approaches allow the identification of etiological structures underlying the covariation of phenotypes, comorbidities, and correlates to shed light on potentially common environmental and genetic influences.30
Apart from using twins to estimate the relative contribution of genes and environment, a discordant twin approach can be used to control for genetic confounding and shared environment.31 Evidently, discordant MZ twins have the same heritability for a disease; however, if the disease is present in only one of the identical twins, it is possible to estimate the extent of environmental risk factors that the affected twin has been exposed to. The discordant MZ twin approach therefore allows to test for “causal” relationships and to identify environmental risk factors for a disease by simultaneously controlling for a range of potentially confounding factors, including genetic factors.31
Molecular genetic methods
Once quantitative genetic studies provide evidence for a genetic influence on a trait, researchers can then make use of a set of molecular methods to try and identify the specific genes associated with the phenotype.32
The development of sophisticated genotyping platforms containing hundreds of thousands of SNPs has allowed the implementation of genome-wide association studies (GWASs) to detect genomic segments associated with particular phenotypes of interest. GWASs allow for the detection of much smaller effects and more refined mapping than classical linkage mapping approaches.52 To carry out a GWAS, researchers use many hundreds to thousands of either population case–control or family samples and randomly scan the entire human to detect small gene variations. Using algorithms, GWAS analysis identifies regions with statistically significant differences in SNP, allele, or genotype frequencies between cases and controls, pointing to their involvement in the trait or disease.32 A similar approach not so frequently used anymore is the candidate gene association study (CGAS).32 CGASs use a selection of genes with a known or inferred biological function whose role indicates that they may predispose to disease or the observed phenotype. Because CGAS looks at a limited number of selected genes, it is cheaper than a GWAS but suffers from needing to be lucky, knowing the biology in advance, and from a lack of knowledge of the exact polymorphism that is causally related to the trait or disease risk.33
Basic principles of epigenetics
Despite the strengths of the presented methods, most disease-associated genetic factors that have been detected by molecular studies so far do not explain as much of the disease risk as might be expected from the estimated h2 fraction. This is also known as missing heritability.34 Epigenetic mechanisms may account for a significant fraction of the missing heritability.35 Epigenetics describes changes in gene expression caused by mechanisms other than differences in the underlying DNA sequence and that can be induced as a response to external environmental factors or stochastic events within the cell.36 These processes can be highly dynamic and are often tissue-specific.37 Epigenetic modifications such as DNA methylation can contribute to alterations in gene expression in a heritable manner without affecting the underlying genomic sequences. Such epigenetic contribution would be systematically missed by conventional DNA sequence–based analyses.
According to our current understanding, heritable traits that determine a phenotype are shaped by an interplay of genetic components and the environment.37 The idea of this interplay is supported by a research of recent years, showing that gene expression is regulated not only by transcription factors but also by the environment – and even by our behavior, emotions, and thoughts. Furthermore, epigenetic patterns can be passed on to the next generation (ie, epigenetic inheritance) and can also be reversed in a single generation, consequently enabling high plasticity of the genome.38 To name a few examples, epigenetic control can be found in memory formation and synaptic plasticity;39 furthermore, epigenetic alterations of the genome have been discovered in Alzheimer’s disease,40 schizophrenia,41 bipolar disorder,40 and major depressive disorder.42 In regard to our phenotype of interest, DNA epigenetic patterns that regulate gene expression profiles could provide the missing link between epigenomic state, chronic pain, and the effect of environmental factors, eventually offering a biological framework for the cognitive–behavioral causes underlying the development of pain and pain chronification.
Results
Quantitative genetic studies on CWP
Recent attempts to disentangle genetic from environmental sources have provided consistent evidence for a genetic influence on CWP. An estimation of the heritability of CWP was first published in 2006. Using a large Swedish twin sample (n=15,950) Kato et al reported a moderate genetic influence on CWP, with a heritability of 54% and 48% in women and men, respectively (Table 1).5 Influences of family environment seemed to be negligible for phenotypic variance. Comparable estimates have been reported in subsequent studies, such as the study by Burri et al (n=3,266, females only), who found a heritability of CWP of 58%,6 or the study by Markkula (n=10,608), who reported a heritability of 51% for a “likely FM group” (Table 1).43
Table 1.
