

SUMMARY
Streptococcus mutans, a dental caries pathogen, can promote systemic infections upon reaching the bloodstream. The two-component system (TCS) VicRKSm of S. mutans regulates the synthesis of and interaction with sucrose-derived exopolysaccharides (EPS), processes associated with oral and systemic virulence. In this study, we investigated the mechanisms by which VicRKSm affects S. mutans susceptibility to blood-mediated immunity. Compared to parent strain UA159, the vicKSm isogenic mutant (UAvic) showed reduced susceptibility to deposition of C3b of complement, low binding to serum IgG, and low frequency of C3b/IgG-mediated opsonophagocytosis by PMN in a sucrose-independent way (p<0.05). RT-qPCR analysis comparing gene expression in UA159 and UAvic revealed that genes encoding putative peptidases of the complement (pepO and smu.399) were up-regulated in UAvic in the presence of serum, although genes encoding murein hydrolases (SmaA and Smu.2146c) or metabolic/surface proteins involved in bacterial interactions with host components (enolase, GAPDH) were mostly affected in a serum-independent way. Among vicKSm-downstream genes (smaA, smu.2146c, lysM, atlA, pepO, smu.399), only pepO and smu.399 were associated with UAvic phenotypes; deletion of both genes in UA159 significantly enhanced levels of C3b deposition and opsonophagocytosis (p<0.05). Moreover, consistent with the fibronectin-binding function of PepO orthologues, UAvic showed increased binding to fibronectin. Reduced susceptibility to opsonophagocytosis was insufficient to enhance ex vivo persistence of UAvic in blood, which was associated with growth defects of this mutant under limited nutrient conditions. Our findings revealed that S. mutans employs mechanisms of complement evasion through peptidases which are controlled by VicRKSm.
Keywords: Streptococcus mutans, two-component system, bacteremia, complement system
INTRODUCTION
Streptococcus mutans plays important functions in the assembly of cariogenic biofilms, which include secretion of glucosyltransferases required for the synthesis of insoluble exopolisaccharides (EPS) from sucrose (EPS)1. Sucrose-derived EPS bound to glucan-binding proteins expressed on S. mutans surface further reduces bacterial susceptibility to blood immunity, thus accounting for the capacity of this microorganism to promote bacteremia2,3. During the processes of host colonization or infection, S. mutans uses two-component systems (TCS) to sense and respond to the environmental challenges. These transductional systems are typically composed by a sensor histidine kinase membrane protein and an intracellular response regulator. Thirteen to 14 TCS as well as the orphan response regulator called CovR (also known as GcrR) were identified in the available genomes of S. mutans4–6. The TCS VicRKSm is of special interest because it regulates functions required for S. mutans cariogenicity and cell wall integrity. VicRKSm-induced genes include those encoding the glucosyltransferases B (GtfB) and C (GtfC) (gtfB and gtfC, respectively) for the synthesis of glucan EPS from sucrose, the glucan-binding protein B (GbpB) (gbpB) and the murein hydrolases LysM and SMU.2146c (lysM and smu2146c respectively), which are involved in S. mutans interactions with EPS7–10. On the other hand, VicRKSm also strongly represses smaA, which encodes the murein hydrolase SmaA10.
The general role of VicRKSm as a modulator of cell division, cell wall biogenesis, and interaction with EPS might explain the essentiality of this TCS for S. mutans viability8,9,11, increasing the interest on VicRKSm as a therapeutic target to control S. mutans infections12,13. However, we have previously observed that deletion of the gene encoding the VicKSm sensor protein (vicKSm) in S. mutans strains impaired bacterial phagocytosis by PMN in samples of human blood14. These findings indicate that VicRKSm downstream genes could be involved in S. mutans evasion to blood-mediated opsonophagocytosis and thus, in systemic virulence. Therefore, it is important to identify gene functions accounting for resistance of vicKSm mutants to opsophagocytosis by PMN.
The aim of this study was to investigate the molecular mechanisms by which deletion of vicKSm affects S. mutans susceptibility to blood-mediated immunity. Here, we assessed the effects of vicK deletion on S. mutans binding to major blood opsonins (C3b of the complement system and IgG antibodies), and on its susceptibility to opsonophagocytosis by PMN isolated from peripheral blood. Also, interactions with plasma fibronectin, and ex vivo persistence in human blood were investigated. We then determined the contribution of VicRKSm downstream genes (smaA, smu.2146c, smu.399 and pepO), known to be involved in functions previously associated with complement evasion in other streptococci, to the vicK mutant phenotypes.
METHODS
Studied strains, culture conditions, oligonucleotides, and construction of mutants
The studied strains are depicted in Table 1. Strains were grown from frozen stocks in Brain Heart Infusion agar (Difco) (37°C; 10% CO2, 24 h). Colonies were then inoculated in BHI, and incubated for 18 h. Inocula of BHI cultures with adjusted absorbance (A550nm) were then transferred to fresh BHI, chemically defined medium (CDM)15 supplemented or not with sucrose (0.01 or 0.1%), or RPMI 1640 (Gibco, Life Technologies, NY, USA). The non-polar isogenic mutants of pepO (UApepO) and smu.399 (UA399) were obtained in strain UA159 by double cross-over recombination with null alleles, which were constructed by PCR-ligation strategy as previously described9,14. Briefly, pepO and smu.399 mutants were obtained by replacing the internal sequences of the encoding regions of pepO (1,432 bp) or smu.399 (405 bp) with an erythromycin resistance gene (amplified from plasmid pVA838). To obtain the complemented strains (+), UApepO and UA399 mutants were transformed with plasmid pDL278 (which harbors a spectinomycin resistance gene) containing the intact copy of the respective deleted gene. Erythromycin (10 μg/ml) and spectinomycin (200 μg/ml) (Merck Labs, Germany) were added to growth medium for maintenance of mutant and complemented strains, respectively. Oligonucleotides used for construction of mutants and transcriptional analyses are shown in Table 2.
Table 1.
Strains used in this study
| Strains | Relevant characteristics | Source or reference |
|---|---|---|
| UA159 | Erms, Specs | ATCC |
| UAvic | ΔvicK::Ermr | 9 |
| UAsmaA | ΔsmaA::Ermr | 10 |
| UA2146c | Δsmu.2146c::Ermr | 10 |
| UA399 | Δsmu.399::Ermr | This study |
| UApepO | ΔpepO::Ermr | This study |
| UAvic+ | UAvic with pDL278::SMU.1516; Specr | 9 |
| UAsmaA+ | UAsmaA with pDL278::smu.609; Specr | 10 |
| UA2146c+ | UA2146c with pDL278::smu.2146c; Specr | 10 |
| UA399+ | UA399 with pDL278::smu.399; Specr | This study |
| UApepO+ | UA2146c with pDL278::smu.2146c; Specr | This study |
Table 2.
