

Abstract
Basal forebrain (BF) cholinergic neurons innervating the cortex regulate cognitive, specifically attentional, processes. Cholinergic atrophy and cognitive decline occur at an accelerated pace in age-related neurodegenerative disorders such as Alzheimer’s disease; however, the mechanism responsible for this phenomenon remains unknown. Here we hypothesized that developmental suppression of nerve growth factor signaling, mediated via tropomyosin-related kinase A (trkA) receptors, would escalate age-related attentional vulnerability. An adeno-associated viral vector expressing trkA shRNA (AAV-trkA) was utilized to knockdown trkA receptors in postnatal rats at an ontogenetic time point when cortical cholinergic inputs mature, and the impact of this manipulation on performance was assessed in animals maintained on an operant attention task throughout adulthood and until old (24 months) age. A within-subject comparison across different time points illustrated a gradual age-related decline in attentional capacities. However, the performance under baseline and distracted conditions did not differ between the AAV-trkA-infused and animals infused with a vector expressing shRNA against the control protein luciferase at any time point. Additional analysis of cholinergic measures conducted at 24 months showed that the capacity of cholinergic terminals to release acetylcholine following a depolarizing stimulus, cortical cholinergic fiber density and BF cholinergic cell size remained comparable between the two groups. Contrary to our predictions, these data indicate that developmental BF trkA disruption does not impact age-related changes in attentional functions. It is possible that life-long engagement in cognitive activity might have potentially rescued the developmental insults on the cholinergic system, thus preserving attentional capacities in advanced age.
Keywords: development, trkA, attention, cholinergic, aging
1. Introduction
Attention constitutes a constellation of cognitive processes responsible for directing awareness to relevant stimuli and filtering out irrelevant information. Attentional impairments are known to constitute performance reductions in other cognitive domains including memory and executive functions in patients suffering from Alzheimer’s disease (AD) (Perry & Hodges, 1999; Rizzo, Anderson, Dawson, & Nawrot, 2000). The basal forebrain (BF) cholinergic system, consisting of cortically-projecting neurons in the nucleus basalis of Meynert and substantia innominate region, mediates essential aspects of attentional information processing (Everitt & Robbins, 1997; Sarter & Parikh, 2005), and cholinergic dysregulation is a common feature of the mild cognitive impairment (MCI) and AD pathology (Bartus, Dean, Beer, & Lippa, 1982; Grothe et al., 2010; Haense et al., 2012; Mesulam, 2004; Perry, Watson, & Hodges, 2000). Thus, it is suggested that disruption in cholinergic signaling may underlie cognitive decline in MCI/AD. Aging is a well-known risk factor for AD. Although a gradual weakening of the cholinergic system and subtle decrements in attentional capacities have been reported in aging (Casu, Wong, De Koninck, Ribeiro-da-Silva, & Cuello, 2002; McGaughy & Sarter, 1995; Mouloua & Parasuraman, 1995; Muir, Fischer, & Björklund, 1999; Schliebs & Arendt, 2011), the mechanisms responsible for the accelerated decline in cholinergic function in sporadic AD are not fully understood.
BF cholinergic neurons depend upon nerve growth factor (NGF) for survival, growth, and maturation (Koliatsos et al., 1991; Mobley et al., 1986; Oosawa, Fujii, & Kawashima, 1999). Substantial evidence indicates that the cholinotrophic effects of NGF are mediated by its binding to the high-affinity tropomyosin kinase A (trkA) receptors (Debeir, Saragovi, & Cuello, 1998; Fagan, Garber, Barbacid, Silos-Santiago, & Holtzman, 1997; Li et al., 1995). Moreover, the loss of trkA receptors has been observed in the postmortem brains of AD subjects (Counts et al., 2004; Ginsberg, Che, Wuu, Counts, & Mufson, 2006; Mufson et al., 2000). Together, these studies support the notion that perhaps deficient trkA signaling in cholinergic neurons may underlie their exceptional vulnerability and lead to cognitive decline in AD (Cuello, Bruno, & Bell, 2007; Mufson, Counts, Perez, & Ginsberg, 2008). However, neurotrophic modulation of the cholinergic system and cognitive capacities has not been thoroughly investigated in the context of aging, which is known to potentially increase the risk of developing AD (Kawas et al., 2000).
We previously found significant attentional impairments and disrupted cholinergic signaling in aged but not adult trkA-suppressed rats (Parikh et al., 2013). In another follow-up study, we observed that cholinergic-attentional capacities were partially rescued by blocking proNGF, a precursor of NGF, in trkA-suppressed aged rats (Yegla & Parikh, 2013). ProNGF is known to induce apoptotic signaling via its interaction with high-affinity p75 receptors and the sortilin complex (Nykjaer et al., 2004), and numerous studies indicated that the expression of this pro-neurotrophin is elevated in aging and AD (Al-Shawi et al., 2007; Al-Shawi et al., 2008; Fahnestock, Michalski, Xu, & Coughlin, 2001; Peng, Wuu, Mufson, & Fahnestock, 2004). Thus, age-related imbalance between proNGF/NGF and trkA/p75 signaling may account for increased vulnerability of the cholinergic system and associated cognitive impairments in AD. Moreover, suppression of trkA signaling per se in young may not impose any inimical effects on the BF cholinergic system. This view is supported by a previous study showing that forebrain-specific targeted deletion of NGF or trkA receptors exerted detrimental effects on BF cholinergic neurons during postnatal development but did not impact learning and memory performance when assessed in either young adult or intermediate-aged mice (Müller et al., 2012).
During the progression of sporadic AD, cortical areas are affected in a stereotypic sequence that recapitulates ontogenetic brain development (Arendt et al., 2017). Although the available evidence supports the idea that perhaps reduced trophic support during ontogenetic development and maturation of the cortical cholinergic input system may escalate into MCI and AD (Sanchez-Ortiz et al., 2012; Sarter and Bruno, 2004), the premise that developmental abrogation of BF trkA signaling would accelerate age-related decline in the cholinergic system and cognitive capacities has never been tested. Here we conducted a longitudinal study to evaluate the life-long impact of developmental trkA suppression on attentional functions. We utilized an adeno-associated viral (AAV) vector that expresses trkA-shRNA to selectively knockdown trkA receptors in the BF during the postnatal phase in rats when cholinergic projections make contact with cortical target regions. The animals were trained in an operant sustained attention task (SAT), and performance was assessed throughout adulthood until old age, at which point cholinergic signaling and morphology were also evaluated. Developmental trkA suppression neither accelerated the emergence, nor exacerbated the presence, of age-related attentional impairments. Moreover, postnatal trkA disruption affected only certain aspects of cholinergic transmission in aged rats. Together, our findings indicate that lifelong cognitive engagement may have compensated for the detrimental effects of developmental trkA knockdown on age-related decline in attention processes and the cholinergic transmission that these cognitive capacities depend on.
2. Materials and Methods
2.1. Subjects
Male Wistar rat pups [post-natal day (PND) 14] were purchased from Charles River Laboratories (Malvern, PA, USA) along with the nursing dams (8 pups/dam). Animals were maintained in a temperature- and humidity-controlled room with a 12:12 light-dark cycle starting at 7AM at Temple University. At PND 18–21, rat pups underwent stereotaxic intracranial infusion of an adeno-associated viral (AAV) vector (see procedure below). Pups were returned to cages and maintained with the dams until weaning at PND 22 following which the animals were group-housed (2 per cage) with food and water ad libitum until the commencement of further experiments. All experimental procedures were conducted in accordance with the National Institute of Health guidelines and were approved by the Institutional Animal Care and Use Committee and the Institutional Biosafety Committee at Temple University.
2.2. Stereotaxic surgeries and experimental design
Rat pups (PND 18–21) were prepared for stereotaxic surgeries to produce knockdown of BF trkA receptors using an adeno-associated viral (AAV) vector-based RNA interference (RNAi) approach. This developmental period was selected because BF cholinergic neurons make contact with NGF-producing target neurons throughout the cortical mantle during postnatal weeks 3–5, and trkA signaling is critical in facilitating proper cortical cholinergic innervation (Dori & Parnavelas, 1989; Fagan et al., 1997; Kiss & Patel, 1992; Mechawar & Descarries, 2001). All surgeries were performed under aseptic conditions. Pups were anesthetized with isoflurane (4–5% for induction and 1–2% maintenance dose) using an anesthesia machine (Surgivet, Dublin, OH, USA) and mounted on a stereotaxic frame (Model 962; David Kopf Instruments, Tujunga, CA, USA). The head of the pups was positioned into the head frame using neonatal rat ear bars containing Zygoma ear cups (David Kopf). An isothermal deltaphase pad (Braintree Scientific, Braintree, MA, USA) was used to consistently maintain animals’ body temperature at 37°C throughout the surgical procedure. A recombinant AAV vector previously designed and validated in our laboratory for in vivo suppression of trkA receptors (AAV-trkA) was used in the present study (Parikh et al., 2013; Yegla & Parikh, 2014). This vector utilizes a synthetic inhibitory B-cell receptor inducible gene BIC-derived RNA (SIBR) expression plasmid to guide the expression of both the small hairpin RNA (shRNA) against the trkA gene and the reporter gene (green fluorescent protein; GFP). Another AAV vector expressing shRNA targeting the gene for firefly luciferase (AAV-luc) served as control for non-specific virus effects. Prior to the surgeries, AAV vectors were desalted using the Hyclone buffer (University of Iowa Vector Core). Rat pups received bilateral infusions of either AAV-trkA or AAV-luc (1.5μL/hemisphere; titer: 0.7–2.8 × 1013 vg/mL) into the basal forebrain (BF), specifically targeting the nucleus basalis magnocellularis/substantia innominata (nBM/SI) region that contain cholinergic neurons (from Bregma; A/P: −1.0; M/L: ± 2.3; D/V: −6.0). All injections were made using a 10μL Hamilton syringe (28 gauge) at a rate of 1 μL/min. After cessation of the injection, the needle was left in place for an additional 4 min to allow complete diffusion of virus particles and then slowly withdrawn. The incision site was stapled and triple antibiotic ointment was applied to the area surrounding the staples. Rat pups were given injections of an antibiotic (Baytril, 0.01mg/kg, s.c.) and an analgesic (buprenorphine, 0.01mg/kg, s.c.) prior to returning to the nursing dam.
