

Preface
Amyotrophic lateral sclerosis (ALS) is a progressive and uniformly fatal neurodegenerative disease. A plethora of genetic factors underlying ALS have now been identified that drive motor neuron degeneration, increase susceptibility to the disease, or influence the rate of progression. Emerging themes include dysfunction in RNA metabolism and protein homeostasis, with specific defects in nucleocytoplasmic trafficking, induction of endoplasmic reticulum stress, and impaired dynamics of ribonucleoprotein bodies such as RNA granules that assemble through the process of liquid-liquid phase separation. Extraordinary recent progress in understanding the biology of ALS provides new grounds for optimism that meaningful therapies for ALS will be identified.
ALS is a prototypical neurodegenerative disease characterized by progressive degeneration of motor neurons in the brain and spinal cord. First described by the neurologist Jean-Martin Charcot, the name reflects both the degeneration of corticospinal motor neurons, whose descending axons in the lateral spinal cord appear scarred (“lateral sclerosis”), and the demise of spinal motor neurons, with secondary denervation and wasting of muscle (“amyotrophy”). Corticospinal neurons make direct or indirect connections onto spinal motor neurons, which in turn innervate skeletal muscles and trigger their contractions (Fig. 1a). This review summarizes clinical and pathological features in ALS and describes how discoveries in ALS genetics have illuminated major themes in the molecular pathophysiology of this disease.
Figure 1. The components of the nervous system impacted in ALS.
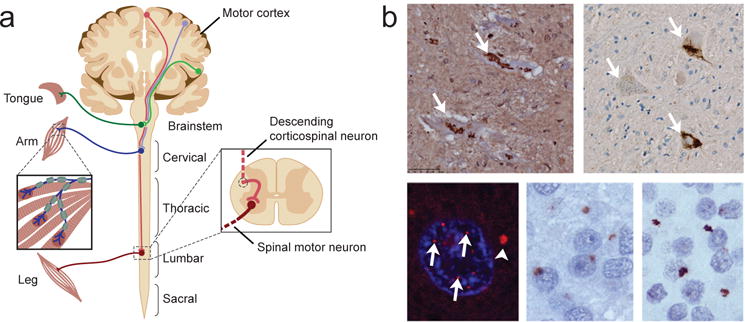
(a) ALS primarily impacts descending corticospinal motor neurons (upper motor neurons) that project from the motor cortex to synapses in the brainstem and spinal cord, and bulbar or spinal motor neurons (lower motor neurons) that project to skeletal muscles. (b) Typical pathology of different ALS subtypes. Clockwise from top left: SOD1 aggregates in spinal motor neurons in SOD1-related familial ALS; TDP-43 redistribution to cytoplasmic inclusions in spinal motor neurons in sporadic ALS; GR dipeptide repeat pathology in the dentate nucleus of C9-ALS/FTD; GA dipeptide repeat pathology in the dentate nucleus of C9-ALS/FTD; RNA foci in the nucleus (arrows) and cytoplasm (arrowhead) of a cortical neuron in C9-ALS/FTD.
ALS is known in the United States as Lou Gehrig’s disease and in the United Kingdom as motor neuron disease. While onset is commonly in mid-adulthood (mean of 55 years of age), ALS may begin as early as the first or second decade or in late life. Like most neurodegenerative diseases, it begins focally and spreads: symptoms that begin as subtle cramping or weakness in the limbs or bulbar muscles progress to paralysis of nearly all skeletal muscles. Some subsets of motor neurons, such as those that innervate extraocular muscles or sphincters, are spared until late in the disease. However, the disease is invariably fatal. Death typically results within 3–5 years, although some forms demonstrate protracted survival.
ALS is an orphan disease, newly afflicting 1–2 individuals per 100,000 each year in most countries; however, the prevalence of ALS is much lower, reflecting the rapid lethality of the disease1. In the United States and United Kingdom, ALS causes more than 1 in 500 adult deaths, a statistic that predicts that more than 15 million people now alive worldwide will succumb to this disease.
The clinical manifestations of ALS
Significant heterogeneity exists within the general rubric of ALS. Distinct clinical subsets are distinguished by involvement of different sets of motor neurons or different regions of the body. Depending on the location of primary pathology, patients may develop weakness with flaccidity and atrophy of the limbs (progressive muscular atrophy, affecting mainly the spinal or lower motor neurons), prominent hyperreflexia and spasticity with increased limb tone but relatively little muscle atrophy (primary lateral sclerosis, affecting corticospinal motor neurons with limited involvement of spinal motor neurons), tongue atrophy with thickness of speech and difficulty swallowing (bulbar ALS, affecting brainstem motor neurons that serve muscles of tongue movement, chewing, swallowing and articulation), or slow and highly dysfunctional speech and swallowing in the absence of tongue atrophy, often accompanied by accentuation of emotional reflexes (pseudobulbar palsy, affecting cortical frontobulbar motor neurons). Importantly, ALS shows clinical overlap with several other adult-onset degenerative disorders, most frequently frontotemporal dementia (FTD), that may constitute a clinical spectrum (see Box).
Box. ALS shows frequent clinical and genetic overlap with related diseases.
Whereas ALS historically had been viewed as a disease with purely motor manifestations, careful observations of ALS patients over the last 30 years has revealed that ALS overlaps clinically, pathologically and genetically with several other degenerative disorders. In particular, it has been appreciated that motor neuron loss may be accompanied by loss of cortical neurons in the frontal and temporal cortices. The clinical correlate is frontotemporal dementia (FTD), an entity that, unlike Alzheimer disease, does not entail major memory loss but instead impairs judgment and executive skills, often leading to behavioral disturbances. About 20% of ALS patients meet the clinical criteria for a concomitant diagnosis of FTD, although as many as 50% of patients experience cognitive impairment. Less frequently ALS occurs concomitantly with Paget’s disease of bone (PDB) or inclusion body myopathy (IBM). Similar to ALS, both FTD and IBM are characterized by inclusions of TDP-43 and related RNA-binding proteins. The relationship between ALS, FTD, PDB and IBM has been extended by genetic evidence, potentially placing them within a continuum of a broader degenerative disorder. The relationship between these diseases is further substantiated by the syndrome multisystem proteinopathy, a genetically heterogeneous, dominantly inherited disorder characterized by ALS/FTD occurring concomitantly with IBM and/or PDB.
Genetic contributions to ALS
About 10% of cases of ALS are transmitted within families, almost always as dominant traits, frequently with high penetrance. The first ALS gene, cytosolic superoxide dismutase or SOD1, was reported in 19932, with more than 50 additional potential ALS genes published since, although validating the causality of specific variants remains a challenge. Applying relatively rigorous criteria, one can generate a list of genes whose mutations are unequivocally implicated in ALS pathogenesis (Table 1). These genes may be grouped loosely into (1) those that alter proteostasis and protein quality control; (2) those that perturb aspects of RNA stability, function and metabolism; and (3) those that disturb cytoskeletal dynamics within the motor neuron axon and distal terminal. The offending mutations are largely missense substitutions, although the genetic lesion in C9ORF72 is an enormous expansion of an intronic hexanucleotide repeat.
Table 1.
