

Abstract
The dopamine transporter (DAT) is the key regulator of dopaminergic transmission and is a target of several xenobiotics, including pesticides and pharmacological agents. Previously, we identified a prominent role for histone deacetylases in the regulation of DAT expression. Here, we utilized a rat dopaminergic cell line (N27) to probe the responsiveness of DAT mRNA expression to inhibitors of histone acetylation. Inhibition of histone deacetylases (HDACs) by valproate, butyrate and Trichostatin A led to a 3 to 10-fold increase in DAT mRNA expression, a 50% increase in protein levels, which were accompanied by increased H3 acetylation levels. To confirm the mechanism of valproate-mediated increase in DAT mRNA, chromatin immunoprecipitation (ChIP) assays were used and demonstrated a significant increase in enrichment of acetylation of histone 3 on lysines 9 and 14 (H3K9/K14ac) in the DAT promoter. Expression of Nurr1 and Pitx3, key regulators of DAT expression, were increased following valproate treatment and Nurr1 binding was enriched in the DAT promoter. Together, these results indicate that histone acetylation and subsequent enhancement of transcription factor binding are plausible mechanisms for DAT regulation by valproate and, perhaps, by other xenobiotics.
Keywords: Dopamine transporter, epigenetics, HDAC, valproate, histone deacetylase, Pitx3, Nurr1
1. Introduction
Mammalian histone deacetylase (HDAC) inhibitors are classified based upon sequence homology, structural differences and expression patterns (Morris and Monteggia, 2013). Class I HDACs (1, 2, 3 and 8) are generally located within the nucleus, whereas Class IIa (HDACs 4, 5, 7 and 9), Class IIb (HDACs 6 and 10) and Class IV (HDAC 11) HDACs are located in the cytoplasm or both (Fischer et al., 2010; Haberland et al., 2009). Although cancer treatment has been the most intensely studied application for HDAC inhibitors, the high expression of many HDAC isoforms within the brain (Broide et al., 2007) suggests that they may be promising targets in the treatment of neurological diseases or disorders. Indeed, drugs possessing HDAC inhibitory properties are currently used to treat bipolar disorder and epilepsy (Casey et al., 2003; Monti et al., 2009; Peterson and Naunton, 2005).
The class I HDAC inhibitor valproate has been implicated in the reduction of Schizophrenia-like behaviors and demonstrated neuroprotective effects in models of neurodegeneration (Kidd and Schneider, 2011; Tremolizzo et al., 2005), which has been linked to potential effects on the dopamine system. Dopaminergic transmission is essential to carry out brain functions such as locomotion, memory, motivated behaviors and cognition (Giros and Caron, 1993; Palmiter, 2008). Disruption in dopaminergic signaling is associated with many neurological disorders such as Parkinson’s disease, Attention-deficit Hyperactivity disorder, Bipolar disorder and Schizophrenia (Damier et al., 1999; Howes and Kapur, 2009; Volkow et al., 2007). The dopamine transporter (DAT) is a transmembrane protein responsible for modulating dopaminergic transmission through selective uptake of the neurotransmitter dopamine (DA). Studies on the regulation of the DAT gene expression identified core promoter features and putative transcription factor binding motifs (Kouzmenko et al., 1997; Sacchetti et al., 1999), including that of the orphan nuclear receptor nuclear receptor related 1 protein (Nr4a2; Nurr1) and paired-like homeobox 3 protein (Pitx3) (Martinat et al., 2006; Riddle and Pollock, 2003; Smits and Smidt, 2006). In addition to transcription factor binding, the DAT promoter lacks a conserved TATA box, suggesting the potential for regulation by histone acetylation, similar to other TATA-less genes (Choi and Kim, 2008).
Work from our laboratory and others has demonstrated an increase of human DAT gene expression following treatment with the histone deacetylase (HDAC) inhibitor valproate in vitro (Wang et al., 2007). However, little is known about the epigenetic modifications and transcription factor binding patterns that may mediate the change in DAT gene expression following treatment with valproate. Histone acetylation is a post-translational modification of chromatin resulting in changes in DNA repair, DNA replication and gene transcription (Groth et al., 2007; Shahbazian and Grunstein, 2007). In this study, we further explore the role of histone acetylation in DAT gene regulation and identify a potential role for the nuclear transcription factor Nurr1 as a mediator of valproate-induced increases in DAT mRNA expression.
2. Materials and Methods
2.1. Chemicals
Cell culture supplies, including Roswell Park Memorial Institute media (RPMI 1640) with L-glutamine, penicillin (100 units/ml)/streptomycin (100 μg/ml), fetal bovine serum (FBS), phosphate-buffered saline (PBS) 1× without calcium and magnesium, trypsin EDTA (0.05% trypsin, 0.53mM EDTA in Hank’s Balanced Salt Solution- without sodium bicarbonate, calcium, and magnesium) 1×, were purchased from Mediatech, Inc. (Herndon, VA). Valproic acid sodium salt (VPA), sodium butyrate (NaB) and Trichostatin A (TSA) were purchased from Sigma-Aldrich (St. Louis, MO). AlamarBlue reagent was purchased from Life Technologies (Grand Island, NY).
2.2. Cell Culture
The immortalized dopaminergic cell line 1RB3AN27 (N27) has been extensively characterized for dopaminergic gene expression and was a generous gift from Dr. Anumantha Kanthasamy (Iowa State University). N27 cells were generated from embryonic day 12 rat mesencephalic tissue and transfected with plasmid vectors carrying T-antigen genes from SV40 for immortalization (Prasad et al., 1994). These cells stably express dopaminergic markers, including tyrosine hydroxylase (TH), NURR1 and DAT (Clarkson et al., 1998). Cells were cultured in RPMI 1640 supplemented with 10% FBS, 50 IU/mL penicillin, 50μg/mL streptomycin and were maintained at 37°C with 5% CO2.
2.3. HDAC Inhibitor Treatment
One day prior to treatment, cells were seeded 2 × 105 cells per well into 6 well plates. For HDAC inhibitor treatment, 25mM stock solutions of valproate and butyrate were made fresh in RPMI 1640 medium, diluted accordingly and added to the cells. A 1μM stock solution of Trichostatin A was prepared in 95% ethanol and diluted in RPMI media before adding to the cells. The final concentration of ethanol was less than 0.1% for all experiments. Following 24 hr of exposure, cells were harvested for RNA, protein and/or chromatin isolation.
2.4. Cell Viability Assay
N27 cells seeded at 6 × 103 cells per well of a 96 well plate were treated the following day with various concentrations of HDAC inhibitors. Twenty four hours later the cell medium was replaced with 10% AlamarBlue (Life Technologies) reagent in 1× RPMI media and incubated for 2 hours at 37°C. The fluorescent signal was quantified with Spectromax M5 microplate reader at 570nm excitation and 585nm emission.