A schematic illustration of the experimental design is shown in Fig. 1. To assess the impact of developmental trkA knockdown on attentional capacities in aging rats, the animals (N=8/vector manipulation) were single-housed at 6 months of age and the operant behavioral training was initiated (see task details below). Rats used for behavioral studies were handled extensively prior to training and were partially water-deprived by restricting access to a 10-min period in the home cage following each behavioral session. On non-training days, water access was increased to a 30-min period. Food was available ad-libitum throughout the duration of behavioral testing. Animals remained on task until 24 months old and the effects of vector manipulation and age on behavioral performance were assessed on a monthly basis starting from month 12 until the completion of the study. At 24 months, animals underwent amperometric recording sessions to evaluate the status of cortical cholinergic signaling followed by immunohistochemical examination of trkA and choline acetyltransferase (ChAT, a marker of cholinergic neurons) expression. A subset of animals (N=4-5/vector manipulation) was perfused at 6 weeks of age to assess the efficiency of the viral vector for developmental trkA suppression (see immunohistochemistry procedure below).
Figure 1. Schematic illustration of the experimental design.

At PND 18–21, rat pups underwent stereotaxic surgeries for the bilateral infusion of the AAV vector expressing the shRNA against either trkA (AAV-trkA) or the control gene luciferase (AAV-luc) into the BF. To evaluate the impact of developmental trkA suppression on attentional capacity throughout aging, rats initiated behavioral training in operant SAT at 6 months and were maintained on the task until 24 months old. Age-related changes in attentional function were assessed specifically from 12 to 24 months old. After completion of the behavioral testing, electrochemical recordings of cholinergic transmission were conducted in the medial PFC from anesthetized rats following which the brain tissues were processed for immunohistochemistry.
2.3. Operant training and testing procedures
Behavioral training and testing was conducted in modular operant conditioning chambers made of clear polycarbonate and mounted on a white polypropylene base (Med Associates Inc., St. Albans, VT, USA). Each chamber contained a front stainless steel panel with three lights (2.8 W each), two retractable levers, and a liquid receptacle attached to a water dispenser in the center of the panel. The back panel of each chamber contained a house light and the floor was composed of a stainless steel grid bar. Operant chambers were enclosed in sound-attenuating cubicles equipped with fans to dampen surrounding noise (Fig 2A). All events including signal delivery, lever presentations, and reward delivery were transmitted using programs written in Medstate notation via SmrtCtrl™ interface on a Dell Optiplex 960 computer.
Figure 2. Behavioral experiments in rats.
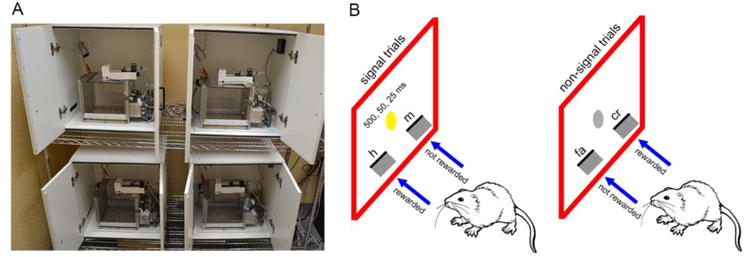
A) Operant conditioning chambers enclosed in sound-attenuating cubicles used for behavioral training and testing. B) A schematic representation of the sustained attention task (SAT). Rats discriminated between signal (illumination of the central panel light) and non-signal (no illumination) trials by exhibiting an appropriate lever press response (either left or right). Animals were pseudo-randomly designated to a lever side for each trial condition. For a left lever assignment on a signal trial, a left lever press was scored as a hit (h) and rewarded but a right lever press was scored as a miss (m) and was not rewarded. In contrast, a right lever press on a non-signal trial was scored as a correct rejection (cr) and rewarded but not a left lever press which was considered a false alarm (fa). The assignment of lever (left or right) for each trial type was counterbalanced across animals within a group. At the final stage of SAT training, each session consisted of a pseudo-randomized sequence of 81 signal and 81 non-signal trials with an inter-trial interval of 9±3s. Signals occurred unpredictably at three durations (500, 50, and 25 ms) and the house light remained illuminated throughout the session.
Rats were trained in an operant sustained attention task (SAT) that encapsulates all elements of signal detection theory to assess attentional capacities (Demeter, Sarter, & Lustig, 2008; McGaughy & Sarter, 1995; St Peters, Demeter, Lustig, Bruno, & Sarter, 2011). Animals were initially autoshaped on a FR-1 schedule of reinforcement for water reward (0.02mL water) to attain the lever press response. During this phase, if the animals pressed the same lever >5 times in succession, the lever ceased to be reinforced until the discrepancy between the two levers was reduced. This step was adopted to deter the development of a side bias. Once the rats made 120 lever presses within a single autoshaping session, they progressed to the next phase of training to learn paradigm contingencies. At this stage, the animals were trained to distinguish between signal (illumination of the central panel light for 1 s) and non-signal (no illumination) trials. Rats were pseudo-randomly designated to a lever side under signal trial conditions and had to respond on the opposite lever for non-signal trials. For example, if a rat was designated to the left lever, on a signal trial, a left lever press was rewarded (i.e., hit) but not a right lever press (i.e., miss). In contrast, a right lever press on a non-signal trial was rewarded (i.e., correct rejection) but left lever press (i.e., false alarm) was not rewarded (see Fig 2B for task illustration). Levers were presented 2 s after a 1s-long signal illumination. If rats did not lever press within 4 s the levers were retracted, and it was deemed an omission. The assignment of lever (left or right) for each trial type was counterbalanced across animals within a group. The inter-trial interval (ITI) was 12 ± 3s. During this training phase, an incorrect response elicited correction trials in which the same trial type was repeated up to 4 sequential times to assist with learning the task contingencies. If the incorrect behavior persisted a forced trial was provided, in which only the correct lever was presented. Rats progressed to the final training stage when they performed 160 trials with ≥70% on hits and correct rejections and ≤20% omissions for three consecutive days.
The final stage of SAT consisted of a pseudo-randomized sequence of 81 signal (27 per signal duration) and 81 non-signal trials (total 162 trials). Each session was divided into three blocks of 54 trials (27 signal trials and 27 non-signal trials). Signals occurred unpredictably at three durations (500, 50, and 25 ms; Fig 2B), with 9 trials/signal duration in each block. Moreover, the ITI was reduced to 9±3s, and the house light remained illuminated throughout the session. These task conditions are known to constrain rats’ behavior for continuous monitoring of the central panel and tax attentional capacity (Demeter et al., 2008). Rats were characterized as acquiring the task once they achieved ≥70% on 500 ms signal duration trials, ≥70% on correct rejections, and ≤10% omissions for three consecutive training sessions. To further assess the impact of aging and developmental trkA knockdown on top-down attentional capacity, rats were tested in a distractor version of SAT (dSAT) at 12 and 24 months of age, respectively. dSAT involved the presentation of a visual distractor (flashing house light at 0.5 Hz) in the second block of the session.
Behavioral performance for each session was evaluated by calculating the proportion of hits (h = hits/hits+misses) for each signal length and the proportion of false alarms (fa = false alarms/false alarms + correct rejections). A composite measure of attention was calculated as a performance score (SAT/dSAT) using the formula, (h−fa)/[2(h+fa)−(h+fa)2], as described earlier (Frey & Colliver, 1973). SAT/dSAT scores ranged from −1 to +1; a score of +1 indicated correct responses on all signal and non-signal trials, 0 indicated inability to discern signal and non-signal trials, and −1 indicated all misses and false alarms in a session. Latency for the lever press response under each performance condition and percentage omissions were also calculated for each session. Performance measures on three consecutive sessions were averaged for each rat at a given age and used for statistical analysis.
2.4. In vivo amperometric recordings
Following the completion of behavioral studies at 24 months of age, rats were prepared for amperometric recordings of cholinergic transmission using enzyme-selective microelectrodes as described earlier (Parikh et al., 2013; Parikh, Man, Decker, & Sarter, 2008; Parikh et al., 2004). Briefly, microelectrode arrays (Quanteon, Nicholasville, KY, USA) consisting of four rectangular platinum recording sites arranged side by side in pairs were coated with choline oxidase (Sigma-Aldrich, St. Louis, MO, USA). The enzyme was immobilized to the bottom pair of recording sites while the upper pair was coated with bovine serum albumin and served as sentinel channels. The electrode channels were electroplated with m-PD (m-phenylenediamine) to enhance the selectivity of choline against electroactive analytes such as ascorbic acid (AA), dopamine (DA) and uric acid. Choline sensitivity and selectivity was tested for all electrodes by conducting an in vitro calibration. Electrodes that exhibited the following calibration criterion were subsequently used for in vivo recordings: choline sensitivity >3 pA/μM, limit of detection (LOD) < 500 nM; selectivity ratio for choline:AA >50, linear response for choline concentration R2 > 0.98, and DA response <3pA.