The genetics of ALS.
| Locus | Gene | Protein | Protein Function | Mutations | Proportion of ALS | Discovery Date | |
|---|---|---|---|---|---|---|---|
| familial | sporadic | ||||||
| 21q22.1 | SOD1 | Cu/Zn superoxide dismutase | Superoxide dismutase | >150 | 20% | 2% | 19932 |
| 2p13 | DCTN1 | Dynactin | Component of dynein motor complex | 10 | 1% | <1% | 200354 |
| 14q11 | ANG | Angiogenin | Ribonuclease | >10 | <1% | <1% | 2006144 |
| q36 | TARDBP | TDP-43 | RNA-binding protein | >40 | 5% | <1% | 200869,145 |
| 16p11.2 | FUS | FUS/TLS | RNA-binding protein | >40 | 5% | <1% | 200970,71 |
| 9p13.3 | VCP | Valosin-containing protein | Ubiquitin segregase | 5 | 1–2% | <1% | 201046 |
| 10p15-p14 | OPTN | Optineurin | Autophagy adaptor | 1 | 4% | <1% | 201044 |
| 9p21-22 | C9ORF72 | C9ORF72 | Possible guanine nucleotide exchange factor | Intronic GGGGCC repeat | 25% | 10% | 20118,79 |
| Xp11.23-Xp13.1 | UBQLN2 | Ubiquilin 2 | Autophagy adaptor | 5 | <1% | <1% | 201142 |
| 5q35 | SQSTM1 | p62 | Autophagy adaptor | 10 | <1% | 201143,146 | |
| 17p13.2 | PFN1 | Profilin-1 | Actin-binding protein | 5 | <1% | <1% | 2012147 |
| 12q13.1 | HNRNPA1 | hnRNPA1 | RNA-binding protein | 3 | <1% | <1% | 201372,73 |
| 5q31.2 | MATR3 | Matrin 3 | RNA-binding protein | 4 | <1% | <1% | 201478 |
| 2q36.1 | TUBA4A | α-tubulin 4a | Microtubule subunit | 7 | <1% | <1% | 2014148 |
| 22q11.23 | CHCHD10 | Coiled-coil-helix-coiled-coil-helix domain containing 10 | Mitochondrial protein of unknown function | 2 | <1% | <1% | 2014149 |
| 12q14.1 | TBK1 | TANK-binding kinase 1 | Regulates autophagy and inflammation | 10 | ? | ? | 2015150 |
Strictly speaking, the term sporadic ALS refers to disease that presents without a family history of the disease, although this term is sometimes mistakenly thought to refer to ALS that occurs without a genetic basis. Technological advances have permitted broad DNA sequencing in sporadic ALS patients and revealed that genetic variants in established ALS genes are not infrequent. For example, it is now evident that 1–3% of sporadic ALS cases reflect missense mutations in SOD13; another 5% or more are caused by intronic expansions in C9ORF724. Pathogenic mutations in other ALS genes (e.g., TARDBP, FUS, HNRNPA1, SQSTM1, VCP, OPTN, PFN1) also have been identified in the sporadic ALS population, although they are rare.
Of immense interest are genetic variants that enhance ALS susceptibility or modify the clinical phenotype, even if the variants do not themselves cause ALS. Robustly confirmed in this category is ATXN2. Large trinucleotide repeat (CAG) expansions in the coding sequence of ATXN2 cause spinocerebellar ataxia type 2, which sometimes displays motor weakness as an early presentation. It is therefore striking that mid-range CAG expansions of ATXN2 increase the risk of developing ALS5. In contrast, variants that reduce expression of axonal guidance gene EPHA4 improve overall survival of ALS patients6.
The pathology of ALS
At autopsy, ALS cases reveal degeneration of motor neurons in the motor cortex, brainstem motor nuclei, and anterior horns in the spinal cord. In the initial phases of the disease, ALS spares the motor neurons that innervate extraocular muscles, as well as those that control bowel and bladder function. As spinal motor neurons degenerate, their target muscles become atrophied. Degeneration of the spinal processes of corticospinal neurons results in scarring in the lateral tracts of the spinal cord. As the disease progresses, affected spinal motor neurons shrink and accumulate rounded or thread-like deposits of aggregated proteins that are collectively referred to as inclusions (Fig. 1b). The cytoplasmic inclusions in ALS often become ubiquitinated; it is now recognized that an early target for ubiquitination is TAR-DNA binding protein (TDP43), which forms the major component of ubiquitinated inclusions in most ALS cases7.
Some additional pathological features are associated with specific genes. For example, cases of ALS caused by microsatellite expansions in C9ORF72 reveal intranuclear RNA foci8, distinctive cytoplasmic inclusions derived from dipeptide repeat proteins (DPRs, discussed below)9,10, as well as p62-positive, largely TDP43-negative neuronal cytoplasmic inclusions predominantly in the cerebellum and hippocampus11. Cases of ALS caused by mutations in SOD1 and FUS are pathologically distinct in that they do not exhibit TDP-43 pathology, but rather inclusions of abnormal SOD1 and FUS proteins, respectively. In addition to these pathological findings in motor neurons, there is abundant evidence for significant pathology in non-neural cell types (e.g., insidious astrogliosis and microgliosis). As reviewed below, it is likely that both forms of non-cell-autonomous cellular reactivity adversely influence progression in ALS.
Pathogenic mechanisms of ALS
The molecular era of discovery in ALS began in 1993 with the identification of dominant mutations in SOD1, the gene encoding an abundant, ubiquitously expressed cytoplasmic superoxide dismutase2. A major antioxidant, the normal function of SOD1 is to catalytically convert highly reactive superoxide (most frequently produced by errors within mitochondria) to either hydrogen peroxide or oxygen.
An important initial realization arose from expression of mutant SOD1 in mice, which demonstrated that motor neuron degeneration is driven by one or more acquired toxicities of the mutant protein12,13 and is independent of dismutase activity14. The >170 ALS-causing mutations now identified (http://alsod.iop.kcl.ac.uk/) lie in virtually every region of the 153-amino acid SOD1 polypeptide. Moreover, while many variants retain partial or full dismutase activity, there is no correlation between reduction in activity and age of disease onset or rapidity of disease progression15. These findings led to the consensus view that disease arises from one or more toxic properties of the many SOD1 mutants and not from reduced dismutase activity.
A sobering reality, however, is that 23 years after the initiating discovery, no consensus has yet emerged as to the primary toxicity of mutant SOD1. Instead, a plethora of toxic mechanisms have been proposed to mediate the degeneration and death of motor neurons (Fig. 2). One prominent finding is that a proportion of each ALS-causing SOD1 mutant fails to fold properly, thus implicating accumulation of misfolded SOD1 as a possible toxic contributor in ALS (Fig. 2a). The misfolded SOD1 forms ubiquitinated cytoplasmic inclusions that can occur early in disease and escalate as disease progresses16.
Figure 2. Schematic representation of the major cellular disease mechanisms implicated in ALS.
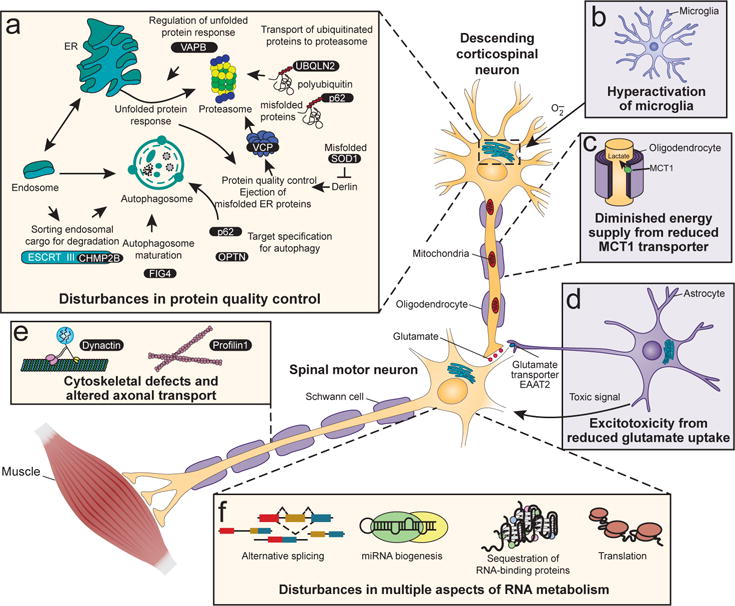
(a) Disturbances in protein quality control and ER stress. (b) Microglial activation and production of extracellular superoxide. (c) Reduced energy supply from oligodendrocytes to the underlying motor axons following reduced levels of the MCT1 lactate transporter. (d) Release from astrocytes of an as-yet unidentified species toxic to motor neurons. (e) Disruption of the cytoskeleton and impaired axonal transport. (f) Disturbance of multiple aspects of RNA metabolism are implicated in ALS.