Oligonucleotides used in this study
| Primer Name | Sequence 5′-3′ | Product size (bp) | Source |
|---|---|---|---|
| 16SRNAF 16SRNAR |
CGGCAAGCTAATCTCTGAAA GCCCCTAAAAGGTTACCTCA |
190 | 17 |
| smu360F smu360R |
CCTAACTCAACTGGTGCTGCT CAGCATTCACTTCATCAACAG |
161 | This study |
| smu630F smu630R |
AGAATGGATGCTCTTGGCTTA GCTGTCATAGGCTGTGTTTCA |
170 | This study |
| smu676F smu676R |
TCGTATGGAAGGTGAAGTC GTAAGAGCCCTGAGATTGAT |
218 | This study |
| smu2036F smu2036R |
TACCCATAGCTTGAGGTGT ACACCAGAACTGCCTTTAG |
253 | This study |
| smu399F smu399R |
GATTGAAGAGTCACCGGATA CCGCTTGTTTAGTCTCTTGA |
242 | This study |
| smu1247F smu1247R |
GACTTCTTCACCTGGTTTG CTCACTCAGATGCTCCAAT |
251 | This study |
| smu609F smu609R |
GGCACAAGGAACCTATCACTTT GCTTTCCAATAACAACATAACGAC |
191 | 10 |
| smu2146F smu2146R |
AATCTGTTCTTGCTCACACTGC ACATTATCAGTTGGTTCAGTTGCT |
145 | 10 |
| smu2147cF smu2147cR |
TTATCAGAGATTGCTTCAACACA CTGAGGTTTCTGCTTCATTTATC |
175 | 10 |
| ermE1-AscI ermE2-XhoI |
TTGGCGCGCCTGGCGGAAACGTAAAAGAAG TTCTCGAGGGCTCCTTGGAAGCTGTCAGT |
998 | 10 |
| smu.399P1 smu.399P2-AscI |
TCTTCTTCACCATTTCTTGC TTGGCGCGCCCGGTGACTCTTCAATCAAAA |
496 | This study |
| smu.399P3-XhoI smu.399P4 |
TTCTCGAGTGTTGAGAGTCATGGAGAGG AAAGCTGCCTGATGGTTACT |
505 | This study |
| smu.2036P1 smu.2036P2-AscI |
TTTACTATCGGCGCTAAGGT TTGGCGCGCCACTGTTTCGGAAAATGTGG |
501 | This study |
| smu.2036P3-XhoI smu.2036P4 |
TTCTCGAGGACGATGGAACTTCACAAAA GATCAAAGGCAATTTACGGG |
490 | This study |
| smu.399C1-PvuI smu.399C2-BamHI |
GGCGATCGTGGATGTTACGTGGACTGT GGGGATCCGGCCATCATAAAGTGCTAAA |
1,883 | This study |
| smu.2036C1-BamHI smu.2036C2-SphI |
GGGGATCCATGCCGCTAATTGCTCAAG GGGCATGCAGTCAATGAAAAAACGCTTGA |
2,750 | This study |
Underlined sequences indicate restriction enzyme linkers.
Volunteers, sera and blood samples
Blood samples were collected by venipuncture in heparin vacuum tubes (BD Vacutainer®) from six health subjects (three males, three females; mean age of 30 years, range 25 to 45 years), who were enrolled in a previous study3, according to standard protocols previously approved by the Ethical Committee of the Piracicaba Dental School, State University of Campinas (proc. n° 031/2012). Serum samples were stored in aliquots at −70°C until use. Serum samples from one volunteer were used as reference. Commercial human serum depleted of C1q and purified human C1q were obtained from Calbiochem (MA, U.S.A.). Heat-inactivated sera (56°C during 20 min) were also used as negative control in preliminary experiments, and showed minimal effects on comparative analyses of C3b deposition between mutant and parent strains.
C3b deposition on S. mutans strains
Deposition of C3b on the surface of serum-treated strains was determined as previously described16 with some modifications3. Briefly, approximately 107 CFU of S. mutans strains harvested (10,000 × g, 4°C) from BHI cultures at mid-log growth phase (A550nm 0.3) were washed twice with PBS (pH 7.4) and suspended in 20% of serum in PBS. After 30 min incubation (37°C), cells were washed twice with PBS-Tween 0.05% (PBST), and incubated on ice (40 min) with fluorescein isothiocyanate (FITC)-conjugated polyclonal goat IgG anti-human C3 antibody (ICN, CA, U.S.A) (1:300 in PBST). Cells were then washed twice with PBST, fixed in 3% paraformaldehyde in PBS and analyzed on a FACSCalibur flow cytometer (BD Biosciences) using forward and side scatter parameters to gate at least 25,000 bacteria. Levels of surface-bound C3b were expressed as the geometric mean fluorescence intensity (MFI) of C3b-positive cells. Control samples included bacteria treated only with PBS instead of serum.
Binding of serum IgG antibodies to S. mutans strains
Levels of serum IgG reactive with S. mutans were determined as previously described3. Briefly, bacterial strains at mid-log phase of growth (A550nm 0.3) were harvested from BHI cultures (500 μl), washed twice with PBS (pH 7.0), and incubated with 20% serum in PBS. Cells were then washed with PBST and incubated (on ice) during 40 min with polyclonal goat IgG anti-human IgG conjugated with FITC (Novus Biological, U.S.A.) (1:900). After two washes with PBST, cells were then harvested by centrifugation and suspended in 3% of paraformaldehyde. Flow cytometry analyses were performed as described before, using forward and side scatter parameters to gate at least 25,000 bacteria. Bacterial samples treated with PBS instead of serum were used as negative controls.
PMN isolation and opsonophagocytic assays
Isolation of human PMN from samples of fresh heparinized blood collected from a reference volunteer were performed as described elsewhere3. Cell viability (>98%) was monitored by trypan blue exclusion and cell purity (>95%) by May-Grunwald Giemsa staining. Bacteria applied in phagocytosis assays were labeled with FITC as previously described3, and aliquots stored overnight in 10% glycerol at −70°C. Bacterial labeling was monitored in a fluorescent microscope (Leica DM LD), and by flow cytometry (FACSCalibur, BD). Aliquots containing 107 CFU of FITC-labeled bacteria were incubated with 20% serum and exposed to 2 × 105 PMN to a multiplicity of infection (MOI) of 200 bacterial per PMN3. After incubation (37°C, 10% CO2, gentle shaking) during 5 or 30 min, reactions were fixed by addition of 100 μl of 3% of paraformaldehyde. PMNs were then analyzed using FACSCalibur (BD Biosciences), and the frequency of phagocytosis expressed as the number of PMN cells with intracellular bacteria, within a total of 10,000 PMN analyzed. To confirm that most of PMN-associated bacteria were internalized, flow cytometry results were compared to light microscopy analysis of samples stained using May-Grunwald-Giemsa, as previously described14. In addition, to confirm that phagocytosis by PMN involved binding to surface C3b or IgG, similar assays were performed with PMN previously incubated (37°C, 30 min) with mouse monoclonal antibodies (MAbs) anti-CD35 (Biolegend, CA, U.S.A.) to block CR1 receptors, or with MAbs anti-CD32 to block IgG2 Fc receptors (eBioscience, CA, U.S.A.)3.
RNA isolation and transcriptional analysis of strains exposed to human serum
Transcriptional analysis of smaA, smu.2146c, lysM, atlA, gapN, gapC, smu.1247 (eno), smu.399 and pepO was performed in strains exposed or not to human serum. Briefly, strains at mid-log phase (A550nm 0.3) in BHI were harvested (6,000 × g, 5 min, 4°C), resuspended in BHI or BHI suplemented with 20% human serum and incubated (37°C ; 5% CO2) during 30 min. Then, cells were harvested, washed with cold saline, and total RNA was purified using RNeasy kit (Qiagen, Germany). Samples were then treated with Turbo DNase (Ambion, USA), as previously described10. The cDNA was obtained from 1 μg of RNA using random primers17 and SuperScript III (Life Technologies, USA), according to the manufacturer’s instruction. Quantitative PCR was performed in StepOne™ Real-Time PCR System (Life Technologies) with cDNA (10 ng), 10 μM of each primer, and 1× Power SYBR® Green PCR Master Mix (Lifetech) in a total volume of 10 μl. The cycling conditions included incubation at 95 °C (10 min), followed by 40 cycles of 95 °C (15 s), optimal temperature for primer annealing (55 – 60 °C, 15 s) (Table 2), and 72 °C (30 s). Ten-fold serial dilutions of genomic DNA (0.003 to 300 ng) were used to generate standard curves for absolute quantification of transcript levels. Melting curves were obtained for each primer set. Results were normalized against S. mutans 16SrRNA gene expression17. Assays were performed in triplicate with three independent RNA samples.
Binding of S. mutans strains to fibronectin
Fibronectin binding assays were performed as previously described18, with modifications19. Briefly, human plasma fibronectin (Sigma-Aldrich) (50 μg/ml) was immobilized in 96-well plates for 18 h at 4°C, washed with PBS and blocked with 5% bovine serum albumin during 1 h at 37°C. Strains from 18 h cultures in BHI were washed twice in PBS (pH 7.2), and resuspended in the same buffer to 1 × 109 CFU/ml. Volumes of 100 μl of these suspensions were transferred to fibronectin-coated wells and incubated (37°C) during 3 h. Afterwards, unbound cells were removed by a series of three washes with PBS. Fibronectin-adherent cells were stained with 0.05% crystal violet, and intensities of staining were measured by spectrophotometry (A575nm) in 7% acetic acid eluates. Wells treated similarly but not added of fibronectin or of bacterial suspensions were used as negative controls.