Summary of study characteristics of existing twin studies conducted on CWP, presented in the chronological order
| Authors | Year | Variable | Cohort | n | h2 |
|---|---|---|---|---|---|
| Kato et al5 | 2006 | CWP | Swedish Twin Registry | 15,950 (53.5% female) | Female: 54% Male: 48% |
| Markkula et al43 | 2009 | FM-like group | Finnish Twin Cohort | 10,608 (54.2% female) | 51% |
| Vehof et al46 | 2014 | CWP | TwinsUK | 4,465 (87.3% female) | 27% |
| Malkin et al59 | 2014 | CWP | TwinsUK | 2,256 (100% female) | 58% |
| Burri et al6 | 2015 | CWP | TwinsUK | 3,266 (100% female) | 58% |
| Momi et al58 | 2015 | CWP | TwinsUK | 4,324 (89.7% female) | 53% |
| Burri et al7 | 2015 | CWP | TwinsUK | 3,266 (100% female) | 71% |
Abbreviations: CWP, chronic widespread pain; FM, fibromyalgia.
As highlighted in the “Introduction” section of this review, CWP is frequently accompanied by a number of symptom-based conditions such as chronic fatigue syndrome, irritable bowel syndrome (IBS), headache, as well as psychiatric comorbidities, including anxiety and depression.44,45 The mechanisms underlying these co-occurrences, however, remain largely unknown, but one way of untangling their etiologic structure is by extending univariate twin analyses to multivariate genetic modeling where the genetic and environmental covariation underlying these phenotypes can be estimated. By doing this, Kato et al found evidence of unmeasured genetic and family environmental factors underlying the comorbidity of all the abovementioned conditions (n=44,897).12 Using the same multivariate approach, the authors further found evidence for two common latent traits underlying CWP and a set of functional somatic syndromes (FSSs). One of these traits loaded heavily on FSS, as well as major depression and general anxiety disorder, whereas the other latent trait loaded only on chronic pain syndromes (CPSs). Similarly, exploring the genetic and environmental factors contributing to CPSs (including CWP, chronic pelvic pain, dry eye disease, and IBS) in a sample of female twins from the TwinsUK registry, Vehof et al reported evidence for an underlying latent variable influencing the pattern of correlation between all the traits and possibly accounting for CPS clustering in certain patients (n=3,075–7,319; depending on the disorder), with this factor showing a strong genetic basis (h2=66%).46
More recently, a series of twin studies conducted by Burri et al have focused on the biopsychosocial correlates of CWP and to what extent these phenotypes share a common etiology with CWP and might therefore represent risk factors for the development of the condition.6,7 Using 3,266 female twins, their first study found a common pathway model with two latent traits to provide the best fit to the data. In other words, the co-occurrence of CWP and depression, as well as the covariation between CWP and its psycho-affective correlates (including anxiety, emotional intelligence, and emotional instability), could be best explained by the two underlying common traits – a more affective one loading heavily on all three psycho-affective correlates, but not on CWP or depression, and a more sensory one loading only on CWP and depression.6 Underlying these two latent traits were two genetic factors, and all variables apart from emotional instability and depression were also affected by genetic and nonshared environmental influences that were specific to them. In a subsequent study on the same female twin sample, the authors explored the genetic environmental association between CWP, fatigue, depression, and dehydroepiandrosterone sulfate (DHEAS) – an endogenous steroid hormone that has been associated with the maintenance of FM symptomatology and fatigue.7 Again, a substantial genetic relationship between CWP, depression, and DHEAS could be detected, supporting earlier study findings indicating that CWP and its related features and correlates are etiologically distinct from each other on an environmental level but also share a strong genetic base that accounts for their clinical co-occurrence. The findings further highlight the complex etiological structure underlying CWP, wherein physical, psychiatric, and premorbid affective traits seem to influence the development of the disorder. Figure 1 illustrates a simplified summary of all the multivariate twin studies on CWP.
Figure 1.
