

Abstract
The spread of multidrug resistance among bacterial pathogens poses a serious threat to public health worldwide. Recent approaches towards combating antimicrobial resistance include repurposing old compounds with known safety and development pathways as new antibacterial classes with novel mechanisms of action. Here we show that an analog of the anticoccidial drug robenidine (4,6-bis(2-((E)-4-methylbenzylidene)hydrazinyl)pyrimidin-2-amine; NCL195) displays potent bactericidal activity against Streptococcus pneumoniae and Staphylococcus aureus by disrupting the cell membrane potential. NCL195 was less cytotoxic to mammalian cell lines than the parent compound, showed low metabolic degradation rates by human and mouse liver microsomes, and exhibited high plasma concentration and low plasma clearance rates in mice. NCL195 was bactericidal against Acinetobacter spp and Neisseria meningitidis and also demonstrated potent activity against A. baumannii, Pseudomonas aeruginosa, Escherichia coli, Klebsiella pneumoniae and Enterobacter spp. in the presence of sub-inhibitory concentrations of ethylenediaminetetraacetic acid (EDTA) and polymyxin B. These findings demonstrate that NCL195 represents a new chemical lead for further medicinal chemistry and pharmaceutical development to enhance potency, solubility and selectivity against serious bacterial pathogens.
Introduction
Infectious diseases particularly lower respiratory infections caused by bacterial and viral pathogens are the third leading cause of death [1] and the second leading cause of disability-adjusted life years, worldwide [2]. Streptococcus pneumoniae (the pneumococcus) and Staphylococcus aureus (‘golden staph’) are two such bacterial pathogens. The pneumococcus accounts for an estimated one million deaths annually in developing countries, and causes a broad spectrum of diseases including pneumonia, meningitis, bacteremia and otitis media (OM), particularly in children under five years of age, the elderly and immunocompromised individuals [3]. Although the current capsular-based pneumococcal vaccines have been effective in preventing pneumococcal sepsis and meningitis in developed countries, the vaccines induce strictly serotype-specific protection against a limited subset of pneumococcal types and furthermore are generally not affordable in developing countries. In addition, there is evident replacement of carriage and disease burden by non-vaccine serotypes following immunization [4, 5]. In developed countries, where effective antimicrobial therapy is readily accessible, deaths from pneumococcal disease occur primarily among people over 60 years of age, with case fatality rates of 10–20% for pneumonia and up to 40% for bacteremia [3]. In contrast, S. aureus is the second most clinically important antibiotic-resistant bacterial pathogen in developed countries behind E. coli, and a major public health concern due to the increasing prevalence of methicillin-resistant S. aureus (MRSA) in hospitals, among hospital workers and within the community [6–9]. Median direct medical costs associated with MRSA sepsis have been estimated at $12,078 US per patient and are approximately1.3 times higher than costs associated with susceptible S. aureus infection [10].
Effective treatment of Gram-positive bacterial infections remains hampered by the alarming increase in prevalence of antibiotic-resistant bacteria, as well as accelerated global spread of bacterial clones resistant to multiple antimicrobial agents [11, 12]. For pneumococcal disease, the problem is compounded by the highly transformable nature of this organism, enhancing genetic exchanges through horizontal gene transfer [13]. Moreover, pneumococcal OM accounts for more antibiotic prescriptions than any other infection [14, 15], driving selection for resistance amongst diverse bacteria, significantly affecting health-care costs in developed countries [$2.88 billion p.a. for USA] [16]. Recently, novel antimicrobials such as third-generation fluoroquinolones (e.g. moxifloxacin) and cyclic lipopeptides (e.g. daptomycin) have been shown to be effective against S. pneumoniae in vitro and in mouse models of infection [17–19]. However, there are reports of increasing frequency of S. pneumoniae resistance to the fluoroquinolones [20]. For MRSA, resistance or reduced susceptibility to multiple agents including vancomycin and daptomycin, is frequently reported in hospital-associated MRSA (HA-MRSA) [21]. Although community-associated MRSA (CA-MRSA) are generally susceptible to most drug classes over time they have acquired additional antimicrobial resistance genes. Furthermore, CA-MRSA are generally more virulent with higher mortality rates compared to HA-MRSA [22, 23]. Certain MRSA clones have recently become host-adapted in animals, furthering the spread of the staphylococcal cassette chromosome mec (SCCmec) within the environment [24]. In some countries, up to 54% of K. pneumoniae cases have been reported as carbapenem-resistant, the current last resort treatment for severe Gram-negative infections [25, 26]. Adding to this dire situation, the transferable colistin resistance determinant (MCR-1) is now widely distributed amongst both animal and human Gram-negative pathogens and commensals, resulting in some Gram-negative pathogens developing pan-resistance to all registered compounds [27, 28]. Therefore, the discovery and development of new broad-spectrum antimicrobials with activity against pan-resistant Enterobacteriaceae causing sepsis is one of the highest global priorities [29–31].
In 2010, the Infectious Diseases Society of America has called for 10 new antimicrobial agents to be registered by 2020 [32]. Since that call, all new registered agents except one (fidaxomicin) have been developed from existing drug classes, against Gram-positive organisms (telavancin, dalbavancin, tedizolid, oritavancin) or Gram-negative organisms (ceftolozane-tazobactam, ceftazidime-avibactam) [33–36]. Robenidine (2,2'-bis[(4-chlorophenyl)methylene] carbonimidic dihydrazide hydrochloride) is an anticoccidial agent which has been used worldwide since the early 1970s to prevent coccidian infections of poultry and rabbits [37]. However, there have been no in-depth in vivo evaluations of this compound or its chemical class as potential antibacterial agents. This presents an attractive prospect for testing the antibacterial activity of robenidine, an agent with known safety and long history of oral use in avian species and an oral LD50 of 150 mg/kg in mice [38], by expanding its chemical space with further medicinal chemistry for potential development as a parenteral or topically administered drug [39–41].
Materials and methods
Antimicrobial agents and medicinal chemistry
Analytical grade aminoguanidine robenidine (NCL812) [42] and two of its analogs (NCL195 and NCL219) were synthesized in house at The University of Newcastle and stored in a sealed sample container out of direct light at 4°C at the study site at the Infectious Diseases Laboratory, Roseworthy campus, The University of Adelaide. Ampicillin (β-lactam broad-spectrum antibiotic) was purchased from Sigma-Aldrich (Australia), while daptomycin (in pharmaceutically active form) was purchased from Cubist. Stock solutions (containing 25.6 mg/ml of each compound in DMSO) were prepared and stored in 1 ml aliquots at -80°C and defrosted immediately prior to use. Ethylenediaminetetraacetic acid (EDTA, Disodium salt, CAS Number: 6381-92-6) was purchased from Chem-Supply Pty Ltd, South Australia and was dissolved in water to 200 mM. Polymyxin B sulfate (CAS Number: 1405-20-5) was purchased from Sigma-Aldrich.
Synthesis of NCL195 (4,6-bis(2-((E)-4-methylbenzylidene)hydrazinyl)pyrimidin-2-amine)
A suspension of 2-amino-4,6-dihydrazinopyrimidine (58.9 mg, 0.380 mmol) and 4-methylbenzaldehyde (0.10 ml, 100 mg, 0.832 mmol, 2.19 eq.) in EtOH (4 ml) was heated at reflux for 16 h. The reaction mixture was cooled to ambient temperature before collecting the pellet-like precipitate, washing with diethyl ether (20 ml). The ‘pellets’ were then crushed and the solid further washed with diethyl ether (10 ml) to afford the pyrimidine (85.8 mg, 63%) as a white ‘fluffy’ powder. 1H NMR ((CD3)2SO, 400 MHz) δ 10.51 (s, 2H), 8.00 (s, 2H), 7.54 (d, J = 8.1 Hz, 4H), .26 (d, J = 8.0 Hz, 4H), 6.27 (s, 1H), 5.78 (s, 2H), 2.34 (s,6H); 13C NMR ((CD3)2SO, 101 MHz) δ 162.8 (2C), 162.6 (2C), 140.1 (2C), 138.4 (2C), 132.5 (2C), 129.4 (4C), 126.0 (4C), 73.3, 21.0 (6C).
Synthesis of NCL219. (E)-N'-((E)-1-(4-(tert-butyl)phenyl)ethylidene)-2-(1-(4-(tert-butyl)phenyl)ethylidene)hydrazine-1-carboximidhydrazide)
A suspension of N,N-diaminoguanidine hydrochloride (47.7 mg, 0.380 mmol) and 4′-(tert-butyl)acetophenone (134 mg, 0.760 mmol, 2 eq.) and in EtOH was heated at reflux for 16 h. The mixture was cooled and diluted with Et2O to effect crystallization. The resulting precipitate was collected and washed with Et2O 920 ml) to afford the carbonimidic dihydrazide (134 mg, 84%) as a yellow crystalline solid. 1H NMR ((CD3)2SO, 400 MHz) δ 11.74 (s, 1H), 8.60 (s, 1H), 7.95 (d, J = 8.6 Hz, 2H), 7.45 (d, J = 8.6 Hz, 2H), 2.42 (s, 3H), 1.31 (s, 9H). 13C NMR ((CD3)2SO, 101 MHz) δ 154.1, 153.3, 152.6, 134.0, 126.8, 125.0, 34.5, 30.9, 14.9.
Bacterial strains and growth conditions
The S. pneumoniae, S. aureus and VRE isolates used in this study are listed in S1, S2 and S3 Tables in Supporting information. Isolates were cultured on Horse Blood Agar (HBA) plates and incubated at 37°C, for 18 h. The pneumococci isolates were incubated in 5% CO2. The NCL812, NCL195 and NCL219 minimum inhibitory concentration (MIC) was performed on all isolates.