Analysis of the production of surface enolase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
Protein extracts from whole cells were obtained as previously described10 with minimal modifications. Briefly, strains at mid-log phase of growth in BHI, were harvested (10,000 × g, 4°C, 3 min), washed twice with cold saline, and 125 μl of ultrapure water with 0.16 g of 0.1 mm Zirconium Beads (Biospec) were added for cell disruption on a Mini-beadbeater (Biospec) (maximum power; 2 cycles of 45 s with 1 min rest on ice). The culture supernatants were also collected, 0.1 mM of phenylmethylsulfonyl fluoride (PMSF) was added for inhibition of serine proteases, and dialyzed at 4 °C against 0.02 M sodium phosphate buffer (pH 6.5) followed by 0.125 M Tris. Afterwards, these samples were 100-fold concentrated by lyophilization20. Protein concentration was determined using a Bradford assay kit (BioRad), according to the manufacturer’s protocol.
Alpha enolase and GAPDH were measured in whole cell extracts and culture supernatants using immuno-dot blot and/or western blot assays performed with polyclonal rabbit IgG antibodies to surface enolase or to extracellular GAPDH of Streptococcus pyogenes, which were kindly provided by Dr. Vijay Pancholi (The Ohio State University College of Medicine, Columbus, OH, U.S.A.). Antibody titers were determined in preliminary western blot assays, using the S. pyogenes strain SF130 (ATCC12344) as control. Equivalent amounts of protein (20 μg) were used in dot blot assays, which were performed as described elsewhere21 with some modifications. Briefly, samples were blotted onto nitrocellulose membranes (BioRad), and blocked (2 h) with PBS with 5% skim milk. After a series of 3 washes with PBS, the membranes were probed with rabbit anti-serum to enolase (1:2000) or to extracellular GAPDH (1:40,000) during 90 min. Membranes were then washed and incubated (RT, 60 min) with goat IgG anti-rabbit IgG conjugated with horseradish peroxidase (Thermo Fisher Scientific). Immune reactions were detected using the SuperSignal West Dura system (Thermo Fisher Scientific). Serially diluted samples of S. pyogenes extracts were used as standard. Autographs were scanned using the Alliance 9.7 documentation system (Uvitec Cambridge) and intensities of the reactions measured using the ImageJ software (http://imagej.nih.gov/ij/). Densitometric measures within a linear range of the S. pyogenes standard curves were expressed as arbitrary units. Experiments were performed in triplicate. For the western blot assays, samples were resolved in 10% SDS-PAGEs gels, transferred to PVDF membranes (Millipore), and immune reactions performed as described before. Protein profiles were monitored in duplicate gels stained with Coomassie blue. As a control for S. mutans secreted proteins, samples of culture supernatants were also probed with anti-GbpB antibody9.
Ex vivo survival of S. mutans strains in human blood
Bacterial survival in human blood was analyzed as previously described3. Briefly, cells from BHI cultures (A550nm 0.3) were harvested (11,000 × g, 2 min), washed twice in PBS, and resuspended in fresh whole human blood (1 ml) collected from the reference volunteer. Samples were then incubated (37°C, 5% CO2, gentle agitation), and aliquots collected at different time points (0.5 to 24 h), serially diluted and plated on BHI agar for determination of bacterial counts (CFU/ml). Aliquots collected just after bacterial suspension in blood (time 0) were used as initial blood counts. Changes in bacterial counts were then calculated in relation to initial counts, to reduce the influence of variations in blood-mediated aggregation between strains in the numbers of CFU per milliliter. Three independent experiments were performed in triplicate.
Statistical analyses
Flow cytometry data (MFI values for C3b and IgG, percentages of PMN with FITC-labeled bacteria) and relative measures of α-enolase and GAPDH were analyzed by comparing means of three independent experiments, using nonparametric Kruskal-Wallis analysis with Dunns’s post hoc test. Transcriptional data were analyzed using ANOVA with post hoc Dunnett’s test. For growth curve experiments and for ex vivo survival in blood, Kruskal-Wallis with Dunns’s post hoc test and correction for repeated measures was used.
RESULTS
Deletion of vicK impairs deposition of C3b, binding to serum IgG and opsonophagocytosis by human PMN from peripheral blood
Previously, we observed that deletion of vicK in UA159 (UAvic) and in LT11 impaired phagocytosis by PMN in samples of human blood14. Because complement-mediated opsonization is crucial for efficient phagocytosis of S. mutans by PMN3, we compared the susceptibility of UA159, UAvic and UAvic+ to C3b deposition. As shown in Fig. 1A, deposition of C3b was significantly impaired in UAvic when strains were treated with a reference serum, or with pooled sera, whereas C3b deposition was completely restored in the complemented mutant UAvic+. Minimal C3b deposition on UA159, UAvic, and UAvic+ was observed in C1q-depleted serum, compatible with previous findings that C3b deposition on S. mutans is dependent of the classical pathway of complement activation. Because specific binding of IgG to the bacterial surface activates the C1 complex of the classical pathway, we also compared levels of IgG binding to the tested strains. As shown in Fig. 1B, the UAvic mutant showed reduced binding to serum IgG whereas the levels of IgG binding to UAvic+ did not significantly differ from the parent strain.
Fig. 1.
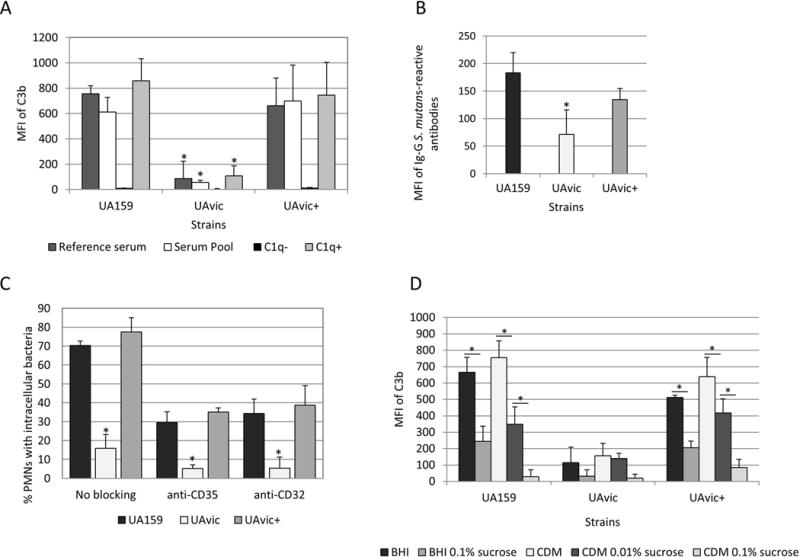
Comparisons of C3b deposition, IgG binding and opsonophagocytosis by PMN between the vicK mutant (UAvic) and parent (UA159) or complemented strain (UAvic+). Intensities of C3b deposition (A) or binding to serum IgG antibodies (B) were determined by flow cytometry (MFI) in strains treated with 20% human serum. A) Levels of surface C3b were measured after bacterial treatment with a reference serum, pools of sera obtained from six volunteers, commercial serum depleted of C1q (C1q−) and C1q− serum supplemented with purified C1q (C1q+). B) Binding to IgG was measured in strains treated with a reference serum. C) Percentages of PMN with internalized bacteria were determined after exposure of PMN isolated from peripheral blood to FITC-labeled bacteria in the presence of 20% serum. PMN treated with MAbs to block CR1 (CD35) or FcɤRIIa (CD32) receptors were used as control. D) Levels of C3b deposition were measured in strains grown in BHI or CMD supplemented or not with 0.01 to 0.1% sucrose. Columns represent means of three independent experiments. Bars indicate standard deviations. Asterisks indicate significant differences in relation to UA159 within the same condition (A-C) or between conditions under horizontal lines (D) (Kruskal-Wallis with post hoc Dunn’s test; p< 0.05).
Consistently with low levels of C3b and IgG binding, the UAvic showed impaired opsonophagocytosis (Fig. 1C). To confirm that the frequencies of phagocytosis measured in our assays involved PMN interactions with surface-bound C3b and/or IgG opsonins, we assessed the influence of blocking PMN receptors for C3b (CR1) and Fc portion of IgG antibodies (FcɤRII) on phagocytosis efficiencies. As shown in Fig. 1C, blocking of either CR1 (CD35) or FcɤRII (CD32) receptors strongly reduced phagocytosis of UA159 (and of UAvic+), and thus reduced differences in the frequencies of phagocytosis between the three tested strains.