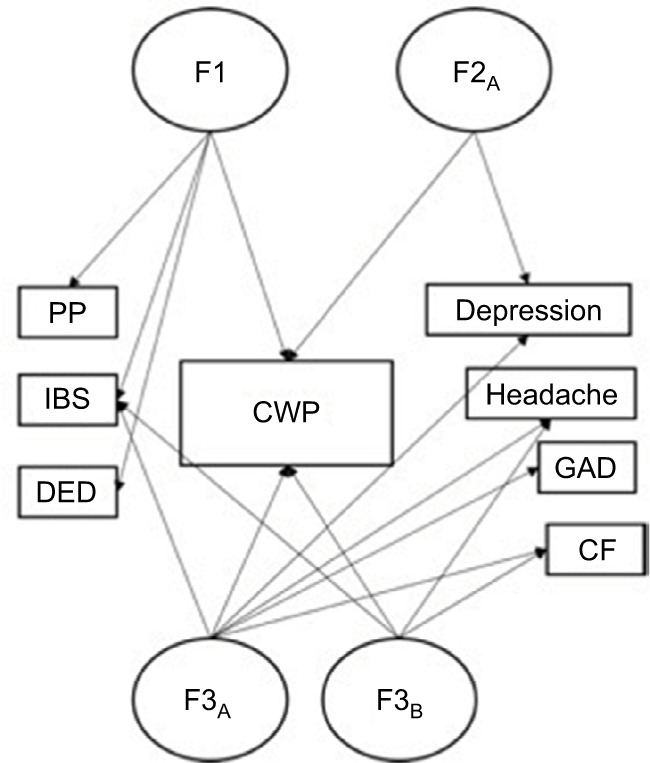
Summary of the three multivariate twin studies exploring the genetic and environmental covariation between CWP and a range of functional somatic syndromes and psychological comorbidities.6,12,46
Notes: In all the three studies, a common pathway model provided the best fit to the data, indicating the existence of latent phenotypes that underlie the covariation between CWP and the other included variables: F1 = latent phenotype identified in Vehof et al’s study,46 F2A = latent phenotype identified in Burri et al’s study;6 F3A+B = latent phenotypes identified in Kato et al’s study.12
Abbreviations: CF, chronic fatigue; CWP, chronic widespread pain; DED, dry eye disease; GAD, generalized anxiety disorder; IBS, irritable bowel syndrome; PP, pelvic pain.
As mentioned earlier in this review, twin studies can be used not only to quantify the genetic contribution but also to explore the environmental basis of individual differences in behavior or traits.31 In the so far only study using this design, Burri et al took a discordant twin approach to explore to what extent previously suggested psychosocial and contextual factors are causal to CWP.8 Using a large sample of DZ (n=360) and MZ (n=266) female twins from the TwinsUK registry, the authors found emotional intelligence to be the strongest independent predictor for CWP once genetic factors were controlled for. Emotional intelligence is said to play an important role in the processing and management of emotional states and, thus, may affect pain perception and pain coping.47 The authors further found evidence for links between CWP and anxiety sensitivity on an individual level but were unable to replicate the findings in the CWP-discordant DZ or MZ twin subsamples – most likely due to lower statistical power because of the reduced sample sizes.
Molecular genetic studies on CWP
Nicholl et al were the first to report an association between a genetic variation in the 5-HTR 2A gene and CWP in two different cohorts within the same study, one being part of the Epidemiology of Function Disorders (EPIFUND) cohort (Table 2).48 Additionally, the research group found SNP associations in the TPH 2 gene with CWP in the so-called discovery cohort (EPIFUND; 164 CWP cases and 172 controls). However, these findings could not be replicated in their validation cohort (203 CWP cases and 929 controls). Another study by Hennings et al (91 patients with unexplained physical symptoms) found an association of SNPs in the serotonin transporter (SLC6A4) gene and somatoform symptoms.49 However, these findings were not supported for CWP or reported number of pain sites in the study conducted by Nicholl et al.48
Table 2.