2.5. Immunofluorescence
Cells were seeded at 25,000 cells/well in 8 well chamber slides. Following treatment, cells were fixed with 10% formalin and then washed with PBS. Cells were then permeabilized using 0.1% Triton X-100 for 10 min at RT. Following this, cells were blocked with 2% BSA for 1 hour and incubated overnight with 1:1000 1:1000 DAT antibody (cat MAB369, Millipore). Cells were then incubated for 1 hour with rabbit anti-rat Alexa Fluor 488 secondary antibody. Images were taken using a Leica DMi8 SPE II confocal microscope using a 40× oil lens (N.A. 1.15) and fluorescence intensity quantified using ImageJ. Specificity was determined by omission or primary or secondary antibodies. In ImageJ, a minimum threshold was set for all imaging, and fluorescence intensity was measured over the same area from multiple images (3 images) from multiple chamber slide wells (4 wells), approximately the same number of cells were measured for all wells.
2.6. RNA Isolation and cDNA Synthesis
RNA was isolated from cells using the Qiagen RNeasy Kit (Qiagen) as described previously (Hossain and Richardson, 2011). RNA concentration was determined using NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). One μg of total RNA was used for cDNA synthesis with the First Strand Synthesis Kit (Invitrogen) as per manufacturer instructions.
2.7. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
qPCR reactions were prepared in 25 μl total volume using SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA); 1:15 diluted cDNA and 1μM of forward and reverse primers as described previously (Green et al., 2015). Primers were designed using the National Center for Biotechnology Information PrimerBLAST application (http://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi; Figure 1). PCR reactions were performed on a ViiA7™ Real-Time PCR system (Applied Biosystems, Carlsbad, CA) at the appropriate annealing temperature for each primer set. Relative gene expression was determined using the SYBR Green technology (Applied Biosystems, Carlsbad, CA). All samples were run in duplicate; β-actin and TATA Binding Protein (TBP) were used as normalization controls for each gene and gave similar results. All primer sets yielded a single PCR product of expected size by agarose gel electrophoresis (data not shown). Relative values were calculated using 2ΔΔCt method as described by Livak and Schmittgen (Livak and Schmittgen, 2001).
Figure 1.
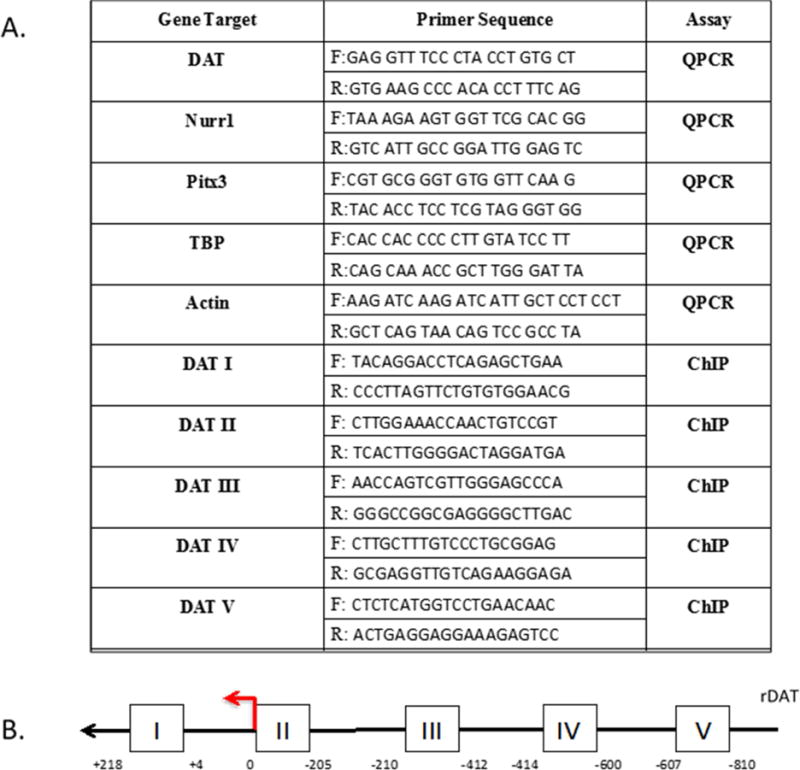
A) Primer sequences for RT-qPCR and ChIP assays B) Schematic view of rat dopamine transporter promoter. The amplicons of each pair of ChIP primers are approximately 200bp each and are Roman numeral labeled accordingly.
2.8. Western Immunoblotting
Nuclear extracts were isolated from N27 cells treated with 1mM valproate using NE-PER™ Kit (Thermo Scientific) as per manufacturer instructions. Protein homogenates were supplemented with 0.1% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Protein concentrations were determined using the Pierce ™ bicinchoninic acid (BCA) assay kit (Thermo Scientific) and 50 μg of protein sample was loaded per lane on a 4–12% Bis-Tris Polyacrylamide Gel (Invitrogen). Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and blocked in 7.5% non-fat milk in 0.1% Tween 20 and Tris buffered saline (TTBS) for 1 hour at room temperature. The membranes were incubated at 4°C overnight with rabbit anti-H3Ac primary antibody (1:2000; cat# 06-599 Millipore), followed by a 1 hour room temperature incubation with anti-rabbit HRP-conjugated secondary antibody. The bound antibody was detected by SuperSignal® West Dura Extended Duration Substrate Kit (Thermo Scientific) and imaged using Alpha Innotech Fluorochem imaging system. The membrane was stripped using Pierce Stripping Buffer (Thermo Scientific) and re-probed with rabbit anti-Histone 3 antibody to confirm equal protein loading.
2.9. Chromatin Immunoprecipitation (ChIP) Assay
N27 dopaminergic cells were plated 3 × 106 cells per 150cm2 flask one day prior to cell treatment and treated with 1 mM valproate for 24 hours. Cells were crosslinked using a final concentration of 1% formaldehyde for 10 minutes. The crosslinking reaction was stopped by adding 10× glycine, to a final concentration of 125 mM, for 5 minutes then washed 3 times in cold PBS supplemented with protease inhibitor cocktail and phenylmethylsulfonyl fluoride (Sigma-Aldrich). Cells were scraped into cold PBS and centrifuged at 2000rpm for 5 minutes at 4°C. Pellets were resuspended in cold cell lysis buffer (10mM Tris pH 8, 10mM NaCl, 3mM MgCl2, 0.5% NP40) supplemented with protease inhibitors for 15 minutes. Nuclei were pelleted at 2000rpm for 5 minutes at 4°C and resuspended in nuclear lysis buffer (1% SDS, 5mM EDTA, 50mM Tris pH8).