Deposition of C3b on UAvic is not influenced by sucrose-derived EPS
The major role of the TCS VicRKSm in cariogenicity of S. mutans seems to be associated with transcriptional activation of genes required for the synthesis of and interaction with EPS derived from sucrose7,10. Because the UAvic mutant has reduced production of sucrose-derived EPS and defects in biofilm formation9,10, it seems likely that reduced susceptibilities of this mutant to C3b deposition and IgG binding were not due to surface-associated EPS or to altered expression of surface components in response to sucrose. To address this issue, we compared levels of C3b deposition between strains grown in sucrose-free CDM and in complex BHI with strains grown in media (CDM and BHI) supplemented with increasing amounts of sucrose. Because sucrose-derived EPS bound to S. mutans blocks C3b deposition3, previous exposure of UA159 and UAvic+ to sucrose reduced levels of C3b deposition on these strains (Fig. 1D). However, UAvic mutant showed low levels of C3b deposition even when grown in sucrose-free CDM (Fig. 1D). Thus, reduced susceptibility of UAvic to C3b deposition does not rely on sucrose-derived EPS or on altered responses to sucrose.
VicRKSm regulates cell surface biogenesis and complement evasion in the presence of serum
The VicRKSm seems to be activated in response to cell wall stress9,22, which might include interactions with host immune components, e.g. pentraxins and complement proteins23,24. Therefore, we investigated the effects of vicKSm inactivation on transcription of downstream cell wall homeostasis genes (smaA, smu.2146c, lysM and atlA)10,25 on cells exposed to BHI supplemented with 20% serum and to unsupplemented BHI at mid- and late-exponential growth. Genes encoding the anchorless surface proteins α-enolase (eno: smu.1247) and the extracellular GAPDH (gapC), were also tested because these proteins are involved in binding to serum or extracellular matrix components26–28. Interestingly, an in silico screening of the genomes of 22 S. mutans strains available in the NCBI database, revealed two genes (smu.399 and pepO) potentially involved in evasion to complement immunity. Smu.399 and pepO were found in all S. mutans strains analyzed, and the promoter regions of both genes include putative VicR consensus motifs30,31, which are shown in Table 3. BLAST analyses of another 20 genes previously implicated in evasion to complement system in other species of streptococci and in Staphylococcus aureus29, did not reveal orthologues in S. mutans strains (data not shown).
Table 3.
Putative binding motifs of VicRSm
| NCBI number | Putative VicR binding motif* | Strand | Position (bp)† |
|---|---|---|---|
| SMU_2036 (pepO) | TGTAAATGATATGaAgC | − | 136 bp |
| TGTGAAGGCATTGgTtAg | − | 109 pb | |
| SMU_399 | TGTTAAAAAAATaTTAA | + | 82 bp |
| TGTTATGGCACTGgTAgC | + | 391 bp |
VicR box: TGTWAHNNNNNTGTWAH; where W is A or T and H is A, T or C; mismatches are indicated by lower cases.
Position from translation start site.
As shown in Figure 2A, at mid-exponential growth, smaA was significantly up-regulated in UAvic compared to UA159 in both conditions, absence (12.9-fold increase; ANOVA with post hoc Dunnett’s test, p<0.01) and presence of serum (3.6-fold increase, p<0.01). At late-log phase, smaA up-regulation achieved significance only in the absence of serum (3.6-fold increase; p<0.05). Consistent with the role of VicRKSm as inducer of smu.2146c10, this gene was significantly down-regulated in the UAvic mutant either in the presence or absence of serum at mid- (13.7 and 10.9-fold decreases respectively in the absence and presence of serum; p<0.01) and late-log (13.0- and 12.2-fold decrease respectively in the absence and presence of serum; p<0.01) growth phases. No significant alterations in transcript levels of the autolysin-encoding genes lysM and altA were detected (data not shown). On the other hand, the UAvic mutant showed increased expression of the metabolic genes only in cells at mid-log phase exposed to BHI (2.6-, 2.4- and 2.2-fold increase for eno, gapC and gapN respectively; p<0.05), suggesting that the presence of serum inhibited transcription of these genes in UAvic (Fig. 2B). Compared to the other functional gene classes (Fig. 2A,B), at mid-log growth phase, genes encoding peptidases of the complement (smu.399 and pepO) were more clearly up-regulated in the presence of serum (2.9- and 2.7-fold increase for smu.399 and pepO, respectively), proportionally to their up-regulation in the absence of serum (3.6- and 3.8-fold increase respectively for smu.399 and pepO) (Fig. 2C). At late-log growth phase, pepO up-regulation was observed only in the presence of serum (3.9-fold up-regulation; p<0.05) (Fig. 2C). Therefore, deletion of vicK in UA159 transcriptionally affects genes involved in cell wall biogenesis (smaA, smu.2146c), metabolic enzymes (eno, gapN, gapC) and putative complement proteases (smu.399 and pepO), but these genes are differently affected by the presence of human serum.
Fig. 2.
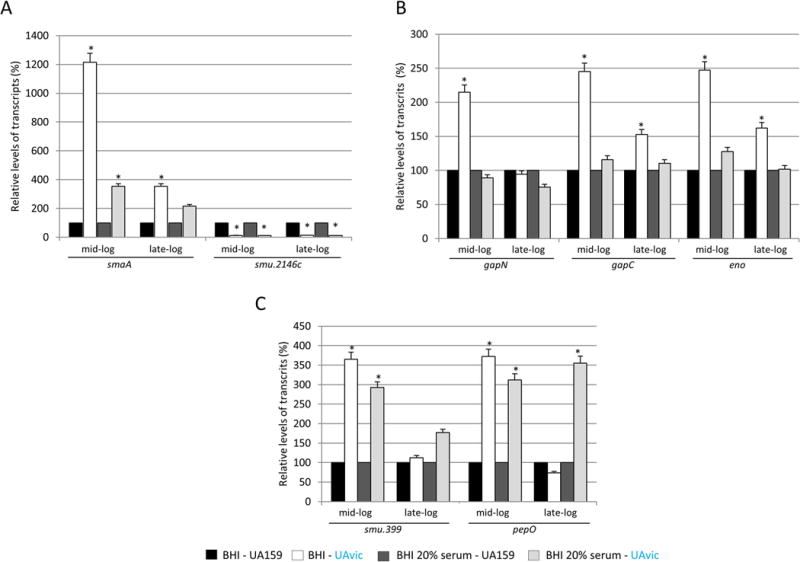
Comparative analysis of transcriptional profiles of vicK mutant (UAvic) with parent strain UA159 in the presence and absence of serum. Strains at mid- and late-log phases of growth were harvested and exposed to BHI or BHI supplemented with 20% human serum before RNA isolation. Relative amounts of transcripts of UA159 at each condition were set to 100% to calculate relative transcript levels obtained with UAvic at the same condition. VicRKSm-downstream genes encoding murein hydrolases (A), metabolic enzymes (B) and peptidases of the complement (C) were analyzed. Columns represent means of three independent experiments; bars represent standard deviations. Asterisks indicate significant differences in relation to parent strain (ANOVA with post hoc Dunnett’s test; * p<0.05).
SmaA, Smu.399 and PepO affect C3b deposition and opsonophagocytosis by human PMN
In S. pneumoniae, murein hydrolases significantly affect bacterial susceptibility to complement immunity and opsonophagocytosis through multiple mechanisms including reduction of cell surface interaction with complement activators (e.g., C-reactive proteins), degradation of C3b linked to the cell wall, and recruitment of fluid phase inhibitors of complement32,33. Thus, reduced susceptibility of UAvic to complement-mediated opsonization could be, at least in part, due to the altered activities of genes smaA and smu2146c. To address this issue, we measured levels of opsonin binding and opsonophagocytosis in the smaA and smu.2146c isogenic mutants. Unexpectedly, although smaA is over-expressed in the UAvic mutant, deletion of smaA significantly reduced S. mutans binding to C3b (52.5% reduction; p<0.05) (Fig. 3A), which was associated with reductions in IgG and opsonophagocytosis (Fig. 3B,C). These phenotypes were restored in the complemented mutant. No significant changes in C3b deposition and phagocytosis were observed in the smu.2146c mutant, although these phenotypes were affected in the complemented mutant UA2146c+ (Fig. 3A,C) Therefore, reduced susceptibility of UAvic to opsonophagocytosis does not involve altered expression of cell wall hydrolases SmaA and SMU.2146c, although SmaA influences S. mutans susceptibilities to C3b deposition and opsonophagocytosis.