The accrual of ubiquitinated SOD1 aggregates in patients with SOD1 mutations is paralleled by accrual of ubiquitinated TDP-43 aggregates in patients with TARDBP mutations (and in the vast majority of patients with sporadic ALS as described in greater detail below), highlighting a correlation between protein aggregation and disease. Nevertheless, as has been demonstrated in other neurodegenerative diseases, large aggregates of the disease-causing mutant SOD1 are not sufficient to drive disease, as their elimination fails to affect any aspect of fatal disease developed in mice17.
Non-cell-autonomous toxicity: the neighborhood matters
Like the other major neurodegenerative diseases, all of the genes whose mutations cause ALS are expressed in multiple cell types. Indeed, it is now clear that ALS arises in part by non-cell-autonomous mechanisms. In other words, disease arises from a combination of mutant damage within motor neurons and their glial partners, rather than from damage exclusively in neurons.
For mutant SOD1, this concept is underscored by studies in mice revealing that high expression levels of mutant SOD1 in all motor neurons is not sufficient for early-onset disease18. Conversely, reduction of mutant SOD1 synthesis in motor neurons is of no benefit in slowing the rate of disease progression after onset even when applied pre-symptomatically19–21. Thus, ALS is a disease not just of the motor neuron but the motor system, which is comprised of the motor neuron and intimately associated cells of several types.
The critical role of glia in ALS
The importance of glial cells in motor neuron degeneration and death emerged from studies in which mutant SOD1 synthesis was silenced in microglia, astrocytes or oligodendrocyte precursors. Microglia, the innate immune cells of the nervous system, become activated in all instances of ALS (Fig. 2b). Synthesis by microglia of mutant SOD1 is a key determinant of rapid disease progression, as determined by selectively silencing the mutant gene within microglia21 or using cell grafting to replace mutant-expressing microglia with normal ones22. Consistent with these findings, inhibiting nuclear factor kappa B (NFκB) suppresses this neuroinflammatory component of microglial toxicity to co-cultured motor neurons23.
An additional mechanism of mutant SOD1 damage produced by microglia is counterintuitive: stimulation of excessive extracellular production of superoxide (O−2)24. Misfolded mutant SOD1 can associate with Rac1, a small GTPase that controls the activation of NADPH oxidase, a complex that produces superoxide (Fig. 2b). Thus, instead of its normal function of removing intracellular superoxide, mutant SOD1 may drive microglia to produce high levels of extracellular superoxide.
Disturbance in microglial function has also recently emerged as a potential contributor to the subtype of ALS associated with mutations in C9ORF72. Recognition that C9ORF72 mutation results in decreased expression levels in ALS patients8 has led to speculation that loss of C9ORF72 protein function may contribute to disease. The protein encoded by C9ORF72 is a probable guanine exchange factor for one or more not-yet-identified G proteins. Its inactivation in mice results in abnormal microglia and age-related neuroinflammation, providing further evidence that non-cell-autonomous, microglial-mediated inflammation may contribute to ALS25–27.
A critical contribution to pathogenesis from mutant SOD1 is driven by oligodendrocytes, the cells that myelinate the axons of upper motor neurons and the initial axonal segments of lower motor neurons. Reduction in mutant SOD1 synthesis early in oligodendrocyte maturation produced a more striking delay in disease onset28 than did similar suppression of mutant synthesis by motor neurons18,21. Oligodendroglia also support motor neuron function by direct supply to the axon of the energy metabolite lactate through the action of the monocarboxylate transporter 1 (MCT1) (Fig. 2c). Mutant SOD1 impairs expression of MCT1 by oligodendrocytes in mouse models of ALS29. Similar reduction in MCT1 accumulation is found in sporadic ALS29, consistent with a non-cell-autonomous role of reduced oligodendroglial energy supply as a general component of ALS pathogenesis.
A third glial cell type, the astrocyte, provides nutrients, buffering ions, recycling of the neurotransmitter glutamate, and the final layer of the blood brain barrier. Selective reduction in mice of mutant SOD1 synthesis by astrocytes slowed disease onset30 or progression20. This delayed progression was accompanied by delayed microglial activation, demonstrating a functional crosstalk between mutant astrocytes and microglia.
One of the early-proposed mechanisms underlying ALS was glutamate excitotoxicity, the excessive firing of motor neurons derived from failure to rapidly remove synaptic glutamate (Fig. 2d). Astrocytes limit motor neuron firing through their rapid recovery of glutamate, a function mediated by the EAAT2 glutamate transporter (Fig. 2d). Loss of EAAT2 has been observed in both SOD1 mutant rodent models16,31 and familial and sporadic ALS patient samples32. The resulting failure to quickly clear synaptic glutamate triggers repetitive action potential firing, a corresponding increase in calcium influx, and endoplasmic reticulum (ER) and mitochondrial stress whose calcium storage abilities become overwhelmed.
Astrocytes also protect motor neurons from excitotoxic damage via release of an unidentified soluble factor(s) that induces motor neurons to upregulate the glutamate receptor subunit GluR233. Incorporation of GluR2 subunits into glutamate receptors reduces their calcium permeability, providing protection from excitotoxicity by decreasing influx of calcium into neurons. Astrocytes expressing mutant SOD1 fail to regulate GluR2 expression in co-cultured neurons, thereby increasing their vulnerability to excitotoxic damage33.
Beyond excitotoxicity, multiple teams have used in vitro co-cultures of motor neurons and astrocytes (or astrocyte-conditioned media) to show that astrocytes expressing ALS-linked mutations produce a diffusible toxicity to motor neurons34–38 (Fig. 4d). There is, however, no consensus as to the identity of the toxic species. Even more provocatively, astrocytes from familial or sporadic ALS patients (produced from autopsy samples by direct isolation of astrocytes38 or isolation of neuronal precursor cells that can subsequently be converted into astrocytic precursors and then into astrocytes37) are toxic to co-cultured normal motor neurons. The latter finding37 is especially provocative as it indicates that neuronal precursor cells in the affected portions of sporadic ALS patient tissue have already acquired damage and this damage is retained following multiple divisions in cell culture and subsequent differentiation. A crucial unsettled controversy remains as to whether toxicity from sporadic ALS-derived astrocytes is37 or is not38 mediated by changes in SOD1.
Figure 4. Three non-exclusive mechanisms have been proposed for C9-ALS/FTD.
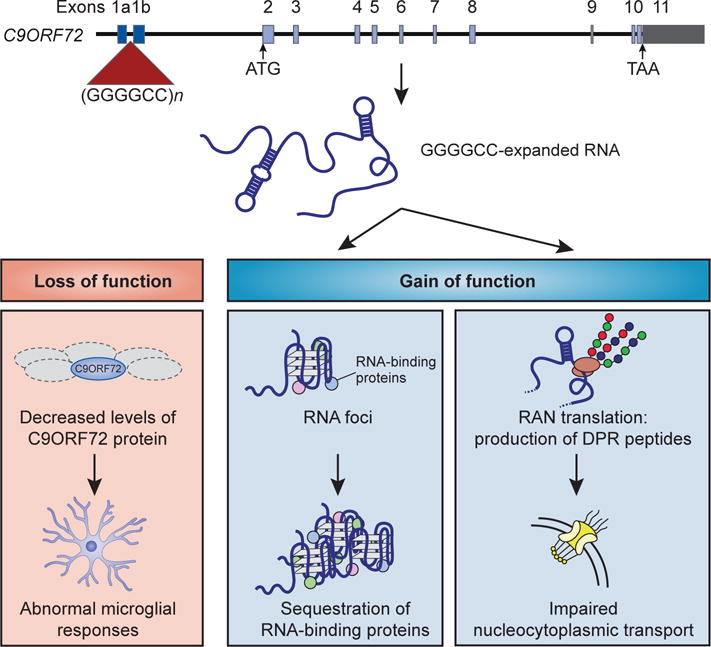
The offending mutation is expansion of a hexanucleotide repeat (GGGGCC) from fewer than 23 to hundreds or thousands in C9-ALS/FTD patients. This mutation results in modest reduction of C9ORF72 protein that appears insufficient to cause disease but may contribute to disease progression. Expression of sense and antisense transcripts containing the expanded repeat likely drive a toxic gain of function. The two major modes of toxic gain of function implicated are (1) toxicity of the mutant transcript, perhaps through sequestration of RNA-binding proteins, and (2) unconventional translation to produce dipeptide repeat proteins, some of which may be toxic.