Pneumococcal serotype-specific capsule production was confirmed on all pneumococcal isolates by the Quellung reaction [43]. The minimum bactericidal concentration (MBC), kill kinetics, TEM and efficacy testing experiments was performed on S. pneumoniae D39 (NCTC7466), a well-documented laboratory strain with well-defined and predictable in vitro characteristics and in vivo pathogenicity profiles [44, 45]. MBCs, kill kinetics and point of resistance assays were performed on S. aureus Xen29 (luminescent ATCC 12600 S. aureus strain).
MIC determination
MICs for NCL812 and its analogs (serial two-fold dilutions commencing at 128 μg/ml) were determined (in duplicate) in round bottom 96-well microtitre trays (Sarstedt 82.1582.001), using the broth micro-dilution method recommended by the Clinical and Laboratory Standards Institute (CLSI) [46]. Luria Bertani (LB) broth (Oxoid, Australia) [supplemented with 3% lysed horse blood and 5% horse serum for S pneumoniae experiments] was used instead of cation-adjusted Mueller-Hinton broth (CAMHB) as it was shown previously that NCL812 can chelate calcium ions [47]. In addition, serial two-fold dilutions of the NCL compounds were performed in 100% DMSO, with 1 μl added to each well, as the compounds are hydrophobic [42]. The MIC for ampicillin or daptomycin against each S. pneumoniae, S. aureus or VRE isolate was determined for each test run as an internal quality control. MIC50, MIC90 and MIC range for NCL812, NCL195 and NCL219 were calculated against each S. pneumoniae, S. aureus or VRE isolate. To assess the potential activity of NCL195 against Gram-negative bacteria, MICs against Acinetobacter spp, E. coli, K. pneumoniae, P. aeruginosa, Proteus spp and Neisseria meningitidis were performed in the presence or absence of 0.08 mM-20 mM EDTA in a standard checkerboard assay [48].
Kill kinetics
Kill kinetic assays on S. pneumoniae D39 were performed (in duplicate) essentially as described for MIC determination, with the exception that the starting concentration of each NCL compound was 64 μg/ml, and the assay was performed using 200 μl volumes in a round bottom 96-well microtitre tray (Sarstedt 82.1582.001). The tray was covered with Breathe-Easy sealing membrane (Z380059, Sigma-Aldrich) and incubated for 16 h overnight at 35°C in an EnSpire Multimode Plate Reader 2300 (PerkinElmer), with data collected every 20 min and 2 sec agitation at 60 rpm between data (A600 nm) acquisitions. The experiment was repeated using the luminescent D39LUX strain on a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek) using IsoplateTM-96 F tray PerkinElmer, 6005020). Kill kinetic assays on S. aureus Xen29 was carried out essentially as described for MIC determination on Cytation 5 Cell Imaging Multi-Mode Reader.
Minimum bactericidal concentration (MBC) determination
In order to determine the MBC of each compound against S. pneumoniae D39 or S. aureus Xen29, 10 μl aliquots from each duplicate well from the MIC assays (starting from the MIC for each compound) was inoculated onto a HBA plate and incubated at 37°C (+ 5% CO2 for S. pneumoniae D39). Plates were examined at 24 and 48 h and the MBC was recorded as the lowest concentration of each test compound at which a 99.9% colony count reduction was observed on the plate [46].
Time-dependent killing assays
Time kill assays were performed (in duplicate) essentially as described previously [42, 49] with slight modifications. Briefly, a few colonies of luminescent S. pneumoniae strain D39LUX or luminescent S. aureus ATCC 12600 (Xen29) from an overnight HBA were emulsified in normal saline and adjusted to A600 nm = 0.10 (equivalent to approx. 5 × 107 c.f.u. per ml) and the bacterial suspension further diluted 1:20 in saline. NCL195 was prepared in 10 ml volumes at 2× and 4× MIC (using 4× MIC of ampicillin and normal saline as controls) in either LB broth (for Xen29) or LB supplemented with 3% lysed horse blood and 5% horse serum (for D39). The compounds were serially diluted in 100% DMSO at 100× the final desired concentration and 100 μl of appropriate concentrations added to each 10 ml preparation. Duplicate cultures were incubated at 37°C (+ 5% CO2 for S. pneumoniae D39), with samples withdrawn at 0, 0.5, 1, 2, 4, 6, 8 and 24 h, serially diluted 10-fold and plated on HBA overnight at 37°C (+ 5% CO2 for S. pneumoniae D39) for bacterial enumeration.
Point of resistance assay
To determine if S. aureus develops resistance to the NCL195, we performed 24 daily sequential culturing of S. aureus Xen29 in 2 ml LB broth the presence of 0.5×MIC, 0.75×MIC, 1×MIC, 1.5×MIC, 2×MIC, and 4×MIC of NCL195, using 1×MIC, 4×MIC and 8×MIC of daptomycin as control (in triplicates), essentially as described by Ling et al.[50]. After the 24th passage, samples were centrifuged at 4,000 × g for 10 min and washed in 50 ml of phosphate buffered saline (PBS) twice to remove any residual antimicrobial, and/or bacterial end products and media. Washed bacteria were resuspended in PBS to A600 nm = 0.1 and MICs were performed as described above.
Effect of NCL812 on S. pneumoniae D39 and S. aureus ATCC29213 cell membrane
Morphological appearance and morphometric analysis of the cell membrane of S. pneumoniae D39 after exposure to either 1 μg/ml, 4 μg/ml or 16 μg/ml of NCL812 was determined using TEM, as described in S1 File (Supplementary Methods). For S. aureus ATCC 29213, macromolecular (DNA, RNA, protein, cell wall, and lipid) synthesis inhibition studies were performed using concentrations of NCL812 that were equivalent to 0, 0.25, 0.5, 1, 2, 4 or 8-fold the MIC value (4 μg/ml), as detailed in S1 File (Supplementary Methods).
Mechanism of action studies
To further examine the mechanism underlying the perturbation of the cell membrane of S. pneumoniae or S. aureus by the NCL compounds, the membrane potential of the cells was measured by fluorescence spectrometry using (DiOC2(3), essentially as described previously[51, 52]. Briefly, D39 cells from an overnight HBA plate were grown in Todd-Hewitt broth supplemented with 1% yeast extract (THY) at 37°C, 5% CO2 until A600 nm = 0.5. The cells were centrifuged at 4,000 × g for 10 min at room temperature, washed twice in one culture volume of 50 mM potassium phosphate buffer (pH 7.0) and resuspended in the same buffer to A600 nm = 10. For the fluorescence assay, 0.2 ml of this suspension was added to 1.8 ml of the same buffer in a quartz cuvette, the mixture was stirred gently for 5 min (with or without addition of 16 μg/ml of test compounds, using 16 μg/ml ampicillin as control). This concentration corresponds to 2×MIC of NCL812 and NCL195, and 4×MIC of NCL219 for S. pneumoniae. The cuvette was then placed in a LS 55 Fluorescence Spectrometer (PerkinElmer) set at Ex. 488 nm/Em. 620 nm, with excitation and emission slit widths at 5 nm and 7 nm, respectively). The background fluorescence of each suspension was followed for 1 min after which DiOC2(3) was added to a final concentration of 10 μM and the fluorescence monitored until it plateaued. Cells were then re-energized with 0.5% glucose and fluorescence further monitored until it plateaued, after which 10 μM of the proton ionophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP) was added and fluorescence followed again until plateaued. A similar procedure was followed using S. aureus cells resuspended in 50 mM potassium phosphate buffer (pH 7.0) to A600 nm = 5, using 16 μg/ml of test compounds, corresponding to 4×MIC of NCL812, 8×MIC NCL195, and 1×MIC of NCL219 for S. aureus. All assays were performed at least twice.
Microsomal stability of NCL812, NCL195 and NCL219
The metabolic stability assay was performed by incubating 0.4 μg/ml of each test compound with human and mouse liver microsomes (Xenotech, Lot# 1210057 and 1310211, respectively) suspended in 0.1 M pH 7.4 phosphate buffer at 37°C and 0.4 mg/ml protein concentration. The metabolic reaction was initiated by the addition of an NADPH-regenerating system (i.e. NADPH is the cofactor required for CYP450-mediated metabolism) and quenched at various time points over a 60 min incubation period by the addition of acetonitrile containing diazepam as internal standard. Control samples (containing no NADPH) were included (and quenched at 2, 30 and 60 min) to monitor for potential degradation in the absence of cofactor. Test compound concentration vs time data were fitted to an exponential decay function to determine the first-order rate constant for substrate depletion. This was used to calculate a degradation half-life (T1/2), in vitro intrinsic clearance (CLINT), microsome-predicted blood clearance (CLblood) and a microsome-predicted hepatic extraction ratio (EH) according to the in vitro half-life method of Obach[53].
Ethics statements
For determination of pharmacokinetic (PK) parameters for NCL812 and NCL195 in mice following systemic exposure, 5-9-week-old male outbred Swiss mice, weighing 21 to 32 g, obtained from Monash Animal Research Platform (MARP), were used. Mice had access to food and water ad libitum throughout the pre- and post-dose sampling period. The Animal Ethics Committee of the Monash Institute of Pharmaceutical Sciences reviewed and approved all animal PK experiments (approval number MIPS.2013.33). The PK studies were conducted in compliance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (7th Edition 2004).