Fig. 3.
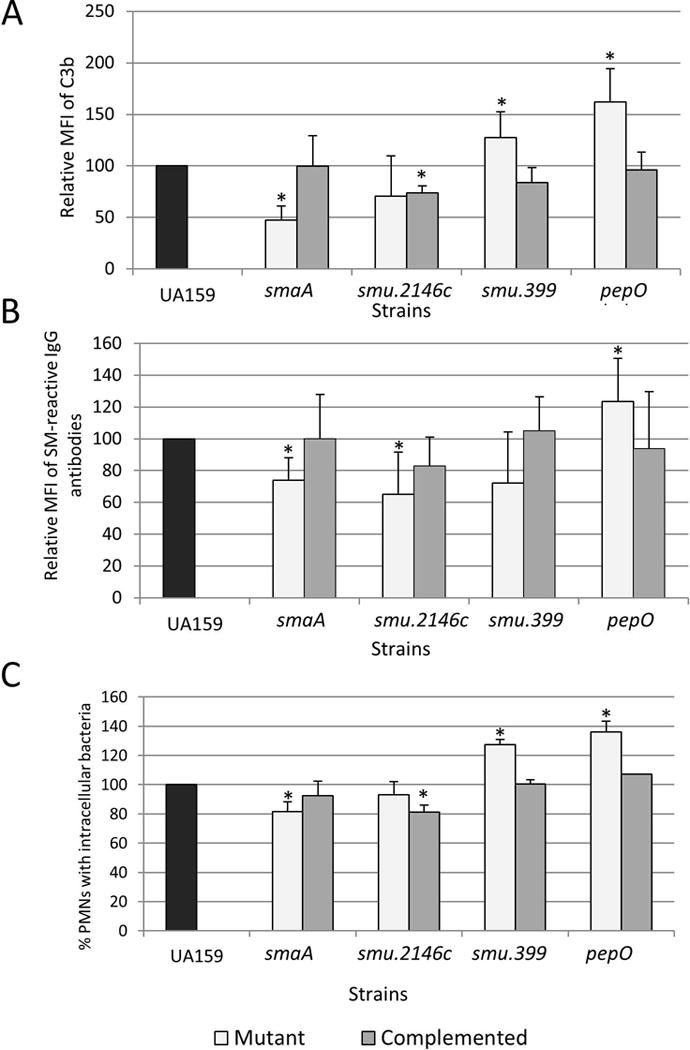
Effects of deletion of VicRSm downstream genes (smaA, smu2146, smu.399 and pepO) on C3b deposition, binding to serum IgG and opsonophagocytosis by PMN. Measures obtained for mutant and complemented strains were expressed in relation to UA159 (set to 100%). Asterisks indicate significant differences in relation to parent strain (Kruskal-Wallis with post hoc Dunn’ test; p<0.05).
On the other hand, deletion of smu.399 and pepO increased deposition of C3b on S. mutans in 27.5 and 62% respectively (p<0.05) (Fig. 3A). Compared to UA159, the smu.399 and pepO mutants had increased susceptibility to opsonophagocytosis (Fig. 3C), although binding to IgG was significantly increased only in the pepO mutant (p<0.05) (Fig. 3B). Importantly, the phenotypes of pepO and smu.399 mutants were restored in the complemented strains (Fig. 3). Thus, pepO and smu.399 account for UAvic resistance to C3b-mediated opsonization.
The vicK mutant has increased binding to human fibronectin
The presence of fibronectin in serum might affect the course of the classical pathway of complement activation because it binds to the collagen-like domain of C1q34. Additionally, PepO of S. pneumoniae functions as a fibronectin-binding protein35 and the gene encoding the PepO orthologue of S. mutans was up-regulated in UAvic (Fig. 2C). Therefore, we compared the ability of UAvic, UA159 and UAvic+ to bind to human plasma fibronectin. As expected, the UAvic showed 5.4-fold increase (p<0.01) in binding to fibronectin (Fig. 4A) whereas the complemented strain displayed wild-type binding levels (Fig. 4A). Thus, our findings show that the TCS VicRKSm modulates the ability of S. mutans to bind fibronectin.
Fig. 4.
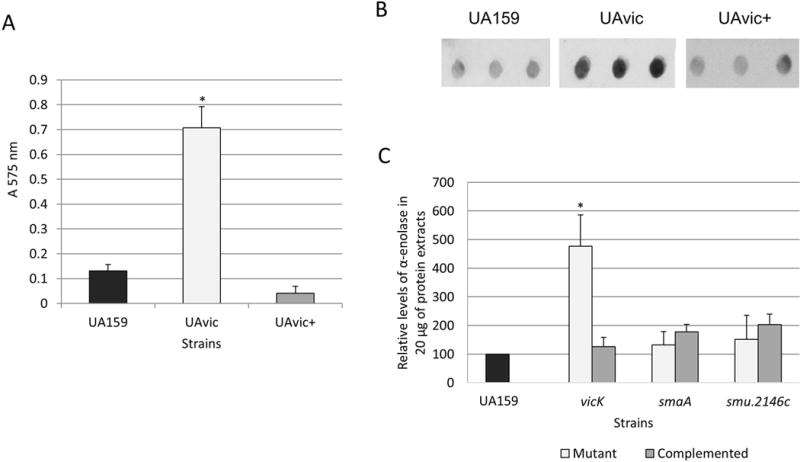
Comparisons of binding to human plasma fibronectin and production of α-enolase between UAvic and parent or complemented strains. A) Fibronectin binding was expressed as the absorbance of crystal violet stain (A575nm) eluted from bacteria bound to immobilized fibronectin. B) Levels of α-enolase produced by strains were measured in whole cell extracts using immuno dot blot assays with polyclonal antibody anti-S. pyogenes α-enolase. C) Densitometric measures of immuno reactions were expressed as the relative levels of α-enolase in relation to parent strain (set as 100%). Columns represent means of three independent experiments; bars represent standard deviations. Asterisks indicate significant differences in relation to parent strain (Kruskal-Wallis with post hoc Dunn’s test; p< 0.05).
The vicK mutant has increased production of α-enolase, but not of extracellular GAPDH
Surface-associated GAPDH and α-enolase promote bacterial binding to fibronectin, plasminogen and/or other host glycoproteins of the extracellular matrix, including collagens and laminin36,37. Although the mechanisms by which these glycolitic enzymes can be associated with the bacterial surface are still unknown28,36, studies in S. pneumoniae revealed that enolase is released in strains with defects in septum formation promoted by altered activities of the VicRKSp-regulated gene pcsB, a gbpB orthologue of S. mutans31,38. Therefore, we compared levels of surface GAPDH and α-enolase in cell lysates and in culture supernatants of strains UAvic, UAsmaA and UA2146c with parent and respective complemented strains. As shown in Fig. 4B and C, when compared to UA159, whole cell extracts of the UAvic mutant at mid-log phase have increased levels of α-enolase (4.8-fold increase; p<0.05), which were restored to parental levels in the UAvic+ complemented strain. Levels of cell-associated α-enolase were not altered in the smaA and smu2146c mutants (Fig. 4C). Alpha-enolase could not be detected in 100-fold concentrated culture supernatants of the strains analyzed, although high levels of GbpB (a secreted and surface-associated protein)21 could be detected in the same samples in parallel immunoassays (data not shown). No significant changes in production of GAPDH were observed between the tested strains (data not shown). Thus, the vicK mutant shows overall increased production of α-enolase, although no significant levels of enolase and GAPDH could be detected in its culture supernatants.