Perhaps most importantly, a non-cell-autonomous contribution of astrocytes to ALS-like disease has been demonstrated in rodents expressing mutant SOD1, in which cervical transplantation of lineage-restricted astrocyte precursors resulted in delayed progression of disease39.
Multiple ALS-linked genes induce ER stress or impair protein degradation machinery
ER stress has been implicated broadly in ALS (Fig. 2a). Initial evidence arose from studies of mutant SOD1, with evidence for misfolded SOD1 binding to the cytoplasmic surface of the integral membrane ER protein derlin-140. This binding led to inhibition of ER-associated degradation (ERAD), the pathway for degradation of misfolded proteins extracted from the ER. Moreover, relieving ER stress delays disease progression in an animal model of disease41.
There is now overwhelming evidence that disruption of the two major protein clearance pathways, the ubiquitin-proteasome system and autophagy, can be central components of disease mechanism in ALS (Fig. 2a). Multiple ALS-causing mutations are in genes whose products are directly involved in protein degradation, including ubiquilin 242 and p62 (encoded by SQSTM1)43, both of which function as adapters to bring polyubiquitinated proteins to the proteasome or autophagosome for degradation. Mutations have also been reported in optineurin44, a proposed receptor for autophagy45, and valosin-containing protein (VCP)46, which functions in ERAD and sorting of endosomal proteins. Additional studies have reported FTD-linked mutations in CHMP2B47, which has been implicated in autophagosome maturation and/or endosomal cargo sorting/degradation. ALS-linked mutations are also found in VAPB48, which functions in the unfolded protein response for delivery of ER-ejected substrates to the proteasome. A preponderance of biochemical evidence from mice expressing mutant SOD1 has demonstrated decreased activities of the proteasome pre-symptomatically in lumbar spinal cords49 or after sustained expression of mutant SOD1 in a cultured neuronal line50.
Axonal disorganization and disrupted transport in ALS
Axonal cytoskeletal disorganization, especially of neurofilaments, has long been a conspicuous feature of both familial and sporadic ALS (Fig. 2e). As the most asymmetric cells in nature (with some axons more than a meter long), motor neurons have a crucial requirement for axonal transport to deliver the components synthesized in the cell bodies to axons and synapses. ALS-linked SOD1 mutants have been demonstrated to slow both anterograde51 and retrograde52,53 routes months before neurodegeneration. Indeed, reduction in retrograde transport by mutation in dynactin54, an activator of the retrograde motor cytoplasmic dynein55, provokes human motor neuron disease.
The peculiar architecture of neurons also presents a challenge for these cells to change local gene expression at the synapse in response to neuronal input or change in the synaptic environment. To meet this challenge, neurons transport all of the necessary components for translation (e.g., mRNA, ribosomes and translation factors) to distal sites for local protein synthesis56. The spatial distribution of mRNAs depends upon proper microtubule-dependent transport of neuronal RNA transport granules and other factors, and is regulated by several RNA-binding proteins associated with ALS, including TDP-43, FUS and hnRNPA1. ALS-causing mutations in TDP-43 impair the axonal transport of RNA granules in Drosophila and cultured neurons, including motor neurons derived from ALS patients57.
A prion-like spread in inherited ALS
Prion-like, templated conversion of a natively folded protein into a misfolded version of itself is now recognized as a prominent feature for cell-to-cell spread of aggregates in neurodegenerative disease. Examples include templated α-synuclein, Aβ, and tau misfolding in Parkinson’s and Alzheimer’s diseases and in chronic brain injury (reviewed in ref58). Evidence for similar templated toxicity has emerged for misfolded SOD159,60, with wild type SOD1 exacerbating toxicity from mutant SOD1 in mice61. Prion-like propagation and disease development initiated focally has been demonstrated after injection of mutant SOD1-containing lysates into mutant SOD1-expressing mice62. This finding replicates the correlation of focal initiation and spreading in both familial and sporadic ALS patients63.
To date, prion-like propagation has not been achieved in rodents without co-existence of a pre-existing, weakly active mutant ALS gene; it is unclear whether this challenges a prion-like spread model for sporadic ALS or raises the possibility that there must be a pre-existing sensitivity in these patients to facilitate such spreading. Coupled with recognition that misfolded mutant SOD1 can be secreted by motor neurons or astrocytes64, potentially by the newly discovered pathway of unconventional secretion of misfolded proteins as an adaptation to proteasome dysfunction65, stochastic focal initiation provides a plausible mechanism for age-dependent disease onset and subsequent spread. With most instances of ALS marked by aggregated TDP-43 rather than SOD1, an unresolved question is whether TDP-43 also exhibits templated misfolding that can spread cell-to-cell.
The intersection of RNA biology with ALS pathogenesis
In 2006 Virginia Lee and colleagues reported mislocalization of the RNA-binding protein TDP-43 from its predominantly nuclear location to ubiquitin-positive cytoplasmic inclusions in affected areas of the brain and spinal cord of ALS patients7. Within just a few years, TDP-43 mislocalization became widely recognized as the hallmark pathology of sporadic and most familial ALS. This seminal discovery has implications beyond ALS, since TDP-43 mislocalization to cytoplasmic inclusions is also the hallmark pathology of tau-negative FTD (roughly half of all FTD cases) and inclusion body myopathy (IBM)7,66, diseases that show genetic overlap with ALS. Moreover, TDP-43 pathology is also found as secondary pathology in a subset of patients with Alzheimer’s and Parkinson’s diseases67,68. The importance of TDP-43 in pathogenesis was cemented by the identification of ALS-causing mutations in this RNA-binding protein69. The subsequent identification of ALS-causing mutations in related RNA-binding proteins, including FUS and hnRNPA1, focused substantial attention on the role of RNA biology in pathogenesis70–73 (Fig. 2f).
TDP-43, FUS and hnRNPA1 are heterogeneous nuclear ribonucleoproteins (hnRNPs), a family of proteins that regulates RNA metabolism at every stage of the RNA life cycle. These three hnRNPs bind to thousands of RNA targets74–77; thus, disturbance in the function of one or more of these proteins has the potential to impact RNA metabolism on a broad scale. Further links between ALS pathogenesis and RNA biology came from the identification of ALS-causing mutations in the RNA-binding protein matrin-378, appreciation of increased risk of ALS in association with certain alleles of the RNA-binding protein ataxin-25, and recognition of RNA-related mechanisms of disease associated with mutations in C9ORF72 (see below)8,79.