Determination of PK parameters for NCL812 and NCL195 after systemic exposure
Plasma concentration vs time profiles were determined for NCL812 and NCL195 after intravenous (IV) administration at a dose of 5 mg/kg, and for NCL195 after IP administration at a dose of 43 mg/kg. For the IV exposure, each compound was administered via a bolus injection into the tail vein (vehicle 20% (v/v) DMSO in PEG400, 1 ml/kg dose volume, n = 8 mice per compound, resulting in average measured concentration of 5.25 and 4.31 mg/ml for NCL812 and NCL195, respectively). The compounds were administered to mice within 2.5 h of preparation. Following administration, blood samples were collected at 5, 15, 30, 120, 240 and 480 min post- dose (n = 2 mice per time point for each compound). For IP administration of NCL195, the formulation was prepared by dissolving solid NCL195 in DMSO (to 20% (v/v) of the final volume) before addition of PEG400, yielding a clear yellow solution that was dosed to mice within 30 min of preparation. The measured concentration of NCL195 in the final formulation was 21.9 mg/ml, resulting in a mean administered dose of 43 mg/kg, and blood samples were collected up to 24 h post-dose.
For both the IV and IP administration, a maximum of two samples were obtained from each mouse, with samples being taken either via submandibular bleed (approximately 120 μl; conscious sampling) or terminal cardiac puncture (0.6 ml; under inhaled Isoflurane anesthesia). No urine samples were collected as mice were housed in bedded cages during the study. In all experiments, mice were observed continuously for the first hour after dosing, then hourly until up to 6–8 hours after dosing. All animals showed normal behaviour (including grooming, eating, drinking, sleeping, alertness) over the post-dose period. Mice were humanely sacrificed by cervical dislocation while under anaesthesia (using gaseous isoflurane) at the end of experiments. Blood was collected directly into polypropylene Eppendorf tubes containing heparin as anti-coagulant, and stabilization cocktail (containing Complete (a protease inhibitor cocktail with EDTA) and potassium fluoride) to minimize the potential for ex vivo degradation of the test compounds in blood/plasma samples. Once collected, blood samples were centrifuged immediately, supernatant plasma was removed, and stored at either -80°C (for the IV exposure) or at -20°C (for the IP exposure) until analysis by liquid chromatography-mass spectrometry (LC-MS). Following protein precipitation with acetonitrile, thawed plasma samples were analyzed by LC-MS (Waters Micromass Quattro Premier coupled to a Waters Acquity UPLC) using positive electrospray ionization operating in MRM mode. PK parameters (apparent half-life, plasma clearance, volume of distribution and area-under-the curve (AUC)) were determined for NCL812 and NCL195 after IV administration using non-compartmental methods (WinNonlin Version 6.3.0.395).
Haemolysis assay
This was performed using fresh donor human red blood cells (RBCs), essentially as described previously [50]. Fresh RBCs were washed in PBS three times at 500 × g for 5 min, and then resuspended 1% (w/v) in PBS. Two microliters of serial 2-fold dilutions of each compound was added into the respective wells, in quadruplicates, in a round bottom 96-well microtitre tray (Sarstedt 82.1582.001), starting at 128 μg/ml for each compound using ampicillin as a control. Thereafter, 198 μL of the 1% RBC solution was added into each well, and the mixture incubated for 1 h at 37°C, with shaking at 100 rpm. Quadruplicate wells containing either 1% Triton X100, or PBS only, served as controls. After incubation, the trays were centrifuged at 1,000 × g for 3 min and 100 μl of supernatant from each well transferred into a new 96-well tray. Absorbance was measured at A450 nm on a Multiskan Ascent 354 Spectrophotometer (Labsystems) and plotted against each dilution. Haemolytic titer was determined as the reciprocal of the dilution at which 50% of erythrocytes were lysed at A450 nm. The experiment was performed twice.
In vitro cytotoxicity assays
In order to ensure minimal off target mammalian cell cytotoxicity, we assayed NCL812, NCL195 and NCL219 for in vitro cytotoxicity using a panel of adherent mammalian cell lines: Caco-2 (human colorectal adenocarcinoma cell line), HEL 299 (non-cancerous human lung fibroblast cell line), Hep G2 (human hepatocellular carcinoma cell line), MDBK (normal bovine kidney cell line) and MCF7 (human mammary gland adenocarcinoma cell line). Cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco Cat No: 12430) supplemented with 10% (vol/vol) FBS and 1% PenStrep (100 U/ml Penicillin and 100 μg/ml Streptomycin) at 37°C, 5% CO2 and passaged ~ every 3 days. Assays were performed in duplicates in flat bottom 96 well tissue culture trays (Sarstedt 83.3924) seeded with either ~2.5 × 104 cells or 5 × 104 cells per well. After 24 h incubation, media was removed, washed once with medium without antibiotics and fresh medium supplemented with 10% (vol/vol) FBS was added. After 2 h, the NCL compounds were diluted in DMSO and added to each well at a concentration of 1% in doubling dilutions starting at the same concentrations used for MIC testing, using wells containing 1% DMSO only as control. Following exposure for 24 h, WST-1 reagent was added to each well at a final concentration of 10%. Absorbance at A450 nm on a Multiskan Ascent 354 Spectrophotometer (Labsystems) was recorded after 1 h of incubation. The IC50 value for each compound against each cell line was determined via non-linear regression (three parameters) using GraphPad Prism v6 software. The robustness of assays was confirmed with selected cell lines (Hep G2 and MDBK) seeded onto a 96–well Microwell Nunclon Delta Surface plate (Thermo Scientific Nunc; Cat No: 136101) at 5 × 104 cells per well. Viability of each cell line in the presence of 2 or 8 μg/ml of each compound was assessed at 1 h intervals for 20 h at 37°C and 5% CO2 on a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek) using the RealTime-Glo MT Cell Viability Assay reagent (Promega). Viability of each cell line in the presence of 2.5, 5, 10 or 20 mM EDTA was also assessed using the same reagent.
Results
In this study, building on our previous exploration of the key structure activity relationship of early stage robenidine (NCL812) analogs against S. aureus and Enterococcus spp.,[42] we evaluated the in vitro and in vivo antibacterial activities of two new analogs in comparison to the parent NCL812 as potential antibacterial agents for both S. pneumoniae and S. aureus infection. We developed a series of analogs through condensation of a range of aldehydes and phenones with diaminoguanidine hydrochloride [42]. Subsequent screening for antibacterial activity led to the identification of two contrasting analogs, NCL219 ((E)-N'-((E)-1-(4-(tert-butyl)phenyl)ethylidene)-2-(1-(4-(tert-butyl)phenyl)ethylidene)hydrazine-1-carboximidhydrazide) and NCL195 (4,6-bis(2-((E)-4-methylbenzylidene)hydrazinyl)pyrimidin-2-amine) with the latter resulting from installation of a 2,4,6-triaminopyrimidine as a guanidyl moiety bioisostere (Fig 1).
Fig 1. Structure activity relationship between NCL812, NCL195 and NCL219.

Installation of a 4-tert-butyl and a C-methyl imine moiety provided NCL219 with considerably enhanced hydrolytic stability while retaining the excellent antimicrobial activity of NCL812, while guanidine to 2,4,6-triaminopyrimindine bioisosteric modification yielded NCL195, which allowed potency and drug-like character enhancement.
NCL compounds are bactericidal against S. pneumoniae, S. aureus and vancomycin-resistant enterococci
To examine the antimicrobial activities of NCL195, NCL219 and NCL812, we carried out MIC and MBC testing of the three compounds against 21 S. pneumoniae isolates representing a broad range of serotypes, and 23 clinical S. aureus isolates of diverse multilocus sequence types. MIC testing of the three compounds was also carried out on 20 vancomycin-resistant enterococcal (VRE) isolates, using Enterococcus faecalis ATCC 29212 as a control strain. For S. pneumoniae, the MIC range for NCL812 and NCL195 were the same (2–8 μg/ml), while the MIC range for NCL219 was 1–8 μg/ml (Table 1). In the presence of 10% foetal bovine serum (FBS), the MIC range for the three compounds remained the same (S1 Table). For S. aureus, the MIC range was 2–8 μg/ml for NCL812 (consistent with previous observation with NCL812 [42]; 1–2 μg/ml for NCL195; and 8 - >64 μg/ml for NCL219 (Table 1). However, in the presence of 10% FBS, the MIC range increased 4-8-fold to 32 μg/ml for NCL812 (again consistent with previous observation with NCL812 [42]; the MIC range increased 4-fold to 4–8 μg/ml for NCL195, while the MIC range remained essentially the same at 4->64 μg/ml for NCL219 (S2 Table). For the VRE isolates, the MIC range for NCL812 and NCL195 were the same (2–4 μg/ml), while the MIC range for NCL219 was 1–2 μg/ml. (Table 1). In the presence of 10% FBS, the MIC range increased 8-16-fold to 32 μg/ml for NCL812, the MIC range increased 4-fold to 16–32 μg/ml for NCL195, while the MIC range increased 8-fold to 8–16 μg/ml for NCL219 (S3 Table). When the MIC determination for NCL195 against S. pneumoniae D39 and S. aureus 49775 was carried out in the presence of human, horse or mouse serum, the MIC remained the same against D39, but a 4-fold increase in MIC was observed against S. aureus 49775, again as seen previously with NCL812 [42].
