Abstract
Intestinal regeneration and tumorigenesis are believed to be driven by intestinal stem cells (ISCs). Elucidating mechanisms underlying ISC activation during regeneration and tumorigenesis can help uncover the underlying principles of intestinal homeostasis and disease including colorectal cancer. Here we show that miR-31 drives ISC proliferation, and protects ISCs against apoptosis, both during homeostasis and regeneration in response to ionizing radiation injury. Furthermore, miR-31 has oncogenic properties, promoting intestinal tumorigenesis. Mechanistically, miR-31 acts to balance input from Wnt, BMP, TGFβ signals to coordinate control of intestinal homeostasis, regeneration and tumorigenesis. We further find that miR-31 is regulated by the STAT3 signaling pathway in response to radiation injury. These findings identify miR-31 as a critical modulator of ISC biology, and a potential therapeutic target for a broad range of intestinal regenerative disorders and cancers.
Research organism: Mouse
eLife digest
Cells lining the inner wall of the gut help to absorb nutrients and to protect the body against harmful microbes and substances. Being on the front line of defense means that these cells often sustain injuries. Specialized cells called intestinal stem cells keep the tissues healthy by replacing the damaged and dying cells.
The intestinal stem cells can produce copies of themselves and generate precursors of the gut cells. They also have specific mechanism to protect themselves from cell death. These processes are regulated by different signals that are generated by the cell themselves or the neighboring cells. If these processes get out of control, cells can easily be depleted or develop into cancer cells. Until now, it remained unclear how intestinal stem cells can differentiate between and respond to multiple and simultaneous signals.
It is known that short RNA molecules called microRNA play an important role in the signaling pathways of damaged cells and during cancer development. In the gut, different microRNAs, including microRNA-31,help to keep the gut lining intact. However, previous research has shown that bowel cancer cells also contain high amounts of microRNA-31.
To see whether microRNA-31 plays a role in controlling the signaling systems in intestinal stem cells, Tian, Ma, Lv et al. looked at genetically modified mice that either had too much microRNA-31 or none. Mice with too much microRNA-31 produced more intestinal stem cells and were able to better repair any cell damage. Mice without microRNA-31 gave rise to fewer intestinal stem cellsand had no damage repair, but were able to stop cancer cells in the gut from growing.
The results showed that microRNA-31 in intestinal stem cells helps the cells to divide and to protect themselves from cell death. It controlled and balanced the different types of cell signaling by either repressing or activating various signals. When Tian et al. damaged the stem cells using radiation, the cells increased their microRNA-31 levels as a defense mechanism. This helped the cells to survive and to activate repair mechanisms. Furthermore, Tian et al. discovered that microRNA-31 can enhance the growth of tumors.
These results indicate that microRNA-31 plays an important role both in repairing gut linings and furthering tumor development. A next step will be to see whether cancer cells use microRNA-31 to protect themselves from chemo- and radiation therapy. This could help scientists find new ways to render cancerous cells more susceptible to existing cancer therapies.
Introduction
The intestinal epithelium is one of the most rapidly renewing tissues, undergoing complete turnover in approximately 3 days (Leblond and Walker, 1956). This rapid turnover protects against insults from bacterial toxins and metabolites, dietary antigens, mutagens, and exposure to DNA damaging agents including irradiation. Upon insult, the rapid intestinal regeneration is particularly important as impaired regeneration can result in epithelial barrier defects that can lead to rapid dehydration and translocation of intestinal microbiota into the bloodstream. The processes of normal tissue turnover and intestinal regeneration are driven by intestinal stem cells (ISCs) that reside at the bottom of crypt and generate the precursors for the specialized differentiated cells (Barker, 2014; Li and Clevers, 2010).
It has been extensively reported that ISC compartment includes two functionally and molecularly distinct stem cell populations (Barker, 2014; Li and Clevers, 2010; Gehart and Clevers, 2015): The active crypt base columnar (CBC) stem cells (Sato et al., 2011), (Barker et al., 2007) and a more dormant, reserve ISC population that reside above the crypt base and exhibit no Wnt pathway activity, also referred as +4 cells due to their position at the crypt (Montgomery et al., 2011; Sangiorgi and Capecchi, 2008; Tian et al., 2011; Takeda et al., 2011; Li et al., 2014; Yan et al., 2012). The CBCs often identified and isolated based on the expression of Lgr5, a Wnt target gene (Barker et al., 2007). During homeostasis, steady-state proliferation of CBCs is driven by extrinsic niche signals – high canonical Wnt activity promotes CBC self-renewal and proliferation (Barker et al., 2007; Miyoshi, 2017) while BMP signals antagonize it (Kosinski et al., 2007). In contrast to the active CBCs, the reserve ISCs represent a slow-cycling population of stem cells that are resistant to high doses of ionizing radiation and appear dispensable for homeostasis (Sangiorgi and Capecchi, 2008; Yousefi et al., 2016). These reserve ISCs are identified through CreERT knockin reporter alleles at the Bmi1 and Hopx loci, as well as by an Tert-CreERT transgene (Montgomery et al., 2011; Sangiorgi and Capecchi, 2008; Tian et al., 2011; Takeda et al., 2011; Li et al., 2014). Reserve ISCs do not have an active Wnt signaling pathway and are refractory to Wnt signals in their resting state (Takeda et al., 2011; Li et al., 2014; Li et al., 2016). Although the activity of the BMP pathway has never been directly examined specifically in reserve ISCs, indirect evidence suggests that it may help to promote their dormancy (Reynolds et al., 2014; He et al., 2004; Kishimoto et al., 2015). During epithelial regeneration upon stresses, reserve ISCs give rise to Wnthigh Lgr5+ CBCs that generate the precursor cells of the specialized differentiated cells (Tian et al., 2011; Takeda et al., 2011; Li et al., 2014). In addition, it has been documented that Lgr5-CreERT- or Bmi1-CreERT-marked cells can act as the cells of origin of intestinal cancer in mice (Sangiorgi and Capecchi, 2008; Barker et al., 2009). However, it remains unclear how ISCs differentially sense and respond to multiple signals under both physiological and pathological conditions, and whether these signals contribute to intestinal tumorigenesis.
MicroRNAs represent a broad class of 18–22 nucleotide noncoding RNAs that negatively regulate the stability and translation of target mRNAs. Mounting evidence indicates that microRNAs play important roles in stress-activated pathways (Leung and Sharp, 2010; Mendell and Olson, 2012; Emde and Hornstein, 2014) and in control of somatic stem cell fate and tumorigenesis (Gangaraju and Lin, 2009; Sun and Lai, 2013; Yi and Fuchs, 2011). Hundreds of microRNAs have been identified in the intestinal epithelium (McKenna et al., 2010). Global ablation of microRNA activity through genetic deletion of the microRNA processing enzyme Dicer demonstrated that microRNAs are critical for homeostasis of intestinal epithelium (McKenna et al., 2010). Recently, numerous reports demonstrate that specific microRNAs play important roles in the complex intestinal immune system and in the epithelium during homeostasis including miR-155, miR-29, miR-122, miR-21, miR-146a and miR-143/145 (Runtsch et al., 2014). Particularly, miR-143/145 are essential for intestinal epithelial regeneration after injury, acting non cell-autonomously in sub-epithelial myofibroblasts (Chivukula et al., 2014), indicating potential importance of microRNA activity in intestinal regeneration.
In the ISC compartment, the function of miR-31 is of a particular interest, as it becomes overexpressed in colorectal cancer (Bandrés et al., 2006; Cottonham et al., 2010; Wang et al., 2009; Yang et al., 2013) and increases during the progression of inflammation-associated intestinal neoplasia (Olaru et al., 2011). In addition, it has been reported that miR-31 is enriched in mammary stem/progenitor cells, suggesting a potential role in somatic stem cells (Ibarra et al., 2007). Here we utilized gain- and loss-of-function mouse models to show that a damage-responsive microRNA, miR-31 drives proliferative expansion of both active and dormant ISCs, and acts as an oncogene promoting intestinal tumorigenesis in different models. Our findings implicated miR-31 as a potential high-value therapeutic target for a broad range of intestinal regenerative disorders and cancers.
Results
MiR-31 expression pattern in intestine under physiologIcal and pathological conditions
Elevated miR-31 expression has been previously observed in colorectal cancers (Bandrés et al., 2006; Cottonham et al., 2010; Wang et al., 2009; Yang et al., 2013), however its expression in normal intestinal epithelium, particularly in ISCs, remains unclear. To begin addressing a potential role for miR-31 in the intestinal epithelium and ISCs, first we examined its expression pattern in intestine. MiR-31 expression levels are the highest in the Lgr5-GFPhighcrypt base columnar stem cells, intermediate in Lgr5-GFPlow transit-amplifying cell population and the lowest in Lgr5-GFPneg populations (Figure 1A). Higher level of miR-31 was also found in Hopx+ reserve ISCs than that in bulk epithelial cells (Figure 1A), based on isolation with Hopx-CreERT;mTmG alleles from mice 15 hr after tamoxifen injection. Consistently, in situ hybridization revealed that miR-31 expression levels are generally higher in the crypts than villi. MiR-31 is predominantly expressed in the epithelial cells of intestinal crypt, including stem cells and transit amplifying cells (Figure 1B). Next, we examined miR-31 expression in response to intestinal injury. Mice were exposed to 12 Gy γ-IR and then miR-31 expression was examined at various timepoints during the recovery phase. MiR-31 levels transiently and markedly drop by 24 hours (coincident with full proliferative arrest/DNA damage response), and then sharply upregulated 48 hours post-γ-IR (during initiation of regenerative proliferation from the radioresistant ISCs), and then return to baseline levels within one week (after full recovery) (Figure 1C). In situ hybridization reveals miR-31 expressing cells to be located in the regenerative foci known to exhibit high Lgr5 expression and Wnt pathway activity (Figure 1D). Together, these data suggest a role for this microRNA in ISC-driven regeneration.
Figure 1. MiR-31 promotes turnover of intestinal epithelial cells.
(A) Schematic picture of intestinal crypt showing Lgr5+ CBCs and Hopx+ cells. qRT-PCR for miR-31 in Lgr5-GFPhigh, Lgr5-GFPlow, Lgr5-GFPneg, Hopx- and Hopx+ sorted intestinal epithelial cells. n = 4 biological replicates. (B) In situ hybridization for miR-31 in the intestinal epithelium. Left panel, representative low magnification image (Scale bar: 200 μm); Middle panels, high magnification images indicated by dashed boxes in left panel; Right panels (Scale bar: 50 μm), miR-31 KO intestinal section used as a negative control (Top) and TRE-miR31 (miR-31 overexpressing) intestinal section used as a positive control (Bottom). (C) qRT-PCR for miR-31 in the intestinal epithelium after exposure to 12 Gy γ-IR at indicated time points. n = 3 biological replicates. (D) In situ hybridization for miR-31 in intestines without γ -IR treatment (non-IR), and intestines 4 days after 12 Gy γ-IR. Arrows, miR-31 positive regenerative foci. Dashes boxes indicate the high magnification images in right panels. Scale bar: 50 μm. (E) Quantification of body weight from M2rtTA and TRE-miR31 mice at the age of 8 weeks before and after Dox treatment for 2 weeks. Quantification of intestine length from M2rtTA and TRE-miR31 mice following 2 week Dox induction. n = 6 biological replicates. ***p<0.001. (F) Representative histologic images showing extension of crypt height in jejunum from TRE-miR31 mice, and quantification of crypt height from M2rtTA and TRE-miR31 intestine. Both M2rtTA and TRE-miR31 mice were treated with Dox for 2 weeks. n = 3 biological replicates. Scale bar: 50 μm. ***p<0.001. (G) Immunohistochemistry for Ki67 and quantification of Ki67+ cells per crypt in M2rtTA andTRE-miR31 jejunum, showing an expanded proliferative zone in TRE-miR31 mice following 2 weeks of Dox induction. n = 3 biological replicates. Scale bar: 50 μm. ***p<0.001. (H) Immunohistochemistry for cleaved-Caspase 3 (Casp3) and quantification of Casp3+ cells in the top of intestinal villi from M2rtTA andTRE-miR31 mice following 2 weeks of Dox induction. n = 3 biological replicates. 60 villi were quantified in each mouse. Scale bar: 100 μm. ***p<0.001. (I) Representative histologic images and quantification of crypt height in intestines from miR-31+/− and miR-31−/− mice at 2 months of age. Brackets mark crypts. Scale bar: 100 μm. n = 3 biological replicates. ***p<0.001.

Figure 1—figure supplement 1. Generation of inducible TRE-miR-31 transgenic mice, constitutive miR-31 KO and conditional miR-31 KO mice.

Figure 1—figure supplement 2. MiR-31 induction promotes crypt expansion.