For in vivo recordings, rats were anesthetized with urethane (1.0–1.25 g/kg, i.p.) and placed in a stereotaxic frame. Enzyme-coated microelectrodes were lowered into the right medial prefrontal cortex (PFC; A/P: +3.0 mm, M/L: −0.7 mm, D/V: −2.7–3.0 mm) of rats using a microdrive (MO-10, Narishige International, East Meadow, NY, USA). Ag/AgCl reference electrodes were implanted into the rostral cortical region of the contralateral hemisphere. Amperometric recordings were conducted at 2 Hz by applying a fixed potential of +0.7V, and data were digitized using a FAST-16 potentiostat (Quanteon). Background currents were stabilized for 60 min following which drugs were locally applied into the PFC using a glass capillary that was attached to the electrode and pulled to an internal tip diameter of 15–20 μm. Depolarization-evoked acetylcholine (ACh) release that mimics the phasic characteristics of cholinergic transmission on a temporal time scale (second-based) was measured by applying brief pulses of potassium (KCl, 70mM; 200nL) as reported earlier (Parikh et al., 2004). To determine the consequences of trkA disruption on tonic changes in cortical ACh release, we applied a high concentration of nicotine (100μM; 200nL) that is known to produce long-lasting (minute-based) changes in cholinergic transmission presumably via low-affinity α7 nicotinic receptor activation (Parikh, Ji, Decker, & Sarter, 2010; Parikh et al., 2008). Pulses of drug solutions were applied at 2–10 psi every 2 min through the capillaries via PTFE tubing connected to a picospritzer (ALA Scientific Instruments, Farmingdale, NY, USA). Choline signals were analyzed with respect to peak signal amplitudes and clearance (T50; time for a signal to be reduced by 50% of peak amplitude). Self-referencing was adopted to eliminate any artifacts on enzyme-coated channels due to background noise levels or drug application by subtracting currents from sentinel channels. Amperometric recording data for KCl- and nicotine-elicited signals were binned at 0.5 s and 2.5 s, respectively. Nicotine-evoked signals were box-car filtered by a moving average of 2–5 data points. All data were expressed as the average of three signals per manipulation per animal.
2.5. Immunohistochemistry and image analysis
Rats were transcardially perfused using 100 mL of ice-cold 0.1 M phosphate-buffered saline (PBS) followed by 200 mL of 4% paraformaldehyde (PFA; pH 7.4–7.6). The brains were removed, post-fixed overnight in PFA, and then transferred to 30% sucrose (in 0.1 M PBS) for 72 h. Coronal sections (50μm thick) were obtained from the rostral-caudal axis of the PFC (range: +3.5mm to +2.5mm A/P) and BF (range: −1.0mm to −1.9mm A/P) using a freezing microtome (SM2000R, Leica, Wetzlar, Germany). Slices were stored in cryoprotectant solution (15% glucose, 30% ethylene glycol, and 0.04% sodium azide in 0.05 M PBS) at −20 °C until further processing.
Vector targeting of BF cholinergic neurons and efficiency of trkA knockdown was assessed using GFP/ChAT double immunostaining and trkA immunohistochemistry as described previously (Parikh et al., 2013; Yegla & Parikh, 2014). Briefly, serial sections from the BF regions were randomly selected and rinsed in 0.5M tris-buffered saline (TBS), blocked with 10% donkey serum and incubated overnight in goat anti-ChAT (1:200; EMD Millipore, Billerica, MA, USA). Sections were rinsed in TBS containing 1% triton X100 (TBST; 3 × 5min), tagged with rhodamine-conjugated donkey anti-goat antibody (1:250; Jackson Immunoresearch Laboratories; West Grove, PA, USA), mounted onto gelatin-coated slides, coverslipped with Prolong Gold Anti-fade with DAPI mounting media (Life Technologies, Grand Island, NY, USA), and visualized for colocalization of GFP protein in ChAT-positive neurons. Parallel sections assessed for trkA expression levels were blocked for endogenous peroxidase using 0.3% H2O2. To eliminate non-specific binding of the primary antibody, slices were blocked in 10% goat serum. Sections were incubated with rabbit anti-trkA antibody (1:1000; EMD Millipore) overnight. Following rinsing with TBST (3 × 5min), sections were incubated with biotinylated goat anti-rabbit IgG (pre-diluted; EMD Millipore) for 2 hrs. The staining was developed by incubating the sections in streptavidin-HRP followed by 3-3′-diaminobenzidine (DAB) and ammonium nickel sulfate. Stained sections were mounted on gelatin-coated slides, air dried, dehydrated, and coverslipped with DPX. To assess the morphology of cholinergic neurons and terminal distribution, serial sections from the BF (nBM/SI region) and medial PFC were stained for ChAT as described earlier (Parikh et al., 2013; Yegla & Parikh, 2014). Free-floating sections were incubated with goat anti-ChAT antibody (1:100; EMD Millipore) and peroxidase staining was developed using HRP-conjugated donkey anti-goat (1:1000 for cell bodies and 1:200 for terminals) and DAB.
All sections were analyzed using a Leica fluorescent microscope equipped with DFC 425C digital camera and Leica Application Suite 3 software (Leica Microsystems Inc., Buffalo Grove, IL, USA). Fluorescent images were captured at 200× using green and red filters to visualize the expression of GFP and ChAT in both hemispheres. Viral vector colocalization with BF cholinergic neurons was calculated as the percentage of ChAT-positive neurons co-expressing GFP out of 100 cholinergic neurons (50/hemisphere). For the analysis of BF trkA expression, images were acquired in the light microscope mode and processed for quantitative image analysis using NIH ImageJ analysis software. Digitized images (400× magnification) were processed in binary mode and threshold levels were adjusted to enhance the visibility of cell bodies. The density of trkA receptors was expressed as percentage of trkA-positive pixels in the analyzed area (325 μm × 245 μm) from both hemispheres. Morphometric analysis of cholinergic neurons involved the measurement of cell size as described previously (Parikh et al., 2013; Yegla & Parikh, 2014). Mean cross-sectional area of ChAT-positive neurons was calculated from randomly selected sampling areas (400× magnification) within the nBM/SI region from both hemispheres. Cell borders were outlined manually and the area (in pixel2 units) was calculated by NIH ImageJ based on the user-defined border. Cross-sectional area was converted from pixels2 to μm2 based on image magnification (scaling factor: 12.09 pixels/μm). TrkA-densities and mean cross-sectional ChAT areas were averaged from both hemispheres and data are expressed as analyses from three sections per animal. PFC ChAT-immunoreactive fiber density was analyzed using a grid counting procedure (Parikh et al., 2013; St Peters et al., 2011; Yegla & Parikh, 2014) from the right prelimbic region (cortical layers III/V), which was also used to measure ACh release following terminal depolarization (see amperometric recordings above). Images were captured at 400× magnification and threshold levels were uniformly adjusted across all sections to maximize visualization of ChAT-positive fibers in Adobe Photoshop CS4 (Adobe Systems Inc., San Jose, CV). Cholinergic fibers were manually traced and counted using a 50 × 50 μm grid for a total area of 40,000 μm2. Average fiber counts were based on three sections per region per animal.
2.6. Statistics
Statistical analyses were performed using SPSS/PC+ version 24.0 (IBM-SPSS, Armonk, NY, USA). Mixed-design analysis of variance (ANOVA) tests were applied to analyze behavioral data with age, signal duration, and/or block as within-subject factors, and trkA manipulation as a between-subject factor. When appropriate, a one-way ANOVA, followed by Bonferroni post hoc test for multiple comparisons, was conducted to determine the source of interaction. Immunohistochemical and amperometric analyses were conducted using a one-way ANOVA to determine the impact of developmental trkA manipulation on trkA density, age, cholinergic morphology and functionality. A cut-off p value of 0.05 was considered statistically significant.
3. Results
3.1. Long-term suppression of BF trkA receptors following postnatal infusion of AAV vector
We first assessed the efficacy of the recombinant AAV vector to suppress trkA receptors in BF neurons, which was injected during the developmental phase when cholinergic neurons make connections with NGF-producing cortical regions and NGF-trkA signaling fine tunes this projection system. Rat pups were infused with the viral vector expressing trkA shRNA or luc shRNA into the BF during postnatal week 3 and brain sections were visualized at 6 weeks old. As shown in Fig 3 (A, B), the vector spread, exemplified by the expression of the reporter protein GFP, into the nBM/SI region and its colocalization with ChAT-positive neurons indicate that our strategy targeted cholinergic neurons, >80% of which express trkA receptors in this region in rats (Sobreviela et al., 1994). BF trkA receptor expression observed at 6 weeks of age significantly declined in AAV-trkA-infused rat pups as compared to pups infused with the control vector (F(1,9)=2.86, p<0.03; Fig 3D, E). These observations are in line with our previous work that utilized the AAV vector-based RNAi approach to target trkA receptors in the nBM/SI region in young and aged rats (Parikh et al., 2013; Yegla & Parikh, 2014). Next, we examined brain slices from animals that have undergone behavioral assessment for 24 months. Assessment of GFP/ChAT colocalization revealed that infection persisted in 47±5% of BF cholinergic neurons, indicating that the vector delivered during the developmental period could maintain stable gene expression in aged rats (Fig 3C). Additionally, quantitative analysis of trkA immunoreactivity revealed a significant reduction of BF trkA protein levels in rats that received AAV-trkA infusions during the postnatal period (F(1,12)=2.17, p<0.05; Fig 3 F, G), demonstrating a sustained decrease in trkA levels for 24 months. It is important to note that although the reduction in trkA receptors were more robust (79% inhibition) during development as compared to reductions noted in aged rats (47% inhibition), the difference appeared to be related to a natural decline of trkA receptors from the post-natal period to adulthood and old age (age effect: 6 weeks vs 24 months; p<0.01), rather than a loss of efficacy of the shRNA (Li et al., 1995).