Phase separation of RNA-binding proteins gives rise to membrane-less organelles
RNA metabolism transpires in complex RNA-protein assemblies, which themselves may coalesce into a variety of membrane-less organelles, such as nucleoli and stress granules. Interestingly, these membrane-less organelles behave as complex liquids that arise by phase separation, a process in which protein-laden RNAs separate from the surrounding aqueous nucleoplasm or cytoplasm in a manner akin to the separation of oil and vinegar80. It was recently discovered that phase separation is mediated by low complexity sequence domains (LCDs) present in RNA-binding proteins such as TDP-43, FUS and hnRNPA181–83 (Fig. 3a–b). Whereas the assembly of membrane-less organelles is biologically advantageous (e.g., stress granule formation), phase transition also presents a risk, since RNA-binding proteins within the liquid phase are highly concentrated, with fibrillization-prone LCDs placed in close proximity. Indeed, ALS-causing mutations are frequently found in the LCDs of TDP-4384, FUS85 and hnRNPA172. As a consequence, these mutations alter the dynamics of membrane-less organelles and also accelerate fibrillization, resulting in the formation of pathological amyloid-like fibrils that deposit in the cell bodies and neuropil81–83,86 (Fig. 3c).
Figure 3. ALS mutations impair the assembly, dynamics and function of membrane-less organelles such as RNA granules.
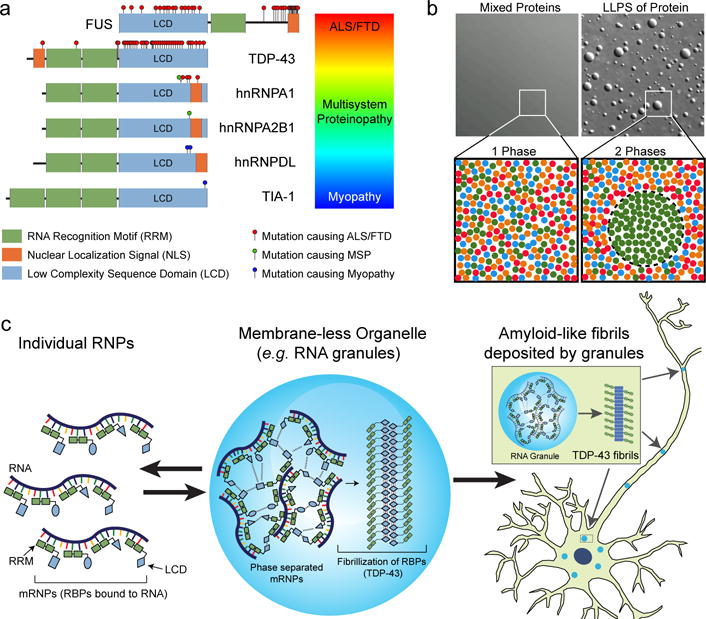
(a) Schematic representation of six hnRNPs that harbor mutations causative of a disease spectrum that ranges from ALS/FTD to myopathy. (b) Phase separation by the RNA-binding protein hnRNPA1. RNA-binding proteins with a LCD can transition from a single mixed phase to two distinct phases, one of which is a concentrated liquid droplet. (c) Phase separation contributes to the assembly, dynamics and liquid properties of membrane-less organelles such as RNA granules. Within RNA granules, the high concentration and close apposition of LCDs risks transition to amyloid-like fibrils, which likely provides the source for pathological deposition of proteins particularly prone to fibrillization, such as TDP-43.
Mutations in the LCDs of at least six different hnRNPs result in a clinicopathological spectrum that ranges from ALS/FTD to IBM (Fig. 3a and Box 1). It should be noted that some disease-causing mutations in RNA-binding proteins do not impact the LCD. For example, some ALS-causing mutations in FUS and hnRNPA1 disturb the nuclear localization sequence (NLS) and result in accumulation of these proteins in the cytoplasm70,71,73. Phase transition by LCD-containing RNA-binding proteins is exquisitely concentration dependent82, and it is likely that increased accumulation of FUS and hnRNPA1 in the cytoplasm as a consequence of mutations impacting their respective NLSs is sufficient to drive excess phase separation, as evidenced by hyperassembly of stress granules in cells derived from patients with the respective mutations72,87.
RNA metabolism defects in ALS
There are multiple potential consequences of disturbed phase transitions by disease-associated RNA-binding proteins, including (1) altered material properties of RNA granules, which can impair the biological processes served by these structures; (2) the generation of oligomers or amyloid-like fibrils that have toxic properties; and (3) a partial or complete loss of the normal function of key RNA-binding proteins88. With respect to this last point, a long-recognized feature of ALS histopathology is redistribution of TDP-43 from the nucleus to the cytoplasm7. Similar redistribution is observed for FUS and hnRNPA1 when disease mutations occur in these genes70,71,73. This redistribution may reflect a cytoplasmic sink produced by poorly dynamic cytoplasmic RNA granules, the deposition of amyloid-like fibrils, defects in nucleocytoplasmic trafficking, and potentially other mechanisms.
The culmination of this redistribution is depletion of RNA-binding proteins from the nucleus that has the potential to cause a relative loss of nuclear function. A long-recognized nuclear function of TDP-43 is regulation of alternative splicing89. Experimental depletion of TDP-43 in rodents was found to alter hundreds of splicing events in the brain, resulting in depletion of several RNAs encoding synaptic proteins75. Additionally, loss of nuclear TDP-43 generates usage of cryptic splice sites90 that, in general, may lower levels of correctly spliced protein-encoding mRNAs. Furthermore, the fact that TDP-43 autoregulates its synthesis75 establishes the possibility of a feed-forward mechanism amplifying the impact of partial loss of TDP-43 function.
Loss of FUS or hnRNPA1 from the adult nervous system produces defects analogous to that of loss of TDP-43, although different subsets of mRNAs are associated with depletion of each of these RNA-binding proteins76,77. An important unanswered question is to what extent does ALS caused by other genetic perturbations, especially C9ORF72-related ALS (see below), also involve disturbances in RNA biology that intersect mechanistically with mutations in TDP-43, FUS and hnRNPA1.
Regulatory RNA biogenesis and function in ALS
Both TDP-43 and FUS are components of macromolecular complexes that generate small non-coding RNAs (microRNAs) that function in RNA silencing. Loss of TDP-43 or FUS results in reduced expression of microRNAs in model systems, including Drosophila models and iPS-derived motor neurons from patients with TDP-43 mutations, suggesting a possible role for altered RNA silencing in ALS91. microRNAs contribute to the maintenance of neuromuscular junctions, suggesting that motor neurons may be especially sensitive to disturbances in microRNA biogenesis92,93. Indeed, global downregulation of microRNAs has been reported in motor neurons from patients with sporadic ALS94, although the role of reduced microRNAs in pathogenesis remains to be established94. Nonetheless, the expression of regulatory RNAs appears to be robustly and consistently altered in the serum of ALS patients, and this may present an opportunity for biomarker development95.
The curious case of C9ORF72-related ALS/FTD
Although the identification of ALS-causing mutations impacting SOD1 and RNA-binding proteins highlighted pathophysiological pathways by which disease might arise, the majority of the genetic burden of ALS remained unaccounted for until 2011. Genetic linkage studies96,97 followed by several large genome-wide association studies98–101 identified a location in the chromosome 9p21 region for a gene whose mutations caused both ALS and FTD. In the course of sequencing the non-coding regions of candidate genes within chromosome 9p21, Rosa Rademakers and colleagues identified pathogenic GGGGCC repeat expansion in C9ORF72 as the basis for C9-ALS/FTD8. In unaffected individuals, the hexanucleotide GGGGCC was present in 2-23 repeats, whereas in affected individuals this was expanded to hundreds or thousands of repeats. Working in parallel, Brian Traynor and colleagues independently discovered pathogenic GGGGCC repeat expansion in C9ORF7279. Similar to numerous other neurodegenerative diseases caused by microsatellite expansion, there appears to be an inverse relationship between repeat expansion length and disease severity102,103. Affected individuals apparently inherit hundreds of repeat copies, but these undergo expansion to 3000–5000 copies in cells of the nervous system104.
A contribution to disease from reduced C9ORF72 function?