Figure 1—figure supplement 3. MiR-31 induction promotes cell proliferation in crypts, and apoptosis at the top of villi.

Figure 1—figure supplement 4. MiR-31 induction impairs cell differentiation.

Figure 1—figure supplement 5. Loss of miR-31 led to shortened crypt.

Figure 1—figure supplement 6. Loss of miR-31 does not affect cell differentiation.

Figure 1—figure supplement 7. Conditional deletion of miR-31 resulted in shortened crypt, reduced proliferation and enhanced apoptosis.

Figure 1—figure supplement 8. MiR-31 promotes cell turnover from crypt to villi.

MiR-31 promotes intestinal epithelial cell turnover along the Crypt-villus axis
To determine the function of miR-31 in the mouse intestine, we generated both gain- and loss- of-function mouse models. MiR-31 gain-of-function was achieved with a targeted, inducible Rosa26-rtTA;TRE-miR-31 mouse model (TRE-miR31) and doxycycline (Dox)-mediated induction of miR-31 in the intestinal epithelium was validated by qRT-PCR (Figure 1—figure supplement 1A,B). For the loss-of-function, we generated constitutive miR-31 null mice using RNA-guided CRISPR/Cas9 nucleases (Figure 1—figure supplement 1C). The 402 bp DNA fragment containing miR-31 was deleted in the knockout (KO) allele (Figure 1—figure supplement 1D), which was validated by sequencing and qRT-PCR (Figure 1—figure supplement 1E). We also generated a Villin-Cre-mediated intestine-specific conditional miR-31 null mice (cKO) using traditional homology-directed gene targeting (Figure 1—figure supplement 1F). The expression of miR-31 was markedly reduced in the cKO intestinal epithelium (Figure 1—figure supplement 1G). The induction of miR-31 in TRE-miR31 intestine and deletion of miR-31 in KO intestine were also confirmed by in situ hybridization (Figure 1—figure supplement 1H).
MiR-31 induction in response to Dox administration in TRE-miR31 mice resulted in a significant reduction in body weight after 2 weeks (Figure 1E) and intestinal lengths were moderately, but significantly shorter than controls (Figure 1E). Dox treatment of TRE-miR31 mice for 2 weeks resulted in expansion of intestinal crypts (Figure 1F). Unexpectedly villus lengths were mildly shortened, and thus the total length of the crypt-villus was not significantly altered in TRE-miR31 mice (Figure 1—figure supplement 2A). The expanded crypts were also found in the TRE-miR31 duodenum and ileum (Figure 1—figure supplement 2B). The length of intestinal crypts in the control M2rtTA mice was not significantly altered at different time points in response to Dox treatment (Figure 1—figure supplement 2C,D). In contrast, crypts were significantly expanded in TRE-miR31 mice after 10 days of Dox treatment, this crypt expansion remained stable for up to 1 year with continuous Dox induction (Figure 1—figure supplement 2C–E). Given that crypt elongation reached maximal levels within 2 weeks of Dox induction, we conducted most of the subsequent assays at this time point. More mitotic cells were found in the TRE-miR31 crypts (Figure 1G and Figure 1—figure supplement 3A,B), while more apoptotic cells were detected at the top of TRE-miR31 villi (Figure 1H and Figure 1—figure supplement 3A,B). The number of Lgr5+ ISCs increased in TRE-miR31 mice after 10 day Dox treatment, while no significant difference was found between them after 7 days of Dox induction (Figure 1—figure supplement 3C,D). In addition, there were fewer differentiated cells including enteroendocrine, goblet and Paneth cells in TRE-miR31 intestine than the controls (Figure 1—figure supplement 4A,B), indicating an impaired cell differentiation. These results suggest that miR-31 induction accelerates the conveyer-belt movement of proliferative cells exiting the cell cycle and progressing into the villi to ultimately be shed into the lumen, which could comprise the differentiation of specialized intestinal cell types.
Next, we examined the consequence of miR-31 loss in both miR-31 germline knockout (KO) and Villin-Cre-driven intestinal epithelial conditional KO (cKO) mice. We followed these mice up to six months. Both miR-31 KO and cKO mice were viable and fertile with no apparent gross phenotypes observed. No differences in the body weight and intestinal length were found between control and miR-31 KO mice (Figure 1—figure supplement 5A), and the transmission of miR-31 knockout alleles generally followed Mendelian ratios (Figure 1—figure supplement 5B). Despite this, loss of miR-31 led to a significant reduction in crypt height with fewer proliferative cells (Figure 1I and Figure 1—figure supplement 5C,D). Interestingly, loss of miR-31 gave rise to a certain number of apoptotic cells throughout the crypt-villus axis, while apoptotic cells are predominantly presented at the tip of control villi and very rare apoptotic cells are presented in crypt-villus axis (Figure 1—figure supplement 5C,D). Deletion of miR-31 also led to increased numbers of enteroendocrine and Paneth cells, while the number of goblet cells remained unaltered in miR-31 KO intestines (Figure 1—figure supplement 6A,B). Moreover, the phenotype of shortened crypts with fewer proliferative cells was also found in cKO intestine (Figure 1—figure supplement 7A,B). Loss of miR-31 gave rise to more apoptotic cells in cKO intestinal epithelium, including in cKO crypts, while cleaved-caspase3+ apoptotic cells were nearly entirely absent from control crypts (Figure 1—figure supplement 7C,D). These results suggest that miR-31 loss functions within intestinal epithelium. We further analyzed DNA synthesis and migration of epithelial cells along the crypt-villus axis after a single pulse of BrdU. Upward movement of BrdU+ cells from crypts to villi was enhanced in TRE-miR31 mice, and this movement was impaired in miR-31−/− mice (Figure 1—figure supplement 8). Taken together, these data indicate that miR-31 functions within the intestinal epithelium to maintain a proper balance between stem cell proliferation, differentiation, and epithelial cell death for optimal intestinal homeostasis.
MiR-31 promotes expansion of Lgr5+ CBC stem cells
Higher expression levels of miR-31 in Lgr5+ CBCs prompted us to examine its effect on their renewal. Lgr5+ ISC frequency was markedly increased in TRE-miR31, and significantly reduced in miR-31−/− and cKO intestine (Figure 2A–C and Figure 2—figure supplement 1A). A 1.5 hr pulse of EdU incorporation demonstrated that the frequency of actively proliferating Lgr5-GFP+/EdU+ cells is higher in TRE-miR31 mice and conversely lower in miR-31−/− mice (Figure 2D). In line with these in vivo findings, miR-31 induction increased the frequency of budding organoids in vitro, and caused more buds per organoid and more elongated crypts (Figure 2E and Figure 2—figure supplement 1B). Furthermore, lineage-tracing assay reveals that miR-31 induction in the intestine increases the height of traced lineages derived from Lgr5-CreERT-marked ISCs (Figure 2F,G and Figure 2—figure supplement 1C). Interestingly, miR-31 induction significantly repressed Hopx expression, while deletion of miR-31 increased it (Figure 2H). Consistently, miR-31 induction in the intestine repressed lineage tracing from Hopx-CreERT-marked reserve ISCs (Figure 2I, and Figure 2—figure supplement 1D,E). In contrast to miR-31 overexpression, deletion of miR-31 within intestinal epithelium induced quiescence (residence in G0) in Lgr5-GFP+ cells concomitant to an increase in apoptosis and a decrease in cycling (G1/S/G2/M) (Figure 2J and Figure 2—figure supplement 1F). In agreement, higher frequency of apoptotic organoids and compromised budding was found in the cKO crypts (Figure 2K), and more apoptotic cells were found inside of the cKO organoids (Figure 2—figure supplement 1G). Taken together, these data strongly indicate that miR-31 promotes proliferative expansion of Lgr5+ CBCs, and concomitantly prevents their apoptosis.
Figure 2. MiR-31 promotes expansion of Lgr5+ CBC stem cells.
(A) Representative FACS profiles and quantification of GFP positive intestinal epithelial cells (Lgr5-GFP+ cells) from an Lgr5-eGFP-CreERT reporter mice crossed with M2rtTA (control) and TRE-miR31 mice. M2rtTA (control) and TRE-miR31 mice were pre-treated with Dox for two weeks. n = 4 biological replicates. ***p<0.001. (B, C) FACS profiles and quantification of Lgr5-GFP+ cells from an Lgr5-eGFP-CreER reporter mice crossed with miR-31+/− (control) and miR-31−/− mice (B), or Vil-Cre (Villin-Cre) and cKO (Vil-Cre;miR-31fl/fl) mice (C). n = 4 biological replicates. ***p<0.001. (D) Assessment of 1.5-hour-pulse EdU incorporation in Lgr5+ CBC cells in M2rtTA, and TRE-miR31 mice following 2 weeks of Dox treatment, and in miR-31+/− and miR-31−/− intestine. ***p<0.001. (E) Crypts purified from M2rtTA and TRE-miR31 mice grown in organoid cultures with Dox. Representative gross images of budding organoids, and quantification of budding and apoptotic organoids at day 7. Scale bar: 500 μm. n = 5 technical replicates. (F) X-gal staining showing lineage tracing events from Lgr5+ ISCs. Lgr5-eGFP-CreERT;R26-LSL-LacZ;TRE-miR31 mice and its control counterpart were pretreated with Dox for 2 weeks, injected with a single dose tamoxifen, and analyzed 2 and 4 days after injection. Scale bar: 100 μm. n = 3 biological replicates. (G) Quantification of the length of LacZ+ cells and LacZ+ units in Panel F. ***p<0.001. (H) qRT-PCR analysis for Hopx in intestines from M2rtTA, TRE-miR31, Vil-Cre and cKO mice. n = 3 biological replicates. **p<0.01; ***p<0.001. (I) Lineage tracing events from Hopx+ ISCs. Hopx-CreERT;mTmG;TRE-miR31 mice and their control counterparts were pretreated with Dox for 2 weeks, injected with a single dose of tamoxifen, and analyzed 15 hr after injection. Hopx-CreERT;R26-LSL-LacZ;TRE-miR31 and their control counterparts were analyzed 4 days after inject with the same treatment. Scale bar: 100 μm. n = 3 biological replicates. (J) Quantification of Cleaved Caspase3+ cells at indicated positions in the intestinal crypts of Vil-Cre and miR-31 cKO mice in Figure 1—figure supplement 7D. n = 3 biological replicates, 50 crypts per sample. (K) Crypts purified from Vil-Cre and miR-31 cKO mice grown in organoid cultures at indicated time points. Quantification of budding organoids and apoptotic organoids, budding number and crypt length. n = 3 biological replicates. ***p<0.001.

Figure 2—figure supplement 1. MiR-31 promotes ISC expansion.

MiR-31 is critical for intestinal epithelial regeneration following irradiation
The dynamic changes of miR-31 expression in response to irradiation prompted us to investigate its function during intestinal epithelial injury repair. Intestinal histology of cKO and control Vil-Cre mice was comparable two hours after 12 Gy γ-IR (Figure 3A). However, by 4 days post-γ-IR, there were significantly fewer regenerative foci and fewer proliferative cells per regenerative focus in cKO mice (Figure 3A). Consistently, intestinal regeneration in response to γ-IR was significantly impaired in miR-31−/− mice (Figure 3—figure supplement 1A,B). Conversely, in the intestine of TRE-miR31 mice pre-treated for 2 weeks with Dox, there were more regenerative foci with higher numbers of proliferative cells than in the control mice (Figure 3—figure supplement 1A,B). These data suggest that miR-31 is important for intestinal epithelial regeneration in response to irradiation.
Figure 3. Loss of miR-31 abrogates epithelial regeneration following irradiation.
(A) Representative images of H&E and/or Ki67 immunohistochemistry from jejunum of irradiated Vil-Cre and cKO mice 2 hrs and 4 days post 12 Gy γ-IR. Quantification of Ki67+ regenerative foci per 1400 μm and No. of Ki67+ cells per regenerative focus. Top panel: n = 6 biological replicates; Scale bar: 200 μm. Middle and bottom panels: n = 5 biological replicates; Scale bar: 50 μm. **p<0.01; ***p<0.001. (B) Immunohistochemistry for Casp3, quantification of the number of Casp3+ cells in intestinal crypts of Vil-Cre and cKO mice 2 and 4 hrs post 12 Gy γ-IR. Quantification of Casp3+ cells at indicated positions in intestinal crypts of Vil-Cre and cKO mice 2 hrs post γ-IR. Scale bar: 50 μm. n = 3 biological replicates, and 50 crypts were quantified in each single mouse. ***p<0.001. (C) qRT-PCR analysis for Lgr5 in intestines from M2rtTA, TRE-miR31, miR-31+/− and miR-31−/− mice 2 hrs, 2 and 4 days post 12 Gy irradiation. M2rtTA and TRE-miR31 mice were pre-treated with Dox for two weeks. n = 3 biological replicates at each time points. *p<0.05; **p<0.01; ***p<0.001. (D) Schematic of Lgr5-eGFP-CreERT;R26-LSL-LacZ lineage tracing experiment after irradiation. X-gal staining showing lineage tracing events from Lgr5+ ISCs. Lgr5-eGFP-CreERT;R26-LSL-LacZ;TRE-miR31 mice and their control counterparts were pretreated with Dox for 2 weeks, injected with a single dose tamoxifen and then immediately exposed to 10 Gy γ-IR, and analyzed 2 and 4 days after γ-IR. Scale bar: 100 μm. n = 3 biological replicates at each time points. (E) Quantification of LacZ+ units and the length of LacZ+ cells in Panel D. (F) Schematic of Hopx-CreERT;R26-LSL-LacZ lineage tracing experiment. Hopx-CreERT;R26-LSL-LacZ;TRE-miR31 and their control counterparts were pretreated with Dox for 2 weeks, then injected with a single dose of tamoxifen, and then irradiated 15 hrs after injection and analyzed 4 days after irradiation. Representative images of LacZ staining in M2rtTA and TRE-miR31 intestine 4 days post 12 Gy γ-IR. Scale bar: 50 μm. Statistics of LacZ+ regenerative foci were shown in Figure 3—figure supplement 1E. n = 3 biological replicates. (G) Schematic of Hopx-CreERT;R26-LSL-LacZ lineage tracing experiment. Representative images of LacZ staining in miR-31+/− and miR-31−/− intestine 4 days post 12 Gy γ-IR. Scale bar: 50 μm. Statistics of LacZ+ regenerative foci. n = 3 biological replicates. (H) Representative images of LacZ (blue) and Ki67 (yellow) immunostaining in miR-31+/− and miR-31−/− intestinal crypts, and statistics of percentage of LacZ+/Ki67+cells in regenerative foci. Scale bar: 25 μm. n = 3 biological replicates. ***p<0.001.