Table 1. MIC range values for NCL812, NCL195 and NCL219 compared to MIC90 values for daptomycin and ampicillin against S. pneumoniae, S. aureus and vancomycin resistant enterococci (VRE).
| Bacterial strain (No of isolates) | MIC range or MIC90 (μg/ml) | ||||
|---|---|---|---|---|---|
| NCL812 | NCL195 | NCL219 | Daptomycin | Ampicillin | |
| S. pneumoniae (21) | 2–8 | 2–8 | 1–8 | 0.5 | 0.5 |
| S. aureus (23) | 2–8 | 1–2 | 8- >64 | 0.5 | >16 |
| VRE (20) a | 2–4 | 2–4 | 1–2 | 2 | 2 |
a University of South Australia strain collection.
NCL195 kills S. aureus without detectable resistance
We next performed a kill kinetic assay for NCL812, NCL195 and NCL219 against S. pneumoniae D39 in a micro-dilution series, with a starting concentration at 64 μg/ml. Our results show that the MBC for the three compounds against S. pneumoniae D39 was 8, 8 and 4 μg/ml, respectively (equivalent to their respective MIC90), confirming that the three compounds are bactericidal against S. pneumoniae. Similar MIC and MBC results were obtained using S. pneumoniae D39. For S. aureus Xen29, the MBC for NCL812, NCL195 and NCL219 was 4, 2 and 16 μg/ml, respectively, confirming their bactericidal activity against another clinically-relevant Gram-positive pathogen. Finally, we interrogated the time-kill profile of NCL195 against either S. pneumoniae D39 or S. aureus Xen29, using 2× and 4× MIC of NCL195, with 4× MIC of ampicillin (or 4× MIC of daptomycin) and normal saline serving as controls. We found that it took 6 h to reduce the test population of D39 below the limit of detection at 2× MIC of NCL195; the time-to-clear was reduced to 4 h using 4× MIC of NCL195. Ampicillin at 4× MIC required 8 h to effect clearance of D39 bacteria below the limit of detection (Fig 2A). For S. aureus Xen29, it took 6 h to clear all Xen29 at 2× and 4× MIC of NCL195, and at 4× MIC of ampicillin (Fig 2B), while it took only 1 h to clear all Xen29 at 4× MIC of daptomycin (Fig 2C). After 24 h, some S. aureus regrowth was observed for NCL195 and ampicillin at the concentrations tested. We confirmed that the rebound growth was neither due to chemical instability of NCL195 or emergence of a resistant population by adding 105 or 103 c.f.u. of S. aureus Xen29 in LB broth containing 1×MIC, 2×MIC and 4×MIC of NCL195 over 24–72 h, using daptomycin at either 1×MIC or 4×MIC as control. In this assay, no regrowth was observed in any of the samples upon plating on antibiotic-free plates after 24 h. However, after 72 h incubation in the presence of antibiotic, a few colonies were obtained from broth containing 105 c.f.u. of Xen29 to which 1×MIC, 2×MIC and 4×MIC of NCL195 was added. We subjected the colonies that grew after 72 h to MIC testing and these returned 1× MIC for NCL195.
Fig 2. Time kill and point of resistance assays.

NCL195 was prepared at 2× and 4× MIC (using 4× MIC of ampicillin (or 4× MIC of daptomycin) and normal saline as controls) in either LB broth supplemented with 3% lysed horse blood and 5% horse serum for S. pneumoniae D39 (A) or LB broth without supplementation for S. aureus Xen29 (B and C). Cultures were incubated statically at 37°C, 5% CO2 (for S. pneumoniae D39) or at 37°C with agitation at 200 rpm (for S. aureus Xen29). Samples withdrawn at indicated times and plated on HBA overnight at 37°C, 5% CO2 (for S. pneumoniae D39) or at 37°C with aeration (for S. aureus Xen29) for bacterial enumeration. For (D), S. aureus Xen29 was grown in 2 ml LB broth the presence of 0.5×MIC, 0.75×MIC, 1×MIC, 1.5×MIC, 2×MIC, and 4×MIC of NCL195, using 1×MIC, 4×MIC and 8×MIC of daptomycin as control.
We then explored the possibility of antimicrobial resistance to NLC195 by performing 24 daily sequential cultures of S. aureus in the presence of 0.5×MIC, 0.75×MIC, 1×MIC, 1.5×MIC, 2×MIC, and 4×MIC of NCL195, using 1×MIC, 4×MIC and 8×MIC of daptomycin as control. We could not find any resistant mutant above 1× MIC for NCL195 over the 24 passages, although resistance to daptomycin increased up to 8×MIC by day 12 of the serial passage (Fig 2D). This was confirmed by MIC testing of the bacteria that grew at the respective MICs.
NCL195 is bactericidal against Gram-negative bacteria
Our recent studies whereby NCL812 was co-administered with a sub-inhibitory concentration of polymyxin B nonapeptide (a compound known to disrupt the outer membrane of Gram-negative bacteria but with negligible antimicrobial activity) demonstrated Gram-negative activity comparable with clofazimine [42]. We also explored the potential activity of NCL195 against Gram-negative bacteria in the presence or absence of EDTA or polymyxin B, agents also known to disrupt the outer membrane of Gram-negative bacteria [54]. Interestingly, NCL195 in the absence of EDTA was bactericidal against 8 A. calcoaceticus isolates (MIC range from 4–32 μg/ml) and one A. anitratus isolate (MIC = 4 μg/ml) (S4 Table) and was also bactericidal against A. baumannii, E. coli, K. pneumoniae and P. aeruginosa (MICs of 0.25–8 μg/ml) in the presence of sub-inhibitory concentrations of EDTA and MICs of 0.25–1 μg/ml in the presence of sub-inhibitory concentrations of polymyxin B (Table 2).
Table 2. Antimicrobial activity of NCL195 against Gram-negative bacteria alone and in combination with EDTA or polymyxin B.
| Bacterial strain | Agent | Combination | |||||
|---|---|---|---|---|---|---|---|
| EDTA (mM) | Polymyxin B (μg/ml) | NCL195 (μg/ml) | EDTA (mM) | NCL 195 (μg/ml) | Polymyxin B (μg/ml) | NCL 195 (μg/ml) | |
| A. baumannii ATCC 12457 | 0.5 | 0.5 | >128 | 0.25 | 4 | 0.125 | 0.5 |
| A. baumannii ATCC 19606 | 1 | 0.5 | >128 | 0.5 | 2 | 0.125 | 0.5 |
| E. coli ATCC 11229 | 2.5 | 0.25 | >128 | 0.625 | 8 | 0.125 | 0.25 |
| E. coli ATCC 25922 | 10 | 0.25 | >128 | 2.5 | 4 | 0.125 | 0.25 |
| K. pneumoniae ATCC 4352 | >30 | 0.25 | >128 | 10 | 4 | 0.125 | 0.5 |
| K. pneumoniae ATCC 33495 | >30 | 0.25 | >128 | 10 | 4 | 0.125 | 0.25 |
| P. aeruginosa ATCC 27853 | 10 | 0.5 | >128 | 5 | 2 | 0.125 | 1 |
| P. aeruginosa PA01 | 2.5 | 0.5 | >128 | 1.25 | 0.25 | 0.125 | 1 |
NCL812 exerts its antibacterial action on the cell membrane of S. pneumoniae and S. aureus
To gain further insight into how NCL compounds exert their antibacterial activity on Gram-positive bacteria, we initially examined the effect on the overall cellular architecture and integrity of S. pneumoniae D39 after 6 h exposure to 16 μg/ml NLC812. Morphometric analysis by transmission electron microscopy (TEM) revealed significant changes to the cell membrane of D39 compared to untreated controls. Treated samples possessed significantly thicker cell membranes (6.43 ± 0.29 nm) compared to untreated samples (4.35 ± 0.24 nm) (p < 0.0001) (Fig 3A; Panels A-D in S1 Fig). The periplasmic space (intracellular space between the cell membrane and the cell wall) of D39 treated with 16 μg/ml NCL812 was also significantly wider (4.54 ±0.096 nm) compared to untreated bacteria (3.91 ± 0.14 nm) (p < 0.001) (Fig 3B; Panels E-H in S1 Fig). Macromolecular (DNA, RNA, protein, cell wall, and lipid) synthesis inhibition studies on S. aureus ATCC 29213 exposed to NCL812 also suggested that NCL812 may interact with the cell membrane (Fig 3C; S2 Fig), causing leakage of vital metabolites (Fig 3D).
Fig 3. NCL812 compounds exert their antibacterial action on the cell membrane of S. pneumoniae and S. aureus.

(A and B), S. pneumoniae strain D39 exposed to 16 μg/ml NCL812 for 6 h exhibited significantly thicker cell membranes compared to untreated samples (A) (p < 0.0001; two-tailed unpaired t-test) and displayed significantly wider periplasmic space compared to untreated samples (B) (p < 0.001; two-tailed unpaired t-test. Data presented are an example from 12 different bacterial cells, each with at least 10 measurements per bacteria for both treated and untreated samples. (C and D) NCL812 affects macromolecular synthesis (c) and ATP release (D) in S. aureus. (E and F), NCL Compounds dissipate the membrane potential of S. pneumoniae and S. aureus. Membrane potential measurements of S. pneumoniae D39 (E) and S. aureus ATCC49775 (F). Bacterial suspensions were exposed to 16 μg/ml NCL812, NCL195, NCL219, or ampicillin (control) for 5 min after which DiOC2(3) was added and the fluorescence monitored until it plateaued. Cells were then re-energized with 0.5% glucose and the establishment of a membrane potential was measured as an increase in fluorescence until it plateaued. The membrane potential was then disrupted by the addition of the proton ionophore (CCCP). Data presented is representative of two experiments. For full description, see Materials and Methods.