Summary of existing molecular genetic studies on CWP presented in chronological order
| Authors | Year | Sample and size | Ethnic origin | Main phenotype | Findings/implicated genes |
|---|---|---|---|---|---|
| Holliday et al50 | 2009 | EPIFUND cohort; n=1,189 | Caucasian (United Kingdom) | CWP | Haplotype frequencies for the “pain-protective” haplotype and allele frequencies for the OPRMI. SNPs did not differ between CWP individuals and controls |
| Hocking et al51 | 2010 | Subjects from a British Cohort Study; n=7,083 | Caucasian (United Kingdom) | Nonpain, nonchronic pain, chronic regional pain, CWP | 1) Association of ADRB2 SNPs and CWP and pain status (nonchronic pain, chronic regional pain) 2) Association of ADRB2 haplotypes and pain status 2) No association of genetic COMT gene variants and reported chronic pain |
| Nicholl et al48 | 2011 | EPIFUND (discovery) cohort; n=336 European Male Aging (validation) cohort; n=1,132 |
Caucasian (United Kingdom) Caucasian (Europe) |
Number of pain sites, CWP | 1) Association of SNPs in TPH2 and serotonin receptor (5-HTR2A) genes and CWP in the discovery cohort 2) Replication of HTR2A association in validation cohort |
| Skouen et al52 | 2012 | Adolescents attending a 17-year cohort review; n=1,004 | Caucasian (Western Australian) | Pain status | 1) Risk factors for chronic pain and number of pain sites: female gender and mental health 2) Association of SNP in ADBR2 gene with chronic musculoskeletal pain |
| Peters et al53 | 2013 | Several European cohorts; n=16,568 | Caucasian (Europe) | CWP | Association of CCT5 and FATA173B genes with 30% higher risk for CWP |
Abbreviations: CWP, chronic widespread pain; EPIFUND, Epidemiological study of functional disorders; SNP, single-nucleotide polymorphism.
Another study by Holliday et al (also EPIFUND; 164 CWP cases and 172 controls) compared a variety of genes related to the HPA axis between individuals with CWP and healthy controls and found several SNPs around the SERPINA6 gene to influence the susceptibility to CWP.50
Hocking et al examined genetic variants of the ADRB2 and the COMT, both of which have been suggested to be likely candidates for chronic pain (n=954–7,611; depending on the SNP).51 Skouen et al followed Hocking’s suit and investigated haplotypes and SNPs of these two candidate genes in an adolescent sample of a 17-year cohort study (n=1,004).52 Both the studies found statistically significant associations between genetic variants across the ADBR2 gene and CWP or chronic pain; however, no COMT polymorphisms were found to be relevant to the predisposition for CWP in either of the studies.
Finally, a GWAS meta-analysis by Peters et al (n=2,788 CWP female cases; n=13,780 female controls) found uncovered genetic variants upstream of the CCT5 and downstream of the FAM173B genes (5p15.2 region), of which for both SNPs the small alleles were associated with a 30% risk increase for CWP (Table 2).53
Epigenetic study on CWP
Epigenetic research into CWP is still in its beginning. So far, only two studies have looked at epigenome-wide DNA methylation alterations in individuals suffering from CWP. The first study was conducted by Burri et al which included a discovery sample consisting of a total of 281 twin individuals from the TwinsUK registry, of which 33 MZ twins discordant for self-reported CWP could be identified.54 Using the Illumina Infinium HumanMethylation 450 DNA Bead-Chip, 40 potential candidates were identified and carried forward for replication in an independent sample consisting of 729 men and 756 women from a subsample of the KORA S4 survey (an independent population-based cohort from Southern Germany). Of these 40 candidates, three CPGs reached significant p-values in the replication cohort, including MDH2, tetranectin (CLEC3B), and HSPB6. Other biologically relevant candidates showing significant associations in the discover cohort – including the COL1A2 and MAOB – could not be replicated. Using a sample of 1,708 MZ and DZ Caucasian adult twins (CWP prevalence of 19.9%), Livshits et al established longitudinally stable methylation bins (lsBINs) by repeated measurements taken ≥3 years apart.55 In this largest EWAS on CWP conducted so far, the authors found the most significant lsBINs associated with CWP to be located within or around highly plausible candidates that have been previously suggested to be involved in the pathogenesis of CWP such as IL17A gene, the ADIPOR2 gene, and the TNFRSF13B gene. Being the first of their kind, preliminary results from these two methylation studies serve as a starting point to encourage further replication in large and independent population-based cohorts and subsequent biological investigation of the top findings to elucidate possible disease mechanisms.