Figure 3—figure supplement 1. MiR-31 is required for intestinal epithelial regeneration in response to γ-IR.

To understand the phenotype resulting from miR-31 modulation, we assayed for apoptotic cells in cKO mice at early stages after irradiation. Loss of miR-31 increased apoptosis in the crypts 2 and 4 hours post-irradiation prior to any overt histological changes (Figure 3B). Quantification of apoptotic cell position analysis reveals that apoptotic events occur with the highest frequently in CBC cells, but are still found in transit-amplifying and +4 zones of cKO crypts, compared to control mice (Figure 3B). Further, flow cytometry for live cell and apoptotic markers within the Lgr5-GFP+ population confirmed higher frequency of late apoptotic Lgr5+ cells (AnnexinV+/7AAD+) and lower frequency of early apoptotic Lgr5+ cells (AnnexinV+/7AAD−) and live Lgr5+ cells (AnnexinV-/7AAD-) in cKO mice, relative to controls (Figure 3—figure supplement 1C). These data suggest that loss of miR-31 increases apoptosis of Lgr5+ cells in response to irradiation. Next, we examined its effect on cell proliferation. Cell cycle analysis indicates that more Lgr5-GFP+ cells resided in G0 relative to G1/S/G2/M in cKO mice 2 hours after γ-IR (Figure 3—figure supplement 1D). In agreement, expression levels of Lgr5 were dramatically up-regulated in TRE-miR31 mice and prominently down-regulated in miR-31−/− mice at multiple time points after irradiation (Figure 3C), and consequently miR-31 induction promoted lineage regeneration from Lgr5+ cells in response to irradiation (Figure 3D,E).
Reserve ISCs, marked either by Bmi1-CreER or Hopx-CreER reporters, have been reported to resist high dose of radiation, being able to replenish the depleted CBC compartment and regenerate the epithelium after irradiation (Sangiorgi and Capecchi, 2008; Tian et al., 2011; Takeda et al., 2011; Yan et al., 2012), (Yousefi et al., 2016). Thus, we examined the response of Hopx-CreER-marked reserve ISCs to 12 Gy γ-IR upon miR-31 induction and deletion. Lineage-tracing assay revealed that miR-31 induction promoted epithelial regeneration from the Hopx+ reserve stem cells (Figure 3F and Figure 3—figure supplement 1E). Conversely, the number and the size of regenerative foci originating from Hopx-CreER;Rosa26-LoxP-Stop-LoxP-LacZ-marked cells were markedly reduced in miR-31−/− mice (Figure 3G). In line with this, the frequency of LacZ+/Ki67+ cells was significantly lower in miR-31−/− mutants compared to controls (Figure 3H). Taken together, miR-31 deficiency-mediated the reduction in proliferation and increase in apoptosis within both CBC and reserve ISC compartments can account for the impaired regeneration of miR-31 null intestine.
MiR-31 activates the Wnt pathway and represses the BMP and TGFβ pathways
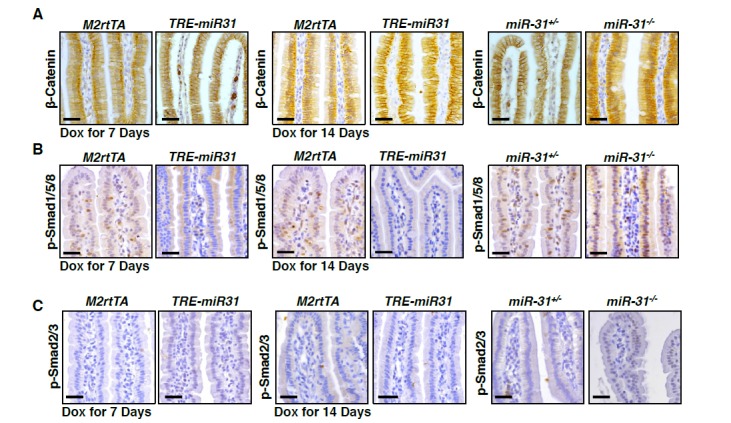
Canonical Wnt pathway activity is a major driving force for self-renewal of CBCs and epithelial regeneration after injury (Clevers et al., 2014), and, thus we examined the effect of miR-31 on Wnt activity. We utilized Axin2-LacZ Wnt reporter mice, which act as a broad readout for canonical Wnt activity, and normally showed its activity to be restricted to the base of crypts in control mice, as expected (Figure 4A) (Davies et al., 2008). In contrast, Wnt pathway activity was strikingly absent from CBCs of miR-31−/− crypts, appearing only faintly above the crypt base in the early TA zones (Figure 4A). Conversely, Wnt activity was expanded in TRE-miR31 crypts (Figure 4A,B). In agreement, the number of nuclear β-Catenin-positive cells was significantly reduced in miR-31−/− intestinal crypts at 2 and 4 months of age (Figure 4—figure supplement 1A). Conversely, they increase in TRE-miR31 crypts 14 days and 2 months after Dox induction (Figure 4—figure supplement 1B). Consistently, the expression levels of Ctnnb1 (encoding β-Catenin) and the Wnt targets, Ccnd1 (encoding Cyclin D1), Myc and Axin2 were significantly reduced in miR-31−/− intestine both at the RNA and protein levels (Figure 4C,D). In contrast, expression levels of the above genes were enhanced in TRE-miR31 intestinal epithelium following 2 weeks of Dox induction (Figure 4E,F). The reduction in Ctnnb1 and Wnt targets was further confirmed in conditional miR-31 KO intestine (Figure 4G). To test whether Wnt activity is directly impacted by miR-31, we analyzed the effects of gain- and loss-of-function of miR-31 on expression of Wnt target genes in HCT116 human colorectal carcinoma cells. Ccnd1, Ctnnb1, Myc and Axin2 were markedly increased in miR-31 over-expressing cells, relative to controls (Figure 4H). Conversely, these genes were downregulated upon miR-31 inhibition (Figure 4H). Considering that HCT116 cells are heterozygous for a β-Catenin gain-of-function mutation at the Gsk3b target site S45 (Ctnnb1+/S45mt) (Ilyas et al., 1997), (Kaler et al., 2012), we examined β-Catenin protein levels. Consistently, β-Catenin was up-regulated in the presence of miR-31 mimics, and down-regulated upon miR-31 inhibition (Figure 4—figure supplement 1C). The Wnt reporter (Topflash/Fopflash) assay using HCT116 cells further confirmed that miR-31 induction enhanced Wnt activity, while inhibition of miR-31 repressed it (Figure 4I). To test the functional relevance of miR-31 potentiation of canonical Wnt activity, we cultured organoids with varying combinations of miR-31 induction and R-spondin, the Lgr5 ligand. Wnt activation by R-spondin is critical for normal organoid growth and budding (Sato et al., 2011). Interestingly, we observed that miR-31 induction via TRE-miR31 was sufficient to maintain crypt organoid growth and budding in the absence of R-spondin (Figure 4J,K) and that the Dox-treated TRE-miR31 organoids can be normally passaged at least five times, similar to the organoids cultured with R-spondin (Figure 4L). Together, these findings demonstrate that miR-31 activates the canonical Wnt signaling in the crypts of small intestine.
Figure 4. MiR-31 activates Wnt pathway activity.
(A) Wnt activity was evaluated by Axin2-LacZ reporter activity in M2rtTA and TRE-miR31 intestine following 2 week Dox induction, and in miR-31+/− and miR-31−/− intestine. Blue, LacZ signals. n = 3 biological replicates. Scale bar: 25 μm. (B) Quantification of LacZ+ cells per crypt in M2rtTA and TRE-miR31 mice. ***p<0.001. (C) qRT-PCR analysis for Ctnnb1 (encoding β-Catenin), Ccnd1 (encoding Cyclin D1), Myc, and Axin2 in miR-31+/− and miR-31−/− intestine. *p<0.05; **p<0.01; ***p<0.001. (D) Western blotting for Cyclin D1, c-Myc and β-Catenin in miR-31+/− and miR-31−/− intestine. GAPDH was used as a loading control. (E) qRT-PCR for Ccnd1, Myc, Axin2 and Ctnnb1 in intestine from M2rtTA and TRE-miR31 mice following 2 weeks of Dox induction. ***p<0.001. (F) Western blotting for Cyclin D1, c-Myc, and β-Catenin in intestine from M2rtTA and TRE-miR31 mice following 2 weeks of Dox induction. (G) qRT-PCR for Ctnnb1, Ccnd1, Myc, and Axin2 in intestine from Vil-Cre and cKO mice. n = 4 biological replicates. **p<0.01; ***p<0.001. (H) qRT-PCR for Axin2, Ccnd1, Myc, and Ctnnb1 in HCT116 colon cancer cells treated with miR-31 inhibitor and negative control (NC, Scramble RNA), as well as miR-31 mimics and negative control (NC, Scramble RNA) for 24 hrs. ***p<0.001. (I) Luciferase activity of TOPflash versus FOPflash in HCT116 cells treated with miR-31 inhibitor and negative control (NC, Scramble RNA), as well as miR-31 mimics and negative control (NC, Scramble RNA) for 24 hrs. n = 3 technical replicates. ***p<0.001. (J) Representative images of organoids cultures from purified M2rtTA and TRE-miR31 crypts at indicated conditions. R; R-Spondin. n = 3 biological replicates. (K) Quantification of budding organoids in Panel J. ***p<0.001. (L) Representative images of organoids cultures from purified M2rtTA and TRE-miR31 crypts at serial passages. M2rtTA organoids were cultured with R-Spondin; TRE-miR31 organoids were cultured with Dox and without R-Spondin. n = 4 biological replicates.

Figure 4—figure supplement 1. MiR-31 activates Wnt signaling pathway.