Chromatin was sheared for 12 cycles (15s on/60s off) to an average length of 300–700 bp using a sonic dismembrator (Fisher Scientific). Insoluble material was pelleted at 14,000rpm for 10 min at 4°C. The protein concentration of each chromatin sample was measured using the BCA assay kit. Sixty μg of chromatin was used for each immunoprecipitation (IP) reaction. IP reactions were performed with 4 μg of each antibody H3K9K14ac (cat # 9441, Cell Signaling), Nurr1 (cat # sc-991, Santa Cruz), Pitx3 (cat # 19307, Santa Cruz), Normal Rabbit IgG (cat# 12-370, Millipore) overnight at 4°C. Magnetic beads (Cell Signaling) were added to each IP reaction and rotated for 3 hours at 4°C. The beads were sequentially washed in low salt buffer (0.1 % SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris pH 8, 150mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris pH 8, 500mM NaCl), LiCl buffer (0.25M LiCl, 1% NP40, 1% Na deoxycholate, 1mM EDTA, 10mM Tris pH 8) and TE buffer (10mM Tris pH 8, 1mM EDTA), respectively. Chromatin was eluted from the beads using elution buffer (1 % SDS, 0.1M NaHCO3) and rotated at 65° C for 30 minutes. Crosslinking was reversed by adding 5M NaCl to each eluate and incubated at 65° C for 5 hours. DNA was treated with RNase A, Proteinase K and purified using Qiagen PCR Purification kit (Qiagen). qPCR was performed to determine relative enrichment of proteins in DAT promoter regions (See Fig. 1 for ChIP primer sequences).
2.10. Statistical Analyses
All data were analyzed using GraphPad Prism 5.0 Software (GraphPad Software, San Diego, CA). All experiments were performed at least three times on three different days. Individual experiments were conducted in duplicate or triplicate and averaged. Data are presented as mean ± SEM and analyzed by One-way ANOVA, Two-way ANOVA or Student’s t-test, where appropriate. Statistical significance was determined at level of p ≤ 0.05.
3. Results
3.1. Dose dependent increase in DAT mRNA expression following HDAC inhibitor treatment
Previously, we and others demonstrated that valproate increases DAT mRNA expression in human SK-N-AS neuroblastoma cells (Wang et al., 2007). We performed a dose response using valproate in rat mesencephalic cells to determine the highest dose, which also resulted in increased DAT expression. Although more sensitive to the dose used previously in SK-N-AS cells, peak expression was reached in N27 cells at 1mM with a 5-fold increase (F= 43.53, df= 5, p< 0.0001) in DAT mRNA (Fig. 2A top figure). To determine whether other HDAC inhibitors had a similar effect, we used additional HDAC inhibitors that targeted histone proteins of different classes. Sodium butyrate, also a Class I HDAC inhibitor, showed up to 4-fold increase (F=16.51, df=5, p<0.0001) in DAT mRNA expression profile following the same concentrations as valproate (Fig 2A middle figure). The more potent Class II HDAC inhibitor, Trichostatin A, showed 10-fold increase (F=9.96, df= 5, p=0.001) in DAT expression at 600nM (Fig. 2A bottom figure).
Figure 2.
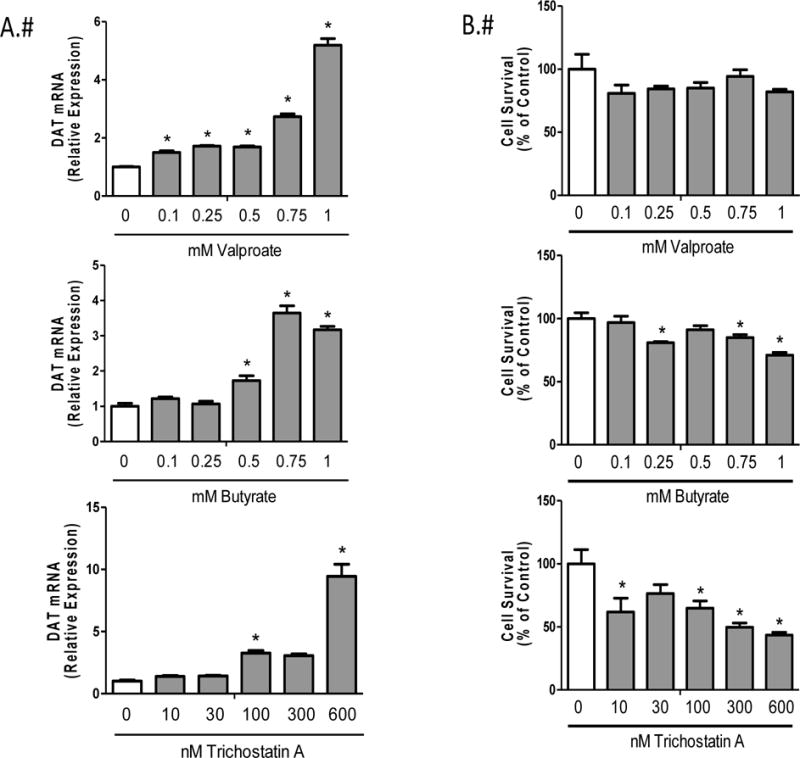
HDAC inhibitor dose response in N27 rat mesencephalic cells A) Relative DAT mRNA expression and B) Cell Viability for valproate, butyrate and Trichostatin A. Data represent mean ± SEM; N=3–4, *p≤0.05 (One-way ANOVA, Dunnet’s post hoc).
3.2. Reduced cell viability with increasing concentrations of butyrate and Trichostatin A
To determine optimal concentrations that induced DAT mRNA without compromising cell viability, an AlamarBlue assay was performed. There was no reduction in cell viability (F=1.61, df=5, p=0.223) in any of the concentrations tested for valproate (Fig 2B top figure). Cell viability was significantly reduced with increasing concentrations of both butyrate (F=9.64, df= 5, p= 0.0003) and Trichostatin A (F=8.47, df=5, p=0.0006) (Fig. 2B middle and bottom figures, respectively). Because of this toxicity, valproate was the only compound used for remaining studies.
3.3. Increased DAT protein following 24 hour valproate treatment
In order to assess whether increased DAT mRNA corresponded to changes in DAT protein, we performed and quantified immunofluorescent staining of DAT in N27 cells following 24 hour exposure to 1 mM valproate. Data indicate a 50 % increase (t=10.23, df= 6, p<0.0001) in DAT protein levels (Figure 3).
Figure 3.