Figure 3. Vector efficiency for long-term trkA suppression.
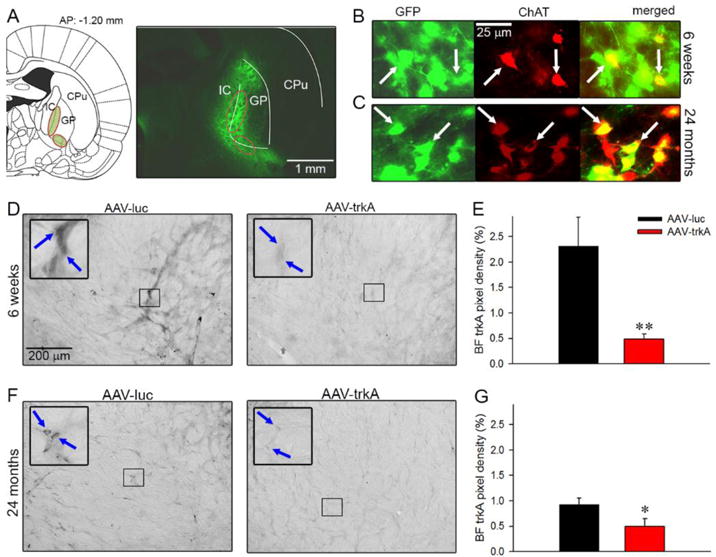
To ensure neuronal infection and vector efficiency during development, a pilot group of rat pups underwent AAV-luc and AAV-trkA injection at PND 18–21 and sections were visualized at 6 weeks. Schematic representation of the AAV vector infusion site targeting the nBM/SI region (highlighted in green) of the BF containing cholinergic neurons (A; left). These neurons are present along the ventromedial wall of the globus pallidus (GP) and project to the entire cortex (IC: internal capsule; CP: caudate putamen). A representative coronal section taken from a 6-week-old rat infused with AAV-trkA vector depicts the infected BF neurons expressing GFP in the nBM/SI region (A; right). (B) Sampled BF area showing GFP-expressing neurons (green), ChAT-positive neurons (red) and colocalization of GFP and ChAT (yellow; merged images) confirm targeting of BF cholinergic neurons by the vector. (C) A representative immunostained section from a 24-month-old aged rat that received AAV-luc infusion during the development. Colocalization of GFP and ChAT illustrated that vector remained stably expressed in BF cholinergic neurons until 24 months. Double labeling is depicted by white arrowheads. BF trkA immunoreactivity from representative sections taken from 6-week-old rats that received postnatal infusions of either AAV-luc or AAV-trkA vector (D; blue arrows point to trkA-positive neurons). The infusion of AAV vector expressing trkA shRNA during PND18-21 produced profound reductions in trkA expression in the nBM/SI region at 6 weeks of age (E). Representative sections depict trkA receptor protein expression in the BF from rats that received AAV vector infusions during the postnatal period and underwent behavioral testing for 24 months (F; blue arrows point to trkA-positive neurons). Significant reduction in BF trkA immunoreactivity was noted in old rats that received developmental infusion of AAV-trkA as compared to the AAV-luc group (G). These data illustrate that our AAV vector strategy to knockdown trkA receptors in rats during development could maintain long-lasting reductions in trkA levels until old age. Data are represented as mean ± SEM. *, ** p<0.05, 0.01 vs AAV-luc.
3.2. Developmental trkA knockdown and SAT performance in aging rats
A total of 16 rat pups infused with AAV vectors (N=8/condition) began SAT training at 6 months. The animals required 38±5 training sessions to attain criterion performance. One AAV-luc-infused rat never reached criterion and was excluded from the study. Average SAT scores for adult AAV-luc- and AAV-trkA-infused animals examined at 9 months of age remained comparable (F(1,10)=0.03, p=0.98). The impact of developmental trkA suppression on attentional capacity in aging animals was assessed by subsequently comparing the performance between AAV-luc and AAV-trkA animals on a bimonthly basis starting from 12 months of age until the completion of the study at 24 months. Behavioral testing was discontinued due to deteriorating health in 4 animals (2/condition) at 16 months of age. At this time point, the performance of these rats was similar to other rats that completed the study (average SAT scores: 0.41±0.09 vs 0.58±0.05; F(1,14)=2.93, p=0.11). The data from these animals were not included in subsequent analyses, comparing performance until 24 months of age. A mixed ANOVA with signal (3-levels) and age (9 month, 12–24 bimonthly time points; 8-levels) as within-subject factors and trkA manipulation (2-levels) as a between-subject factor was used to analyze attentional performance. This analysis revealed a significant main effect of signal (F(2,126)=130.55; p<0.001), a main effect of age (F(7,126)=12.87; p<0.001), and a significant interaction between the two factors (F(14,126)=2.24; p<0.02) on SAT scores. However, developmental trkA suppression did not affect attentional performance (main effect: F(1,9)=0.08; p=0.78), and no 3-way interaction between age, signal duration and the developmental trkA manipulation on performance was observed (F(14,126)=1.47; p=0.13). To determine the source of interaction between signal duration-dependent performance and age, a one-way repeated-measures ANOVA was conducted on SAT scores for each signal. A consistent decline in attentional performance with rising age was observed in both AAV-luc and AAV-trkA-infused rats for all signal durations (all F(7,63)>7.82; p<0.001; Fig 4A–C). Post-hoc comparisons revealed that deficits in SAT scores arose at 24 months on trials with 50 ms and 25 ms signal durations, respectively (both p<0.03 vs 9-month). Response latencies increased with age (main effect: F(7,63)=7.24; p<0.001; see Fig. 3D for multiple comparisons), and these data are in accordance with human studies that show decrements in reaction times in attentional tasks in older adults (Sparrow, Begg, & Parker, 2006; Tam, Luedke, Walsh, Fernandez-Ruiz, & Garcia, 2015). Like SAT scores, response latencies remained comparable between the AAV-luc and AAV-trkA animals (F(1,9)=0.10; p=0.75), and no interaction between age and vector infusion was observed on this behavioral measure (F(7,63)=0.37; p=0.91). Aging rats omitted more trials (main effect of age: F(7,63)=2.41; p<0.04). However, omissions did not differ by vector infusion (F(1,9)=0.05; p=0.83) and the age × vector infusion interaction remained insignificant (F(7,63)=0.95; p=0.47). Because the rate of omissions remained very low throughout the behavioral testing of animals (range: 0% – 9.8%), and SAT scores are not confounded by this measure (see Methods), the age effect is indicative of subtle changes in motivational states to perform the task, which does not impact the composite index of attentional performance.
Figure 4. Developmental BF trkA suppression and attentional performance in aging rats.
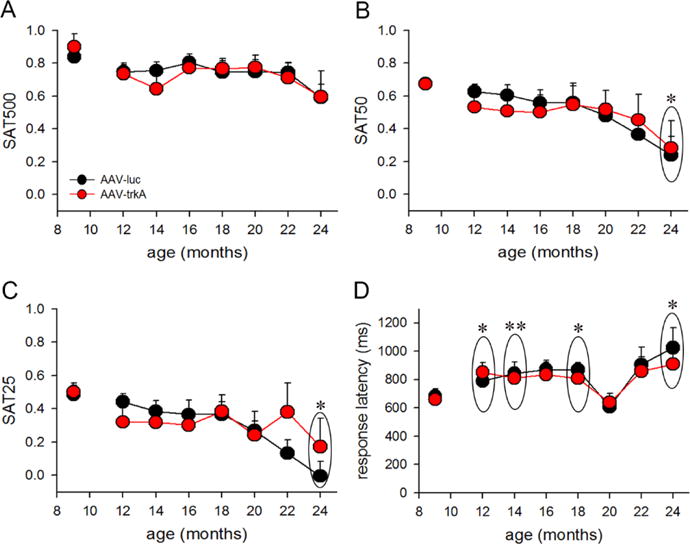
A within-subject comparison of SAT scores for 500ms (A), 50ms (B) and 25ms (C) in aging rats that received AAV-luc and AAV-trkA into the BF during development. A general deficit in the ability to detect signals was noted with rising age in both groups (main effect: p<0.001) and this effect interacted with signal duration (p<0.02). Post hoc analyses show that attentional deficits were observed at 24 months as compared to 9 months on trials with shorter signal durations (50ms and 25ms). (D) In general, response latency increased with aging in both AAV-luc and AAV-trkA groups (p<0.001), with a post-hoc revealing significantly slower responding occurring at 12, 14, 18, and 24 months old in comparison to 9 months old. Data are represented as mean ± SEM. *p<0.05, **p<0.01.