Deletion of vicK result in diminished survival of S. mutans in human blood, and reduced growth in poor nutrient medium
To persist in the bloodstream, bacteria must survive the host defences and to undergo physiological changes to adapt to nutrient limitations present in blood39. Therefore, we assessed the capacities of the UAvic mutant to persist in human blood and to grow in poor nutrient medium (RPMI). As shown in Fig. 5A, over time, the numbers of viable UAvic recovered from blood in the survival assays were overall lower than that observed for UA159 and UAvic+, with 2 and 24 h incubation period showing statistically significant reductions compared to parent and complemented strains (p<0.05). Survival of UAvic+ in blood was very similar to the parent strain. Comparisons of the growth curves of the studied strains in rich medium (BHI) revealed no alterations in growth rate in UAvic compared to UA159 (Fig. 5B). However, the UAvic showed significant reduced growth rate in RPMI, when compared to UA159 and UAvic+ (Fig. 5C). The growth rate of UAvic+ was slower in both media when compared to UA159, probably because when growing UAvic+, the media were supplemented with spectinomycin to maintain the plasmid expressing vicKSm. Thus, the vicK mutant has a diminished capacity to persist in human blood compared to parent and complemented strains, which is associated with its defects in adaptation to limited nutrient conditions.
Fig. 5.
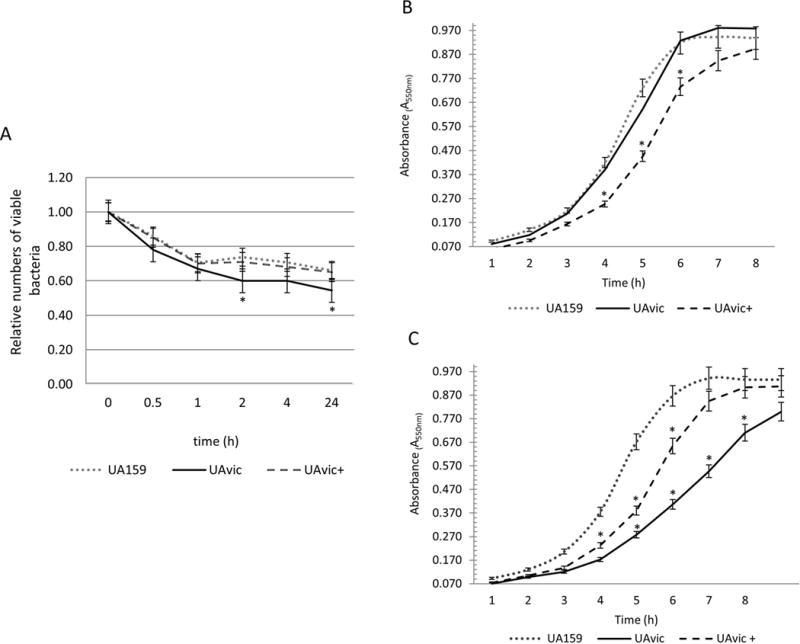
Comparisons of ex vivo survival in human blood and growth at different nutritional conditions. A) Numbers of viable bacteria (log cfu/ml) were expressed in relation to initial counts in blood suspension (time 0). Growth curves in BHI (B) and in poor nutrient RPMI (C). Dots represent means of triplicate of one representative experiment. Bars indicate standard deviations. Differences in relation to parent strain at each time-point were tested using Kruskal-Wallis with post hoc Dunn’s test with correction for repeated measures (*p<0.05).
DISCUSSION
Streptococcus mutans is an important species of viridans streptococci involved in cardiovascular diseases40,41. Mechanisms of systemic virulence of S. mutans remain to be elucidated, but seems to involve expression of different genes in serotype- and strain-specific ways3,40–43. Previously, we observed that deletion of vicK and covR, known to coordinate the expression of virulence genes involved in S. mutans cariogenicity7,10,15, reduced S. mutans phagocytosis by PMN in samples of human blood in the serotype c strain UA15914. Inactivation of covR was later shown to promote S. mutans resistance to complement opsonization and survival in blood through up-regulation of gbpC and epsC (CovR-repressed genes), genes required for S. mutans binding to sucrose-derived EPS3. Strains isolated from systemic infections express reduced levels of covR and stably bind to EPS, which in turn, functions as an anti-opsonic capsule3. However, binding to EPS does not entirely explain diversity in susceptibility to complement immunity among strains3. In addition, the mechanisms involved in the reduced susceptibility of the vicK mutant to opsonophagocytosis by PMN remained unknown, because this mutant is defective in the synthesis of EPS and in binding to these polymers7,9,10, which should increase its susceptibility to complement-mediated opsonization. In the present study, we show that the vicK mutant is resistant to C3b deposition and has reduced binding to serum IgG antibodies, explaining its low susceptibility to phagocytosis in blood. Low levels of C3b deposition in UAvic were observed even when this strain was grown in sucrose-free medium (Fig. 1D), implying that S. mutans expresses additional proteins to evade complement deposition, besides proteins involved in binding to EPS.
Streptococcal pathogens, e.g. S. pyogenes and S. pneumoniae, typically apply a diverse array of mechanisms to evade complement immunity, including the production of EPS capsule, secretion of proteases for degradation of complement components, surface binding to fluid phase inhibitors of the complement system or to other host proteins which indirectly inhibit complement activation29,44,45. Because the vicK mutant in UA159 is defective in binding to sucrose-derived EPS9,10, but resistant to C3b deposition, analysis of this strain was important for the screening of EPS-independent mechanisms of complement evasion. Thus, we identified 4 genes (smaA, smu2146c, smu.399 and pepO) that were transcriptionally affected in the UAvic mutant in the presence of serum. The genes encoding peptidases of the complement (smu.399 and pepO) were more clearly altered in response to serum in relation to unsupplemented BHI, when compared to genes encoding murein hydrolases (smaA and smu.2146c) and to the metabolic genes (eno, gapC and gapN). Similarly to our results, peptidases of complement are also up-regulated in Streptococcus agalactiae exposed to 10% of serum46. It is worth noting that BHI might include stimulatory components also found in serum, because this complex medium is rich in tissue and immune host factors. It could explain, at least in part, the effects of BHI on transcription of smu.399 and pepO observed in cells at mid-exponential growth. Transcriptional changes in smaA and smu.2146c in either the presence or absence of serum might also reflect multiple biological functions of these genes in response to different stimuli.
Phenotypic analyses of smaA- and smu.2146c-defective strains did not explain the resistance of UAvic to C3b deposition. Moreover, S. mutans expresses other murein hydrolases, which are down-regulated at transcriptional and/or post-transcriptional levels in vicKSm mutants, including LysM10 and AtlA25. However, deletion of lysM does not affect complement opsonization3, and deletion of atlA increases S. mutans susceptibility to phagocytosis by PMN47. These findings suggest that increased resistance of the UAvic strain to C3b deposition and opsonophagocytosis is not associated with altered functions of the murein hydrolases investigated so far. However, here we show that S. mutans expresses peptidases of the complement (Smu.399 and PepO) that are negatively-regulated by VicRKSm. Deletion of smu.399 and pepO significantly increases C3b deposition and opsonophagocytosis of S. mutans (Fig. 3), suggesting that these peptidases may be potential targets to control systemic infections by this species.
In S. pneumoniae, PepO (PepOSp) binds to C4b-binding protein (C4BP), a fluid phase inhibitor of the classical pathway of the complement system48, the major pathway of complement activation on S. mutans3. Aminoacid sequence of PepOSm shows 79 and 88% of similarity with PepO orthologues expressed by S. pneumoniae35 and S. pyogenes49, respectively. Secreted PepOSp further promotes degradation of C3b through binding to plasminogen and its activation to plasmin. PepOSp also binds to fibronectin, which is present in soluble form in serum and saliva, and is involved in a large number of physiological processes, including formation of vegetations on injured cardiac endothelium, and atheromatose50,51. The enhanced capacity of UAvic to bind to fibronectin is therefore compatible with the up-regulation of pepOSm in UAvic, and with resistance of this mutant to opsonophagocytosis. In S. mutans strain GS5, binding to fibronectin was associated with resistance to opsonophagocytosis and increased survival in the bloodstream, but this property was associated with the expression of atlA47, whose transcription was not significantly altered in UAvic (data not shown). In addition, S. mutans expresses other surface proteins that could contribute to fibronectin binding37, but their role in UAvic phenotype remains to be elucidated. Studies are under way to define the role of PepOSm in systemic infections by S. mutans.