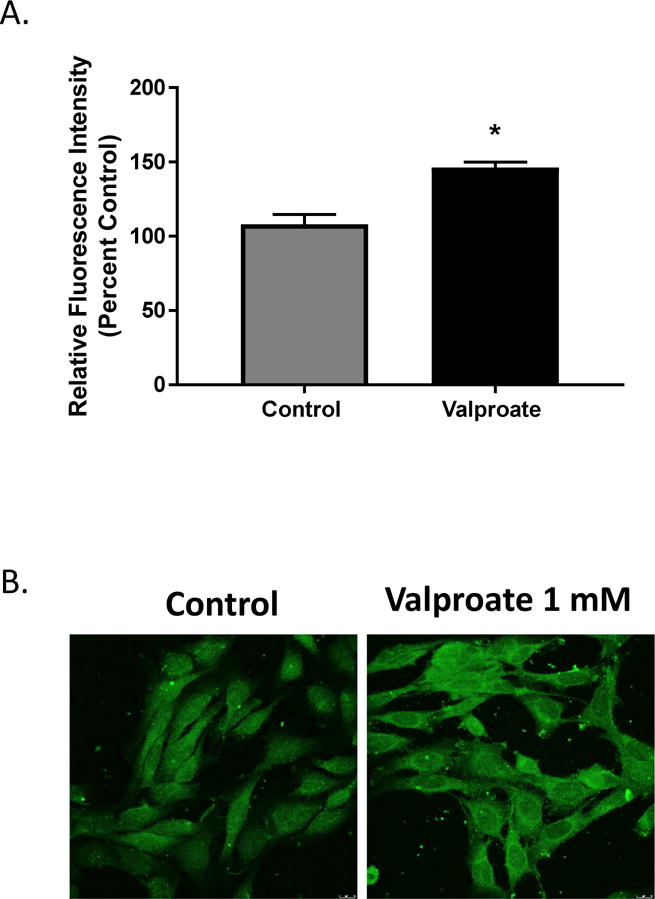
A) Quantification of DAT protein in N27 cells following 24 h exposure to 1mM valproate. Data represented as mean ± SEM, *p≤0.05 percent to control, t-test, N=4. B) Representative images of cells taken at 40×, scale bar represents 20 microns.
3.4. Increased acetylated histone 3 protein in N27 cells following valproate treatment
Dose response and cell viability assays demonstrated 1mM valproate to be the optimal concentration. To determine whether histone proteins were altered following valproate treatment, western immunoblotting was performed on N27 cell nuclear lysates. Results confirmed over 10-fold increase (F=65940, df=2, p value <0.0001) in acetylated histone 3 protein compared to untreated cells (Fig. 4A).
Figure 4.
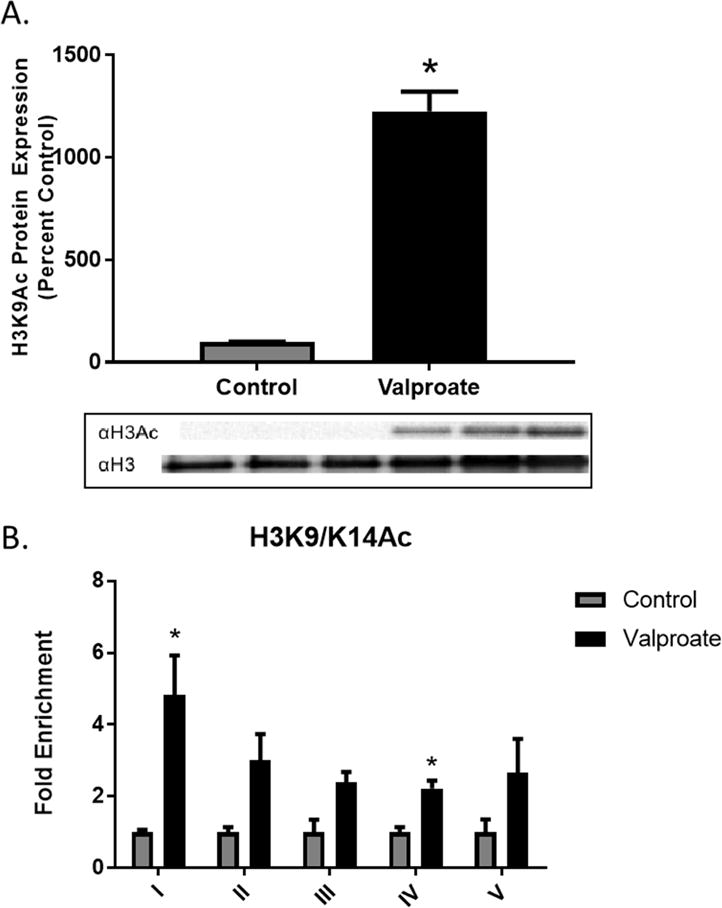
A) Acetylated histone 3 protein in N27 cell lysates following 24 h exposure to 1mM valproate. Data represent mean ± SEM, *p≤0.05 compared to control, t-test, N=3. Chromatin Immunoprecipitation (ChIP) of acetylated Histone 3 at lysines 9 and 14 (H3K9/K14ac) in DAT promoter regions. Data were normalized to input, IgG and represented as enrichment relative to control (N=3).
3.5. Histone protein enrichment in DAT promoter following valproate treatment in N27 cells
The known regulatory region of DAT includes an 18-kb promoter (Zhou et al., 2014) and the core promoter, spanning between −236 and +44 relative to the transcription start site, has been previously demonstrated to drive gene expression of DAT (Sacchetti et al., 1999). The lack of a conserved TATA and CAAT box suggests the presence of other regulatory elements, which may contribute to regulation of gene expression. The responsiveness of DAT mRNA induction following treatment of SK-N-AS neuroblastoma cells to histone deacetylase inhibitors (Green et al., 2015) further suggests a potential role for an epigenetic mechanism. To determine the role of histone acetylation and transcription factors in the role of DAT regulation, we performed chromatin immunoprecipitation (ChIP). Regions I-V represent 200bp fragments of the rat DAT promoter (−810 to +218; Fig. 1B). Co-immunoprecipitation of DAT with H3K9/K14ac showed increased enrichment within regions I (t=3.471, df=4, p=0.0256) and IV (t=5.031, df=3, p=0.0151) of the DAT promoter following 24-hour valproate treatment (Fig. 4B).
3.6. Transcription factor mRNA expression and enrichment in DAT
The transcription factors Nurr1 and Pitx3 are essential for the development and maintenance of midbrain dopaminergic neurons (Lee et al., 2010). Binding of Nurr1 in the DAT promoter has been shown to enhance DAT expression (Sacchetti et al., 2001). Previous studies established Pitx3 as an essential regulator of Nurr1-mediated transcription in midbrain dopaminergic cells (Yi et al., 2014). The mRNA expression of Nurr1 (t=4.912, df=2, p=0.039) and Pitx3 (t=6.587, df=2, p=0.0223) increase 4 and 6-fold, respectively, following 1mM valproate treatment of N27 cells (Figs. 5A and 6A). Immunoprecipitation of Nurr1 showed significant enrichment in region 1(t=4.344, df=3, p=0.0225) of the DAT promoter (Fig. 5B). There were no significant changes in Pitx3 enrichment (Fig. 6B).
Figure 5.