Discussion
The limited number of quantitative genetic studies focusing on CWP has consistently pointed toward around half of the variance in the development and maintenance of CWP being due to genetic factors.5–7 Overall, the study findings have emphasized the strong genetic contribution to CWP but have equally highlighted the importance of lifetime experiences in the etiology of the disorder. No existing studies have found evidence that indicates shared environmental factors such as upbringing to contribute to the susceptibility to CWP. According to the findings from a series of multivariate twin studies, CWP further shares a common genetic and environmental etiology with a range of co-occurring psycho-affective factors, including depression, anxiety, emotional intelligence, and stability, as well as with other chronic pain and symptom-based syndromes, including IBS, chronic fatigue, headache, and pelvic pain.6,7,46
As it is the case with any studies of this and similar nature, several limitations have to be considered when interpreting the results and should be addressed in future projects aiming at unraveling the genetic basis of CWP and related disorders. First, in all the twin studies discussed in this review, the assessment of CWP relied on self-report measures without additional information from clinicians, consequently increasing the risk of bias and limiting the extrapolation of findings to clinical samples. Second, all the samples consisted either entirely or predominantly of Caucasian females, impeding the generalization of the results to other ethnic groups as well as to the male population. Third, gene–environment interactions were not considered in the majority of studies, implying that genetic and environmental contributions to CWP are a first approximation. Finally, most of the quantitative and molecular findings presented in this review are the first to be reported and therefore demand replication and validation.
Molecular studies on somatoform symptoms and CWP provide evidence for specific molecular markers but show inconsistent results and mostly remain to be replicated. Nevertheless, certain candidates have been implicated, which point toward the involvement of the serotoninergic system in CWP,49 in addition to the HPA axis (eg, SERPINA6 gene).50 Furthermore, two of the presented studies reported the association of the ADRB2 gene, but no relation to the COMT gene, which is involved in pain sensitivity and CWP.51,52 A review and meta-analysis by Tammimäki and Männistö, however, found evidence that one allele of an SNP of the COMT gene does raise the risk for CWP, but not for other chronic pain conditions.56 In addition, two other genes (OPRM1 and GCH1) that did not show any association in an earlier study could then, along with the COMT gene, be connected to CWP in the before-mentioned genome-GWAS meta-analysis conducted by Peters et al.53 They found that the candidate SNPs with the strongest associations were located in the OPRM1, CGH1, and COMT gene regions. OPRM1, CGH1, and COMT being the most studied pain genes according to the authors.53 Although initially reported otherwise, the COMT gene does seem to contribute to CWP.
Again, a set of limitations should be considered that might explain these inconclusive and inconsistent results. First of all, careful attention should be paid to the definition and ascertainment of the phenotype, which should be standardized to allow a comparison between studies. This was not the case for CWP nor for the somatoform pain symptom as the studies used different assessment criteria and instruments. Second, most of the molecular studies relied on rather small sample sizes, therefore substantially reducing the statistical power needed for the detection of gene variants with rather small effect sizes.57 Third, significant findings may also have occurred due to population stratification.57 Population stratification occurs if allele frequencies differ between participants due to diverse ethnicity. These significant results may disappear if corrected for ethnicity; therefore, samples should be ethnically homogenous.57 All study samples consisted exclusively of Caucasian individuals, but ethnic heterogeneity within the sample was not always controlled for and therefore Caucasian origin could only be assumed to be most likely. Forth, even though multiple genes are suggested to interact in CWP, the majority of the discussed molecular studies used a candidate gene approach rather than focusing on a broad spectrum of genes, therefore limiting the ability to discover potentially new susceptibility genes, as well as disabling the control for gene–gene interaction. Finally, most studies did not correct for multiple testing.
Future studies, especially replication studies, should examine large and independent samples across different ages and ethnic groups and adjust for confounding variables and multiple testing. Novel genetic studies should further control for gene–gene and gene–environment interactions where indicated. The consideration of gene–environment interaction will also pave the way for epigenetic research. Additionally, longitudinal studies should be conducted to determine causal relationships.