3.3. Attentional capacities under distracting conditions
To assess the consequences of developmental trkA suppression on attentional capacity under conditions of high cognitive load in aging rats, a visual distractor was presented during the second block of the test session (dSAT) when rats were 12 and 24 months old, respectively. A mixed-factor repeated-measures ANOVA with block (3-levels) and age (2-levels) as within-subject variables displayed a main effect of block (F(2,18)=23.44; p<0.001), a main effect of age (F(1,18)=6.98; p<0.03), and a significant interaction between the two factors F(2,18)=4.48; p<0.04). One-way ANOVA showed that at 12 months of age distractor presentation robustly diminished performance, as noted by a main effect of block (F(2,18)=22.28; p<0.01), with a post-hoc analysis demonstrating deficits arising in block 2 (i.e., distractor presentation) compared to block 1 (p<0.001) and block 3 (p<0.03; Fig 5A). However, at 24 months old, this main effect of block induced by the distractor presentation was no longer observed (F(2,18)=1.21, p=0.33; Fig 5B). This shift may be attributed to the overall reduction in attentional performance under baseline conditions at 24 months (Block 1: F(1,20)=18.74; p<0.001; 12 vs 24 months) rather than impaired ability to cope up with a distractor challenge (Block 3: F(1,20)=3.36; p=0.08; 12 vs 24 months). Surprisingly, AAV-trkA-infused rats performed similarly to AAV-luc-infused rats under distracting conditions [main effect for trkA manipulation as a between-subject factor: (F(1,9)=0.18, p=0.68)] and the three-way interaction remained insignificant (block × age × trkA manipulation: F(2,18)=2.44, p=0.12). Because baseline attentional capacity was affected in both groups at 24 months, we cannot rule out that a floor effect might have contributed to the lack of performance differences in the distractor session.
Figure 5. Attentional performance under distracting conditions.
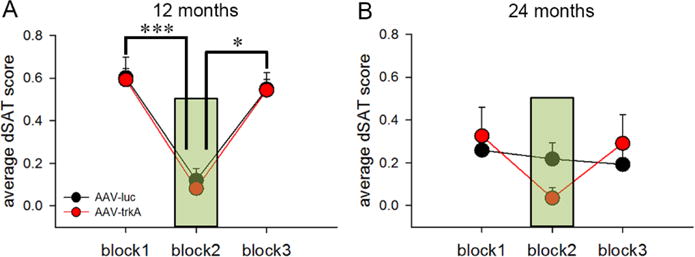
Attentional performance under conditions of higher cognitive load was assessed in a dSAT testing session that consisted of the presentation of a visual distractor (flashing house light at 0.5 Hz) during the 2nd block of trials. The presentation of trials in blocks 1 and 3 remained similar to the SAT session. (A) Average dSAT scores show robust impairments in attentional performance during block 2 (depicted as green shaded area) as compared to performance in the pre- and post-distractor blocks (blocks 1 and 3) at 12 months of age. (B) However, at 24 months of age, the performance remained similar across all 3 blocks. The impact of the distractor was not prominent due to an overall reduction in attentional capacity at 24 months vs. 12 months. Developmental trkA suppression did not impact dSAT performance at either 12 or 24 months (p=0.68). Data are represented as mean ± SEM. *p<0.05, ***p<0.001.
3.4. Impact of life-long suppression of trkA receptors on cholinergic function in attention task-performing rats
The status of cholinergic transmission was determined by in vivo monitoring of ACh dynamics in the medial PFC from aged rats after the completion of behavioral testing. Potassium and nicotine pulses were locally applied to generate choline spikes that represent hydrolyzed choline from newly released ACh events and mimic phasic and tonic characteristics of cholinergic transmission based on its temporal time-scale (Parikh et al., 2010; Parikh et al., 2008; Parikh et al., 2004). Average second-based increases in extracellular choline levels recorded following terminal depolarization in the medial PFC are depicted in Fig 6A. The amplitudes of depolarization-evoked cholinergic signals did not differ between the AAV-trkA- and AAV-luc-infused aged rats (F(1,7)=1.04, p=0.33; Fig 6B). Moreover, the clearance of choline signals remained unaffected by the developmental trkA manipulation (t50: F(1,7)=0.66, p=0.53; Fig 6C). As expected, local application of nicotine produced a robust increase in cholinergic transmission that lasted hundreds of seconds in aged rats infused with the control vector (see population traces in Fig 6D). Interestingly, developmental trkA suppression produced a drastic reduction in the amplitudes on nicotine-evoked choline signals (F(1,7)=2.32, p<0.05; Fig 6E). Additionally, the T50 values of these signals remained significantly lower as compared to the AAV-luc-infused rats (F(1,7)=2.57, p<0.05; Fig 6F).
Figure 6. Impact of life-long trkA suppression on cholinergic function.
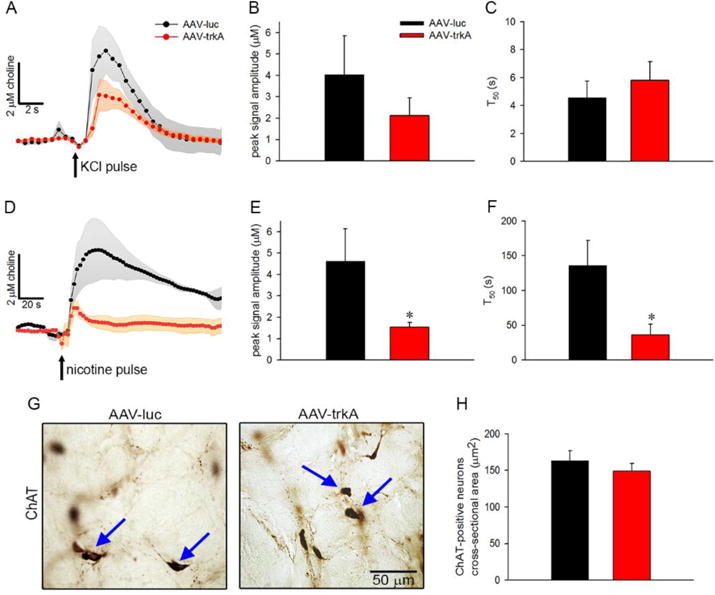
In vivo recordings of prefrontal cholinergic transmission were conducted to evaluate the impact of developmental trkA reductions on the functional capacity of cortically-projecting BF cholinergic neurons in aging. (A) Population traces depicting choline spikes, following brief depolarizing pulses of potassium in the medial PFC, of 24-month-old rats infused with either AAV-luc or AAV-trkA into the BF during development. The transient increases in extracellular choline levels reflect phasic ACh release and occur as a consequence of rapid hydrolysis of ACh by acetylcholinesterase. Peak amplitudes (B) and clearance (C) of depolarization-evoked cholinergic signals remained unaffected by the developmental AAV vector manipulation. (D) Population traces depicting tonic increases in cholinergic transmission following local application of nicotine in 24-month-old rats. Developmental trkA knockdown reduced peak signal amplitudes (E) and T50 (F) of nicotine-evoked cholinergic signals. (G) Representative coronal sections depicting ChAT-immunopositive neurons (marked by blue arrowheads) from the nBM/SI region of aged rats infused with either AAV-luc or AAV-trkA vector during development. (H) The cross-sectional area of cholinergic neurons in aged rats remained unaffected by the developmental vector manipulation. Data are represented as mean ± SEM. *p<0.05.
The impact of developmental trkA knockdown was also examined on the morphology of BF cholinergic neurons and cortical cholinergic terminal density using ChAT immunohistochemistry in the postmortem brains of aged rats. As depicted in Fig 6G and H, the mean cross-sectional area of BF ChAT-positive neurons did not differ between the AAV-luc- and AAV-trkA-infused rats (F(1,12)=0.82, p=0.43). Moreover, densitometric analysis of ChAT-positive fibers sampled from the prelimbic region of the medial PFC exhibited no significant differences in fiber counts (AAV-luc: 121.54±14.39; AAV-trkA: 112.39 ± 4.54; F(1,12)=0.61, p=0.56). Collectively, these data indicate that although certain aspects of cholinergic transmission were affected by developmental trkA suppression in aged rats engaged in an attention task for the full extent of their lives, the integrity of the cortical cholinergic input system was not impacted.
4. Discussion
The cholinotrophic effects of NGF-trkA signaling during development are well established (Hartikka & Hefti, 1988; Johnston, Rutkowski, Wainer, Long, & Mobley, 1987; Sanchez-Ortiz et al., 2012). Moreover, reduced trkA receptor density observed in AD subjects has been shown to correlate with deteriorating cognitive performance (Counts et al., 2004; Mufson et al., 2008). Because BF cholinergic signaling is critical for attentional performance (Parikh & Sarter, 2008; Sarter & Parikh, 2005), we hypothesized that developmental suppression of trkA signaling would escalate age-related attentional vulnerability by producing cholinergic disruption. To test this premise, we utilized an AAV vector-based RNAi approach to knockdown BF trkA during the developmental maturation of the corticopetal cholinergic circuit and examined the interactions between age-related changes in attentional function and trkA disruption using a longitudinal study design. In spite of developmental reductions in BF trkA receptor expression levels, AAV-trkA-infused rats exhibited comparable decline in attentional capacity with rising age as compared to control animals. Although nicotinic cholinergic transmission in aging was impacted by developmental trkA knockdown, the morphology of BF cholinergic neurons, prefrontal cholinergic innervation, and depolarization-evoked ACh release in aged rats remained unaffected.