Three non-exclusive mechanisms by which expanded GGGGCC repeats may cause C9-ALS/FTD have been proposed (Fig. 4). Decreased C9ORF72 expression levels in C9-ALS/FTD patients104,105 has led to speculation that loss of C9ORF72 protein may contribute to disease. The function of the C9ORF72 protein is poorly understood, but it contains a conserved DENN domain and can function as a guanine nucleotide exchange factor for several Rab proteins in experimental systems106. In cultured cells and in zebrafish, depletion of endogenous C9orf72 may exacerbate the toxicity of aggregation-prone proteins such as polyglutamine-expanded ataxin-2107. However, knockdown of endogenous C9orf72 mRNA with antisense oligonucleotides was well tolerated in mice and did not result in behavioral or motor impairments108. Furthermore, conditional knockout of C9orf72 in mouse brain did not cause appreciable motor neuron or other degenerative phenotypes, with no evidence of hallmark ALS/FTD pathologies109. Mice with complete C9orf72 knockout in all tissues also developed and aged normally without motor neuron disease, but, interestingly, developed progressive splenomegaly and lymphadenopathy25–27,110. These mice were found to have abnormal macrophages and microglia, as well as age-related neuroinflammation25,110 and signs of autoimmunity27, raising the possibility of a non-cell-autonomous inflammatory contribution to C9-ALS/FTD.
Nevertheless, the dominant inheritance pattern of C9-ALS/FTD, the absence of ALS/FTD patients with null alleles or missense mutations in C9ORF72, and the absence of neurodegeneration in C9orf72 knockout mice argue against loss of function of C9ORF72 as the sole driver of disease. Indeed, most empirical evidence points to gain-of-toxic-function as the primary mechanism driving neurodegeneration in C9-ALS/FTD. For example, AAV-mediated delivery to the brain of a construct expressing expanded GGGGCC repeats elicits neurodegeneration111, although the nature of the toxic species in C9-ALS/FTD remains unclear.
Toxic gain of function from accumulation of repeat-containing RNA
The initial description of the C9ORF72 mutation included evidence that RNA foci containing the GGGGCC repeat accumulate in the brain and spinal cord of C9-ALS/FTD patients8 (Fig. 1b). Soon thereafter it was appreciated that C9ORF72 is bidirectionally transcribed, and that both sense- (GGGGCC) and antisense- (CCCCGG) containing foci accumulate in affected cells112–114. The accrual of pathological RNA foci in C9-ALS/FTD is reminiscent of pathological RNA foci observed in myotonic dystrophy types 1 and 2 and fragile X tremor ataxia syndrome, which are also caused by pathological repeat expansions in non-coding regions102. In these diseases, the accumulated repeat-containing RNA sequesters RNA-binding proteins involved in splicing, leading to defects in RNA splicing that underlie some aspects of pathogenesis102. Similarly, a number of RNA-binding proteins bind to expanded GGGGCC and GGCCCC repeats in vitro, and rare co-localization with RNA foci has been observed for several of these proteins in patient tissue115–119.
Simple model systems have illustrated functional consequences of sequestration of some hexanucleotide repeat-binding proteins, for example Purα and RANGAP, but the contribution of these interactions to disease remains uncertain118,120. Notably, GGGGCC (but not CCCCGG) repeats can adopt a stable secondary structure known as a G-quadruplex, which may contribute to the persistence of this RNA species as well as its ability to reach distal neurites and associate with RNA-binding proteins in transport granules and potentially interfere with local translation121–123.
Toxic gain of function from accumulation of dipeptide repeats
Substantial evidence has now accrued implicating a second mode of gain-of-toxic-function in C9-ALS/FTD; specifically, toxicity from DPR proteins produced by repeat-associated non-AUG (RAN) translation. This type of unconventional translation occurs in the absence of an initiating AUG codon124 and may rely upon secondary structure formed by repeat-expanded RNA124,125. In C9-ALS/FTD, this unconventional translation occurs in all reading frames from both sense and antisense transcripts, and results in the production of five DPR proteins: glycine-alanine (GA) and glycine-arginine (GR) from sense GGGGCC transcripts, proline-arginine (PR) and proline-alanine (PA) from antisense GGCCCC transcripts, and glycine-proline (GP) from both sense and antisense transcripts112–114,126. It is now appreciated that all of these DPRs are produced in C9-ALS/FTD patients and account for the ubiquitin- and p62-positive, but TDP-43-negative neuronal cytoplasmic and intranuclear inclusions that are found widely in the brain and spinal cord112,114,117,126,127 (Fig. 1b).
Issues of when, where, and how much expression of each DPR species occurs in patient brain remain to be clarified. Multiple reports have described DPR deposition in C9-ALS/FTD patient brain, and in some instances an inverse relationship between the burden of DPRs and neurodegeneration has been described126,128,129. These studies have been based on examination of post-mortem brain at end-stage disease and have relied upon detection of large inclusions by immunohistochemistry, an approach that is likely to under-represent the pathological burden of soluble DPRs. Nevertheless, the apparent discrepancy between the burden of DPR deposition (greatest in the cerebellum) and the severity of neurodegeneration (greatest in the motor cortex and spinal cord) need to be resolved in order to understand the role of DPRs in disease.
Experiments conducted in model systems have provided evidence that at least some DPR species are toxic in cultured cells and animal models of disease, although the DPR expression levels used to produce short-term toxicity have sometimes been quite high. The arginine-containing peptides (GR and PR) in particular appear to be most toxic. For example, the addition of GR or PR peptides to cultured cells results in their uptake and accumulation in nucleoli, defects in RNA processing, and subsequent cell death130. Similarly, independent expression of each of the five DPR species in cultured neurons revealed GR and PR to be very toxic, whereas similar levels of PA, GA and PG were well tolerated. Consistent with these observations, Drosophila engineered to express each of the five DPR species have consistently shown that GR and PR are very toxic in neuronal tissue, whereas GA is modestly toxic and GP and PA appear to be non-toxic131–133. That said, other investigations have reported toxicity associated with GA expression in cell culture134–136 and in AAV-mediated delivery to mouse brain136, albeit in the latter instance the GA-mediated toxicity accompanied its accumulation to just under 2% of total brain protein. Additionally, the efforts reported thus far to model DPR toxicity have used short repeat (<100) lengths. Unsettled too is how the properties of those short DPRs compare with the RAN translation products in patients, which may be much larger.
A defect in nucleocytoplasmic trafficking
Whereas the nature of the toxic gain of function remains an open question, there is converging evidence that one downstream consequence of the C9ORF72 mutation is impaired nucleocytoplasmic trafficking. A comprehensive, unbiased screen for genetic modifiers of GGGGCC repeat-expanded toxicity in Drosophila identified 18 genes centered on the nuclear pore complex and nucleocytoplasmic trafficking133. A separate unbiased screen for genetic modifiers of PR toxicity in yeast also identified numerous genes encoding components of the nuclear pore complex and effectors of nucleocytoplasmic trafficking137. A third study focused on the nucleocytoplasmic transport factor RANGAP, which binds GGGGCC RNA. In these experiments, genes encoding RANGAP and other nucleocytoplasmic transport factors were identified as modifiers of GGGGCC repeat-expanded toxicity in Drosophila120. Consistent with these results, morphological abnormalities were found in nuclear envelope architecture in cell and animal models of disease as well as in the brain of C9-ALS/FTD patients. Moreover, defects in nucleocytoplasmic transport of RNA and proteins were found in induced pluripotent stem cell (iPSC)-derived neurons from C9-ALS/FTD patients120,133,137.
Approaches to therapy for C9-ALS/FTD
The relative contribution of the various proposed modes of toxicity to the eventual development of C9-ALS/FTD remains an important unanswered question that will influence strategies for therapeutic intervention. For example, efforts are underway to impede RAN translation with small molecules, but the success of such an approach will depend on the role of DPRs in disease. Irrespective of the primary basis for toxic gain of function, however, the mutant C9ORF72 gene itself presents an attractive target for therapeutic intervention. For example, antisense oligonucleotides are able to reverse pathological features present in iPSC-derived neurons108,138,139 or fibroblasts108 from C9-ALS/FTD patients. Indeed, iPSC-derived neuronal and glial cells may prove to be a useful model system in which to develop approaches to mitigate mutant C9ORF72-related toxicity even before the basis of toxicity is fully elucidated.