To further investigate cell membrane perturbation by the NCL compounds, the ability of S. pneumoniae and S. aureus cells to establish a membrane potential (Δψ) when challenged with 16 μg/ml NCL812, NCL195 or NCL219 was determined using the fluorescent membrane potential probe 3,3-diethyloxacarbocyanine iodide (DiOC2 (3)). We have previously used this probe to measure the magnitude and stability of Δψ in bacterial cells and proteoliposomes [51]. Bacterial cells were energized by the addition of glucose to establish a proton motive force (negative and basic inside the cell). This led to an increase in fluorescence associated with aggregation of the DiOC2 (3). When S. pneumoniae and S. aureus were pre-incubated with the NCL compounds, a large reduction in the magnitude of the generated membrane potential compared to that of the untreated cells and cells in the presence of ampicillin was observed (Fig 3E and 3F).
Stability of NCL compounds when incubated with liver microsomes
The metabolic stability of NCL812, NCL195 and NCL219 was evaluated in both human and mouse liver microsomes. NCL812 and NCL195 showed low rates of degradation in both species of liver microsomes (EH values <0.22), with degradation rates for each compound being broadly comparable between species. We did not observe measurable degradation of any of the compounds in control (non-cofactor) incubations in either species suggesting that there was no major cofactor independent metabolism contributing to their overall rates of metabolism. However, NCL219 displayed high rates of degradation in both species of liver microsomes (EH values 0.79 and 0.72, respectively; Table 3).
Table 3. Physicochemical and metabolism parameters for NCL812, NCL195 and NCL219 in human and mouse liver microsomes.
| Properties | Compound | |||||
|---|---|---|---|---|---|---|
| NCL812 | NCL195 | NCL219 | ||||
| Physicochemical Parameters | ||||||
| Mol Wta | 334.20 | 359.43 | 405.59 | |||
| PSAa (Å2) | 72.6 | 100.6 | 72.6 | |||
| FRBa | 4 | 6 | 6 | |||
| H-Bond Donora | HBD = 3 | HBA = 6 | HBD = 4 | HBA = 7 | HBD = 3 | HBA = 5 |
| pKaa | 5.0; imine (italics) | 1.2; amine (bold) | 4.9 / 4.3; amine (italics) | 2.2; pyrimidine (bold) | 5.2; imine (bold) | 2.2 amine (italics) |
| LogDpH 7. 4b | 4.5 | 4.5 | >5.3d | |||
| Solubilityc (μg/ml) | pH 2.0 = 6.3–12.5 | pH 6.5 = <1.6 | pH 2.0 = 3.1–6.3 | pH 6.5 = <1.6 | pH 2.0 = 0.78–1.6 | pH 6.5 = 0.78–1.6 |
| Metabolism parameters | ||||||
| Species | Human | Mouse | Human | Mouse | Human | Mouse |
| T1/2 (min) | >247 | >247 | >247 | >247 | 18 | 14 |
| CLINT (μl/min/mg protein) | <7 | <7 | <7 | <7 | 94 | 121 |
| Predicted CLblood (ml/min/kg) | <5 | <16 | <5 | <16 | 16 | 87 |
| Predicted EH | <0.22 | <0.13 | <0.22 | <0.13 | 0.79 | 0.72 |
a Calculated using ChemAxon (JChem for Excel).
b Estimated using a gradient HPLC chromatographic method.
c Kinetic solubility estimated via nephelometry.
d the retention times were greater than the most lipophilic standard compound, therefore LogD values are reported as > 5.3.
Mol Wt = molecular weight; PSA = polar surface area; HBD/HBA = hydrogen bond donor / acceptor; pKa is colour coded to the atoms highlighted in each structure. Clint = in-vitro intrinsic clearance; EH = predicted in-vivo hepatic extraction ratio.
NCL compounds exhibit high plasma concentration and low plasma clearance rates
To examine if NCL812 and NCL195 had appropriate pharmacokinetic (PK) profiles in vivo for potential therapeutic use, we evaluated key PK parameters including maximum plasma concentration (Cmax) and apparent terminal elimination half-lives in the plasma of mice after systemic exposure. Our analysis of the PK parameters for NCL812 and NCL195 after IV administration at a dose of 5 mg/kg indicated both compounds exhibited high plasma concentrations (Fig 4A and 4B) and long apparent terminal elimination half-lives, with NCL812 showing a higher overall exposure [AUC0-inf 27.9 h*μM (S5 Table)]. Furthermore, the estimated plasma clearance values for NCL812 and NCL195 were low (9.5 and 16.3 ml/min/kg, respectively), consistent with the in vitro metabolic stability in both human and mouse liver microsomes, and with the microsome-predicted blood clearance values (assuming a blood-to-plasma partitioning ratio close to 1). However, parenteral administration of NCL812 resulted in adverse neurological signs requiring the mice to be anaesthetized for the remainder of the experiment.
Fig 4. NCL812 and NCL195 exhibit high plasma concentration and low plasma clearance rates.
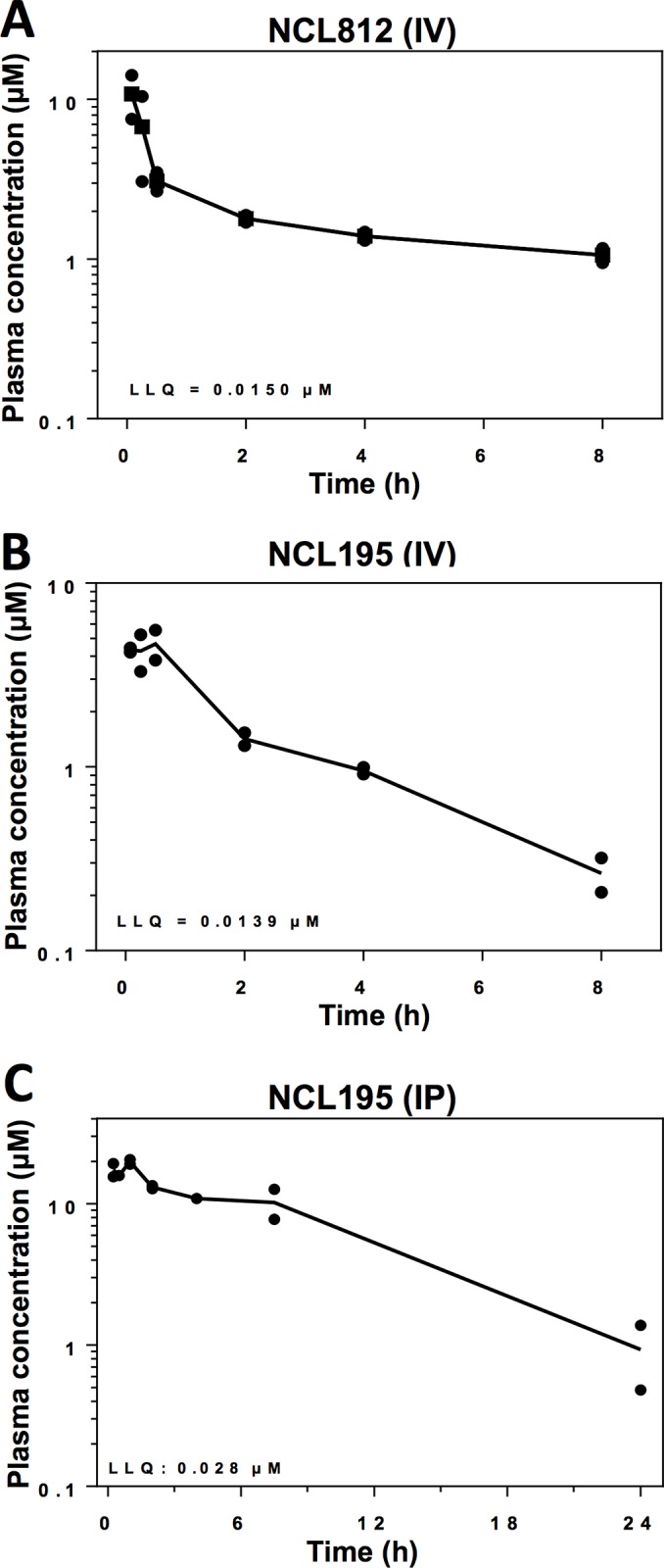
Pharmacokinetic parameters for NCL812 (A) and NCL195 (B) after IV administration at a dose of 5 mg/kg to male CD1 mice (n = 8 per compound), indicating that both compounds exhibited high plasma concentrations and long apparent terminal elimination half-lives. (C), Pharmacokinetic parameters for NCL195 after IP administration at a dose of 43 mg/kg to male CD1 mice (n = 2 per time point), indicating that NCL195 was rapidly absorbed after dosing, and plasma concentrations remained above 3–4 μg/ml for the initial 7.5 h post-dose period.
We then evaluated the PK parameters of NCL195 after intraperitoneal (IP) administration to Swiss mice at a dose of 43 mg/kg. The plasma concentration-time profile (Fig 4C) indicated NCL195 was rapidly absorbed after dosing, and plasma concentrations remained above 3–4 μg/ml for the initial 7.5 h post-dose period, falling to 0.2–0.5 μg/ml between 7.5 and 24 h post-dose. No adverse reactions or compound-related side effects were observed in any of the mice following IP administration of NCL195 at a dose of 43 mg/kg.