AAV vectors allow efficient neuronal infection and maintenance of gene expression for a long period of time in rodents (Ivanova & Pan, 2009; Reimsnider, Manfredsson, Muzyczka, & Mandel, 2007). Moreover, quantitative analysis of trkA immunoreactivity revealed significant reductions of BF trkA protein levels in rats that received AAV-trkA infusions during the postnatal period, and these reductions persisted until 24 months of age. This suggests that our AAV vector-based RNAi approach produced stable and long-lasting expression of trkA shRNA from the SIBR plasmid in the cholinergic neurons that mostly express trkA receptors in the BF region. It is important to note that ~45–50% reduction in trkA receptor levels was maintained in these rats, and this degree of trkA loss has been reported in mild to moderate AD patients exhibiting cognitive impairments (Counts, Che, Ginsberg, & Mufson, 2011; Counts et al., 2004). We also previously observed attentional impairments in aged rats that received a vector manipulation, producing a similar magnitude of trkA reduction (Parikh et al., 2013; Yegla & Parikh, 2014). Furthermore, the same strategy was employed in the present study to produce developmental suppression of trkA receptors, and trkA knockdown persisted in aged rats, which was at a comparable level to our earlier work. Together, these findings dismiss the possibility that the lack of performance differences noted between the aging rats infused with AAV-trkA and AAV-luc vector during the postnatal period could have resulted from either insufficient shRNA expression or trkA degradation due to the vector.
As noted previously, the integrity of the BF cholinergic system depends upon NGF-trkA signaling (Fagan et al., 1997; Huang & Reichardt, 2003; Li et al., 1995). Moreover, accelerated age-related cellular changes including the production of amyloid-β, which is also observed in AD and exerts a negative effect on cholinergic neurons, are predicted to occur due to a shift in trkA receptor signaling from neurotrophic to apoptotic signaling. This shift is mediated by the p75 receptor, a low-affinity NGF receptor (Costantini et al., 2005; 2006; Kar, 2004). We previously reported that suppression of trkA signaling further exacerbated age-related reductions in cholinergic soma size and terminal densities (Parikh et al., 2013). Additionally, depolarization-evoked ACh release, which mimics temporal characteristics of phasic cholinergic transients critical for the detection of attention-demanding cues, declined in the PFC of trkA knockdown aged rats, and this effect was partially rescued by blockade of p75 signaling with a proNGF antibody (Yegla & Parikh, 2014). Together, these findings illustrated that the aging cholinergic system is exceptionally vulnerable to trkA dysregulation. In contrast to our earlier studies, persistent suppression of trkA receptors from early development into old age did not affect the integrity of the cortical cholinergic input system and the ability of cholinergic synapses to produce cholinergic transients in the PFC, as compared to aged-matched controls with intact trkA receptors in the present study. Moreover, attentional performance mirrored this pattern, with no differences emerging throughout aging in trkA-knockdown and control rats. Tonic cortical cholinergic activity is known to reflect the status of top-down attentional capacity (Sarter & Paolone, 2011). Although tonic increases in ACh release following local application of nicotine in the PFC was attenuated in AAV-trkA-infused rats, the dSAT performance, which recruits top-down mechanisms in aging, remained unaffected with the developmental trkA suppression. Together, these findings indicate that postnatal trkA knockdown affected only certain aspects of cholinergic transmission in aging and that these disruptions were insufficient to impact attentional capacities. Because nicotinic cholinergic signaling is implicated in a wide range of behaviors including spatial and contextual learning and working memory (Levine et al., 2006; Raybuck and Gould, 2010; Terry et al., 2000), the long-term effects of developmental trkA disruption on cognitive domains beyond attention remains to be seen.
It is plausible that extensive practice effects may have diluted the effect of developmental trkA suppression on attentional capacities. However, the persistent presence of a vigilance decrement into old age, reflected as signal duration-dependent performance effects, in our study (see Results) argues against the development of any habitual responding. This is consistent with a previous longitudinal study that reported vigilance decrements until 31 months following lifelong SAT training in rats (Burk, Herzog, Porter, & Sarter, 2002). In addition, we did observe a progressive decline in attentional capacities with age in both the developmentally-suppressed trkA and developmentally-intact trkA rats, which presumably would have resulted from a natural decline in trkA signaling. Thus, it seems unlikely that prolonged training of animals might have contributed to a lack of performance effects between these two groups of rats.
In the past decade, the concept of cognitive reserve has emerged, positing that engagement in mental/cognitive activity exerts neuroprotective effects and enhances the capacity for functional compensation (Cheng, 2016; Stern, 2009; Wilson et al., 2013). In rats, performance in a spatial learning task elicited age-specific shifts in redox homeostasis, with young-adult animals demonstrating reductions in oxidative stress while aged rats displayed increased antioxidant capacity in several brain regions including the frontal cortex, olfactory bulb, pons and medulla oblongata (Krivova, Zaeva, & Grigorieva, 2015). Moreover, cognitive training produced functional and enzymatic changes, such as greater functional connectivity between task-dependent brain regions and increased frontal and hippocampal ChAT levels (López et al., 2014; Nakamura & Ishihara, 1989; Suo et al., 2016). In the present study, all animals that underwent developmental trkA manipulation were performing SAT from young adulthood until old age. Thus, it is possible that constant recruitment of BF cortical cholinergic inputs through lifelong engagement in attentional activity might have bolstered this circuit to withstand some of the insult produced through reduced trkA signaling during development. Additionally, persistent cognitive activity could plausibly have minimized the impact of developmental trkA disruption by strengthening the dynamic interactions between BF cholinergic neurons and neuromodulatory systems such as the hypothalamic orexin/hypocretin system, that has also been suggested to regulate attentional functions (Chieffi et al., 2017; Fadel and Burk, 2010; Villano et al., 2017). At the molecular level, one potential player that could mediate circuit-level plasticity could be the brain-derived neurotrophic factor that is known to exert neuroprotective effects on cholinergic neurons and whose expression increases with persistent cortical activity (Galloway, Woo, & Lu, 2008; Nonomura, Nishio, Lindsay, & Hatanaka, 1995; Widmer, Knüsel, & Hefti, 1993). Lastly, in alignment with the cognitive reserve idea, activation of compensatory non-cholinergic networks may explain the lack of dSAT performance differences between trkA-knockdown and trkA-intact aged rats despite alterations in tonic cholinergic signaling. In light of current evidence, these interpretations remain speculative, and future experiments on the neurobiological underpinnings of cognitive reserve and how this neuroadaptive mechanism protects attentional functions from insults that increase the vulnerability of the cholinergic system are warranted.
Cholinergic responsivity is also known to be altered during normal aging (Decker, 1987; Parikh et al., 2013; Sarter & Bruno, 1998; Schliebs & Arendt, 2011), and the gradual yet modest declines in the ability to detect signals with rising age noted in the present study may have resulted from progressive age-related cholinergic deficits as suggested earlier (Baxter & Chiba, 1999; McGaughy & Sarter, 1995; Muir et al., 1999). However, this study remains limited in providing insights on whether sustained cognitive activity minimizes the detrimental effects of normal aging on cholinergic-attentional capacities. This would require an age-matched nonperforming control group of animals that received similar manipulations during development and were assessed for attentional performance and the status of their cholinergic signaling in old age.
In conclusion, our results indicate that postnatal reductions in BF trkA receptors did not impact cholinergic integrity and attentional capacities in aging rats that had received cognitive training throughout life. Although disruption in NGF-trkA signaling is known to drive cholinergic pathology in AD, recruitment of brain compensatory mechanisms seems to counter the detrimental effects of early cholinergic compromise on attentional functions in advanced age. Our findings provide strong support to the idea that reserve mechanisms orchestrated by cognitively stimulating environments may have the potential to either minimize or delay the progression of AD (Stern, 2013). More research is needed to delineate specific neurobiological mechanisms that provide resilience to cognitive capacities in pathological aging.
Highlights.
AAV-mediated RNAi approach produced life-long reduction in trkA receptors
Developmental trkA knockdown did not impact attention in aged rats
Knockdown of trkA reduced nicotine- but not depolarization-evoked ACh release
Life-long cognitive activity could have preserved attentional capacities
Acknowledgments
We thank Dr. David L. Turner (University of Michigan) for the initial design and construction of AAV vectors. We also thank Adnan Mookhtiar, Jasmine Forde and Surbhi Joshi for assisting us with behavioral training and immunohistochemistry.
Funding Sources
This work was supported by grants from the National Institute on Aging (AG029592 and AG046580).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
None.