UAvic shows defects in septum division associated with reduced expression of GbpB9, an essential protein involved in S. mutans binding to EPS and in cell wall division9,52. Down-regulation of the GbpB orthologue (PcsB) in S. pneumoniae affects cell wall division31 and promotes release of enolase38. Enolase on S. pneumoniae surface binds to C4BP to inhibit complement activation53. Therefore, up-regulation of enolase in UAvic could contribute to UAvic resistance to C3b deposition. Although we could not detect enolase in the culture supernatants of the studied strains, it is possible that extracellular enolase could be associated with the S. mutans cell wall.
The essentiality of VicRSm regulator for S. mutans viability is not entirely understood7,9,10. In addition, vicKSm-defective strains show increased sensitivity to oxidative, pH and osmotic stresses9,11,25,54. VicRKSm is also responsive to nutritional changes11, and here we show that the vicKSm mutant is defective in metabolic adaptation to limited nutrient conditions. All these defects should expectedly reduce the capacity of vicKSm-defective strains to persist in blood, because metabolic adaptation to limited nutrient conditions and resistance to oxidative and pH stresses present in blood are crucial for bacterial survival in the bloodstream39,55. It is thus possible that the increased resistance of UAvic to opsonophagocytosis may have counterbalanced the UAvic deficiencies in stress response and/or adaptation to blood nutrient limitations, resulting in limited changes in curves of UAvic persistence in blood.
In summary, in this study we showed that deletion of vicK reduces S. mutans susceptibility to phagocytosis by PMN by impairing C3b deposition and surface binding to serum IgG, in a way independent of the production of sucrose-derived EPS or of the expression of murein hydrolases encoded by smaA and smu2146c. Two novel genes expressed by S. mutans under regulation of VicRKSm were identified and were shown to contribute to S. mutans resistance to C3b deposition (pepO and smu.399), establishing that this bacterium expresses multiple factors associated with complement immunity evasion. Although deletion of vicKSm result in increased resistance to opsonophagocytosis, it does not contribute to S. mutans survival in human blood, which is, at least in part, associated with defects in adaption to nutrient limitations.
Acknowledgments
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; grant numbers 2012/50966-6 and 2015/12940-3). LAA was supported by FAPESP (fellowship numbers 2012/04222-5 and 2015/07237-1). THD was supported by a fellowship of Coordenação de Pessoal de Nível Superior (CAPES). EHC was supported by FAPESP (fellowship number 2009/50547-0) and CAPES-PNPD (2013). JA was supported by NIDCR (DE022559). We thank Dr. Vijay Pancholi for the gift of the antibodies to S. pyogenes α-enolase and anti-extracellular GAPDH.
Footnotes
PROFESSOR RENATA DE OLIVEIRA MATTOS-GRANER (Orcid ID : 0000-0001-8309-8135)
References
- 1.Bowen WH, Koo H. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res. 2011;45:69–86. doi: 10.1159/000324598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munro CL, Macrina FL. Sucrose-derived exopolysaccharides of Streptococcus mutans V403 contribute to infectivity in endocarditis. Mol Microbiol. 1993;8:133–142. doi: 10.1111/j.1365-2958.1993.tb01210.x. [DOI] [PubMed] [Google Scholar]
- 3.Alves LA, Nomura R, Mariano FS, et al. CovR regulates Streptococcus mutans susceptibility to complement immunity and survival in blood. Infect Immun. 2016 doi: 10.1128/IAI.00406-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ajdic D, McShan WM, McLaughlin RE, et al. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci U S A. 2002;99:14434–14439. doi: 10.1073/pnas.172501299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmer SR, Miller JH, Abranches J, et al. Phenotypic heterogeneity of genomically-diverse isolates of Streptococcus mutans. PLoS One. 2013;8:e61358. doi: 10.1371/journal.pone.0061358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornejo OE, Lefebure T, Bitar PD, et al. Evolutionary and population genomics of the cavity causing bacteria Streptococcus mutans. Mol Biol Evol. 2013;30:881–893. doi: 10.1093/molbev/mss278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senadheera MD, Guggenheim B, Spatafora GA, et al. A VicRK signal transduction system in Streptococcus mutans affects gtfBCD, gbpB, and ftf expression, biofilm formation, and genetic competence development. J Bacteriol. 2005;187:4064–4076. doi: 10.1128/JB.187.12.4064-4076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ayala E, Downey JS, Mashburn-Warren L, Senadheera DB, Cvitkovitch DG, Goodman SD. A biochemical characterization of the DNA binding activity of the response regulator VicR from Streptococcus mutans. PLoS One. 2014;9:e108027. doi: 10.1371/journal.pone.0108027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duque C, Stipp RN, Wang B, et al. Downregulation of GbpB, a component of the VicRK regulon, affects biofilm formation and cell surface characteristics of Streptococcus mutans. Infect Immun. 2011;79:786–796. doi: 10.1128/IAI.00725-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stipp RN, Boisvert H, Smith DJ, Hofling JF, Duncan MJ, Mattos-Graner RO. CovR and VicRK regulate cell surface biogenesis genes required for biofilm formation in Streptococcus mutans. PLoS One. 2013;8:e58271. doi: 10.1371/journal.pone.0058271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senadheera DB, Cordova M, Ayala EA, et al. Regulation of bacteriocin production and cell death by the VicRK signaling system in Streptococcus mutans. J Bacteriol. 2012;194:1307–1316. doi: 10.1128/JB.06071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe T, Igarashi M, Okajima T, et al. Isolation and characterization of signermycin B, an antibiotic that targets the dimerization domain of histidine kinase WalK. Antimicrob Agents Chemother. 2012;56:3657–3663. doi: 10.1128/AAC.06467-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eguchi Y, Kubo N, Matsunaga H, Igarashi M, Utsumi R. Development of an antivirulence drug against Streptococcus mutans: repression of biofilm formation, acid tolerance, and competence by a histidine kinase inhibitor, walkmycin C. Antimicrob Agents Chemother. 2011;55:1475–1484. doi: 10.1128/AAC.01646-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Negrini TC, Duque C, Vizoto NL, et al. Influence of VicRK and CovR on the interactions of Streptococcus mutans with phagocytes. Oral Dis. 2012;18:485–493. doi: 10.1111/j.1601-0825.2011.01896.x. [DOI] [PubMed] [Google Scholar]
- 15.Biswas I, Drake L, Biswas S. Regulation of gbpC expression in Streptococcus mutans. J Bacteriol. 2007;189:6521–6531. doi: 10.1128/JB.00825-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuste J, Sen A, Truedsson L, et al. Impaired opsonization with C3b and phagocytosis of Streptococcus pneumoniae in sera from subjects with defects in the classical complement pathway. Infect Immun. 2008;76:3761–3770. doi: 10.1128/IAI.00291-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stipp RN, Goncalves RB, Hofling JF, Smith DJ, Mattos-Graner RO. Transcriptional analysis of gtfB, gtfC, and gbpB and their putative response regulators in several isolates of Streptococcus mutans. Oral Microbiol Immunol. 2008;23:466–473. doi: 10.1111/j.1399-302X.2008.00451.x. [DOI] [PubMed] [Google Scholar]
- 18.Sato Y, Okamoto K, Kagami A, Yamamoto Y, Igarashi T, Kizaki H. Streptococcus mutans strains harboring collagen-binding adhesin. J Dent Res. 2004;83:534–539. doi: 10.1177/154405910408300705. [DOI] [PubMed] [Google Scholar]
- 19.Aviles-Reyes A, Miller JH, Simpson-Haidaris PJ, Hagen FK, Abranches J, Lemos JA. Modification of Streptococcus mutans Cnm by PgfS contributes to adhesion, endothelial cell invasion, and virulence. J Bacteriol. 2014;196:2789–2797. doi: 10.1128/JB.01783-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mattos-Graner RO, Smith DJ, King WF, Mayer MP. Water-insoluble glucan synthesis by mutans streptococcal strains correlates with caries incidence in 12- to 30-month-old children. J Dent Res. 2000;79:1371–1377. doi: 10.1177/00220345000790060401. [DOI] [PubMed] [Google Scholar]