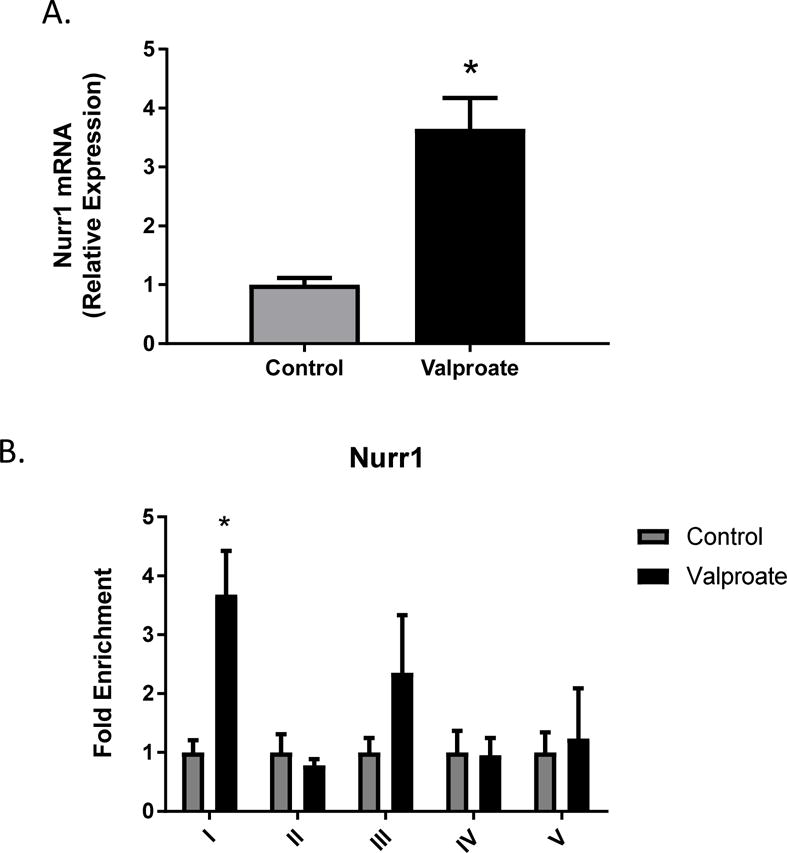
Orphan nuclear receptor Nurr1 A) mRNA expression and B) fold enrichment in the DAT promoter following 1mM valproate treatment in N27 cells. Data represented as mean ± SEM; N=3–4, *p≤0.05 (t-test). For mRNA expression, data were first normalized to β-actin, then normalized to the control treatment group. For ChIP assay, data were normalized to input, IgG and represented as enrichment relative to control.
Figure 6.
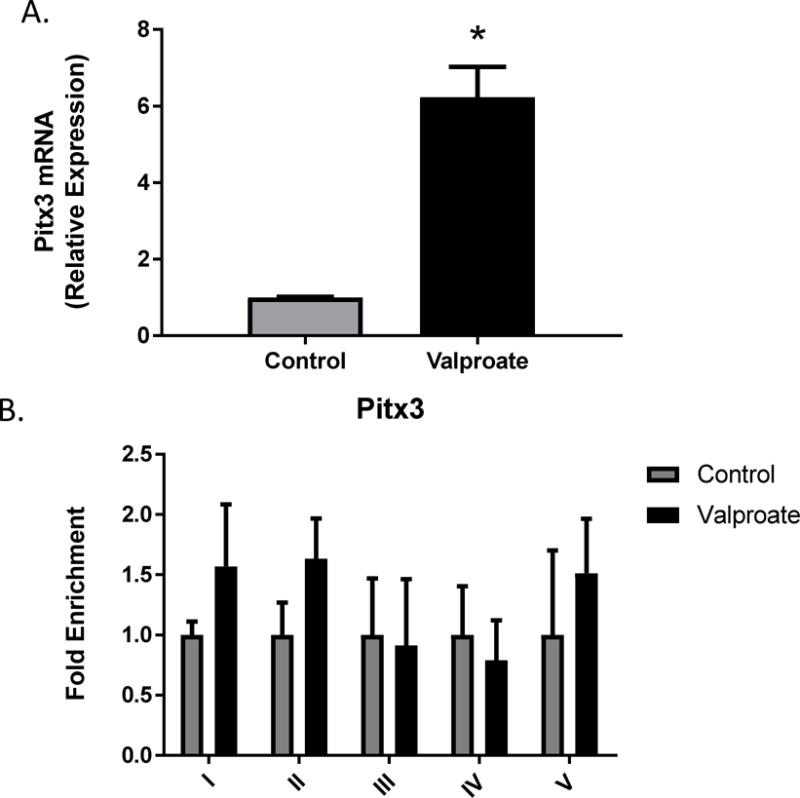
Pitx3 A) mRNA expression and B) fold enrichment in the DAT promoter binding following 1mM valproate treatment in N27 cells. Data represented as mean ± SEM; N=3–4, *p≤0.05 (t-test). For mRNA expression, data were first normalized to β-actin, then normalized to the control treatment group. For ChIP assay, data were normalized to input, IgG and represented as enrichment relative to control.
4. Discussion
Here, we report that HDAC inhibitors are effective dose-dependent inducers of DAT gene expression in vitro. These findings confirmed previous studies showing valproate treatment increased DAT mRNA (Bence et al., 2012; Wang et al., 2007), and extended them by defining a molecular mechanism responsible for this effect. Previous studies found global changes in gene expression following HDAC inhibitor treatment and gene regulation through acetylation of other non-histone proteins (Bantscheff et al., 2011; Fass et al., 2013; Marinova et al., 2009; Perisic et al., 2010). The direct epigenetic modifications that occur on the DAT gene following HDAC inhibitor treatment have not yet been identified. Using HDAC inhibitors as model compounds to manipulate DAT gene expression, we identified a potential role of histone acetylation and enhanced Nurr1 transcription factor binding in the transcriptional regulation of the DAT following valproate treatment.
Valproate (valproic acid) was first identified as an anti-epileptic drug in the 1960’s and is used today with broader therapeutic applications (Nalivaeva et al., 2009). A known mechanism of action of valproate is inhibition of Class I and IIa HDACs (Dokmanovic et al., 2007). Previous studies with valproate in human neuroblastoma cells showed over 4-fold increase in DAT mRNA with concentrations at 5mM (Wang et al., 2007), whereas we observed that doses over 2mM produced cell death. Similar results were obtained following treatment with the Class I/IIa inhibitor sodium butyrate and the more potent Class I/II inhibitor Trichostatin A. Because reduced cell viability was observed with increasing concentrations of butyrate and Trichostatin A, we focused on valproate for the remainder of the mechanistic studies. Valproate and sodium butyrate target the same HDAC classes within a similar IC50 range (Gottlicher et al., 2001; Marks et al., 2001; Rodriquez et al., 2006). Therefore, the cytotoxicity observed with sodium butyrate suggests that alternate targets other than HDACs may mediate this effect.
In addition to inhibiting HDACs, valproate has a number of targets that could mediate its effects on DAT expression (Novy et al., 2011). To verify histone acetylation was being targeted, we evaluated the most well studied and modified histone, H3 (Xie et al., 2012). Western immunoblotting showed a significant increase in H3 acetylation following valproate treatment. In addition to the common H3 acetylated histone marker, H3K9/K14ac is generally associated with open chromatin structure and increased gene expression (Roy et al., 2014). The hyperacetylated regions I and IV within the DAT promoter may be more promising in identifying additional transcription factors and other markers involved in the regulation of DAT. Although valproate has been observed to induce DNA demethylation in some promoters (Manev and Uz, 2002; Tremolizzo et al., 2002), others have not observed this effect (Balasubramanian et al., 2015). Further, the DAT promoter is highly unmethylated (over 95%) in rat primary neurons (He et al., 2011) and we found no significant effect of DNA methyltransferase inhibitors (RG108 and 5-aza-2-deoxycytidine) on DAT expression in SK-N-AS cells (Green et al., 2015) or in N27 cells (data not shown). Therefore, it is unlikely that DNA demethylation by valproate contributed to the effects observed in our experiments reported here.