BMP and TGFβ pathways are known to inhibit the canonical Wnt pathway, inhibiting proliferation and promoting intestinal progenitor differentiation (Reynolds et al., 2014; He et al., 2004; Furukawa et al., 2011). We thus examined the effects of miR-31 on BMP and TGFβ signals. BMP-specific Smad1/5/8 and TGFβ-specific Smad2/3 phosphorylation were significantly increased in miR-31−/− intestine (Figure 5A and Figure 5—figure supplement 1A), and downregulated in TRE-miR31 intestine (Figure 5A and Figure 5—figure supplement 1B), suggesting an inhibitory effect of miR-31 on BMP and TGFβ signaling pathways. Consistently, we observed a significant increase on the expression of BMP target genes including Id1, Id2, Id3, Msx1, Msx2 and Junb and TGFβ target genes Cdkn1c (p57), Cdkn1a (p21), Cdkn2a (p16), and Cdkn2b (p15) in miR-31−/− intestine (Figure 5B). Conversely, BMP and TGFβ targets were repressed upon forced expression of miR-31 in TRE-miR31 intestine following 2 weeks of Dox induction (Figure 5C). The upregulation of BMP and TGFβ targets was further confirmed upon conditional miR-31 deletion in cKO intestine (Figure 5D,E). BMP-specific Smad1/5/8 and TGFβ-specific Smad2/3 phosphorylation were also increased in miR-31 cKO cultured organoids (Figure 5—figure supplement 1C). Further, we examined the effect of miR-31 on BMP and TGFβ signaling in HCT116 colorectal cancer cells. These cells carry biallelic mutations in the Tgfbr2 gene, but still express functional TGFBR2 proteins and respond to TGFβ (de Miranda et al., 2015). In line with the in vivo findings, we found down-regulation of p-Smad2/3 and p-Smad1/5/8 in HCT116 cells treated with miR-31 mimics, and their up-regulation in cells treated with miR-31 inhibitor (Figure 5—figure supplement 1D). Luciferase assays using BMP- and TGFβ-responsive luciferase reporters, BRE-Luc and CAGA-Luc, respectively, revealed that inhibition of miR-31 resulted in significant increases in luciferase activities, and that miR-31 mimics decreased them (Figure 5F,G). More importantly, increasing concentrations of the BMP inhibitor Noggin in organoid culture was able to rescue the budding defect in miR-31 cKO organoids in a dose-dependent manner (Figure 5H,I). Together, these data suggest that miR-31 promotes ISC proliferation possibly through repressing BMP and TGFβ signaling pathways in a cell-autonomous manner.
Figure 5. MiR-31 represses BMP/TGFβ signaling pathways.
(A) Western blotting for p-Smad1/5/8 and p-Smad2/3 in miR-31+/−, miR-31−/−, M2rtTA and TRE-miR31 intestine. Both M2rtTA and TRE-miR31 mice were treated with DOX for 2 weeks. β-Tubulin was used as a loading control. (B) qRT-PCR analysis for BMP downstream genes, Id1, Id2, Id3, Msx-1, Msx-2 and Junb, and TGFβ downstream genes, Cdkn1c (p57), Cdkn1a (p21), Cdkn2a (p16), Cdkn2b (p15) and Cdkn1b (p27) in miR-31+/− and miR-31−/− intestine. *p<0.05; **p<0.01; ***p<0.001. (C) qRT-PCR analysis for BMP downstream genes, Id1, Id2, Id3, Msx-1, Msx-2 and Junb, and TGFβ downstream genes, Cdkn1c, Cdkn1a, Cdkn2a, Cdkn2b and Cdkn1b in M2rtTA and TRE-miR31 intestine following 2 weeks of Dox induction. **p<0.01; ***p<0.001. (D) qRT-PCR analysis for TGFβ downstream genes, Cdkn1c, Cdkn1a, Cdkn2a, Cdkn2b and Cdkn1b in intestine from Vil-Cre and cKO mice. *p<0.05; **p<0.01; ***p<0.001. (E) qRT-PCR analysis for BMP downstream genes, Id1, Id2, Id3, Msx2 and Junb in Vil-Cre and cKO intestine. **p<0.01; ***p<0.001. (F and G) HEK293T cells were transfected with CAGA- or BRE- luciferase reporter vector, combined with scramble RNA (negative control, NC) or anti-miR-31 (miR-31 inhibitors) (F), or scramble RNA (negative control, NC) and miR-31 mimics (G) for 24 hrs and then harvested for luciferase activity determination. n = 3 technical replicates. **p<0.01; ***p<0.001. (H) Quantification of organoid forming efficiency (budding organoids per 100 crypts) after Vil-Cre or cKO crypts cultured with noggin at indicated concentrations for 4 days. n = 3 technical replicates. (I) Representative images of organoids from Vil-Cre and cKO crypts cultured with noggin at indicated concentrations (100, 200, 400, 600 and 800 ng/mL) for 4 Days in Panel H.

Figure 5—figure supplement 1. MiR-31 represses BMP and TGFβ signaling pathways.

Identification of direct targets of miR-31
To understand how miR-31 regulates Wnt, BMP and TGFβ pathways, we analyzed miR-31 binding sites in 3’UTRs of transcripts encoding for regulators of these pathways. Genes containing miR-31 binding sites include Wnt antagonists Axin1, Gsk3b, and Dkk1, along with transcripts containing BMP/TGFβ signaling pathway components such as Smad3, Smad4, Bmpr1a and Tgfbr2 (Figure 6—figure supplement 1A). The expression of Axin1, Gsk3b, Dkk1, Smad3, Smad4, Bmpr1a and Tgfbr2 was significantly upregulated in miR-31−/− intestine (Figure 6A) and remarkably downregulated in TRE-miR31 intestine following Dox induction (Figure 6B), suggesting that they are negatively regulated by miR-31. The upregulation of these putative target genes was further confirmed in conditional miR-31 KO intestine (Figure 6C). Axin1, Gsk3b, Dkk1, Bmpr1a and Smad4 were selected for further validation at protein level (Figure 6D,E and Figure 6—figure supplement 2A–C) and in organoids cultured from miR-31 cKO mice (Figure 6—figure supplement 3A). This effect was further confirmed in HCT116 cells with miR-31 modulation (Figure 6—figure supplement 3B). Next, we validated the direct repression of target transcripts by miR-31 activity using WT-3’UTR-luciferase constructs for Axin1, Gsk3b, Dkk1, Bmpr1a, Smad3 and Smad4. Mutation of the miR-31 3’UTR binding site in these constructs abrogated this repression (Figure 6F and Figure 6—figure supplement 1B). Furthermore, RNA crosslinking, immunoprecipitation, and RT-PCR (CLIP-PCR) assays with Ago2 antibodies confirmed that transcripts of Axin1, Dkk1, Gsk3b, Smad3, Smad4 and Bmpr1a were highly enriched in Ago2 immunoprecipitates, and that increasing miR-31 activity augmented their enrichment (Figure 6G), providing evidence that miR-31 directly binds to these transcripts. Taken together, these findings indicate that Axin1, Gsk3b, Dkk1, Smad3, Smad4, and Bmpr1a transcripts are the direct targets of miR-31. Next, we asked whether these targets functionally contribute to impaired regeneration in miR-31−/− mice. Derepression of these target transcripts was observed in miR-31−/− intestine after irradiation (Figure 6H,I). As a consequence, Wnt activity was reduced, while the BMP and TGFβ activities were increased in miR-31−/− intestine, evidenced by β-Catenin, p-Smad1/5/8 and p-Smad2/3 immunohistochemistry assays (Figure 6J). Considering that intestinal regeneration following irradiation requires Wnt hyperactivity (Davies et al., 2008), and that BMP activity counterbalances Wnt signaling (He et al., 2004), our findings suggest that miR-31 is an important amplifier of Wnt signaling during intestinal regeneration.
Figure 6. Identification of miR-31 target genes in intestinal epithelium.
(A, B) qRT-PCR analysis for Axin1, Gsk3b, Dkk1, Smad3, Bmpr1a, Smad4 and Tgfbr2 in miR-31+/− and miR-31−/− intestine (A), as well as M2rtTA and TRE-miR31 intestine following 2 weeks of Dox induction (B). **p<0.01; ***p<0.001. (C) qRT-PCR analysis for Axin1, Bmpr1a, Dkk1, Gsk3b, Smad3, and Smad4 in Vil-Cre and cKO intestine. **p<0.01; ***p<0.001. (D) Western blotting for Axin1, Gsk3β, Dkk1, Smad4, and Bmpr1a in miR-31+/− and miR-31−/− intestine. β-Tubulin was used as a loading control, which is identical with Figure 5A. n = 3 biological replicates. (E) Western blotting for Axin1, Gsk3β, Dkk1, Bmpr1a and Smad4 in M2rtTA and TRE-miR31 intestine following 2 weeks of Dox induction. β-Tubulin was used as a loading control. n = 3 biological replicates. (F) Ratio of luciferase activity of miR-31 mimics versus scramble RNA in wild type and mutant 3’UTR constructs based on 3 independent experiments. *p<0.05; **p<0.01; ***p<0.001. (G) RNA crosslinking, immunoprecipitation, and qRT-PCR (CLIP-PCR) assay for Dkk1, Axin1, Gsk3b, Smad3, Smad4 and Bmpr1a upon Ago2 antibody immunoprecipitates in response to miR-31 mimics and scramble RNA (NC). IgG was used as a negative control. (H) qRT-PCR analysis for Axin1, Gsk3b, Dkk1, Smad3, Bmpr1a, Smad4 andTgfbr2 in miR-31+/− and miR-31−/− intestine 4 days post 12 Gy γ-IR. n = 3 biological replicates. *p<0.05; **p<0.01; ***p<0.001. (I) Immunohistochemistry for Axin1, Gsk3β and Dkk1 in miR-31+/− and miR-31−/− intestinal crypts 4 days post 12 Gy γ-IR. Scale bar: 25 μm. (J) Immunohistochemistry for p-Smad2/3, p-Smad1/5/8 and β-Catenin in miR-31+/− and miR-31−/− intestinal crypts 4 days post 12 Gy γ-IR. Scale bar: 25 μm.

Figure 6—figure supplement 1. Identification of miR-31 target genes.

Figure 6—figure supplement 2. Identification of miR-31 target genes.

Figure 6—figure supplement 3. Identification of miR-31 target genes.

MiR-31 contributes to tumor growth through Wnt activation and TGFβ and BMP repression
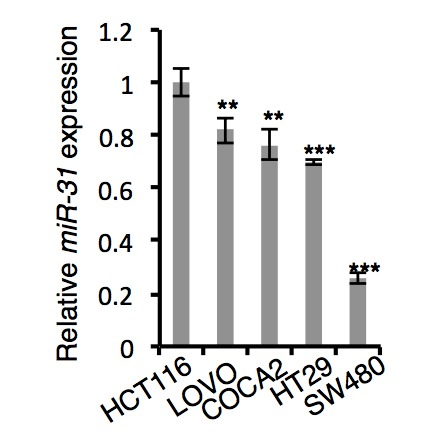
Given that miR-31 promotes proliferation and inhibits apoptosis in the ISCs, it is plausible that miR-31 may function in intestinal tumorigenesis. Supporting this notion, miR-31 has been found to be upregulated in human colorectal cancers and in colitis (Bandrés et al., 2006; Cottonham et al., 2010; Wang et al., 2009; Yang et al., 2013). We tested the role of miR-31 in intestinal tumorigenesis and observed that miR-31 mimics promoted proliferation of HCT116, SW480 and LOVO colon cancer cells in vitro (Figure 7—figure supplement 1A). Conversely, inhibition of miR-31 with anti-miR-31 abrogated growth of these cells (Figure 7—figure supplement 1A). We further performed xenograft assays using miR-31 mimics- and inhibitor-treated HCT116 cells. Thirty days after grafting, tumor volume and weight were increased in miR-31 mimic-treated tumors, and markedly reduced in miR-31 knockdown tumors (Figure 7A). The decrease in tumor size from miR-31 inhibition coincided with the reduction in Ki67+ and Cyclin D1+ proliferating cells (Figure 7B and Figure 7—figure supplement 1B), and correlated with reduced Wnt activity and increased BMP and TGFβ activities (Figure 7—figure supplement 1B). To verify these findings in more physiologically relevant settings, we examined tumor formation in the AOM-DSS (Azoxymethane-Dextran Sodium Sulfate) model of the inflammation-driven colorectal adenocarcinoma (De Robertis et al., 2011). In comparison with the controls, we observed a marked decrease in both tumor size and number in miR-31−/− mice (Figure 7C), along with a concomitant reduction in proliferating cells (Figure 7D,E), and reduced Wnt pathway and increased BMP and TGFβ activity (Figure 7D,F). This tumor-promoting effect of miR-31 in mice became even more evident when miR-31 was deleted in Vil-Cre;Apcflox/+ mice. Intestinal adenomas form in this mouse model upon loss of heterozygosity at the Apc locus, which is relevant to human disease in that spontaneous loss of Apc is found in the vast majority of human colorectal cancer (Kinzler et al., 1991; Nagase et al., 1992). Loss of miR-31 in this animal model remarkably reduced tumor burden (Figure 7G), which was associated with decreased Wnt activity, enhanced BMP and TGFβ signaling, and decreased proliferating cells (Figure 7H–J and Figure 7—figure supplement 1C). Correspondingly, the miR-31 targets Axin1, Dkk1, Gsk3β, Smad4 and Bmpr1a were up-regulated in the miR-31 null tumors (Figure 7—figure supplement 1D). Together, these data demonstrate that miR-31 plays an oncogenic role in intestinal and colorectal tumorigenesis by mediating activation of Wnt and repression of BMP and TGFβ signaling pathways.
Figure 7. MiR-31 promotes tumor growth in vivo.
(A) Gross appearance of tumors of HCT116 colorectal cancer cell xenograft 30 days post transplantation. HCT116 colorectal cancer cells were transfected with mimics-NC or miR-31 mimics, and inhibitor-NC or anti-miR-31 (inhibitor) for 36 hrs before xenograft. NC-mimics, n = 5; miR-31 mimics, n = 5; NC-inhibitor, n = 4; anti-miR-31, n = 5. Quantification of tumor volume and tumor weight at indicated conditions. **p<0.01; ***p<0.001. Scale bar: 1 cm. (B) Quantification of Ki67+ and Cyclin D1+ cells in NC-inhibitor and miR-31 inhibitor treated tumors in Figure 7—figure supplement 1B. ***p<0.001. (C) Representative photograph of distal colon resected from WT and miR-31−/− mice at the end of AOM-DSS protocol. Frequency and tumor size of inflammation-driven colorectal adenomas in mice treated with the AOM-DSS protocol, with or without miR-31 deletion. n = 6 mice per group, *p<0.05; **p<0.01. (D) H&E, and immunohistochemistry for Ki67, β-Catenin and p-Smad1/5/8 in adenomas of WT and miR-31−/− mice resulting from AOM-DSS treatment. Scale bar: 100 μm. (E) Quantification of Ki67+ cells in Panel D. ***p<0.001. (F) Western blotting for p-Smad2/3, p21, Axin1, β-Catenin, Cyclin D1 in adenomas of WT and miR-31−/− mice resulting from AOM-DSS treatment. β-Tubulin was used as a loading control. (G) Representative photograph of intestine resected from Vil-Cre;Apcfl/+ and Vil-Cre;Apcfl/+;miR-31−/− mice at 6 months of age. Arrows point to tumors. Quantification of tumor number and tumor volume in intestines from these mice. n = 6 biological replicates. ***p<0.001. (H) Representative histology of intestine resected from Vil-Cre;Apcfl/+ and Vil-Cre;Apcfl/+;miR-31−/− mice at 6 months of age. Arrows point to tumors. Scale bar: 2.5 mm. (I) Immunohistochemistry for β-Catenin and quantification of nuclear β-Catenin positive cells in Vil-Cre;Apcfl/+ and Vil-Cre;Apcfl/+;miR-31−/− tumors. (Black, Vil-Cre;Apcfl/+; Blue, Vil-Cre;Apcfl/+;miR-31−/−). n = 6 biological replicates. Scale bar: 50 μm. ***p<0.001. (J) Immunohistochemistry for p-Smad2/3, p-Smad1/5/8 and Ki67 in Vil-Cre;Apcfl/+ and Vil-Cre;Apcfl/+;miR-31−/− tumors. Scale bar: 50 μm.