- 21.Mattos-Graner RO, Jin S, King WF, Chen T, Smith DJ, Duncan MJ. Cloning of the Streptococcus mutans gene encoding glucan binding protein B and analysis of genetic diversity and protein production in clinical isolates. Infect Immun. 2001;69:6931–6941. doi: 10.1128/IAI.69.11.6931-6941.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tremblay YD, Lo H, Li YH, Halperin SA, Lee SF. Expression of the Streptococcus mutans essential two-component regulatory system VicRK is pH and growth-phase dependent and controlled by the LiaFSR three-component regulatory system. Microbiology. 2009;155:2856–2865. doi: 10.1099/mic.0.028456-0. [DOI] [PubMed] [Google Scholar]
- 23.Bottazzi B, Garlanda C, Salvatori G, Jeannin P, Manfredi A, Mantovani A. Pentraxins as a key component of innate immunity. Curr Opin Immunol. 2006;18:10–15. doi: 10.1016/j.coi.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 24.van Kessel KP, Bestebroer J, van Strijp JA. Neutrophil-Mediated Phagocytosis of Staphylococcus aureus. Front Immunol. 2014;5:467. doi: 10.3389/fimmu.2014.00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn SJ, Burne RA. Effects of oxygen on biofilm formation and the AtlA autolysin of Streptococcus mutans. J Bacteriol. 2007;189:6293–6302. doi: 10.1128/JB.00546-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pancholi V, Fischetti VA. alpha-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J Biol Chem. 1998;273:14503–14515. doi: 10.1074/jbc.273.23.14503. [DOI] [PubMed] [Google Scholar]
- 27.Pancholi V, Fischetti VA. A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J Exp Med. 1992;176:415–426. doi: 10.1084/jem.176.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge J, Catt DM, Gregory RL. Streptococcus mutans surface alpha-enolase binds salivary mucin MG2 and human plasminogen. Infect Immun. 2004;72:6748–6752. doi: 10.1128/IAI.72.11.6748-6752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dubrac S, Bisicchia P, Devine KM, Msadek T. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol Microbiol. 2008;70:1307–1322. doi: 10.1111/j.1365-2958.2008.06483.x. [DOI] [PubMed] [Google Scholar]
- 31.Ng WL, Tsui HC, Winkler ME. Regulation of the pspA virulence factor and essential pcsB murein biosynthetic genes by the phosphorylated VicR (YycF) response regulator in Streptococcus pneumoniae. J Bacteriol. 2005;187:7444–7459. doi: 10.1128/JB.187.21.7444-7459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramos-Sevillano E, Urzainqui A, Campuzano S, et al. Pleiotropic effects of cell wall amidase LytA on Streptococcus pneumoniae sensitivity to the host immune response. Infect Immun. 2015;83:591–603. doi: 10.1128/IAI.02811-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramos-Sevillano E, Moscoso M, Garcia P, Garcia E, Yuste J. Nasopharyngeal colonization and invasive disease are enhanced by the cell wall hydrolases LytB and LytC of Streptococcus pneumoniae. PLoS One. 2011;6:e23626. doi: 10.1371/journal.pone.0023626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bing DH, Almeda S, Isliker H, Lahav J, Hynes RO. Fibronectin binds to the C1q component of complement. Proc Natl Acad Sci U S A. 1982;79:4198–4201. doi: 10.1073/pnas.79.13.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agarwal V, Kuchipudi A, Fulde M, Riesbeck K, Bergmann S, Blom AM. Streptococcus pneumoniae endopeptidase O (PepO) is a multifunctional plasminogen- and fibronectin-binding protein, facilitating evasion of innate immunity and invasion of host cells. J Biol Chem. 2013;288:6849–6863. doi: 10.1074/jbc.M112.405530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pancholi V, Chhatwal GS. Housekeeping enzymes as virulence factors for pathogens. Int J Med Microbiol. 2003;293:391–401. doi: 10.1078/1438-4221-00283. [DOI] [PubMed] [Google Scholar]
- 37.Nobbs AH, Lamont RJ, Jenkinson HF. Streptococcus adherence and colonization. Microbiol Mol Biol Rev. 2009;73:407–450. doi: 10.1128/MMBR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giefing-Kroll C, Jelencsics KE, Reipert S, Nagy E. Absence of pneumococcal PcsB is associated with overexpression of LysM domain-containing proteins. Microbiology. 2011;157:1897–1909. doi: 10.1099/mic.0.045211-0. [DOI] [PubMed] [Google Scholar]
- 39.Samant S, Lee H, Ghassemi M, et al. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008;4:e37. doi: 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakano K, Nemoto H, Nomura R, et al. Serotype distribution of Streptococcus mutans a pathogen of dental caries in cardiovascular specimens from Japanese patients. J Med Microbiol. 2007;56:551–556. doi: 10.1099/jmm.0.47051-0. [DOI] [PubMed] [Google Scholar]
- 41.Nakano K, Nomura R, Matsumoto M, Ooshima T. Roles of oral bacteria in cardiovascular diseases–from molecular mechanisms to clinical cases: Cell-surface structures of novel serotype k Streptococcus mutans strains and their correlation to virulence. J Pharmacol Sci. 2010;113:120–125. doi: 10.1254/jphs.09r24fm. [DOI] [PubMed] [Google Scholar]
- 42.Nakano K, Lapirattanakul J, Nomura R, et al. Streptococcus mutans clonal variation revealed by multilocus sequence typing. J Clin Microbiol. 2007;45:2616–2625. doi: 10.1128/JCM.02343-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abranches J, Miller JH, Martinez AR, Simpson-Haidaris PJ, Burne RA, Lemos JA. The collagen-binding protein Cnm is required for Streptococcus mutans adherence to and intracellular invasion of human coronary artery endothelial cells. Infect Immun. 2011;79:2277–2284. doi: 10.1128/IAI.00767-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 45.Blom AM, Hallstrom T, Riesbeck K. Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol Immunol. 2009;46:2808–2817. doi: 10.1016/j.molimm.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 46.Gleich-Theurer U, Aymanns S, Haas G, Mauerer S, Vogt J, Spellerberg B. Human serum induces streptococcal c5a peptidase expression. Infect Immun. 2009;77:3817–3825. doi: 10.1128/IAI.00826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung CJ, Zheng QH, Shieh YH, Lin CS, Chia JS. Streptococcus mutans autolysin AtlA is a fibronectin-binding protein and contributes to bacterial survival in the bloodstream and virulence for infective endocarditis. Mol Microbiol. 2009;74:888–902. doi: 10.1111/j.1365-2958.2009.06903.x. [DOI] [PubMed] [Google Scholar]
- 48.Agarwal V, Sroka M, Fulde M, Bergmann S, Riesbeck K, Blom AM. Binding of Streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. J Biol Chem. 2014;289:15833–15844. doi: 10.1074/jbc.M113.530212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilkening RV, Chang JC, Federle MJ. PepO, a CovRS-controlled endopeptidase, disrupts Streptococcus pyogenes quorum sensing. Mol Microbiol. 2016;99:71–87. doi: 10.1111/mmi.13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rohwedder I, Montanez E, Beckmann K, et al. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol Med. 2012;4:564–576. doi: 10.1002/emmm.201200237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore KJ, Fisher EA. The double-edged sword of fibronectin in atherosclerosis. EMBO Mol Med. 2012;4:561–563. doi: 10.1002/emmm.201200238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mattos-Graner RO, Porter KA, Smith DJ, Hosogi Y, Duncan MJ. Functional analysis of glucan binding protein B from Streptococcus mutans. J Bacteriol. 2006;188:3813–3825. doi: 10.1128/JB.01845-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Agarwal V, Hammerschmidt S, Malm S, Bergmann S, Riesbeck K, Blom AM. Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J Immunol. 2012;189:3575–3584. doi: 10.4049/jimmunol.1102934. [DOI] [PubMed] [Google Scholar]
- 54.Deng DM, Liu MJ, ten Cate JM, Crielaard W. The VicRK system of Streptococcus mutans responds to oxidative stress. J Dent Res. 2007;86:606–610. doi: 10.1177/154405910708600705. [DOI] [PubMed] [Google Scholar]
- 55.Gerlini A, Colomba L, Furi L, et al. The role of host and microbial factors in the pathogenesis of pneumococcal bacteraemia arising from a single bacterial cell bottleneck. PLoS Pathog. 2014;10:e1004026. doi: 10.1371/journal.ppat.1004026. [DOI] [PMC free article] [PubMed] [Google Scholar]