The orphan nuclear receptor Nurr1 and the paired-like homeobox gene Pitx3 are among the transcription factors involved in the development and maintenance of dopaminergic neurons (Riddle and Pollock, 2003; Smits and Smidt, 2006). Previous studies reported that Nurr1 and Pitx3 target genes involved in dopaminergic metabolism and regulation (Baffi et al., 1999; Hwang et al., 2009; Zetterstrom et al., 1997). Studies evaluating loss of Pitx3 demonstrate significant reduction in DAT mRNA expression and protein levels (Hwang et al., 2003; Smidt et al., 2004). Nurr1 knockout experiments show no expression of DAT and other dopaminergic genes suggesting a more dominate role of Nurr1 in DAT regulation (Jankovic et al., 2005; Smits et al., 2003). Valproate treatment significantly increased DAT mRNA expression in N27 rat mesencephalic cells, which was accompanied by significant increases in Nurr1 and Pitx3 mRNA expression. This is consistent with recent studies in which Nurr1 expression was increased following valproate treatment in a variety of cell types (Almutawaa et al., 2014; Yoshikawa et al., 2013). Although long assumed to regulate the DAT through different mechanistic pathways (Chung et al., 2005), growing evidence suggests that several transcription factors generally work together to maintain and regulate DAT expression in dopamine neurons (Martinat et al., 2006; Wallen et al., 1999).
He and co-workers used ChIP-qPCR to demonstrate enrichment of Nurr1 within the promoter of the DAT and increased DAT gene expression in neural progenitor cells from the ventral mesencephalon following depolarization with KCl (He et al., 2011). In that study, sites I and V of the rat DAT promoter were demonstrated to be enriched for Nurr1 following depolarization. Here, we determined that region I of the DAT promoter showed significant enrichment of Nurr1, whereas no change in binding was observed for region V. This difference may relate to the fact that He and co-workers did not observe increased H3 acetylation in any of the promoter regions following depolarization, but rather increased H3K4 methylation indicative of a different mechanism of relaxing chromatin and allowing enhanced binding of transcription factors.
In contrast to Nurr1 and in spite of significantly increased Pitx3 mRNA following valproate treatment, no significant enrichment for Pitx3 was observed in any of the DAT promoter regions. This is relatively consistent with a previous study, which has demonstrated that Nurr1 target genes were upregulated following HDAC inhibitor treatment in the absence of Pitx3 (Jacobs et al., 2009). It should be noted that although Nurr1 has been thought to directly bind to the DAT promoter through the NGFI-B responsive element (NBRE)(Murphy et al., 1996). Some studies suggest that, although Nurr1 may be immunoprecipitated at the DAT, it does not bind at the conserved NBRE site (Sacchetti et al., 2001) and that a Nurr1-interacting protein may be responsible for the effects on DAT expression (Luo et al., 2008). More recently, Foxa2 has been demonstrated to serve an important role in dopamine neuron development and increase DAT expression by driving an activator complex with Nurr1 that results in relaxed chromatin structure and increased H3 acetylation (Yi et al., 2014), similar to that observed here following valproate treatment. Further investigation will be required to directly assess the role of direct or indirect Nurr1 binding to the promoter region of the DAT following treatment with valproate.
Altered DAT levels adversely affect dopaminergic transmission and have been associated with several neurological disorders. Valproate has been shown to affect dopamine-related behaviors in several in vivo model systems (Tremolizzo et al., 2005; van Enkhuizen et al., 2013). Indeed, a previous study using an in vivo knock down model of the DAT showed that neurobehavioral alterations were attenuated following valproate treatment (Ralph-Williams et al., 2003), which might be the result of increasing DAT expression. More recently, valproate has been shown to be neuroprotective in animal models of Parkinson’s disease (Carriere et al., 2014; Kidd and Schneider, 2011). Although several potential mechanisms for this neuroprotection are possible, the ability of valproate to increase Nurr1 expression and binding to promoter regions of catecholamine genes, including the DAT, tyrosine hydroxylase and the vesicular monoamine transporter 2 (Smits et al., 2003) may result in maintenance of these phenotypic markers of dopamine neurons during neurodegeneration. Moreover, Nurr1 has recently been demonstrated to play an important role in dampening neuroinflammation through its action in microglia (Fan et al., 2009; Saijo et al., 2009), which would be expected to be protective in Parkinson’s disease. Additional studies will be required to determine the relative roles of these potential mechanisms in the neuroprotective effects of valproate.
5. Conclusions
The work presented in this manuscript is novel because we provide a direct mechanism by which valproate increases DAT expression that links the disparate individual observations identified above. Our results indicate that valproate treatment results in enhanced histone acetylation at the DAT promoter in cultured rat N27 cells. In addition to these findings, increased expression of the transcription factor Nurr1 and enhanced binding of Nurr1, or proteins associated with Nurr1, to the DAT promoter appears to be primarily responsible for the pharmacological induction of DAT by valproate. Taken together, these data provide a potential mechanism by which valproate exerts its effect on dopamine-related behaviors and for some of its neuroprotective effects in models of Parkinson’s disease.
Highlights.
Valproate increases DAT mRNA and Protein
Valproate increases Nurr1 and Pitx3 mRNA
DAT increases result from increased H3K9 and Nurr1 enrichment in the DAT promoter
Acknowledgments
This work was supported in part by the following NIH Grants: R01ES015991, R01ES021800, R01ES026057, R01GM104037, P30ES005022, U01NS079249, T32007148 and R21ES013828. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or any other funding source.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they have no competing financial interests or other conflicts of interest.