Conclusion
Genetic epidemiologic research on CWP has provided promising insights regarding the genetic etiological structure of pain and its cosymptomatology, which can potentially be translated and extrapolated to other chronic pain conditions, as well as can set a direction for research to come. In addition to contributing explanatory value to the biological mechanisms underlying CWP, these results might also serve as a template for further genetic studies on chronic pain and help detect genetic commonalities and distinctions between the various chronic pain disorders.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Arthritis Rheum. 1990;33:160–172. doi: 10.1002/art.1780330203. [DOI] [PubMed] [Google Scholar]
- 2.Lee J, Ellis B, Price C, Baranowski AP. Chronic widespread pain, including fibromyalgia: a pathway for care developed by the British Pain Society. Br J Anaesth. 2014;112:16–24. doi: 10.1093/bja/aet351. [DOI] [PubMed] [Google Scholar]
- 3.Macfarlane GJ, Pye SR, Finn JD, et al. Investigating the determinants of international differences in the prevalence of chronic widespread pain: evidence from the European Male Ageing Study. Ann Rheum Dis. 2009;68:690–695. doi: 10.1136/ard.2008.089417. [DOI] [PubMed] [Google Scholar]
- 4.Buskila D, Neumann L, Hazanov I, Carmi R. Familial aggregation in the fibromyalgia syndrome. Semin Arthritis Rheum. 1996;26:605–611. doi: 10.1016/s0049-0172(96)80011-4. [DOI] [PubMed] [Google Scholar]
- 5.Kato K, Sullivan PF, Evengård B, Pedersen NL. Importance of genetic influences on chronic widespread pain. Arthritis Rheum. 2006;54:1682–1686. doi: 10.1002/art.21798. [DOI] [PubMed] [Google Scholar]
- 6.Burri A, Ogata S, Vehof J, Williams F. Chronic widespread pain: clinical comorbidities and psychological correlates. Pain. 2015;156:1458–1464. doi: 10.1097/j.pain.0000000000000182. [DOI] [PubMed] [Google Scholar]
- 7.Burri A, Ogata S, Livshits G, Williams F. The association between chronic widespread musculoskeletal pain, depression and fatigue is genetically mediated. PLoS One. 2015;10:e0140289. doi: 10.1371/journal.pone.0140289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burri A, Lachance G, Williams F. A discordant monozygotic-twin approach to potential risk factors for chronic widespread pain in females. Twin Res Hum Genet. 2015;18:188–197. doi: 10.1017/thg.2015.7. [DOI] [PubMed] [Google Scholar]
- 9.Sommer C, Häuser W, Gerhold K, et al. Etiology and pathophysiology of fibromyalgia syndrome and chronic widespread pain. Schmerz. 2008;22:267–282. doi: 10.1007/s00482-008-0672-6. German. [DOI] [PubMed] [Google Scholar]
- 10.Chang MH, Hsu JW, Huang KL, et al. Bidirectional association between depression and fibromyalgia syndrome: a nationwide longitudinal study. J Pain. 2015;16:895–902. doi: 10.1016/j.jpain.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Benjamin S, Morris S, McBeth J, Macfarlane GJ, Silman AJ. The association between chronic widespread pain and mental disorder: a population-based study. Arthritis Rheum. 2000;43:561–567. doi: 10.1002/1529-0131(200003)43:3<561::AID-ANR12>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 12.Kato K, Sullivan PF, Evengård B, Pedersen NL. Chronic widespread pain and its comorbidities: a population-based study. Arch Intern Med. 2006;166:1649–1654. doi: 10.1001/archinte.166.15.1649. [DOI] [PubMed] [Google Scholar]
- 13.Posthuma D, De Geus EJ, Baaré WF, Hulshoff Pol HE, Kahn RS, Boomsma DI. The association between brain volume and intelligence is of genetic origin. Nat Neurosci. 2002;5:83–84. doi: 10.1038/nn0202-83. [DOI] [PubMed] [Google Scholar]
- 14.Jeanmonod P, von Känel R, Maly FE, Fischer JE. Elevated plasma C-reactive protein in chronically distressed subjects who carry the A allele of the TNF-alpha-308 G/A polymorphism. Psychosom Med. 2004;66:501–506. doi: 10.1097/01.psy.0000130903.78444.7d. [DOI] [PubMed] [Google Scholar]
- 15.van den Akker EL, Koper JW, van Rossum EF, et al. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med. 2008;168:33–39. doi: 10.1001/archinternmed.2007.41. [DOI] [PubMed] [Google Scholar]
- 16.Wüst S, Federenko I, Hellhammer DH, Kirschbaum C. Genetic factors, perceived chronic stress, and the free cortisol response to awakening. Psychoneuroendocrinology. 2000;25:707–720. doi: 10.1016/s0306-4530(00)00021-4. [DOI] [PubMed] [Google Scholar]
- 17.Plomin R. Behavioral Genetics. 6th ed. New York: Worth; 2013. [Google Scholar]
- 18.Zhang TY, Meaney MJ. Epigenetics and the environmental regulation of the genome and its function. Annu Rev Psychol. 2010;61:439–466. doi: 10.1146/annurev.psych.60.110707.163625. [DOI] [PubMed] [Google Scholar]
- 19.Champagne FA. Epigenetic influence of social experiences across the lifespan. Dev Psychobiol. 2010;52:299–311. doi: 10.1002/dev.20436. [DOI] [PubMed] [Google Scholar]
- 20.Web of Science [database on the Internet] Thomson Reuters; 2016. [Google Scholar]
- 21.MEDLINE [database on the Internet]. National Center for Biotechnology Information.