References
- Al-Shawi R, Hafner A, Chun S, Raza S, Crutcher K, Thrasivoulou C, et al. Prongf, sortilin, and age-related neurodegeneration. Ann N Y Acad Sci. 2007;1119:208–215. doi: 10.1196/annals.1404.024. [DOI] [PubMed] [Google Scholar]
- Al-Shawi R, Hafner A, Olsen J, Olson J, Chun S, Raza S, et al. Neurotoxic and neurotrophic roles of prongf and the receptor sortilin in the adult and ageing nervous system. Eur J Neurosci. 2008;27(8):2103–2114. doi: 10.1111/j.1460-9568.2008.06152.x. [DOI] [PubMed] [Google Scholar]
- Arendt T, Stieler J, Ueberham U. Is sporadic Alzheimer’s disease a developmental disorder? J Neurochem. 2017 doi: 10.1111/jnc.14036. in press. [DOI] [PubMed] [Google Scholar]
- Baxter MG, Chiba AA. Cognitive functions of the basal forebrain. Curr Opin Neurobiol. 1999;9(2):178–183. doi: 10.1016/s0959-4388(99)80024-5. [DOI] [PubMed] [Google Scholar]
- Burk JA, Herzog CD, Porter MC, Sarter M. Interactions between aging and cortical cholinergic deafferentation on attention. Neurobiol Aging. 2002;23(3):467–477. doi: 10.1016/s0197-4580(01)00315-3. [DOI] [PubMed] [Google Scholar]
- Casu MA, Wong TP, De Koninck Y, Ribeiro-da-Silva A, Cuello AC. Aging causes a preferential loss of cholinergic innervation of characterized neocortical pyramidal neurons. Cereb Cortex. 2002;12(3):329–337. doi: 10.1093/cercor/12.3.329. [DOI] [PubMed] [Google Scholar]
- Cheng ST. Cognitive reserve and the prevention of dementia: The role of physical and cognitive activities. Curr Psychiatry Rep. 2016;18(9):85. doi: 10.1007/s11920-016-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieffi S, Carotenuto M, Monda V, Valenzano A, Villano I, Precenzano F, Tafuri D, Salerno M, Filippi N, Nuccio F, Ruberto M, De Luca V, Cipolloni L, Cibelli G, Mollica MP, Iacono D, Nigro E, Monda M, Messina G, Messina A. Orexin System: The Key for a Healthy Life. Front Physiol. 2017;8:357. doi: 10.3389/fphys.2017.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, Weindruch R, Della Valle G, Puglielli L. A TrkA-to-p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem J. 2005;391:59–67. doi: 10.1042/BJ20050700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, Scrable H, Puglielli L. An aging pathway controls the TrkA-to-p75NTR receptor switch and amyloid beta-peptide generation. EMBO J. 2006;25:1997–2006. doi: 10.1038/sj.emboj.7601062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Che S, Ginsberg SD, Mufson EJ. Gender differences in neurotrophin and glutamate receptor expression in cholinergic nucleus basalis neurons during the progression of alzheimer’s disease. J Chem Neuroanat. 2011;42(2):111–117. doi: 10.1016/j.jchemneu.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Reduction of cortical trka but not p75(ntr) protein in early-stage alzheimer’s disease. Ann Neurol. 2004;56(4):520–531. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- Cuello AC, Bruno MA, Bell KF. Ngf-cholinergic dependency in brain aging, mci and alzheimer’s disease. Curr Alzheimer Res. 2007;4(4):351–358. doi: 10.2174/156720507781788774. [DOI] [PubMed] [Google Scholar]
- Debeir T, Saragovi HU, Cuello AC. Trka antagonists decrease ngf-induced chat activity in vitro and modulate cholinergic synaptic number in vivo. J Physiol Paris. 1998;92(3–4):205–208. doi: 10.1016/s0928-4257(98)80011-9. [DOI] [PubMed] [Google Scholar]
- Decker MW. The effects of aging on hippocampal and cortical projections of the forebrain cholinergic system. Brain Res. 1987;434(4):423–438. doi: 10.1016/0165-0173(87)90007-5. [DOI] [PubMed] [Google Scholar]
- Demeter E, Sarter M, Lustig C. Rats and humans paying attention: Cross-species task development for translational research. Neuropsychology. 2008;22(6):787–799. doi: 10.1037/a0013712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dori I, Parnavelas JG. The cholinergic innervation of the rat cerebral cortex shows two distinct phases in development. Exp Brain Res. 1989;76(2):417–423. doi: 10.1007/BF00247899. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu Rev Psychol. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- Fadel J, Burk JA. Orexin/hypocretin modulation of the basal forebrain cholinergic system: role in attention. Brain Res. 2010;1314:1120123. doi: 10.1016/j.brainres.2009.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM. A role for trka during maturation of striatal and basal forebrain cholinergic neurons in vivo. J Neurosci. 1997;17(20):7644–7654. doi: 10.1523/JNEUROSCI.17-20-07644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in alzheimer’s disease. Mol Cell Neurosci. 2001;18(2):210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Frey PW, Colliver JA. Sensitivity and responsivity measures for discrimination learning. 1973;4:327–342. Learning and Motivation. [Google Scholar]
- Galloway EM, Woo NH, Lu B. Persistent neural activity in the prefrontal cortex: A mechanism by which bdnf regulates working memory? Prog Brain Res. 2008;169:251–266. doi: 10.1016/S0079-6123(07)00015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75ntr gene expression in single cholinergic basal forebrain neurons mark the progression of alzheimer’s disease. J Neurochem. 2006;97(2):475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- Grothe M, Zaborszky L, Atienza M, Gil-Neciga E, Rodriguez-Romero R, Teipel SJ, et al. Reduction of basal forebrain cholinergic system parallels cognitive impairment in patients at high risk of developing alzheimer’s disease. Cereb Cortex. 2010;20(7):1685–1695. doi: 10.1093/cercor/bhp232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haense C, Kalbe E, Herholz K, Hohmann C, Neumaier B, Krais R, et al. Cholinergic system function and cognition in mild cognitive impairment. Neurobiol Aging. 2012;33(5):867–877. doi: 10.1016/j.neurobiolaging.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Hartikka J, Hefti F. Comparison of nerve growth factor’s effects on development of septum, striatum, and nucleus basalis cholinergic neurons in vitro. J Neurosci Res. 1988;21(2–4):352–364. doi: 10.1002/jnr.490210227. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: Roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Ivanova E, Pan ZH. Evaluation of the adeno-associated virus mediated long-term expression of channelrhodopsin-2 in the mouse retina. Mol Vis. 2009;15:1680–1689. [PMC free article] [PubMed] [Google Scholar]
- Johnston MV, Rutkowski JL, Wainer BH, Long JB, Mobley WC. Ngf effects on developing forebrain cholinergic neurons are regionally specific. Neurochem Res. 1987;12(11):985–994. doi: 10.1007/BF00970927. [DOI] [PubMed] [Google Scholar]
- Kar S. Interactions between beta-amyoid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatr Neurosci. 2004;29:427–441. [PMC free article] [PubMed] [Google Scholar]
- Kawas C, Gray S, Brookmeyer R, Fozard J, Zonderman A. Age-specific incidence rates of Alzheimer’s disease: the Baltimore Longitudinal Study of Aging. Neurology. 2000;54:2072–2077. doi: 10.1212/wnl.54.11.2072. [DOI] [PubMed] [Google Scholar]
- Kiss J, Patel AJ. Development of the cholinergic fibres innervating the cerebral cortex of the rat. Int J Dev Neurosci. 1992;10(2):153–170. doi: 10.1016/0736-5748(92)90043-y. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Applegate MD, Knüsel B, Junard EO, Burton LE, Mobley WC, et al. Recombinant human nerve growth factor prevents retrograde degeneration of axotomized basal forebrain cholinergic neurons in the rat. Exp Neurol. 1991;112(2):161–173. doi: 10.1016/0014-4886(91)90066-l. [DOI] [PubMed] [Google Scholar]
- Krivova NA, Zaeva OB, Grigorieva VA. Effect of a water-maze procedure on the redox mechanisms in brain parts of aged rats. Front Aging Neurosci. 2015;7:29. doi: 10.3389/fnagi.2015.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacol. 2006;184(3–4):523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Holtzman DM, Kromer LF, Kaplan DR, Chua-Couzens J, Clary DO, et al. Regulation of trka and chat expression in developing rat basal forebrain: Evidence that both exogenous and endogenous ngf regulate differentiation of cholinergic neurons. J Neurosci. 1995;15(4):2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López ME, Aurtenetxe S, Pereda E, Cuesta P, Castellanos NP, Bruña R, et al. Cognitive reserve is associated with the functional organization of the brain in healthy aging: A meg study. Front Aging Neurosci. 2014;6:125. doi: 10.3389/fnagi.2014.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughy J, Sarter M. Behavioral vigilance in rats: Task validation and effects of age, amphetamine, and benzodiazepine receptor ligands. Psychopharmacology (Berl) 1995;117(3):340–357. doi: 10.1007/BF02246109. [DOI] [PubMed] [Google Scholar]
- Mechawar N, Descarries L. The cholinergic innervation develops early and rapidly in the rat cerebral cortex: A quantitative immunocytochemical study. Neuroscience. 2001;108(4):555–567. doi: 10.1016/s0306-4522(01)00389-x. [DOI] [PubMed] [Google Scholar]
- Mesulam M. The cholinergic lesion of alzheimer’s disease: Pivotal factor or side show? Learn Mem. 2004;11(1):43–49. doi: 10.1101/lm.69204. [DOI] [PubMed] [Google Scholar]
- Mobley WC, Rutkowski JL, Tennekoon GI, Gemski J, Buchanan K, Johnston MV. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Brain Res. 1986;387(1):53–62. doi: 10.1016/0169-328x(86)90020-3. [DOI] [PubMed] [Google Scholar]