References
- Almutawaa W, Kang NH, Pan Y, Niles LP. Induction of neurotrophic and differentiation factors in neural stem cells by valproic acid. Basic Clin Pharmacol Toxicol. 2014;115:216–221. doi: 10.1111/bcpt.12201. [DOI] [PubMed] [Google Scholar]
- Baffi JS, Palkovits M, Castillo SO, Mezey E, Nikodem VM. Differential expression of tyrosine hydroxylase in catecholaminergic neurons of neonatal wild-type and Nurr1-deficient mice. Neuroscience. 1999;93:631–642. doi: 10.1016/s0306-4522(99)00124-4. [DOI] [PubMed] [Google Scholar]
- Balasubramanian D, Deng AX, Doudney K, Hampton MB, Kennedy MA. Valproic acid exposure leads to upregulation and increased promoter histone acetylation of sepiapterin reductase in a serotonergic cell line. Neuropharmacology. 2015;99:79–88. doi: 10.1016/j.neuropharm.2015.06.018. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Bence M, Koller J, Sasvari-Szekely M, Keszler G. Transcriptional modulation of monoaminergic neurotransmission genes by the histone deacetylase inhibitor trichostatin A in neuroblastoma cells. J Neural Transm. 2012;119:17–24. doi: 10.1007/s00702-011-0688-4. [DOI] [PubMed] [Google Scholar]
- Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- Carriere CH, Kang NH, Niles LP. Neuroprotection by valproic acid in an intrastriatal rotenone model of Parkinson’s disease. Neuroscience. 2014;267:114–121. doi: 10.1016/j.neuroscience.2014.02.028. [DOI] [PubMed] [Google Scholar]
- Casey DE, Daniel DG, Wassef AA, Tracy KA, Wozniak P, Sommerville KW. Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology. 2003;28:182–192. doi: 10.1038/sj.npp.1300023. [DOI] [PubMed] [Google Scholar]
- Choi JK, Kim YJ. Epigenetic regulation and the variability of gene expression. Nat Genet. 2008;40:141–147. doi: 10.1038/ng.2007.58. [DOI] [PubMed] [Google Scholar]
- Chung S, Hedlund E, Hwang M, Kim DW, Shin BS, Hwang DY, Kang UJ, Isacson O, Kim KS. The homeodomain transcription factor Pitx3 facilitates differentiation of mouse embryonic stem cells into AHD2-expressing dopaminergic neurons. Mol Cell Neurosci. 2005;28:241–252. doi: 10.1016/j.mcn.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Clarkson ED, Rosa FG, Edwards-Prasad J, Weiland DA, Witta SE, Freed CR, Prasad KN. Improvement of neurological deficits in 6-hydroxydopamine-lesioned rats after transplantation with allogeneic simian virus 40 large tumor antigen gene-induced immortalized dopamine cells. Proc Natl Acad Sci U S A. 1998;95:1265–1270. doi: 10.1073/pnas.95.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain. 1999;122(Pt 8):1437–1448. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- Fan X, Luo G, Ming M, Pu P, Li L, Yang D, Le W. Nurr1 expression and its modulation in microglia. Neuroimmunomodulation. 2009;16:162–170. doi: 10.1159/000204229. [DOI] [PubMed] [Google Scholar]
- Fass DM, Reis SA, Ghosh B, Hennig KM, Joseph NF, Zhao WN, Nieland TJ, Guan JS, Kuhnle CE, Tang W, Barker DD, Mazitschek R, Schreiber SL, Tsai LH, Haggarty SJ. Crebinostat: a novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology. 2013;64:81–96. doi: 10.1016/j.neuropharm.2012.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–617. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14:43–49. doi: 10.1016/0165-6147(93)90029-j. [DOI] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AL, Hossain MM, Tee SC, Zarbl H, Guo GL, Richardson JR. Epigenetic Regulation of Dopamine Transporter mRNA Expression in Human Neuroblastoma Cells. Neurochem Res. 2015;40:1372–1378. doi: 10.1007/s11064-015-1601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XB, Yi SH, Rhee YH, Kim H, Han YM, Lee SH, Lee H, Park CH, Lee YS, Richardson E, Kim BW, Lee SH. Prolonged membrane depolarization enhances midbrain dopamine neuron differentiation via epigenetic histone modifications. Stem Cells. 2011;29:1861–1873. doi: 10.1002/stem.739. [DOI] [PubMed] [Google Scholar]
- Hossain MM, Richardson JR. Mechanism of pyrethroid pesticide-induced apoptosis: role of calpain and the ER stress pathway. Toxicol Sci. 2011;122:512–525. doi: 10.1093/toxsci/kfr111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bull. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang DY, Ardayfio P, Kang UJ, Semina EV, Kim KS. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Brain Res Mol Brain Res. 2003;114:123–131. doi: 10.1016/s0169-328x(03)00162-1. [DOI] [PubMed] [Google Scholar]
- Hwang DY, Hong S, Jeong JW, Choi S, Kim H, Kim J, Kim KS. Vesicular monoamine transporter 2 and dopamine transporter are molecular targets of Pitx3 in the ventral midbrain dopamine neurons. J Neurochem. 2009;111:1202–1212. doi: 10.1111/j.1471-4159.2009.06404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs FM, van Erp S, van der Linden AJ, von Oerthel L, Burbach JP, Smidt MP. Pitx3 potentiates Nurr1 in dopamine neuron terminal differentiation through release of SMRT-mediated repression. Development. 2009;136:531–540. doi: 10.1242/dev.029769. [DOI] [PubMed] [Google Scholar]
- Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog Neurobiol. 2005;77:128–138. doi: 10.1016/j.pneurobio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Kidd SK, Schneider JS. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience. 2011;194:189–194. doi: 10.1016/j.neuroscience.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzmenko AP, Pereira AM, Singh BS. Intronic sequences are involved in neural targeting of human dopamine transporter gene expression. Biochem Biophys Res Commun. 1997;240:807–811. doi: 10.1006/bbrc.1997.7754. [DOI] [PubMed] [Google Scholar]
- Lee HS, Bae EJ, Yi SH, Shim JW, Jo AY, Kang JS, Yoon EH, Rhee YH, Park CH, Koh HC, Kim HJ, Choi HS, Han JW, Lee YS, Kim J, Li JY, Brundin P, Lee SH. Foxa2 and Nurr1 synergistically yield A9 nigral dopamine neurons exhibiting improved differentiation, function, and cell survival. Stem Cells. 2010;28:501–512. doi: 10.1002/stem.294. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Manev H, Uz T. DNA hypomethylating agents 5-aza-2′-deoxycytidine and valproate increase neuronal 5-lipoxygenase mRNA. Eur J Pharmacol. 2002;445:149–150. doi: 10.1016/s0014-2999(02)01711-9. [DOI] [PubMed] [Google Scholar]
- Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, Leeds P, Chuang DM. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem. 2009;111:976–987. doi: 10.1111/j.1471-4159.2009.06385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Martinat C, Bacci JJ, Leete T, Kim J, Vanti WB, Newman AH, Cha JH, Gether U, Wang H, Abeliovich A. Cooperative transcription activation by Nurr1 and Pitx3 induces embryonic stem cell maturation to the midbrain dopamine neuron phenotype. Proc Natl Acad Sci U S A. 2006;103:2874–2879. doi: 10.1073/pnas.0511153103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr Mol Pharmacol. 2009;2:95–109. doi: 10.2174/1874467210902010095. [DOI] [PubMed] [Google Scholar]