- 22.PubMed [database on the Internet]. National Center for Biotechnology Information.
- 23.Embase [database on the Internet] Elsevier; 2016. [Google Scholar]
- 24.Google Scholar [database on the Internet]. Google.
- 25.Rechercheportal [database on the Internet]. Zentralbibliothek Zürich/Hauptbibliothek der Universität Zürich.
- 26.Griffiths AJ, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM. An Introduction to genetic analysis. New York: W. H. Freeman and Company; 2012. [Google Scholar]
- 27.Posthuma D. Multivariate genetic analysis. In: Kim YK, editor. Handbook of Behavior Genetics. New York: Springer; 2009. pp. 47–59. [Google Scholar]
- 28.Fisher RA. The genesis of twins. Genetics. 1991;4:489–499. doi: 10.1093/genetics/4.5.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillespie NA, Zhu G, Heath AC, Hickie IB, Martin NG. The genetic aetiology of somatic distress. Psychol Med. 2000;30:1051–1061. doi: 10.1017/s0033291799002640. [DOI] [PubMed] [Google Scholar]
- 30.Rijsdijk FV, Sham PC. Analytic approaches to twin data using structural equation models. Brief Bioinform. 2002;3:119–133. doi: 10.1093/bib/3.2.119. [DOI] [PubMed] [Google Scholar]
- 31.Vitaro F, Brendgen M, Arseneault L. The discordant MZ-twin method: one step closer to the holy grail of causality. Int J Behav Dev. 2009;33:376–382. [Google Scholar]
- 32.Risch NJ. Searching for genetic determinants in the new millennium. Nature. 2000;405:847–856. doi: 10.1038/35015718. [DOI] [PubMed] [Google Scholar]
- 33.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 34.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trerotola M, Relli V, Simeone P, Alberti S. Epigenetic inheritance and the missing heritability. Hum Genomics. 2015;9:17. doi: 10.1186/s40246-015-0041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russo VEA, Martienssen RA, Riggs AD. Epigenetic Mechanisms of Gene Regulation. New York: Cold Spring Harbor Laboratory Press; 1996. [Google Scholar]
- 37.Bell JT, Spector TD. A twin approach to unravelling epigenetics. Trends Genet. 2011;27:116–125. doi: 10.1016/j.tig.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005;7:103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim MS, Akhtar MW, Mahgoub M, et al. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci. 2012;32:10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwok JB. Role of epigenetics in Alzheimer’s and Parkinson’s disease. Epigenomics. 2010;2:671–682. doi: 10.2217/epi.10.43. [DOI] [PubMed] [Google Scholar]
- 41.Dempster EL, Pidsley R, Schalkwyk LC, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Labonté B, Turecki G. The epigenetics of depression and suicide. In: Petronis A, Mill J, editors. Brain, Behavior and Epigenetics. Berlin Heidelberg: Springer; 2011. [Google Scholar]
- 43.Markkula RA, Kalso EA, Kaprio JA. Predictors of fibromyalgia: a population-based twin cohort study. BMC Musculoskelet Disord. 2016;17:29. doi: 10.1186/s12891-016-0873-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aaron LA, Buchwald D. Chronic diffuse musculoskeletal pain, fibromyalgia and co-morbid unexplained clinical conditions. Best Pract Res Clin Rheumatol. 2003;17:563–574. doi: 10.1016/s1521-6942(03)00033-0. [DOI] [PubMed] [Google Scholar]
- 45.Bradley LA. Psychiatric comorbidity in fibromyalgia. Curr Pain Headache Rep. 2005;9:79–86. doi: 10.1007/s11916-005-0042-3. [DOI] [PubMed] [Google Scholar]