NCL195’s haemolytic activity and cytotoxicity to mammalian cell lines
We evaluated the toxicity profile of the three compounds in a panel of normal and carcinoma mammalian cell lines representing different tissue types in order to assess the potential of off-target or dose-limiting toxicity using the WST-1 Cell Proliferation Assay reagent (Roche). The results of the in vitro toxicity measurements show IC50 values of >4 μg/ml for NCL812, IC50 values of >7 μg/ml for NCL219, while the cytotoxicity profile for NCL195 was the most favorable, with IC50 values of >10 μg/ml against all the cell lines tested (S6 Table). Real-time cell viability measurements using Hep G2 and MDBK cell lines also show no measurable effect on cell viability for NCL195 at either 2 or 8 μg/ml up to 20 h post-treatment (Fig 5). However, NCL812 affected viability at 8 μg/ml, while NCL219 had a demonstrable effect on viability at 2 μg/ml.
Fig 5. NCL195 demonstrates limited cytotoxicity to mammalian cell lines.

Real-time cell viability measurements for Hep G2 (A, B, C) and MDBK (D, E, F) cells after treatment with 2 or 8 μg/ml NCL812, NCL195 or NCL219. Cell viability was measured every 60 min for 20 h at 37°C and 5% CO2 on a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek) using the RealTime-Glo MT Cell Viability Assay reagent (Promega). Data are means (± s.e.m.) relative light units (RLU) for each treatment per time point (in duplicate).
Haemolytic activity of NCL812, NCL195 and NCL219 against human RBCs showed that the 50% haemolysis titre (HC50) for NCL195 was >128 μg/ml (the highest concentration used) and is comparable to that for ampicillin, indicating that NCL195 is well tolerated by RBCs. In contrast, the HC50 for NCL812 and NCL219 were 24 and 6 μg/ml, respectively, showing some level of toxicity to human RBCs.
Discussion
Bacterial pathogens have evolved numerous strategies to adapt to, and thrive in, different environments that they encounter during infection. A major challenge to treatment and control of bacterial pathogens is the emergence and rapid global spread of multidrug-resistant clones that are refractory to last line antimicrobial therapy. To address this problem, we have examined the anticoccidial agent robenidine (NCL812) as a parent scaffold for developing a new antimicrobial class with a potentially novel mechanism of action. Two analogs of NCL812, NCL219 and NCL195, were viewed as promising candidates with the installation of a 4-tert-butyl and a C-methyl imine moiety providing NCL219 with considerably enhanced hydrolytic stability while retaining the excellent antimicrobial activity of NCL812. Guanidine to 2,4,6-triaminopyrimindine bioisosteric modification to yield NCL195 allowed potency and drug-like character enhancement. To evaluate their potential as antibacterial agents, we assessed their metabolic stability as well as their pharmacokinetic and safety profiles in a series of in vitro and in vivo analyses. Furthermore, we assessed in vitro efficacy against a range of S. pneumoniae, S. aureus and VRE isolates, and examined if activity was extended to Gram-negative bacteria with and without the presence of sub-inhibitory concentrations of EDTA or polymyxin B.
Our preliminary in vitro and in vivo studies demonstrated NCL812 and NCL195 possess a number of desirable antimicrobial characteristics such as high metabolic stability, prolonged plasma concentration and low plasma clearance rates and fast-acting concentration-dependent bactericidal activity that targets the cell membrane of S. pneumoniae and S. aureus. NCL195 showed the most favourable in vitro cell cytotoxicity data of the three NCL compounds tested across a range of cell lines, including Caco-2 cells. NCL195’s in vitro toxicity data was better across all cell lines to that of the parent compound robenidine (NCL812), an oral anticoccidial drug already approved for veterinary use, and showed no haemolysis of human red blood cells at the highest concentration tested (128 μg/ml). It is possible that in vitro cytotoxicity of the NCL compounds is not predictive of in vivo toxicity, in agreement with reports by others who note the importance of in vitro responses but conclude that the true profile of compound toxicity can only be determined in vivo [55–58]. However, NCL812 did elicit adverse clinical signs when administered intravenously to mice at a dose rate of 5mg/kg. Importantly, no compound attributable adverse effects were noted in mice treated with NCL195 which has undergone a major structural modification in the aminoguanidine core compared to NCL812, though further investigation of in vivo safety and further medicinal chemistry development is considered desirable and necessary before considering proof of concept in vivo efficacy trials in bioluminescent sepsis models.
We previously obtained preliminary data supporting our hypothesis that the site of action of robenidine analogs is present in the Gram-negative bacteria [42], and similar results were obtained for NCL195 in the present study against N. meningitidis and Gram-negative ESKAPE pathogens such as Acinetobacter spp. Importantly, NCL195 in the presence of sub-inhibitory concentrations of EDTA or polymyxin B was active against a variety of additional ESKAPE pathogens. Overall, these are desirable characteristics driving further exploration of robenidine analogs as a novel antimicrobial class to treat acute bacterial infections. The finding that the MICs of NCL195 against strains of S. aureus and VREs were increased 4-fold in the presence of serum suggests a high protein binding, which may be one of the factors allowing enhanced stability and plasma lifetime without necessarily reducing its effectiveness in vivo [59]. As the NCL compounds possess a mechanism of action that targets the cell membrane, they could be more effective than other bactericidal concentration-dependent antimicrobials that have intracellular targets, such as fluoroquinolones and aminoglycosides. Morphometric analysis of the cell membrane and periplasmic space of D39 treated with NCL812 for 6 h showed the cell membrane and periplasmic space was larger in treated samples, compared to control samples. Previous findings with daptomycin [60, 61] and telavancin [62] have described disruption of cell membrane integrity. Our fluorescence membrane potential measurements provide confirmation that the NCL compounds permeabilize the cytoplasmic membrane of S. pneumoniae and S. aureus, thereby hindering the establishment and maintenance of essential energy source for cell functioning.
In a comparative resistance selection study of NCL195 and daptomycin, no resistance to NCL195 developed above the MIC for S. aureus over 24 daily serial passages, whereas resistance to daptomycin developed by day 5, and increased up to 8×MIC by day 12 of the serial passage. Together, these findings demonstrate that NCL195 has potential for further pharmaceutical and medicinal chemistry development to increase solubility, reduce plasma binding and toxicity as well as enhance in vitro and in vivo potency against leading bacterial pathogens.
Supporting information
(A-D); Comparative visual differences between the cell membranes of treated (A and B) and untreated (C and D) S. pneumoniae D39 samples. The cell membranes of D39 cells exposed to 16 μg/ml NCL812 (A and B) for 6 h were visually thicker compared to untreated D39 grown for 6 h (C and D). Length markers represent the cell membrane thickness. (E-H); Comparative visual differences in the periplasmic space of treated (E and F) and untreated (G and H) S. pneumoniae D39 samples. The periplasmic space width of D39 cells exposed to 16 μg/ml NCL812 for 6 h (E and F) was visually larger compared to untreated D39 grown for 6 h (G and H). Length markers represent the thickness of the periplasmic space. Measurements are representative of 12 bacterial cells for each treatment.
(DOC)
NCL812 inhibited DNA (A), RNA (B), protein (C), cell wall (D), and lipid (E) pathways in exponentially growing culture of S. aureus, suggesting that NCL812 may interact with the cell membrane. Data are means ± s.e.m. values from triplicate samples for each treatment.
(DOC)
Each MIC test was performed in duplicate. ND = Not determined.
(DOCX)
Each MIC test was performed in duplicate. MLST = Multi-locus Sequence Type; CA = Community Acquired; HA = Healthcare-Associated; MRSA = Methicillin-Resistant S. aureus; EMRSA = Epidemic Methicillin-Resistant S. aureus; MSSA = Methicillin-Sensitive S. aureus; PVL = Panton-Valentine Leukocidin status; ND = Not determined.
(DOCX)
Each MIC test was performed in duplicate. a Porcine VRE isolates were obtained from The University of South Australia collection. b E. faecalis ATCC 29212 was used as a control. ND = Not determined.
(DOCX)
(DOCX)
a Terminal elimination phase was not well defined, value is an approximation only. 8 mice were used per compound.
(DOCX)
Data presented are mean IC50 values from duplicate samples from one experiment. Each experiment was performed twice.
(DOCX)
(DOCX)
Acknowledgments
We wish to thank Ms Ruth Williams of the Centre for Advanced Microscopy and Microanalysis at Adelaide Microscopy, The University of Adelaide for assistance with TEM. We thank Ms Jan Bell of the Australian Centre for Antimicrobial Resistance Ecology for the supply of Acinetobacter strains. We also wish to thank Ms Rumana Mowla, Ms Amanda Ruggero and Ms Krishna Kathawala at the University of South Australia for assistance with mechanism of action studies, technical assistance, and drug formulation, respectively.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by an Australian Research Council (ARC; arc.gov.au) Linkage grant (LP110200770) to D.J.T., A.M. and S.W.P. with Neoculi Pty Ltd as the Partner Organization. Additionally, it was funded in part by a grant from the National Health and Medical Research Council of Australia (NHMRC; nhmrc.gov.au) Program Grant 565526 to J.C.P., Project Grant 627142 to J.C.P. and A.D.O., and University of South Australia and the Sansom Institute of Health Research fund to H.V. Micromyx LLC provided support in the form of salary for D.L.S., but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of all authors are articulated in the ‘author contributions’ section.
References
- 1.World Health Organization Media Centre; The top 10 causes of death: Fact sheet No 310. 2014.
- 2.Murray CJ, Barber RM, Foreman KJ, Abbasoglu Ozgoren A, Abd-Allah F, Abera SF, et al. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990–2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145–91. Epub 2015/09/01. doi: 10.1016/S0140-6736(15)61340-X ; PubMed Central PMCID: PMC4673910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization Media Centre; Pneumonia: Fact sheet No 331. 2015.