Therapeutic efforts will also be aided by the recent development of transgenic mouse models expressing human C9ORF72 with ~450 hexanucleotide repeats that recapitulate aspects of the molecular pathology26,140–142, neuropsychological deficits142,26 and the motor phenotype142 of C9-ALS/FTD. Particularly promising is the finding that pathological abnormalities can be reversed, and development of neuropsychological deficits delayed, by single-dose infusion of an antisense oligonucleotide that induces catalytic degradation of hexanucleotide-containing RNAs, without exacerbating reduction in RNAs encoding the C9ORF72 protein26.
Looking forward
Clearly there has been dramatic recent progress in defining the genetic topography and molecular biology of ALS. There is little doubt that the pace of discovery will continue or accelerate in at least three domains.
First, it is certain that our understanding of the genetic basis of ALS will continue to evolve. Programs are already in place to accumulate thousands of fully sequenced whole ALS genomes. More ALS genes are likely to be defined, both via conventional Mendelian genetics and through enhanced association studies that identify increased burdens of rare variants, including those in non-coding DNA. In parallel, enhanced scoring and recording of quantifiable clinical parameters will permit definition of genetic variants that modify the phenotype of ALS. The existence of extensive ALS genome databases will for the first time allow comprehensive studies of epistasis, characterizing the interactions of multiple genes to perturb motor neuron viability.
Second, while the last two decades have witnessed extraordinary progress in understanding familial ALS, it is likely that insights will be acquired to elucidate sporadic ALS. One view is that all sporadic ALS will ultimately be shown to reflect multiple genetic determinants. Alternatively, there is increasing interest in exogenous factors that might trigger sporadic neurodegeneration. It has been proposed that atypical infections or activation of endogenous retroviruses143 may be critical. While external environmental factors have been elusive in ALS, there is new interest in the intrinsic environment represented by the microbiome.
Third, and perhaps most importantly, there will be significant achievements in the development of ALS therapies. Though daunting, the complexity of the molecular pathology of ALS is promising as a roadmap to defining therapeutic targets. Moreover, for categories of ALS arising from well-defined genetic defects, advances in gene silencing and editing technologies will permit personalized therapeutic programs. When combined with improved methods of delivering therapy to the central nervous system, these approaches will lead to strategies to attenuate the lethal course of this disease.
Acknowledgments
We apologize for the many fine studies we were unable to cite because of space limitations. The authors gratefully acknowledge the artwork and editorial assistance provided by Natalia Nedelsky and Hong-Joo Kim, and the provision of histopathology images from John Ravits and Shahram Saberi.
JPT receives support from the Howard Hughes Medical Institute, NINDS, ALSAC, Target ALS, and the ALS Association. RHB receives support from the NINDS, the ALS Association, ALSFindingACure, ALSOne, the Angel Fund, and Project ALS. DWC receives salary support from the Ludwig Cancer Research and is supported by funding from NINDS (R01 NS27036), the ALS Association, and Target ALS. JPT is a consultant for Inception Biosciences. DWC is a consultant for Ionis Pharmaceuticals.
References
- 1.Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 2.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. This is the first report of a gene defect causing ALS. [DOI] [PubMed] [Google Scholar]
- 3.Gamez J, et al. Mutational analysis of the Cu/Zn superoxide dismutase gene in a Catalan ALS population: should all sporadic ALS cases also be screened for SOD1? J Neurol Sci. 2006;247:21–28. doi: 10.1016/j.jns.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Cooper-Knock J, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. 2012;135:751–764. doi: 10.1093/brain/awr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elden AC, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. This is the first report of a relationship between microsatellite expansion in ataxin-2 and ALS susceptibility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Hoecke A, et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med. 2012;18:1418–1422. doi: 10.1038/nm.2901. doi:nm.2901 [pii] 10.1038/nm.2901. [DOI] [PubMed] [Google Scholar]
- 7.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 8.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. This is one of two reports (see also ref 79) that first described pathogenic microsatellite expansion in C9orf72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ash PE, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. doi:S0896-6273(13)00135-9 [pii] 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mori K, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. This is one of two reports (see also ref 9) that identified DPR pathology in C9-ALS/FTD tissue. [DOI] [PubMed] [Google Scholar]
- 11.Al-Sarraj S, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 12.Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 13.Wong PC, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 14.Bruijn LI, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. This study demonstrated that ALS-causing mutant SOD1 generates a toxicity that is independent of its dismutase activity. [DOI] [PubMed] [Google Scholar]
- 15.Cleveland DW, Laing N, Hurse PV, Brown RH., Jr Toxic mutants in Charcot’s sclerosis. Nature. 1995;378:342–343. doi: 10.1038/378342a0. [DOI] [PubMed] [Google Scholar]
- 16.Bruijn LI, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. doi:S0896-6273(00)80272-X [pii] [DOI] [PubMed] [Google Scholar]
- 17.Parone PA, et al. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci. 2013;33:4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamanaka K, et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci U S A. 2008;105:7594–7599. doi: 10.1073/pnas.0802556105. doi:0802556105 [pii] 10.1073/pnas.0802556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ralph GS, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 20.Yamanaka K, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. doi:nn2047 [pii] 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boillee S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. doi:312/5778/1389 [pii] 10.1126/science.1123511. This report showed that expression of mutant SOD1 in microglia accelerates progression of ALS, confirming a role for non-cell autonomous events in motor neuron degeneration in ALS. [DOI] [PubMed] [Google Scholar]
- 22.Beers DR, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frakes AE, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harraz MM, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Rourke JG, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351:1324–1329. doi: 10.1126/science.aaf1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang J, et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 2016;90:535–550. doi: 10.1016/j.neuron.2016.04.006. This report showed that expression of mutant SOD1 in microglia accelerates progression of ALS, confirming a role for non-cell autonomous events in motor neuron degeneration in ALS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burberry A, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8:347ra393. doi: 10.1126/scitranslmed.aaf6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang SH, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee Y, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet. 2011;20:286–293. doi: 10.1093/hmg/ddq463. [DOI] [PubMed] [Google Scholar]
- 31.Howland DS, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 33.Van Damme P, et al. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci U S A. 2007;104:14825–14830. doi: 10.1073/pnas.0705046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchetto MC, et al. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–657. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Nagai M, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haidet-Phillips AM, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. This report demonstrated that motor neuron viability is compromised by astrocytes derived at post-mortem from both familial and sporadic ALS cases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Re DB, et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lepore AC, et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11:1294–1301. doi: 10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishitoh H, et al. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22:1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 42.Deng HX, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fecto F, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 44.Maruyama H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- 45.Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson JO, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parkinson N, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B) Neurology. 2006;67:1074–1077. doi: 10.1212/01.wnl.0000231510.89311.8b. [DOI] [PubMed] [Google Scholar]
- 48.Chen HJ, et al. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J Biol Chem. 2010;285:40266–40281. doi: 10.1074/jbc.M110.161398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kabashi E, Agar JN, Taylor DM, Minotti S, Durham HD. Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004;89:1325–1335. doi: 10.1111/j.1471-4159.2004.02453.x. [DOI] [PubMed] [Google Scholar]
- 50.Urushitani M, Kurisu J, Tsukita K, Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. doi:1211 [pii] [DOI] [PubMed] [Google Scholar]
- 51.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 52.Chen XJ, et al. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. doi:27/52/14515[pii]10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perlson E, et al. A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J Neurosci. 2009;29:9903–9917. doi: 10.1523/JNEUROSCI.0813-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puls I, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. doi:10.1038/ng1123ng1123 [pii] [DOI] [PubMed] [Google Scholar]