- 46.Vehof J, Zavos HM, Lachance G, Hammond CJ, Williams FM. Shared genetic factors underlie chronic pain syndromes. Pain. 2014;155:1562–1568. doi: 10.1016/j.pain.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Bishop MD, Craggs JG, Horn ME, George SZ, Robinson ME. Relationship of intersession variation in negative pain-related affect and responses to thermally-evoked pain. J Pain. 2010;11:172–178. doi: 10.1016/j.jpain.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicholl BI, Holliday KL, Macfarlane GJ, et al. Association of HTR2A polymorphisms with chronic widespread pain and the extent of musculoskeletal pain: results from two population-based cohorts. Arthritis Rheum. 2011;63:810–818. doi: 10.1002/art.30185. [DOI] [PubMed] [Google Scholar]
- 49.Hennings A, Zill P, Rief W. Serotonin transporter gene promoter polymorphism and somatoform symptoms. J Clin Psychiatry. 2009;70(11):1536–1539. doi: 10.4088/JCP.08m04613. [DOI] [PubMed] [Google Scholar]
- 50.Holliday KL, Nicholl BI, Macfarlane GJ, Thomson W, Davies KA, McBeth J. Genetic variation in the hypothalamic-pituitary-adrenal stress axis influences susceptibility to musculoskeletal pain: results from the EPIFUND study. Ann Rheum Dis. 2010;69:556–560. doi: 10.1136/ard.2009.116137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hocking LJ, Smith BH, Jones G, Reid DM, Strachan DP, Macfarlane GJ. Genetic variation in the beta2-adrenergic receptor but not catechol-amine-O-methyltransferase predisposes to chronic pain: Results from the 1958 British birth cohort study. Pain. 2010;149:143–151. doi: 10.1016/j.pain.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 52.Skouen JS, Smith AJ, Warrington NM, et al. Genetic variation in the beta-2 adrenergic receptor is associated with chronic musculoskeletal complaints in adolescents. Eur J Pain. 2012;16:1232–1242. doi: 10.1002/j.1532-2149.2012.00131.x. [DOI] [PubMed] [Google Scholar]
- 53.Peters MJ, Broer L, Willemen HL, et al. Genome-wide association study meta-analysis of chronic widespread pain: evidence for involvement of the 5p15.2 region. Ann Rheum Dis. 2013;72:427–436. doi: 10.1136/annrheumdis-2012-201742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burri A, Marinova Z, Robinson MD, et al. Are epigenetic factors implicated in chronic widespread pain? PLoS One. 2016;11(11):e0165548. doi: 10.1371/journal.pone.0165548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Livshits G, Malkin I, Freidin MB, et al. Genome-wide methylation analysis of a large population sample shows neurological pathways involvement in chronic widespread musculoskeletal pain. Pain. 2017;158(6):1053–1062. doi: 10.1097/j.pain.0000000000000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tammimäki A, Männistö PT. Catechol-O-methyltransferase gene polymorphism and chronic human pain: a systematic review and meta-analysis. Pharmacogenet Genomic. 2012;22:673–691. doi: 10.1097/FPC.0b013e3283560c46. [DOI] [PubMed] [Google Scholar]
- 57.Limer KL, Nicholl BI, Thomson W, et al. Exploring the genetic susceptibility of chronic widespread pain: the tender points in genetic association studies. Rheumatology (Oxford) 2008;47:572–577. doi: 10.1093/rheumatology/ken027. [DOI] [PubMed] [Google Scholar]
- 58.Momi SK, Fabiane SM, Lachance G, Livshits G, Williams FN. Neuropathic pain as part of chronic widespread pain: environmental and genetic influences. Pain. 2015;156:2100–2106. doi: 10.1097/j.pain.0000000000000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malkin I, Williams FMK, LaChance G, et al. Low back and common widespread pain share common genetic determinants. Ann Hum Genet. 2014;78:357–366. doi: 10.1111/ahg.12074. [DOI] [PubMed] [Google Scholar]