- 4.Olarte L, Barson WJ, Barson RM, Lin PL, Romero JR, Tan TQ, et al. Impact of the 13-Valent Pneumococcal Conjugate Vaccine on Pneumococcal Meningitis in US Children. Clin Infect Dis. 2015;61(5):767–75. Epub 2015/05/15. doi: 10.1093/cid/civ368 . [DOI] [PubMed] [Google Scholar]
- 5.Weinberger DM, Malley R, Lipsitch M. Serotype replacement in disease after pneumococcal vaccination. Lancet. 2011;378(9807):1962–73. Epub 2011/04/16. doi: 10.1016/S0140-6736(10)62225-8 ; PubMed Central PMCID: PMC3256741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calfee DP. Crisis in hospital-acquired, healthcare-associated infections. Annu Rev Med. 2012;63:359–71. Epub 2011/10/25. doi: 10.1146/annurev-med-081210-144458 . [DOI] [PubMed] [Google Scholar]
- 7.Turnidge JD, Kotsanas D, Munckhof W, Roberts S, Bennett CM, Nimmo GR, et al. Staphylococcus aureus bacteraemia: a major cause of mortality in Australia and New Zealand. Med J Aust. 2009;191(7):368–73. Epub 2009/10/08. . [DOI] [PubMed] [Google Scholar]
- 8.Knox J, Van Rijen M, Uhlemann AC, Miller M, Hafer C, Vavagiakis P, et al. Community-associated methicillin-resistant Staphylococcus aureus transmission in households of infected cases: a pooled analysis of primary data from three studies across international settings. Epidemiol Infect. 2015;143(2):354–65. Epub 2014/04/26. doi: 10.1017/S0950268814000983 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hart J, Christiansen KJ, Lee R, Heath CH, Coombs GW, Robinson JO. Increased EMRSA-15 health-care worker colonization demonstrated in retrospective review of EMRSA hospital outbreaks. Antimicrob Resist Infect Control. 2014;3(1):7 Epub 2014/03/05. doi: 10.1186/2047-2994-3-7 ; PubMed Central PMCID: PMC3944736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thampi N, Showler A, Burry L, Bai AD, Steinberg M, Ricciuto DR, et al. Multicenter study of health care cost of patients admitted to hospital with Staphylococcus aureus bacteremia: Impact of length of stay and intensity of care. Am J Infect Control. 2015;43(7):739–44. Epub 2015/03/15. doi: 10.1016/j.ajic.2015.01.031 . [DOI] [PubMed] [Google Scholar]
- 11.World Health Organization. Antimicrobial resistance: global report on surveillance. 2014.
- 12.ECDC. Antimicrobial resistance surveillance in Europe 2014. Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net). Stockholm: ECDC; 2015. 2015. doi: 10.2900/23549 PubMed PMID: 26607473. [Google Scholar]
- 13.Leung MH, Oriyo NM, Gillespie SH, Charalambous BM. The adaptive potential during nasopharyngeal colonisation of Streptococcus pneumoniae. Infect Genet Evol. 2011;11(8):1989–95. Epub 2011/09/20. doi: 10.1016/j.meegid.2011.09.002 . [DOI] [PubMed] [Google Scholar]
- 14.Palmu AA, Jokinen J, Nieminen H, Rinta-Kokko H, Ruokokoski E, Puumalainen T, et al. Effect of pneumococcal Haemophilus influenzae protein D conjugate vaccine (PHiD-CV10) on outpatient antimicrobial purchases: a double-blind, cluster randomised phase 3–4 trial. Lancet Infect Dis. 2014;14(3):205–12. Epub 2013/11/30. doi: 10.1016/S1473-3099(13)70338-4 . [DOI] [PubMed] [Google Scholar]
- 15.Vaz LE, Kleinman KP, Lakoma MD, Dutta-Linn MM, Nahill C, Hellinger J, et al. Prevalence of Parental Misconceptions About Antibiotic Use. Pediatrics. 2015;136(2):221–31. Epub 2015/07/22. doi: 10.1542/peds.2015-0883 ; PubMed Central PMCID: PMC4516948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmed S, Shapiro NL, Bhattacharyya N. Incremental health care utilization and costs for acute otitis media in children. Laryngoscope. 2014;124(1):301–5. Epub 2013/05/08. doi: 10.1002/lary.24190 . [DOI] [PubMed] [Google Scholar]
- 17.Henken S, Bohling J, Martens-Lobenhoffer J, Paton JC, Ogunniyi AD, Briles DE, et al. Efficacy profiles of daptomycin for treatment of invasive and noninvasive pulmonary infections with Streptococcus pneumoniae. Antimicrob Agents Chemother. 2010;54(2):707–17. Epub 2009/11/18. doi: 10.1128/AAC.00943-09 ; PubMed Central PMCID: PMC2812129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoshino K, Inoue K, Murakami Y, Kurosaka Y, Namba K, Kashimoto Y, et al. In vitro and in vivo antibacterial activities of DC-159a, a new fluoroquinolone. Antimicrob Agents Chemother. 2008;52(1):65–76. Epub 2007/10/17. doi: 10.1128/AAC.00853-07 ; PubMed Central PMCID: PMC2223876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones RN, Fritsche TR, Sader HS. Antimicrobial activity of DC-159a, a new fluoroquinolone, against 1,149 recently collected clinical isolates. Antimicrob Agents Chemother. 2008;52(10):3763–75. Epub 2008/06/25. doi: 10.1128/AAC.00294-08 ; PubMed Central PMCID: PMC2565897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynch JP 3rd, Zhanel GG. Streptococcus pneumoniae: epidemiology, risk factors, and strategies for prevention. Semin Respir Crit Care Med. 2009;30(2):189–209. Epub 2009/03/20. doi: 10.1055/s-0029-1202938 . [DOI] [PubMed] [Google Scholar]
- 21.Smith JR, Yim J, Raut A, Rybak MJ. Oritavancin combinations with beta-lactams against multidrug-resistant Staphylococcus aureus and vancomycin-resistant enterococci. Antimicrob Agents Chemother. 2016. Epub 2016/02/03. doi: 10.1128/AAC.03006-15 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De la Calle C, Morata L, Cobos-Trigueros N, Martinez JA, Cardozo C, Mensa J, et al. Staphylococcus aureus bacteremic pneumonia. Eur J Clin Microbiol Infect Dis. 2016. Epub 2016/01/19. doi: 10.1007/s10096-015-2566-8 . [DOI] [PubMed] [Google Scholar]
- 23.David MZ, Daum RS. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev. 2010;23(3):616–87. Epub 2010/07/09. doi: 10.1128/CMR.00081-09 ; PubMed Central PMCID: PMC2901661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groves MD, Crouch B, Coombs GW, Jordan D, Pang S, Barton MD, et al. Molecular Epidemiology of Methicillin-Resistant Staphylococcus aureus Isolated from Australian Veterinarians. PLoS One. 2016;11(1):e0146034 Epub 2016/01/07. doi: 10.1371/journal.pone.0146034 ; PubMed Central PMCID: PMC4703204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghafur A. Call for global action to halt the superbug. Med J Aust. 2013;198(5):251 Epub 2013/03/19. . [DOI] [PubMed] [Google Scholar]
- 26.Prasad S, Smith P. Meeting the threat of antibiotic resistance: Building a new frontline defence. Occasional Paper Series. Canberra, Office of the Chief Scientist. 7. 2013.
- 27.Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2015. Epub 2015/11/26. doi: 10.1016/S1473-3099(15)00424-7 . [DOI] [PubMed] [Google Scholar]
- 28.Falgenhauer L, Ghosh H, Doijad S, Yao Y, Bunk B, Sproer C, et al. Genome analysis of the carbapenem- and colistin-resistant Escherichia coli isolate NRZ14408 reveal horizontal gene transfer pathways towards pan-resistance and enhanced virulence. Antimicrob Agents Chemother. 2017. Epub 2017/01/18. doi: 10.1128/aac.02359-16 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bush K. Investigational Agents for the Treatment of Gram-Negative Bacterial Infections: A Reality Check. ACS Infectious Diseases. 2015;1:509–11. doi: 10.1021/acsinfecdis.5b00100 [DOI] [PubMed] [Google Scholar]
- 30.Perez F, El Chakhtoura NG, Papp-Wallace K, Wilson BM, Bonomo RA. Treatment Options for Infections Caused by Carbapenem-resistant Enterobacteriaceae: Can We Apply “Precision Medicine” to Antimicrobial Chemotherapy? Expert Opinion on Pharmacotherapy. 2016; doi: 10.1517/14656566.2016.1145658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poirel L, Kieffer N, Liassine N, Thanh D, Nordmann P. Plasmid-mediated carbapenem and colistin resistance in a clinical isolate of Escherichia coli. Lancet Infect Dis. 2016. Epub 2016/01/18. doi: 10.1016/S1473-3099(16)00006-2 . [DOI] [PubMed] [Google Scholar]
- 32.IDSA. Infectious Diseases Society of America. The 10 X ‘20 Initiative: Pursuing a Global Commitment to Develop 10 New Antibacterial Drugs by 2020. Clinical Infectious Diseases. 2010;50:1081–3. doi: 10.1086/652237 [DOI] [PubMed] [Google Scholar]
- 33.Bush K. New antimicrobial agents for Gram-negative pathogens in pipelines. International Journal of Antimicrobial Agents. 2015;45(Supplement 2), S10. [Google Scholar]
- 34.Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline in 2013. J Antibiot (Tokyo). 2013;66(10):571–91. Epub 2013/09/05. doi: 10.1038/ja.2013.86 . [DOI] [PubMed] [Google Scholar]
- 35.Eliopoulos G. New antimicrobial agents for Gram-positive pathogens in pipelines. International Journal of Antimicrobial Agents. 2015;45(Supplement 2), S5. [Google Scholar]
- 36.Silver LL. New Targets for Antibacterial Compounds. Antibiotics: Current Innovations and Future Trends Sánchez S. and Demain A. L. Portland, Oregon, Caister Academic Press; 2015. [Google Scholar]
- 37.Kantor S, Kennett RL Jr., Waletzky E, Tomcufcik AS. 1,3-Bis(p-chlorobenzylideneamino)guanidine hydrochloride (robenzidene): new poultry anticoccidial agent. Science. 1970;168(3929):373–4. Epub 1970/04/17. . [DOI] [PubMed] [Google Scholar]
- 38.EFSA. Scientific Opinion on safety and efficacy of Cycostat 66G (robenidine hydrochloride) for rabbits for breeding and fattening. EFSA Journal. 2011;9(3):2102. [Google Scholar]
- 39.Brown D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat Rev Drug Discov. 2015;14(12):821–32. Epub 2015/10/24. doi: 10.1038/nrd4675 . [DOI] [PubMed] [Google Scholar]
- 40.Chong CR, Sullivan DJ Jr. New uses for old drugs. Nature. 2007;448(7154):645–6. Epub 2007/08/10. doi: 10.1038/448645a . [DOI] [PubMed] [Google Scholar]
- 41.O’Neill J. Securing new drugs for future generations: The pipeline of antibiotics. Review on Antimicrobial Resistance, O’Neill Commission. British Society for Antimicrobial Chemotherapy, May 2015. p1-41. 2015.