Figure 7—figure supplement 1. MiR-31 promotes tumor growth.

STAT3 signaling pathway mediated miR-31 expression in response to irradiation
Lastly, we asked how radiation injury induces miR-31 expression. We analyzed a 2 kb region upstream of the transcription start site of the miR-31 gene locus for the potential binding sites of transcription factors using the JASPAR database and identified one STAT3 and two NF-κB binding sites (Figure 8A). Interestingly, the STAT3 and NF-κB signaling pathways were shown to be activated in response to γ-IR, evidenced by p-STAT3 and p65 levels, respectively (Figure 8B,C). The activation of the STAT3 pathway occurred mainly in the regenerative foci where miR-31 is highly induced, while NF-κB was more prominently activated in villi where little miR-31 is present and not in the regenerative foci (Figure 8D). This suggested a link between STAT3 activity and miR-31 upon irradiation. To verify whether active STAT3 signaling could induce miR-31 expression, mICc12 intestinal epithelial cells were treated with IL-6, a known activator of the STAT3 signaling. Indeed, miR-31 expression was significantly induced upon IL-6 treatment (Figure 8E), concomitant with the activation of the STAT3 pathway (Figure 8F). In contrast, inhibition of STAT3 signaling with Stattic prominently dampened miR-31 induction response to IL-6 treatment (Figure 8G), and reduced STAT3 signaling (Figure 8H). This inhibitory effect on miR-31 expression was further validated using Stat3 siRNA (Figure 8I,J). Importantly, miR-31 was induced by IL-6 in the organoid cultures, indicating that this is an epithelial cell-autonomous mechanism (Figure 8K). Luciferase reporter assays reveal that IL-6 is able to induce its activity, while mutation of the p-STAT3 binding site blocked it (Figure 8L). Furthermore, Chromatin Immunoprecipitation (ChIP) assays show that p-STAT3 is recruited to its binding site on the miR-31 promoter (Figure 8M). Thus, our data strongly suggest that STAT3 activity potentiates miR-31 induction to promote crypt regeneration in response to radiation injury.
Figure 8. The STAT3 pathway mediates the induction of miR-31 caused by γ-IR.

(A) The schematic diagram showed two potential p65 binding sites and one p-STAT3 binding site in the miR-31 promoter. (B) qRT-PCR analysis for Rela, Ikk-b, IL-1, IL-6, IL-18, Tnf and Stat3 in the intestinal epithelium 4 days after exposure to 12 Gy γ-IR, relative to non-irradiated controls. n = 3 biological replicates. *p<0.05, **p<0.01, ***p<0.001. (C) Western blotting for STAT3, p-STAT3, p65 and p-p65 in the intestinal epithelium 4 days after exposure to 12 Gy γ-IR, relative to non-irradiated controls. n = 3 biological replicates. (D) Immunohistochemistry for p-STAT3 and p65 in control and the intestinal epithelium 4 days after exposure to 12 Gy γ-IR. n = 3 biological replicates. Scale bar: 25 μm. (E) qRT-PCR for miR-31 in mouse intestinal epithelial cell line (mICc12) in response to IL-6 with concentrations of 20, 40, 80, 100 and 150 ng/mL. n = 3 technical replicates. **p<0.01; ***p<0.001. (F) Western blotting for STAT3 and p-STAT3 in mICc12 cells in response to 40 ng/mL IL-6. (G) qRT-PCR analysis for miR-31 in mICc12 cells treated with IL-6 and STAT3 inhibitor, Stattic. **p<0.01; ***p<0.001. (H) Western blotting for p-STAT3 in mICc12 cells treated with IL-6 and Stattic. (I) qRT-PCR analysis for miR-31 in mICc12 cells treated with Stat3 siRNA. ***p<0.001. (J) Western blotting for STAT3 and p-STAT3 in mICc12 cells treated with STAT3 siRNA. (K) qRT-PCR analysis for miR-31 in cultured organoids treated with IL-6. n = 4 technical replicates. ***p<0.001. (L) Luciferase activity in lysates of mICc12 cells transfected with luciferase reporter plasmids of pGL3-basic empty vector (basic), wild type miR-31 promoter or mutant promoter with mutation of p-STAT3 binding sites. ***p<0.001. (M) Chromatin immunoprecipitation (ChIP) assay carried out on mICc12 cells using antibodies against p-STAT3 and Histone 3. The antibody against Histone 3 was used as a positive control. The enrichment of p-STAT3 binding to miR-31 promoter was quantified using qPCR. ***p<0.001.
Discussion
The intestinal epithelium is one of the most rapidly renewing tissues (Leblond and Walker, 1956). Those Lgr5+ CBC stem cells residing at the base of crypts maintain the proliferative capacity necessary to meet this demands of high-turnover tissue, which is driven by activation of the canonical Wnt pathway, as well as repression of BMP signaling (Li and Clevers, 2010), (Li et al., 2014), (Kosinski et al., 2007). Wnt pathway activity and BMP inhibition are believed to be the niche for cycling CBCs. However, it is largely unknown how those Lgr5+ CBCs integrate the signals of Wnt antagonists and activators of BMP and TGFβ. Here we show that the miR-31 activates Wnt signaling by directly repressing a cohort of Wnt antagonists Dkk1, Axin1 and Gsk3b, and represses BMP/TGFβ signaling by directly inhibiting activators of the pathways, Smad3, Smad4 and Bmpr1a, pointing to an important role of miR-31 acting as a rheostat to integrating niche signals sensed by cycling CBCs. In agreement with this point, our in vivo analysis demonstrated that miR-31 induction increases the number of Lgr5+ CBCs whereas miR-31 deletion reduces CBC frequency. Niche Wnt signals likely originate from sub-epithelial telocytes whose presence is required for CBC activity, and possibly to a lesser extent from Paneth cells, who secrete Wnt ligands but are dispensable for CBC activity (Durand et al., 2012; Aoki et al., 2016; Sato et al., 2011; Kim et al., 2012; San Roman et al., 2014; Kabiri et al., 2014). BMP antagonists noggin and gremlin are similarly secreted by sub-mucosal tissues below the crypts (Kosinski et al., 2007), repressing the BMP signaling in CBCs. Thus, sub-epithelial mesenchyme constitutes an extrinsic niche for cycling ISCs. In contrast to secretory signals from an extrinsic niche, miR-31 appears to be an intrinsic coordinator of these extrinsic niche signals, supporting canonical Wnt and represses BMP/TGFβ signals within CBCs. Thus, we identify miR-31 as a cell-autonomous post-transcriptional regulator of the ISC niche, maintaining proliferative capacity of cycling CBC cells. In addition, we also noticed that miR-31 loss resulted in an increased apoptosis in CBC cells, suggesting the importance of miR-31 in maintaining cell survival. The molecular mechanism by which miR-31 protects against apoptosis warrants future study.
The response to high dose of γ-IR can be separated into two distinct stages. First, within 24 hours, the majority of CBCs die via apoptosis and subsequent mitotic death, caused by residual misrepaired and unrepaired of DNA double-strand breaks (Hua et al., 2012). Next, between 24 hours and 4 days after γ-IR, rare surviving CBCs and quiescent reserve ISCs enter the cell cycle and form regenerative foci that produce mitotically active Lgr5+ cells that repair lost epithelium (Yousefi et al., 2016; Hua et al., 2012). We assume that reserve ISCs also undergo the same process, although lack of direct evidence. In line with this, miR-31 is dramatically reduced within the first 24 hours post γ-IR, most likely due to loss of CBCs. Loss of miR-31 led to an marked increase in apoptosis in both CBCs and +4 cells 2 hours post-γ-IR. Based on our data, we conclude that during the first stage miR-31 acts as an anti-apoptotic factor, protecting CBCs and reserve ISCs against apoptosis. During the second stage, the surviving stem cells start proliferating to repopulate the depleted intestinal epithelium. The surviving stem cells are relatively damage-resistant (Tian et al., 2011; Takeda et al., 2011; Li et al., 2014; Yousefi et al., 2016; Ritsma et al., 2014), a property attributed to their quiescence, a state likely maintained by BMP/TGFβ signaling and inactivation of Wnt signaling (Li et al., 2014; Yousefi et al., 2016; He et al., 2004). We show that miR-31 is prominently induced at the regenerative foci 36 hr post-γ-IR and that miR-31 activates Wnt, and represses BMP/TGFβ activities. This points to the potential importance of miR-31 in activating the surviving ISCs. Given BMP/TGFβ inhibiting ability of miR-31, we speculate that the homeostatic insensitivity of reserve ISCs to Wnt ligands (Yan et al., 2012) results from their having active BMP and TGFβ pathways, that must be suppressed for cells to become competent to respond to Wnt ligands. Our findings suggest that miR-31 functions as an activator of dormant reserve ISCs. We also want to mention that the expression patterns of Bmi1 and Hopx are not specific to +4 position, as both of these transcripts are found non-specifically throughout the crypt base (Li et al., 2014; Muñoz et al., 2012; Itzkovitz et al., 2011). This means that miR-31-activated stem cells represent a complex population including +4 cells, surviving Lgr5+ cells, and those TA cells dedifferentiated in response to irradiation. Taken together, our findings suggest that miR-31 functions as the anti-apoptotic factor in ISCs during the early post-γ-IR stage, and, potentially, serves as the cell-intrinsic activator of surviving ISCs regenerative foci promoting regeneration. Future studies will be needed to comprehensively test this idea.
Many reports have showed that miR-31 is overexpressed in CRC tissues (Bandrés et al., 2006; Cottonham et al., 2010; Wang et al., 2009) and increases in progressively during progression from normal to inflammatory bowl disease (IBD) to IBD-related neoplasia (Olaru et al., 2011). We demonstrate that miR-31 promotes tumor development using several models, including cancer cells xenografting, AOM- and DSS- induced inflammation-driven tumors, and Apc-loss driven tumors, characterized by activated Wnt, and repressed BMP/TGFβ signalings. Indeed, several reports showed that miR-31 is overexpressed in colorectal cancer (CRC) tissues (Bandrés et al., 2006; Cottonham et al., 2010; Wang et al., 2009). Wnt signaling is aberrantly up-regulated in CRCs, which due primarily to mutations in the Wnt antagonist APC (Novellasdemunt et al., 2015). Our current study suggests that miR-31 up-regulation might also contribute to Wnt activation in CRCs. In addition, decreased BMP and TGFβ signaling is also often found in CRCs (Bellam and Pasche, 2010; Hardwick et al., 2008), and can be the consequence of miR-31 upregulation. As such, our data suggests that miR-31 acts as the oncogenic microRNA in CRCs. Moreover, tight association between miR-31 induction and STAT3 pathway activation in intestinal tissues is worth noting. Our molecular data suggest direct activation of miR-31 expression by STAT3 signaling pathway. Indeed, many reports showed that constitutive activation of STAT3 is frequently detected in primary human colorectal carcinoma (Kusaba et al., 2005; Corvinus et al., 2005) and contributes to invasion, survival, and growth of colorectal cancer cells (Tsareva et al., 2007; Lin et al., 2005). Therefore, our current study suggests a signaling pathway involving STAT3, miR-31 and WNT/BMP/TGFβ that promotes colorectal tumorigenesis.
In summary, we propose a model in which miR-31 functions as a cell-intrinsic master modulator of the intestinal stem cell niche signaling during normal homeostasis, regeneration and tumorigenesis (Figure 9). During homeostasis, miR-31 functions to integrate niche signals, supporting canonical Wnt activity and represses BMP/TGFβ signaling pathways within cycling CBC stem cells. MiR-31 is stress inducible and plays an important role in epithelial regeneration. In response to high dose of γ-IR, miR-31 is markedly induced via STAT3 signaling pathway, and appears capable of regulating the activation state of a population of dormant, radiation resistant reserve ISCs during regeneration. Further, we demonstrate that miR-31 acts as an oncomiR in promoting tumor growth.
Figure 9. The miR-31 working model in intestinal epithelial regeneration and tumorigenesis.