- Mouloua M, Parasuraman R. Aging and cognitive vigilance: Effects of spatial uncertainty and event rate. Exp Aging Res. 1995;21(1):17–32. doi: 10.1080/03610739508254265. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of alzheimer’s disease: Therapeutic implications. Expert Rev Neurother. 2008;8(11):1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Ma SY, Cochran EJ, Bennett DA, Beckett LA, Jaffar S, et al. Loss of nucleus basalis neurons containing trka immunoreactivity in individuals with mild cognitive impairment and early alzheimer’s disease. J Comp Neurol. 2000;427(1):19–30. doi: 10.1002/1096-9861(20001106)427:1<19::aid-cne2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Muir JL, Fischer W, Björklund A. Decline in visual attention and spatial memory in aged rats. Neurobiol Aging. 1999;20(6):605–615. doi: 10.1016/s0197-4580(99)00098-6. [DOI] [PubMed] [Google Scholar]
- Müller M, Triaca V, Besusso D, Costanzi M, Horn JM, Koudelka J, et al. Loss of ngf-trka signaling from the cns is not sufficient to induce cognitive impairments in young adult or intermediate-aged mice. J Neurosci. 2012;32(43):14885–14898. doi: 10.1523/JNEUROSCI.2849-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Ishihara T. Region selective increase in activities of cns cholinergic marker enzymes during learning of memory tasks in aged rats. Pharmacol Biochem Behav. 1989;34(4):805–810. doi: 10.1016/0091-3057(89)90278-5. [DOI] [PubMed] [Google Scholar]
- Nonomura T, Nishio C, Lindsay RM, Hatanaka H. Cultured basal forebrain cholinergic neurons from postnatal rats show both overlapping and non-overlapping responses to the neurotrophins. Brain Res. 1995;683(1):129–139. doi: 10.1016/0006-8993(95)00357-v. [DOI] [PubMed] [Google Scholar]
- Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, et al. Sortilin is essential for prongf-induced neuronal cell death. Nature. 2004;427(6977):843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- Oosawa H, Fujii T, Kawashima K. Nerve growth factor increases the synthesis and release of acetylcholine and the expression of vesicular acetylcholine transporter in primary cultured rat embryonic septal cells. J Neurosci Res. 1999;57(3):381–387. [PubMed] [Google Scholar]
- Parikh V, Howe WM, Welchko RM, Naughton SX, D’Amore DE, Han DH, et al. Diminished trka receptor signaling reveals cholinergic-attentional vulnerability of aging. Eur J Neurosci. 2013;37(2):278–293. doi: 10.1111/ejn.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Ji J, Decker MW, Sarter M. Prefrontal beta2 subunit-containing and alpha7 nicotinic acetylcholine receptors differentially control glutamatergic and cholinergic signaling. J Neurosci. 2010;30(9):3518–3530. doi: 10.1523/JNEUROSCI.5712-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Man K, Decker MW, Sarter M. Glutamatergic contributions to nicotinic acetylcholine receptor agonist-evoked cholinergic transients in the prefrontal cortex. J Neurosci. 2008;28(14):3769–3780. doi: 10.1523/JNEUROSCI.5251-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Pomerleau F, Huettl P, Gerhardt GA, Sarter M, Bruno JP. Rapid assessment of in vivo cholinergic transmission by amperometric detection of changes in extracellular choline levels. Eur J Neurosci. 2004;20(6):1545–1554. doi: 10.1111/j.1460-9568.2004.03614.x. [DOI] [PubMed] [Google Scholar]
- Parikh V, Sarter M. Cholinergic mediation of attention: Contributions of phasic and tonic increases in prefrontal cholinergic activity. Ann N Y Acad Sci. 2008;1129:225–235. doi: 10.1196/annals.1417.021. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased prongf levels in subjects with mild cognitive impairment and mild alzheimer disease. J Neuropathol Exp Neurol. 2004;63(6):641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Hodges JR. Attention and executive deficits in alzheimer’s disease. A critical review. Brain. 1999;122(Pt 3):383–404. doi: 10.1093/brain/122.3.383. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Watson P, Hodges JR. The nature and staging of attention dysfunction in early (minimal and mild) alzheimer’s disease: Relationship to episodic and semantic memory impairment. Neuropsychologia. 2000;38(3):252–271. doi: 10.1016/s0028-3932(99)00079-2. [DOI] [PubMed] [Google Scholar]
- Raybuck JD, Gould TJ. The role of nicotinic acetylcholine receptors in the medial prefrontal cortex and hippocampus in trace fear conditioning. Neurobiol Learn Mem. 2010;94(3):353–363. doi: 10.1016/j.nlm.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimsnider S, Manfredsson FP, Muzyczka N, Mandel RJ. Time course of transgene expression after intrastriatal pseudotyped raav2/1, raav2/2, raav2/5, and raav2/8 transduction in the rat. Mol Ther. 2007;15(8):1504–1511. doi: 10.1038/sj.mt.6300227. [DOI] [PubMed] [Google Scholar]
- Rizzo M, Anderson SW, Dawson J, Nawrot M. Vision and cognition in alzheimer’s disease. Neuropsychologia. 2000;38(8):1157–1169. doi: 10.1016/s0028-3932(00)00023-3. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ortiz E, Yui D, Song D, Li Y, Rubenstein JL, Reichardt LF, et al. Trka gene ablation in basal forebrain results in dysfunction of the cholinergic circuitry. J Neurosci. 2012;32(12):4065–4079. doi: 10.1523/JNEUROSCI.6314-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Bruno JP. Age-related changes in rodent cortical acetylcholine and cognition: Main effects of age versus age as an intervening variable. Brain Res Brain Res Rev. 1998;27(2):143–156. doi: 10.1016/s0165-0173(98)00003-4. [DOI] [PubMed] [Google Scholar]
- Sarter M, Bruno JP. Developmental origins of age-related decline in cortical cholinergic function and associated cognitive abilities. Neurobiol Aging. 2004;25(9):1127–1139. doi: 10.1016/j.neurobiolaging.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Sarter M, Paolone G. Deficits in attentional control: Cholinergic mechanisms and circuitry-based treatment approaches. Behav Neurosci. 2011;125(6):825–835. doi: 10.1037/a0026227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci. 2005;6(1):48–56. doi: 10.1038/nrn1588. [DOI] [PubMed] [Google Scholar]
- Schliebs R, Arendt T. The cholinergic system in aging and neuronal degeneration. Behav Brain Res. 2011;221(2):555–563. doi: 10.1016/j.bbr.2010.11.058. [DOI] [PubMed] [Google Scholar]
- Sobreviela T, Clary DO, Reichardt LF, Brandabur MM, Kordower JH, Mufson EJ. Trka-immunoreactive profiles in the central nervous system: Colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J Comp Neurol. 1994;350(4):587–611. doi: 10.1002/cne.903500407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow WA, Begg RK, Parker S. Aging effects on visual reaction time in a single task condition and when treadmill walking. Motor Control. 2006;10(3):201–211. doi: 10.1123/mcj.10.3.201. [DOI] [PubMed] [Google Scholar]
- St Peters M, Demeter E, Lustig C, Bruno JP, Sarter M. Enhanced control of attention by stimulating mesolimbic-corticopetal cholinergic circuitry. J Neurosci. 2011;31(26):9760–9771. doi: 10.1523/JNEUROSCI.1902-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve. Neuropsychologia. 2009;47(10):2015–2028. doi: 10.1016/j.neuropsychologia.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve: Implications for assessment and intervention. Folia Phoniatr Logop. 2013;65(2):49–54. doi: 10.1159/000353443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo C, Singh MF, Gates N, Wen W, Sachdev P, Brodaty H, et al. Therapeutically relevant structural and functional mechanisms triggered by physical and cognitive exercise. Mol Psychiatry. 2016;21(11):1645. doi: 10.1038/mp.2016.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam A, Luedke AC, Walsh JJ, Fernandez-Ruiz J, Garcia A. Effects of reaction time variability and age on brain activity during stroop task performance. Brain Imaging Behav. 2015;9(3):609–618. doi: 10.1007/s11682-014-9323-y. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Hernandez CM, Buccafusco JJ, Gattu M. Deficits in spatial learning and nicotinic-acetylcholine receptors in older, spontaneously hypertensive rats. Neuroscience. 2000;101(202):357–368. doi: 10.1016/s0306-4522(00)00377-8. [DOI] [PubMed] [Google Scholar]
- Villano I, Messina A, Valenzano A, Moscatelli F, Esposito T, Monda V, Esposito M, Precenzano F, Carotenuto M, Viggiano A, Chieffi S, Cibelli G, Monda M, Messina G. Basal forebrain cholinergic system and orexin neurons: effects on attention. Front Behav Neurosci. 2017;11:10. doi: 10.3389/fnbeh.2017.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer HR, Knüsel B, Hefti F. Bdnf protection of basal forebrain cholinergic neurons after axotomy: Complete protection of p75ngfr-positive cells. Neuroreport. 1993;4(4):363–366. doi: 10.1097/00001756-199304000-00005. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Boyle PA, Yu L, Barnes LL, Schneider JA, Bennett DA. Life-span cognitive activity, neuropathologic burden, and cognitive aging. Neurology. 2013;81(4):314–321. doi: 10.1212/WNL.0b013e31829c5e8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegla B, Parikh V. Effects of sustained prongf blockade on attentional capacities in aged rats with compromised cholinergic system. Neuroscience. 2013 doi: 10.1016/j.neuroscience.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegla B, Parikh V. Effects of sustained prongf blockade on attentional capacities in aged rats with compromised cholinergic system. Neuroscience. 2014;261:118–132. doi: 10.1016/j.neuroscience.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]