- 42.Abraham R, Stevens A, Young KA, Russell C, Qvist A, Khazandi M, et al. Robenidine analogues as Gram positive antibacterial agents. Journal of Medicinal Chemistry. 2016. Epub 2016/01/15. doi: 10.1021/acs.jmedchem.5b01797 . [DOI] [PubMed] [Google Scholar]
- 43.Berry AM, Paton JC. Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect Immun. 2000;68(1):133–40. Epub 1999/12/22. ; PubMed Central PMCID: PMC97112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahdi LK, Ogunniyi AD, LeMessurier KS, Paton JC. Pneumococcal virulence gene expression and host cytokine profiles during pathogenesis of invasive disease. Infect Immun. 2008;76(2):646–57. Epub 2007/11/28. IAI.01161-07 [pii] doi: 10.1128/IAI.01161-07 ; PubMed Central PMCID: PMC2223468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogunniyi AD, LeMessurier KS, Graham RMA, Watt JM, Briles DE, Stroeher UH, et al. Contributions of pneumolysin, pneumococcal surface protein A (PspA), and PspC to pathogenicity of Streptococcus pneumoniae D39 in a mouse model. Infect Immun. 2007;75(4):1843–51. doi: 10.1128/IAI.01384-06 ISI:000245421400036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Tenth Edition. CLSI Document M07-A10. Clinical and Laboratory Standards Institute 2015;35(2):1–87.
- 47.Rozengart EV, Saakov VS. The chelating ability of the anticoccidial drug 1,3-bis(p-chlorobenzilideneamino)guanidine: the complexes with Ca2+ and La3+. Dokl Biochem Biophys. 2002;385:219–23. Epub 2002/12/05. . [DOI] [PubMed] [Google Scholar]
- 48.Hamoud R, Zimmermann S, Reichling J, Wink M. Synergistic interactions in two-drug and three-drug combinations (thymol, EDTA and vancomycin) against multi drug resistant bacteria including E. coli. Phytomedicine. 2014;21(4):443–7. doi: 10.1016/j.phymed.2013.10.016 . [DOI] [PubMed] [Google Scholar]
- 49.Klugman KP, Capper T. Concentration-dependent killing of antibiotic-resistant pneumococci by the methoxyquinolone moxifloxacin. J Antimicrob Chemother. 1997;40(6):797–802. Epub 1998/02/14. . [DOI] [PubMed] [Google Scholar]
- 50.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517(7535):455–9. Epub 2015/01/07. doi: 10.1038/nature14098 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Venter H, Shilling RA, Velamakanni S, Balakrishnan L, Van Veen HW. An ABC transporter with a secondary-active multidrug translocator domain. Nature. 2003;426(6968):866–70. Epub 2003/12/20. doi: 10.1038/nature02173 . [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Mowla R, Guo L, Ogunniyi AD, Rahman T, De Barros Lopes MA, et al. Evaluation of a series of 2-napthamide derivatives as inhibitors of the drug efflux pump AcrB for the reversal of antimicrobial resistance. Bioorg Med Chem Lett. 2017. Epub 2017/01/29. doi: 10.1016/j.bmcl.2017.01.042 . [DOI] [PubMed] [Google Scholar]
- 53.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27(11):1350–9. Epub 1999/10/26. . [PubMed] [Google Scholar]
- 54.Vaara M. Agents that increase the permeability of the outer membrane. Microbiol Rev. 1992;56(3):395–411. Epub 1992/09/01. ; PubMed Central PMCID: PMCPMC372877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKim JM Jr. Building a tiered approach to in vitro predictive toxicity screening: a focus on assays with in vivo relevance. Comb Chem High Throughput Screen. 2010;13(2):188–206. Epub 2010/01/08. doi: 10.2174/138620710790596736 ; PubMed Central PMCID: PMC2908937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans SM, Casartelli A, Herreros E, Minnick DT, Day C, George E, et al. Development of a high throughput in vitro toxicity screen predictive of high acute in vivo toxic potential. Toxicol In vitro. 2001;15(4–5):579–84. Epub 2001/09/22. . [DOI] [PubMed] [Google Scholar]
- 57.Roberts J, Bingham J, McLaren AC, McLemore R. Liposomal Formulation Decreases Toxicity of Amphotericin B in vitro and in vivo. Clin Orthop Relat Res. 2015;473(7):2262–9. Epub 2015/03/26. doi: 10.1007/s11999-015-4232-y ; PubMed Central PMCID: PMC4457755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garle MJ, Fentem JH, Fry JR. In vitro cytotoxicity tests for the prediction of acute toxicity in vivo. Toxicol In vitro. 1994;8(6):1303–12. Epub 1994/12/01. . [DOI] [PubMed] [Google Scholar]
- 59.Lee BL, Sachdeva M, Chambers HF. Effect of protein binding of daptomycin on MIC and antibacterial activity. Antimicrob Agents Chemother. 1991;35(12):2505–8. Epub 1991/12/01. ; PubMed Central PMCID: PMC245421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cotroneo N, Harris R, Perlmutter N, Beveridge T, Silverman JA. Daptomycin exerts bactericidal activity without lysis of Staphylococcus aureus. Antimicrob Agents Chemother. 2008;52(6):2223–5. Epub 2008/04/02. doi: 10.1128/AAC.01410-07 ; PubMed Central PMCID: PMC2415783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Silverman JA, Perlmutter NG, Shapiro HM. Correlation of daptomycin bactericidal activity and membrane depolarization in Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47(8):2538–44. Epub 2003/07/25. doi: 10.1128/AAC.47.8.2538-2544.2003 ; PubMed Central PMCID: PMC166110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Higgins DL, Chang R, Debabov DV, Leung J, Wu T, Krause KM, et al. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2005;49(3):1127–34. Epub 2005/02/25. doi: 10.1128/AAC.49.3.1127-1134.2005 ; PubMed Central PMCID: PMC549257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A-D); Comparative visual differences between the cell membranes of treated (A and B) and untreated (C and D) S. pneumoniae D39 samples. The cell membranes of D39 cells exposed to 16 μg/ml NCL812 (A and B) for 6 h were visually thicker compared to untreated D39 grown for 6 h (C and D). Length markers represent the cell membrane thickness. (E-H); Comparative visual differences in the periplasmic space of treated (E and F) and untreated (G and H) S. pneumoniae D39 samples. The periplasmic space width of D39 cells exposed to 16 μg/ml NCL812 for 6 h (E and F) was visually larger compared to untreated D39 grown for 6 h (G and H). Length markers represent the thickness of the periplasmic space. Measurements are representative of 12 bacterial cells for each treatment.
(DOC)
NCL812 inhibited DNA (A), RNA (B), protein (C), cell wall (D), and lipid (E) pathways in exponentially growing culture of S. aureus, suggesting that NCL812 may interact with the cell membrane. Data are means ± s.e.m. values from triplicate samples for each treatment.
(DOC)
Each MIC test was performed in duplicate. ND = Not determined.
(DOCX)
Each MIC test was performed in duplicate. MLST = Multi-locus Sequence Type; CA = Community Acquired; HA = Healthcare-Associated; MRSA = Methicillin-Resistant S. aureus; EMRSA = Epidemic Methicillin-Resistant S. aureus; MSSA = Methicillin-Sensitive S. aureus; PVL = Panton-Valentine Leukocidin status; ND = Not determined.
(DOCX)
Each MIC test was performed in duplicate. a Porcine VRE isolates were obtained from The University of South Australia collection. b E. faecalis ATCC 29212 was used as a control. ND = Not determined.
(DOCX)
(DOCX)
a Terminal elimination phase was not well defined, value is an approximation only. 8 mice were used per compound.
(DOCX)
Data presented are mean IC50 values from duplicate samples from one experiment. Each experiment was performed twice.
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.