Materials and methods
Animal experiments
All mouse experiment procedures and protocols were evaluated and authorized by the Regulations of Beijing Laboratory Animal Management and strictly followed the guidelines under the Institutional Animal Care and Use Committee of China Agricultural University (approval number: SKLAB-2011-04-03).
Mouse strains
To generate TRE-miR-31 transgenic mice, the mmu-miR-31 sequence was amplified using the following primers: Forward 5’-CTCGGATCCTGTGCATAACTGCCTTCA-3’ (BamHI site was added), and Reverse 5’-CACAAGCTTGAAGTCAGGGCGAGACAGAC-3’ (HindIII site was added), and was inserted into pTRE2 vector (Clontech) to generate a pTRE2-miR31 construct. TRE-miR31 transgenic mice were produced using standard protocols and crossed with Rosa26-rtTA mice which harboring the modified reverse tetracycline transactivator (M2rtTA) targeted to and under transcriptional control of the Rosa26 locus. Constitutive miR-31−/− mice were generated using CRISPR/Cas9 approach at the Nanjing Animal Center, and 402 bp DNA fragment containing miR-31 was deleted to produce the null allele. Conditional miR-31 KO allele was generated at the Shanghai Model Animal Center, the first exon (14806–15522) of miR-31 was targeted with flanking LoxP sites resulting in the 2 LoxP locus. Villin-Cre (Vil-Cre) mice were purchased from the National Resource Center of Model Mice (stock number:T000142). mTmG, Lgr5-eGFP-CreERT, Apc floxed, and Rosa26-LSL-lacZ mice were obtained from Jackson Laboratories (stock number: 007576, 008875, 009045 and 009427). Hopx-CreERT mice were obtained from John Epstein laboratory. Axin2-LacZ mice were obtained from Yi Zeng laboratory.
Cell culture
HCT116, SW480 and LOVO human colorectal cancer cell lines are purchased from American Type Culture Collection (ATCC) and the mouse mICc12 intestinal epithelial cell line was obtained from the Institute of Interdisciplinary Research (Fudan University, Shanghai, China) who originally obtained them from Dr A Vandervalle (Institut National de la Santé et de la Recherche Médicale, Faculté X, Paris, France). They were confirmed to come from a mouse cell line by Beijing Microread Genetics Co., Ltd using STR profiling. No cell lines are on the list of commonly misidentified cell lines. We have tested for mycoplasma contamination using a Mycoplasma Detection Kit, and no mycoplasma contamination was detected in any of the cultures. These cell lines were cultured in DMEM/F12 medium. The sequence of miR-31 inhibitor is 5’-AGCUAUGCCAGCAUCUUGCCU-3’. The sequence of Scramble RNA is 5’-CAGUACUUUUGUGUAGUACAA-3’. The Sequence of miR-31 mimics:
5’-AGGCAAGAUGCUGGCAUAGCU-3’
3’-CUAUGCCAGCAUCUUGCCUUU-5’
The sequence of negative control for miR-31 mimics:
5’-UUCUCCGAACGUGUCACGUUU-3’
3’-ACGUGACACGUUCGGAGAAUU-5’.
Doxycycline induction and isolation of intestinal epithelium
For the induction, 2 mg/mL Dox (Doxycycline hyclate, Sigma) was added to the drinking water along with 1% w/v sucrose. Mice were induced at 8 weeks of age. To isolate intestinal epithelial cells, mouse intestine was dissected longitudinally and rinsed three times with ice-cold 1x DPBS, then cut into 2–4 mm long pieces, incubated in 1x DPBS containing 2 mM EDTA and 0.2 mM DTT for 30 min at 4°C on a rotating platform. Suspended cells were then collected folowing gentle vortexing. To isolate intestinal crypts, rinsed small intestine was cut-opened and and villi were scraped using coverslip glass, the technique which left the crypts attached. Crypts were then detached after tissue incubation in 1x DPBS with 2 mM EDTA for 30 min at 4°C with gentle vortexing. Isolated crypts were counted and pelleted as previously described (Sato et al., 2009).
Flow cytometry
Dissected intestine was incubated with 5 mM EDTA and 1.5 mM DTT in HBSS for 30 min at 4°C. Single cell suspension was produced following Dispase (BD Biosciences) treatment and passing cells through 40 μm cell strainer. Flow cytometry analysis was performed using BD LSR Fortessa cell analyzer (BD Biosciences). PI-negative cells were selected, then gated for single cells based on the forward-scatter height vs. forward-scatter width (FSC-H vs. FSC-W) and side-scatter height vs. side-scatter width (SSC-H vs. SSC-W) profiles. The size of the nozzle for all sorting runs was 100 μm (20 psi). Lgr5-eGFP+ cells were quantified by flow cytometry in TRE-miR31;Lgr5-eGFP-CreERT and M2rtTA;Lgr5-eGFP-CreERT mice after two weeks of Dox treatment. Lgr5-eGFP+ cells in miR-31+/−;Lgr5-eGFP-CreERT and miR-31−/−;Lgr5-eGFP-CreERT mice were quantified using the same method.
Crypt organoid culture
Crypt culture was performed as previously described in Sato et al. (2009). A total of 500 isolated crypts from TRE-miR31, M2rtTA, Vil-Cre and Vil-Cre;miR31fl/fl (cKO) mice were mixed with 80 μL of matrigel (BD Bioscience) and plated in 24-well plates. After matrigel polymerization, 500 μL of crypt culture medium [advanced DMEM/F12 (Gibco), 2 mM Glutamax (Invitrogen), 100 U/mL penicillin, 100 μg/mL streptomycin (Sigma), 1 mM N-acetyl cysteine (Sigma), B27 supplement (Invitrogen), N2 supplement (Invitrogen), 50 ng/mL mouse, EGF (Peprotech), 100 ng/mL mouse Noggin (Peprotech) and 10% human R-spondin-1 (Peprotech)] was added to M2rtTA, Vil-Cre and Vil-Cre;miR-31fl/fl small intestine crypt cultures. For TRE-miR31 culture, human R-spondin-1 was removed from the medium, and instead 2 μg/mL of Dox was added.
Hybridization
For miR-31 in situ hybridizations, digoxigenin (DIG)-labeled probes (Exiqon) were used following the manufacturer’s protocol. Both DIG-labeled miR-31 and scrambled probes (Exiqon) were hybridized at 61°C. U6 probe was used as the positive control. In situ signals were detected by staining with Anti-DIG-AP antibody (Roche) and developed using BM purple substrate (Roche).
Quantitative RT-PCR
Total RNA was isolated from total mouse small intestinal epithelial cells using TRIzol reagent (Life Technologies) according to the manufacturer’s instructions. Each RNA sample was reverse transcribed with the M-MLV Reverse Transcriptase (Sigma) using Oligo (dT) primers. Real-time PCR was performed using the LightCycler 480 SYBR Green I master mix on a LightCycler 480 real-time PCR system (Roche). qRT-PCR primers were follows:
Axin1-forward: 5’- TTCTGGGTTGAGGAAGCAGC −3’; Axin1-reverse: 5’- GATTAGGGGCTGGATTGGGT-3’;
Axin2-forward: 5’- GGCTAGCTGAGGTGTCGAAG −3’; Axin2 -reverse: 5’- GCCAGTTTCTTTGGCTCTTT −3’;
Ctnnb1-forward: 5’- TCCTAGCTCGGGATGTTCAC −3’; Ctnnb1 -reverse: 5’- TTCTGCAGCTTCCTTGTCCT −3’;
Bmpr1a-forward: 5’- GCTGTCATCATCTGTTGTCCTGG −3’; Bmpr1a-reverse: 5’- CATTACCACAAGGGCTACACCACC −3’;
Myc-forward: 5’- CTACTCGTCGGAGGAAAG −3’; Myc-reverse: 5’- ACTAGACAGCATGGGTAAG −3’;
Ccnd1-forward: 5’- TGGTGAACAAGCTCAAGTGG −3’; Ccnd1-reverse: 5’- GGCGGATTGGAAATGAACT −3’;
Dkk1-forward: 5’- TCCGAGGAGAAATTGAGGAA −3’; Dkk1-reverse: 5’- CCTGAGGCACAGTCTGATGA −3’;
Gsk3b-forward: 5’- CCAACAAGGGAGCAAATTAGAGA −3’; Gsk3b-reverse: 5’- GGTCCCGCAATTCATCGAAA −3’;
Id1-forward: 5’- ACCCTGAACGGCGAGATC −3’; Id1-reverse: 5’- GCGGTAGTGTCTTTCCCAGA −3’;
Id2-forward: 5’- CTACTCGTCGGAGGAAAG −3’; Id2 -reverse: 5’- ACTAGACAGCATGGGTAAG −3’;
Id3-forward: 5’- TCCGGAACTTGTGATCTCCA −3’; Id3-reverse: 5’- GTAAGTGAAGAGGGCTGGGT −3’;
Junb-forward: 5’- CGGATGTGCACGAAAATGGA −3’; Junb-reverse: 5’- GACCCTTGAGACCCCGATAG −3’;
Msx1-forward: 5’- CAGAGTCCCCGCTTCTCC −3’; Msx1-reverse: 5’- CTGAGCGAGCTGGAGAATTC −3’;
Msx2-forward: 5’- TTCACCACATCCCAGCTTCT −3’; Msx2-reverse: 5’- TTCAGCTTTTCCAGTTCCGC −3’;
Cdkn2b-forward: 5’- GCCCAATCCAGGTCATGATG −3’; Cdkn2b-reverse: 5’- TCACACACATCCAGCCGC −3’;
Cdkn2a-forward: 5’- AGAGCTAAATCCGGCCTCAG −3’; Cdkn2a -reverse: 5’- CTCCCTCCCTCCTTCTGCT −3’;
Cdkn1a-forward: 5’- ATCACCAGGATTGGACATGG −3’; Cdkn1a -reverse: 5’- CGGTGTCAGAGTCTAGGGGA −3’;
Cdkn1b-forward: 5’- GGGGAACCGTCTGAAACATT −3’; Cdkn1b -reverse: 5’- AGTGTCCAGGGATGAGGAAG −3’;
Cdkn1c-forward: 5’- GTTCTCCTGCGCAGTTCTCT −3’; Cdkn1c -reverse: 5’- GAGCTGAAGGACCAGCCTC −3’;
Smad3-forward: 5’- ACAGGCGGCAGTAGATAACG −3’; Smad3-reverse: 5’- AACGTGAACACCAAGTGCAT −3’;
Smad4-forward: 5’- GGCTGTCCTTCAAAGTCGTG −3’; Smad4-reverse: 5’- GGTTGTCTCACCTGGAATTGA −3’;
Tgfbr2-forward: 5’- TTGTTGAGACATCAAAGCGG −3’; Tgfbr2-reverse: 5’- ATAAAATCGACATGCCGTCC −3’;
Rela-forward: 5’- agataccaccaagacccacc-3’; Rela-reverse: 5’- ggtgaccagggagattcgaa −3’;
Ikkb-forward: 5’-agaagtacaccgtgaccgtt-3’;Ikkb-reverse: 5’-gggaagggtagcgaacttga-3’;
IL-1-forward: 5’- tacctgtgtctttcccgtgg-3’; IL-1-reverse: 5’- ttgttcatctcggagcctgt-3’;
IL-6-forward: 5’- gccagagtccttcagagaga-3’; IL-6-reverse: 5’-ggtcttggtccttagccact-3’;
IL-18-forward: 5’- gtctaccctctcctgtaagaaca-3’; IL-18-reverse: 5’- tggcaagcaagaaagtgtcc-3’;
Tnf-forward: 5’- aatggcctccctctcatcag-3’; Tnf-reverse: 5’- cccttgaagagaacctggga-3’;
Stat3-forward: 5’- tgacatggatctgacctcgg-3’; Stat3-reverse: 5’- tgcccagattgcccaaagat −3’;
For quantification of microRNA expression, mature miR-31 was quantified using TaqMan microRNA assays according to the manufacturer’s instructions. U6 snRNA was used as the internal control (Applied Biosystems).
Histology, immunofluoresence, and immunochemistry
Intestines were rinsed with 1x DPBS, fixed in 10% formalin, paraffin-embedded and sectioned at 5 μm. Sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry, antigen retrieval was performed by heating slides in 0.01 M citrate buffer (pH 6.0) in a microwave. Sections were then immunostained using ABC peroxidase method (Vector labs) with diaminobenzidine (DAB) as the enzyme substrate and hematoxylin as the counterstain. For immunofluorescence staining, paraffin sections were microwave pretreated in 0.01 M citrate buffer (pH 6.0), and incubated with primary antibodies, then incubated with secondary antibodies (Invitrogen) and counterstained with DAPI in the mounting medium (Vector labs). The following antibodies were used: anti-Ki67 (1:150, Leica), anti-GFP (1:200, Abcam), anti-Axin1 (1:100, Cell Signaling), anti-Gsk3β (1:2000, Abcam), anti-Dkk1 (1:50, Santa Cruz), anti-β-Catenin (1:500, Sigma), anti-BrdU (1:50, Abcam), anti-cleaved Caspase3 (1:100, Cell Signaling), anti-p-Smad1/5/8 (1:200, Cell Signaling), anti-p-Smad2/3 (1:200, Cell Signaling), anti-CyclinD1 (1:50, Abcam), anti-p65 (1:400, Cell Signaling), anti-p-STAT3 (1:800, Cell Signaling).
Dual luciferase activity assays
To generate reporter constructs for luciferase assays, 300–600 bp fragments in length containing predicted miR-31 target site in the 3’UTRs of Axin1, Dkk1, Bmpr1a, Gsk3b, Smad3 and Smad4 were cloned into the psiCHECK-2 vector (Promega) between the XhoI and NotI sites immediately downstream of the Renilla luciferase gene. To generate reporters with mutant 3’UTRs, nucleotides in the target site complementary to the sequence of the miR-31 seed region sequence were mutated using QuikChange Site-Directed Mutagenesis kit according to the manufacturer’s protocol (Stratagene).