- Morris MJ, Monteggia LM. Unique functional roles for class I and class II histone deacetylases in central nervous system development and function. Int J Dev Neurosci. 2013;31:370–381. doi: 10.1016/j.ijdevneu.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy EP, Dobson AD, Keller C, Conneely OM. Differential regulation of transcription by the NURR1/NUR77 subfamily of nuclear transcription factors. Gene Expr. 1996;5:169–179. [PMC free article] [PubMed] [Google Scholar]
- Nalivaeva NN, Belyaev ND, Turner AJ. Sodium valproate: an old drug with new roles. Trends Pharmacol Sci. 2009;30:509–514. doi: 10.1016/j.tips.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Novy J, Patsalos PN, Sander JW, Sisodiya SM. Lacosamide neurotoxicity associated with concomitant use of sodium channel-blocking antiepileptic drugs: a pharmacodynamic interaction? Epilepsy Behav. 2011;20:20–23. doi: 10.1016/j.yebeh.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Palmiter RD. Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci. 2008;1129:35–46. doi: 10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perisic T, Zimmermann N, Kirmeier T, Asmus M, Tuorto F, Uhr M, Holsboer F, Rein T, Zschocke J. Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology. 2010;35:792–805. doi: 10.1038/npp.2009.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson GM, Naunton M. Valproate: a simple chemical with so much to offer. J Clin Pharm Ther. 2005;30:417–421. doi: 10.1111/j.1365-2710.2005.00671.x. [DOI] [PubMed] [Google Scholar]
- Prasad KN, Carvalho E, Kentroti S, Edwards-Prasad J, Freed C, Vernadakis A. Establishment and characterization of immortalized clonal cell lines from fetal rat mesencephalic tissue. In Vitro Cell Dev Biol Anim. 1994;30A:596–603. doi: 10.1007/BF02631258. [DOI] [PubMed] [Google Scholar]
- Ralph-Williams RJ, Paulus MP, Zhuang X, Hen R, Geyer MA. Valproate attenuates hyperactive and perseverative behaviors in mutant mice with a dysregulated dopamine system. Biol Psychiatry. 2003;53:352–359. doi: 10.1016/s0006-3223(02)01489-0. [DOI] [PubMed] [Google Scholar]
- Riddle R, Pollock JD. Making connections: the development of mesencephalic dopaminergic neurons. Brain Res Dev Brain Res. 2003;147:3–21. doi: 10.1016/j.devbrainres.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Rodriquez M, Aquino M, Bruno I, De Martino G, Taddei M, Gomez-Paloma L. Chemistry and biology of chromatin remodeling agents: state of art and future perspectives of HDAC inhibitors. Curr Med Chem. 2006;13:1119–1139. doi: 10.2174/092986706776360905. [DOI] [PubMed] [Google Scholar]
- Roy D, Paul A, Roy A, Ghosh R, Ganguly P, Chaudhuri S. Differential acetylation of histone H3 at the regulatory region of OsDREB1b promoter facilitates chromatin remodelling and transcription activation during cold stress. PLoS One. 2014;9:e100343. doi: 10.1371/journal.pone.0100343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchetti P, Brownschidle LA, Granneman JG, Bannon MJ. Characterization of the 5′-flanking region of the human dopamine transporter gene. Brain Res Mol Brain Res. 1999;74:167–174. doi: 10.1016/s0169-328x(99)00275-2. [DOI] [PubMed] [Google Scholar]
- Sacchetti P, Mitchell TR, Granneman JG, Bannon MJ. Nurr1 enhances transcription of the human dopamine transporter gene through a novel mechanism. J Neurochem. 2001;76:1565–1572. doi: 10.1046/j.1471-4159.2001.00181.x. [DOI] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Smidt MP, Smits SM, Bouwmeester H, Hamers FP, van der Linden AJ, Hellemons AJ, Graw J, Burbach JP. Early developmental failure of substantia nigra dopamine neurons in mice lacking the homeodomain gene Pitx3. Development. 2004;131:1145–1155. doi: 10.1242/dev.01022. [DOI] [PubMed] [Google Scholar]
- Smits SM, Ponnio T, Conneely OM, Burbach JP, Smidt MP. Involvement of Nurr1 in specifying the neurotransmitter identity of ventral midbrain dopaminergic neurons. Eur J Neurosci. 2003;18:1731–1738. doi: 10.1046/j.1460-9568.2003.02885.x. [DOI] [PubMed] [Google Scholar]
- Smits SM, Smidt MP. The role of Pitx3 in survival of midbrain dopaminergic neurons. J Neural Transm Suppl. 2006:57–60. doi: 10.1007/978-3-211-45295-0_10. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, Carboni G, Ruzicka WB, Mitchell CP, Sugaya I, Tueting P, Sharma R, Grayson DR, Costa E, Guidotti A. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci U S A. 2002;99:17095–17100. doi: 10.1073/pnas.262658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremolizzo L, Doueiri MS, Dong E, Grayson DR, Davis J, Pinna G, Tueting P, Rodriguez-Menendez V, Costa E, Guidotti A. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- van Enkhuizen J, Geyer MA, Kooistra K, Young JW. Chronic valproate attenuates some, but not all, facets of mania-like behaviour in mice. Int J Neuropsychopharmacol. 2013;16:1021–1031. doi: 10.1017/S1461145712001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Newcorn J, Telang F, Solanto MV, Fowler JS, Logan J, Ma Y, Schulz K, Pradhan K, Wong C, Swanson JM. Depressed dopamine activity in caudate and preliminary evidence of limbic involvement in adults with attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2007;64:932–940. doi: 10.1001/archpsyc.64.8.932. [DOI] [PubMed] [Google Scholar]
- Wallen A, Zetterstrom RH, Solomin L, Arvidsson M, Olson L, Perlmann T. Fate of mesencephalic AHD2-expressing dopamine progenitor cells in NURR1 mutant mice. Exp Cell Res. 1999;253:737–746. doi: 10.1006/excr.1999.4691. [DOI] [PubMed] [Google Scholar]
- Wang J, Michelhaugh SK, Bannon MJ. Valproate robustly increases Sp transcription factor-mediated expression of the dopamine transporter gene within dopamine cells. Eur J Neurosci. 2007;25:1982–1986. doi: 10.1111/j.1460-9568.2007.05460.x. [DOI] [PubMed] [Google Scholar]
- Xie W, Ames RS, Li H. A cell-based high-throughput screening assay to measure cellular histone h3 lys27 trimethylation with a modified dissociation-enhanced lanthanide fluorescent immunoassay. J Biomol Screen. 2012;17:99–107. doi: 10.1177/1087057111422378. [DOI] [PubMed] [Google Scholar]
- Yi SH, He XB, Rhee YH, Park CH, Takizawa T, Nakashima K, Lee SH. Foxa2 acts as a co-activator potentiating expression of the Nurr1-induced DA phenotype via epigenetic regulation. Development. 2014;141:761–772. doi: 10.1242/dev.095802. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Samata B, Ogura A, Miyamoto S, Takahashi J. Systemic administration of valproic acid and zonisamide promotes differentiation of induced pluripotent stem cell-derived dopaminergic neurons. Front Cell Neurosci. 2013;7:11. doi: 10.3389/fncel.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Michelhaugh SK, Schmidt CJ, Liu JS, Bannon MJ, Lin Z. Ventral midbrain correlation between genetic variation and expression of the dopamine transporter gene in cocaine-abusing versus non-abusing subjects. Addict Biol. 2014;19:122–131. doi: 10.1111/j.1369-1600.2011.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]