- 55.Gill SR, et al. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J Cell Biol. 1991;115:1639–1650. doi: 10.1083/jcb.115.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 57.Alami NH, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grad LI, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S A. 2011;108:16398–16403. doi: 10.1073/pnas.1102645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Munch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A. 2011;108:3548–3553. doi: 10.1073/pnas.1017275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng HX, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ayers JI, Fromholt SE, O’Neal VM, Diamond JH, Borchelt DR. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol (Berl) 2016;131:103–114. doi: 10.1007/s00401-015-1514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009;73:805–811. doi: 10.1212/WNL.0b013e3181b6bbbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Urushitani M, et al. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- 65.Lee JG, Takahama S, Zhang G, Tomarev SI, Ye Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol. 2016 doi: 10.1038/ncb3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salajegheh M, et al. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. 2009;40:19–31. doi: 10.1002/mus.21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lippa CF, et al. Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome. Arch Neurol. 2009;66:1483–1488. doi: 10.1001/archneurol.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chanson JB, et al. TDP43-positive intraneuronal inclusions in a patient with motor neuron disease and Parkinson’s disease. Neurodegener Dis. 2010;7:260–264. doi: 10.1159/000273591. [DOI] [PubMed] [Google Scholar]
- 69.Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. This is the first report of a germline TDP-43 mutation as a cause of familial ALS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. This is one of two reports (see also ref70) that first identified mutations in the FUS/TLS gene causing familial ALS. [DOI] [PubMed] [Google Scholar]
- 72.Kim HJ, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. This is the first report of a germline hnRNPA1 mutation as a cause of familial ALS, and the first report that the consequence of mutation in the low complexity sequence domain is promotion of higher order protein assembly. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu Q, et al. Whole-exome sequencing identifies a missense mutation in hnRNPA1 in a family with flail arm ALS. Neurology. 2016;87 doi: 10.1212/WNL.0000000000003256. in press. [DOI] [PubMed] [Google Scholar]
- 74.Tollervey JR, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polymenidou M, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lagier-Tourenne C, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huelga SC, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012;1:167–178. doi: 10.1016/j.celrep.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johnson JO, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17:664–666. doi: 10.1038/nn.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hyman AA, Weber CA, Julicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- 81.Patel A, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 82.Molliex A, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. This study demonstrated that low complexity sequence domains undergo phase separation and promote the assembly of membrane-less organelles. It is also one of two reports (see also ref81) to first show that pathological fibrils arise from these liquid assemblies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin Y, Protter DS, Rosen MK, Parker R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell. 2015;60:208–219. doi: 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shang Y, Huang EJ. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016 doi: 10.1016/j.brainres.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murakami T, et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron. 2015;88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lim SM, et al. Directly converted patient-specific induced neurons mirror the neuropathology of FUS with disrupted nuclear localization in amyotrophic lateral sclerosis. Mol Neurodegener. 2016;11:8. doi: 10.1186/s13024-016-0075-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buratti E, Baralle FE. The molecular links between TDP-43 dysfunction and neurodegeneration. Adv Genet. 2009;66:1–34. doi: 10.1016/S0065-2660(09)66001-6. [DOI] [PubMed] [Google Scholar]
- 90.Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349:650–655. doi: 10.1126/science.aab0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Z, et al. Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS ONE. 2013;8:e76055. doi: 10.1371/journal.pone.0076055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Valdez G, Heyer MP, Feng G, Sanes JR. The role of muscle microRNAs in repairing the neuromuscular junction. PLoS ONE. 2014;9:e93140. doi: 10.1371/journal.pone.0093140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Williams AH, et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326:1549–1554. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Emde A, et al. Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: a new mechanism for ALS. EMBO J. 2015;34:2633–2651. doi: 10.15252/embj.201490493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cloutier F, Marrero A, O’Connell C, Morin P., Jr MicroRNAs as potential circulating biomarkers for amyotrophic lateral sclerosis. J Mol Neurosci. 2015;56:102–112. doi: 10.1007/s12031-014-0471-8. [DOI] [PubMed] [Google Scholar]
- 96.Morita M, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 97.Vance C, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 98.Shatunov A, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9:986–994. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Laaksovirta H, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Van Deerlin VM, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van Es MA, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- 102.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Suh E, et al. Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta Neuropathol. 2015;130:363–372. doi: 10.1007/s00401-015-1445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van Blitterswijk M, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. The Lancet Neurology. 2013;12:978–988. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Waite AJ, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35:1779 e1775–1779 e1713. doi: 10.1016/j.neurobiolaging.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Webster CP, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016 doi: 10.15252/embj.201694401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sellier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016;35:1276–1297. doi: 10.15252/embj.201593350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110:E4530–4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Koppers M, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78:426–438. doi: 10.1002/ana.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Atanasio A, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204. doi: 10.1038/srep23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chew J, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–1154. doi: 10.1126/science.aaa9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gendron TF, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mori K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126:881–893. doi: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- 114.Zu T, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110:E4968–4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Haeusler AR, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lee YB, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mackenzie IR, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013;126:859–879. doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- 118.Xu Z, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cooper-Knock J, et al. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 2015;130:63–75. doi: 10.1007/s00401-015-1429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang K, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Taylor JP. Neurodegenerative diseases: G-quadruplex poses quadruple threat. Nature. 2014;507:175–177. doi: 10.1038/nature13067. [DOI] [PubMed] [Google Scholar]
- 122.Schweizer Burguete A, et al. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife. 2015;4:e08881. doi: 10.7554/eLife.08881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ishiguro A, Kimura N, Watanabe Y, Watanabe S, Ishihama A. TDP-43 binds and transports G-quadruplex-containing mRNAs into neurites for local translation. Genes Cells. 2016;21:466–481. doi: 10.1111/gtc.12352. [DOI] [PubMed] [Google Scholar]
- 124.Zu T, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kearse MG, et al. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol Cell. 2016;62:314–322. doi: 10.1016/j.molcel.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Davidson YS, et al. Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2014;2:70. doi: 10.1186/2051-5960-2-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gomez-Deza J, et al. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol Commun. 2015;3:38. doi: 10.1186/s40478-015-0218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mackenzie IR, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015;130:845–861. doi: 10.1007/s00401-015-1476-2. [DOI] [PubMed] [Google Scholar]
- 129.Schludi MH, et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015;130:537–555. doi: 10.1007/s00401-015-1450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kwon I, et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mizielinska S, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wen X, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Freibaum BD, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yamakawa M, et al. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet. 2015;24:1630–1645. doi: 10.1093/hmg/ddu576. [DOI] [PubMed] [Google Scholar]
- 135.Zhang YJ, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128:505–524. doi: 10.1007/s00401-014-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang YJ, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19:668–677. doi: 10.1038/nn.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jovicic A, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Donnelly CJ, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.O’Rourke JG, et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron. 2015;88:892–901. doi: 10.1016/j.neuron.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Peters OM, et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron. 2015;88:902–909. doi: 10.1016/j.neuron.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu Y, et al. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. 2016;90:521–534. doi: 10.1016/j.neuron.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 143.Brown RH, Jr, Al-Chalabi A. Endogenous retroviruses in ALS: A reawakening? Sci Transl Med. 2015;7:307fs340. doi: 10.1126/scitranslmed.aad3533. [DOI] [PubMed] [Google Scholar]
- 144.Greenway MJ, et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- 145.Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 146.Teyssou E, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125:511–522. doi: 10.1007/s00401-013-1090-0. [DOI] [PubMed] [Google Scholar]
- 147.Wu CH, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488:499–503. doi: 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Smith BN, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84:324–331. doi: 10.1016/j.neuron.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bannwarth S, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014;137:2329–2345. doi: 10.1093/brain/awu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Freischmidt A, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]