293T cells were seeded in 96-well plates one day before transfection. 10 ng of each reporter construct was co-transfected with miR-31 mimics or scramble RNA at a final concentration of 50 nM into 293T cells using Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen). After 24 hr, firefly and renilla luciferase activities were measured with the Dual-Glo luciferase assay system according to the manufacturer’s instructions (Promega) and then be calculated using this formula (WT-mimics/WT-mimics NC) /(MUT-mimics/MUT-mimic NC).
The primers used for amplifying 3’-UTRs of candidate target genes of miR-31 were as follows:
Dkk1-forward: 5’-GCGCTCGAGTGGGCTTGAATTTGGTAT-3’; Dkk1-reverse:5’-TTAGCGGCCGCGTCCCGACTATCCTGTGA-3’;
Smad3-forward: 5’-CCGCTCGAGCACCACACCGAATGAATG-3’; Smad3-reverse: 5’-ATAAGAATGCGGCCGCTGGCAATCCTTTACCATAGC-3’;
Gsk3b-forward: 5’-TTAGCGGCCGCTCAGTTTCACAGGGTTAT-3’; Gsk3b-reverse: 5’-GCGCTCGAGACAAAGGCATTCAAGTAG-3’;
Axin1-forward: GCCTCGAGTCAGTCAGGTGGACAGCC; Axin1-reverse:TAGCGGCCGCACACGGACACTTGGAAGG;
Bmpr1a-forward: GCCTCGAGAATTAAACAATTTTGAGGGAG; Bmpr1a-reverse: TTGCGGCCGCCTACAGTTACAAGGTGGAT;
Smad4-forward: 5’- TTACTCCTAGCAGCACCC −3’; Smad4-reverse: 5’-CAGTTGTCGTCTTCCCTC-3’;
Western blotting
For western blotting assay, intestinal epithelial tissues were lysed in lysis buffer (Beyotime, China) with 1% PMSF (Phenylmethylsulfonyl fluoride). After quantification using a BCA protein assay kit (Beyotime, China), 30 μg of total protein was separated by 10% SDS-PAGE under denaturing conditions and transferred to PVDF membranes (GE Healthcare). Membranes were blocked in 5% nonfat dry milk in incubation buffer and incubated with primary antibodies, followed by incubation with the secondary antibody and chemiluminescent detection system (Pierce). The primary antibodies were: anti-GAPDH (Sigma), anti-β-Tubulin (Sigma), anti-CyclinD1 (Santa Cruz), anti-c-Myc (Santa Cruz), anti-β-Catenin (Sigma), anti-Dkk1 (Santa Cruz), anti-Gsk3β (Abcam), anti-Axin1 (Cell Signaling), anti-p-Smad2/3 (Cell Signaling), anti-p21(Santa Cruz), anti-Smad4 (Santa Cruz), anti-p-Smad1/5/8 (Cell Signaling), anti-Bmpr1a (Abcam), anti-p65 (Cell Signaling), anti-STAT3 (Cell Signaling), anti-p-p65 (Cell Signaling), anti-p-STAT3 (Cell Signaling).
Irradiation injury
For irradiation, 2-month-old adult mice were subjected to 12 Gy γ-IR and executed at appointed time.
Establishment of the AOM-DSS mouse model
Seven week-old control and miR-31−/− mice were intraperitoneally injected with AOM (Sigma-Aldrich,) at 10 mg/kg body weight. One week after AOM injection, mice were treated with the so-called DSS cycle, comprised of two steps in which mice were fed with 2.5% (w/v) DSS (molecular weight 36,000–50,000, MP Biomedicals) for 7 days followed by 14 days of normal water feeding. Mice were subjected to a total of three DSS cycles. After treatment, mice were sacrificed and distal colon tissues were collected and tumor number and volume were evaluated.
Luciferase assay for miR-31 promoter activity
The transcript of primary miR-31 is located at Chromosome 4, NC_000070.6 (88910557..88910662, complement) in the mouse genome. The upstream 2 kb region of transcript start site (TSS) was identified as the miR-31 promoter in this study, which is located at Chromosome 4, NC_000070.6 (88910663..88912663) and was cloned into the pGL3-Basic reporter constructs. The binding site of STAT3 is located at 88911572–88911582. The binding site 1 and 2 of p65 are located at 88912038–88912048 and 88912409–88912419, respectively.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed according to the manufacturer’s protocol with minor modifications, using Simple-ChIP enzymatic chromatin immunoprecipitation kit (Cell Signaling Technology). The sonicated nuclear fractions were divided for input control and for overnight incubated at 4°C with p-STAT3 or the positive control with H3, negative control with IgG. The recruited genomic DNA from the ChIP assays was quantified by qPCR with primers specific to p-Stat3 binding elements of the miR-31 promoter regions. Primers were as follows: p-STAT3-binding site forward: 5’-TCCAGGCAAGAAAGTGAGGG −3’; p-STAT3- binding site reverse: 5’- TGAGTAACAGTGCAACAGAGC-3’.
Apoptosis analysis
The 21nt oligonucleotide miR-31 inhibitor (5-AGCUAUGCCAGCAUCUUGCCU-3) or negative control Scramble RNA (5-CAGUACUUUUGUGUAGUACAA-3) were transfected into HCT116 cells with or without CHIR99021 (GSK3β inhibitor). The apoptotic cells were evaluated by FITC-Annexin V/PI staining (BD PharMingen) and analyzed by FACS (Becton, Dickinson).
RNA crosslinking, immunoprecipitation, and qRT-PCR (CLIP-PCR) assay
CLIP-PCR assay performed as previously described with modification (Wang et al., 2015). Cells were treated with scramble RNA or miR-31 inhibitor, and then harvested after being irradiated at 400 mJ/cm2 twice. They were then re-suspended in PXL buffer with RNAsin (Promega) and RQ1 DNAse (Promega), and spun at 15000 rpm for 30 min. Supernatant was collected. Protein A Dynabeads (Dynal, 100.02, Thermo Fisher) and goat anti-rabbit IgG (Jackson ImmunoResearch,) or Ago2 antibody were incubated for 4 hr at 4°C with rotation. The supernatant was added to the beads for 2–4 hr at 4°C. Beads were then washed twice and digested with Proteinase K (4 mg/ml) for 20 min at 37°C. RNA was then extracted using Trizol Reagent (Invitrogen) and quantified by qRT-PCR.
Statistical analysis
All analyses were performed in triplicate or greater and the means obtained were used for independent t-tests. Asterisks denote statistical significance (*p<0.05; **p<0.01; ***p<0.001). All data are reported as mean ±SD. Means and standard deviations from at least three independent experiments are presented in all graphs.
Acknowledgement
We are grateful to Bogi Andersen for editing the manuscript, and Yeguang Chen for providing the Apc floxed mice. ZY is supported by the National Natural Science Foundation of China (No. .81772984, 81572614, 31271584); Beijing Nature Foundation Grant (5162018); the Major Project for Cultivation Technology (2016ZX08008001, 2014ZX08008001); Basic Research Program (2015QC0104, 2015TC041, 2016SY001, 2016QC086); SKLB Open Grant (2015SKLB6-16). JS is supported by the National Natural Science Foundation of China (No. 31370830 and 11675134) and the 111 Project (No. B16029). MVP is supported by the NIH NIAMS grants R01-AR067273, R01-AR069653, and Pew Charitable Trust grant. TA is supported by the NIAMS/NIH grant R01 AR061474-01.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Zhengquan Yu, Email: zyu@cau.edu.cn.
Roel Nusse, Stanford University, United States.
Funding Information
This paper was supported by the following grants:
National Institutes of Health R01 AR061474-01 to Thomas Andl.
National Institutes of Health R01-AR067273 to Maksim V Plikus.
National Institutes of Health R01-AR069653 to Maksim V Plikus.
National Natural Science Foundation of China 31370830 to Jinyue Sun, Jianwei Shuai.
National Natural Science Foundation of China 11675134 to Jinyue Sun, Jianwei Shuai.
National Natural Science Foundation of China 81572614 to Zhengquan Yu.
National Natural Science Foundation of China 31271584 to Zhengquan Yu.
National Natural Science Foundation of China 81772984 to Zhengquan Yu.
Additional information
Competing interests
No competing interests declared.
Author contributions
Data curation, Software, Formal analysis, Validation, Investigation, Methodology, Writing—original draft, Project administration.
Data curation, Formal analysis, Validation, Investigation, Methodology.
Data curation, Formal analysis, Validation, Investigation, Methodology.
Formal analysis, Methodology.
Formal analysis.
Formal analysis, Investigation, Methodology.
Formal analysis, Methodology.
Data curation.
Formal analysis, Writing—review and editing.
Investigation, Writing—review and editing.
Formal analysis, Writing—review and editing.
Investigation, Methodology, Writing—review and editing.
Formal analysis, Writing—review and editing.
Formal analysis, Writing—review and editing.
Conceptualization, Resources, Data curation, Formal analysis, Supervision, Funding acquisition, Validation, Investigation, Writing—original draft, Project administration, Writing—review and editing.
Ethics
Animal experimentation: All mouse experiment procedures and protocols were evaluated and authorized by the Regulations of Beijing Laboratory Animal Management and strictly followed the guidelines under the Institutional Animal Care and Use Committee of China Agricultural University (approval number: SKLAB-2011-04-03).
Additional files
References
- Aoki R, Shoshkes-Carmel M, Gao N, Shin S, May CL, Golson ML, Zahm AM, Ray M, Wiser CL, Wright CV, Kaestner KH. Foxl1-expressing mesenchymal cells constitute the intestinal stem cell niche. Cellular and Molecular Gastroenterology and Hepatology. 2016;2:175–188. doi: 10.1016/j.jcmgh.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandrés E, Cubedo E, Agirre X, Malumbres R, Zárate R, Ramirez N, Abajo A, Navarro A, Moreno I, Monzó M, García-Foncillas J. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Molecular Cancer. 2006;5:29. doi: 10.1186/1476-4598-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nature Reviews Molecular Cell Biology. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- Bellam N, Pasche B. Tgf-beta signaling alterations and colon cancer. Cancer treatment and research. 2010;155:85–103. doi: 10.1007/978-1-4419-6033-7_5. [DOI] [PubMed] [Google Scholar]
- Chivukula RR, Shi G, Acharya A, Mills EW, Zeitels LR, Anandam JL, Abdelnaby AA, Balch GC, Mansour JC, Yopp AC, Maitra A, Mendell JT. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell. 2014;157:1104–1116. doi: 10.1016/j.cell.2014.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346:1248012. doi: 10.1126/science.1248012. [DOI] [PubMed] [Google Scholar]
- Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huber LA, Zatloukal K, Beug H, Ohlschläger P, Schütz A, Halbhuber KJ, Friedrich K. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7:545–555. doi: 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottonham CL, Kaneko S, Xu L. miR-21 and miR-31 converge on TIAM1 to regulate migration and invasion of colon carcinoma cells. Journal of Biological Chemistry. 2010;285:35293–35302. doi: 10.1074/jbc.M110.160069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PS, Dismuke AD, Powell AE, Carroll KH, Wong MH. Wnt-reporter expression pattern in the mouse intestine during homeostasis. BMC Gastroenterology. 2008;8:57. doi: 10.1186/1471-230X-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Miranda NF, van Dinther M, van den Akker BE, van Wezel T, ten Dijke P, Morreau H. Transforming Growth Factor β Signaling in Colorectal Cancer Cells With Microsatellite Instability Despite Biallelic Mutations in TGFBR2. Gastroenterology. 2015;148:e1428. doi: 10.1053/j.gastro.2015.02.052. [DOI] [PubMed] [Google Scholar]
- De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. Journal of carcinogenesis. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand A, Donahue B, Peignon G, Letourneur F, Cagnard N, Slomianny C, Perret C, Shroyer NF, Romagnolo B. Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1) PNAS. 2012;109:8965–8970. doi: 10.1073/pnas.1201652109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emde A, Hornstein E. miRNAs at the interface of cellular stress and disease. The EMBO Journal. 2014;33:1428–1437. doi: 10.15252/embj.201488142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Sato T, Katsuno T, Nakagawa T, Noguchi Y, Tokumasa A, Yokote K, Yokosuka O, Saito Y. Smad3 contributes to positioning of proliferating cells in colonic crypts by inducing EphB receptor protein expression. Biochemical and Biophysical Research Communications. 2011;405:521–526. doi: 10.1016/j.bbrc.2011.01.045. [DOI] [PubMed] [Google Scholar]
- Gangaraju VK, Lin H. MicroRNAs: key regulators of stem cells. Nature Reviews Molecular Cell Biology. 2009;10:116–125. doi: 10.1038/nrm2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehart H, Clevers H. Repairing organs: lessons from intestine and liver. Trends in Genetics. 2015;31:344–351. doi: 10.1016/j.tig.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Hardwick JC, Kodach LL, Offerhaus GJ, van den Brink GR. Bone morphogenetic protein signalling in colorectal cancer. Nature Reviews Cancer. 2008;8:806–812. doi: 10.1038/nrc2467. [DOI] [PubMed] [Google Scholar]
- He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, Mishina Y, Li L. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nature Genetics. 2004;36:1117–1121. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- Hua G, Thin TH, Feldman R, Haimovitz-Friedman A, Clevers H, Fuks Z, Kolesnick R. Crypt base columnar stem cells in small intestines of mice are radioresistant. Gastroenterology. 2012;143:1266–1276. doi: 10.1053/j.gastro.2012.07.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra I, Erlich Y, Muthuswamy SK, Sachidanandam R, Hannon GJ. A role for microRNAs in maintenance of mouse mammary epithelial progenitor cells. Genes & Development. 2007;21:3238–3243. doi: 10.1101/gad.1616307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. PNAS. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzkovitz S, Lyubimova A, Blat IC, Maynard M, van Es J, Lees J, Jacks T, Clevers H, van Oudenaarden A. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nature Cell Biology. 2011;14:106–114. doi: 10.1038/ncb2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabiri Z, Greicius G, Madan B, Biechele S, Zhong Z, Zaribafzadeh H, Edison, Aliyev J, Wu Y, Bunte R, Williams BO, Rossant J, Virshup DM. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development. 2014;141:2206–2215. doi: 10.1242/dev.104976. [DOI] [PubMed] [Google Scholar]
- Kaler P, Augenlicht L, Klampfer L. Activating mutations in β-catenin in colon cancer cells alter their interaction with macrophages; the role of snail. PLoS One. 2012;7:e45462. doi: 10.1371/journal.pone.0045462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Escudero S, Shivdasani RA. Intact function of Lgr5 receptor-expressing intestinal stem cells in the absence of Paneth cells. PNAS. 2012;109:3932–3937. doi: 10.1073/pnas.1113890109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- Kishimoto M, Fukui T, Suzuki R, Takahashi Y, Sumimoto K, Okazaki T, Sakao M, Sakaguchi Y, Yoshida K, Uchida K, Nishio A, Matsuzaki K, Okazaki K. Phosphorylation of Smad2/3 at specific linker threonine indicates slow-cycling intestinal stem-like cells before reentry to cell cycle. Digestive Diseases and Sciences. 2015;60:362–374. doi: 10.1007/s10620-014-3348-3. [DOI] [PubMed] [Google Scholar]
- Kosinski C, Li VS, Chan AS, Zhang J, Ho C, Tsui WY, Chan TL, Mifflin RC, Powell DW, Yuen ST, Leung SY, Chen X. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. PNAS. 2007;104:15418–15423. doi: 10.1073/pnas.0707210104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusaba T, Nakayama T, Yamazumi K, Yakata Y, Yoshizaki A, Nagayasu T, Sekine I. Expression of p-STAT3 in human colorectal adenocarcinoma and adenoma; correlation with clinicopathological factors. Journal of Clinical Pathology. 2005;58:833–838. doi: 10.1136/jcp.2004.023416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CP, Walker BE. Renewal of cell populations. Physiological reviews. 1956;36:255–276. doi: 10.1152/physrev.1956.36.2.255. [DOI] [PubMed] [Google Scholar]
- Leung AK, Sharp PA. MicroRNA functions in stress responses. Molecular Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327:542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Nakauka-Ddamba A, Tobias J, Jensen ST, Lengner CJ. Mouse Label-Retaining Cells Are Molecularly and Functionally Distinct From Reserve Intestinal Stem Cells. Gastroenterology. 2016;151:298–310. doi: 10.1053/j.gastro.2016.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Yousefi M, Nakauka-Ddamba A, Jain R, Tobias J, Epstein JA, Jensen ST, Lengner CJ. Single-cell analysis of proxy reporter allele-marked epithelial cells establishes intestinal stem cell hierarchy. Stem Cell Reports. 2014;3:876–891. doi: 10.1016/j.stemcr.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Lai R, Chirieac LR, Li C, Thomazy VA, Grammatikakis I, Rassidakis GZ, Zhang W, Fujio Y, Kunisada K, Hamilton SR, Amin HM. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. The American journal of pathology. 2005;167:969–980. doi: 10.1016/S0002-9440(10)61187-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna LB, Schug J, Vourekas A, McKenna JB, Bramswig NC, Friedman JR, Kaestner KH. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology. 2010;139:1654–1664. doi: 10.1053/j.gastro.2010.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H. Wnt-expressing cells in the intestines: guides for tissue remodeling. Journal of Biochemistry. 2017;161:19–25. doi: 10.1093/jb/mvw070. [DOI] [PubMed] [Google Scholar]
- Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk ME, Henderson DE, Baffour-Awuah NY, Ambruzs DM, Fogli LK, Algra S, Breault DT. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. PNAS. 2011;108:179–184. doi: 10.1073/pnas.1013004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S, Myant K, Versteeg R, Sansom OJ, van Es JH, Barker N, van Oudenaarden A, Mohammed S, Heck AJ, Clevers H. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent '+4' cell markers. The EMBO Journal. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H, Miyoshi Y, Horii A, Aoki T, Petersen GM, Vogelstein B, Maher E, Ogawa M, Maruyama M, Utsunomiya J. Screening for germ-line mutations in familial adenomatous polyposis patients: 61 new patients and a summary of 150 unrelated patients. Human Mutation. 1992;1:467–473. doi: 10.1002/humu.1380010603. [DOI] [PubMed] [Google Scholar]
- Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. American Journal of Physiology - Cell Physiology. 2015;309:C511–C521. doi: 10.1152/ajpcell.00117.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaru AV, Selaru FM, Mori Y, Vazquez C, David S, Paun B, Cheng Y, Jin Z, Yang J, Agarwal R, Abraham JM, Dassopoulos T, Harris M, Bayless TM, Kwon J, Harpaz N, Livak F, Meltzer SJ. Dynamic changes in the expression of MicroRNA-31 during inflammatory bowel disease-associated neoplastic transformation. Inflammatory Bowel Diseases. 2011;17:221–231. doi: 10.1002/ibd.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A, Wharton N, Parris A, Mitchell E, Sobolewski A, Kam C, Bigwood L, El Hadi A, Münsterberg A, Lewis M, Speakman C, Stebbings W, Wharton R, Sargen K, Tighe R, Jamieson C, Hernon J, Kapur S, Oue N, Yasui W, Williams MR. Canonical Wnt signals combined with suppressed TGFβ/BMP pathways promote renewal of the native human colonic epithelium. Gut. 2014;63:610–621. doi: 10.1136/gutjnl-2012-304067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritsma L, Ellenbroek SIJ, Zomer A, Snippert HJ, de Sauvage FJ, Simons BD, Clevers H, van Rheenen J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature. 2014;507:362–365. doi: 10.1038/nature12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runtsch MC, Round JL, O'Connell RM. MicroRNAs and the regulation of intestinal homeostasis. Frontiers in Genetics. 2014;5:347. doi: 10.3389/fgene.2014.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Roman AK, Jayewickreme CD, Murtaugh LC, Shivdasani RA. Wnt secretion from epithelial cells and subepithelial myofibroblasts is not required in the mouse intestinal stem cell niche in vivo. Stem Cell Reports. 2014;2:127–134. doi: 10.1016/j.stemcr.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nature Genetics. 2008;40:915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Sun K, Lai EC. Adult-specific functions of animal microRNAs. Nature Reviews Genetics. 2013;14:535–548. doi: 10.1038/nrg3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334:1420–1424. doi: 10.1126/science.1213214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsareva SA, Moriggl R, Corvinus FM, Wiederanders B, Schütz A, Kovacic B, Friedrich K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia. 2007;9:279–291. doi: 10.1593/neo.06820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CJ, Zhou ZG, Wang L, Yang L, Zhou B, Gu J, Chen HY, Sun XF. Clinicopathological significance of microRNA-31, -143 and -145 expression in colorectal cancer. Disease Markers. 2009;26:27–34. doi: 10.1155/2009/921907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Li N, Yousefi M, Nakauka-Ddamba A, Li F, Parada K, Rao S, Minuesa G, Katz Y, Gregory BD, Kharas MG, Yu Z, Lengner CJ. Transformation of the intestinal epithelium by the MSI2 RNA-binding protein. Nature Communications. 2015;6:6517. doi: 10.1038/ncomms7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR, Sangiorgi E, Capecchi MR, Kuo CJ. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. PNAS. 2012;109:466–471. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang MH, Yu J, Chen N, Wang XY, Liu XY, Wang S, Ding YQ. Elevated microRNA-31 expression regulates colorectal cancer progression by repressing its target gene SATB2. PLoS One. 2013;8:e85353. doi: 10.1371/journal.pone.0085353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Fuchs E. MicroRNAs and their roles in mammalian stem cells. Journal of Cell Science. 2011;124:1775–1783. doi: 10.1242/jcs.069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi M, Li N, Nakauka-Ddamba A, Wang S, Davidow K, Schoenberger J, Yu Z, Jensen ST, Kharas MG, Lengner CJ. Msi RNA-binding proteins control reserve intestinal stem cell quiescence. The Journal of Cell Biology. 2016;215:401–413. doi: 10.1083/jcb.201604119. [DOI] [PMC free article] [PubMed] [Google Scholar]