

Abstract
Early-onset mitochondrial encephalomyopathy is a rare disorder that presents in the neonatal period with lactic acidosis, hypotonia, and developmental delay. Sequence variants in the nuclear-encoded gene FBXL4 have been previously demonstrated to be a cause of early-onset mitochondrial encephalomyopathy in several unrelated families. We have identified a pair of siblings with mutations in FBXL4 who each presented in the neonatal period with hyperammonemia, low plasma levels of aspartate, low urine levels of tricarboxylic acid cycle intermediates suggesting a defect in anaplerosis, and cerebellar hypoplasia in addition to lactic acidosis and other classic signs of mitochondrial encephalomyopathy. After initial clinical stabilization, both subjects continued to have episodic exacerbations characterized by lactic acidosis and hyperammonemia. Previously reported cases of FBXL4 mutations are reviewed and compared with these affected siblings. These two new cases add to the spectrum of disease caused by mutations in FBLX4 and suggest possible benefit from anaplerotic therapies.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2016_17) contains supplementary material, which is available to authorized users.
Keywords: Anaplerosis, Aspartate, FBXL4, Hyperammonemia, Lactic acidosis, Mitochondrial encephalomyopathy
Introduction
Mitochondria are maternally derived and function to produce adenosine triphosphate (ATP) via oxidative phosphorylation. Abnormalities in mitochondrial function can lead to a variety of diseases including mitochondrial encephalomyopathy. Early-onset forms of encephalomyopathy present in the neonatal period and are characterized by lactic acidosis, hypotonia, developmental delay, and progressive neurologic disease.
Genetic variants linked to mitochondrial disease can be found in either the nuclear or mitochondrial DNA. Some mitochondrial diseases are characterized by mitochondrial DNA (mtDNA) depletion, with affected cells having less total mtDNA per cell. Diseases with mtDNA depletion are often caused by defects in a single gene such as POLG, a nuclear-encoded DNA polymerase essential for mitochondrial DNA replication (Tang et al. 2011; El-Hattab and Scaglia 2013).
More recently, mutations in the nuclear gene FBXL4 were demonstrated to be a cause of mitochondrial encephalomyopathy in several patients who had defective oxidative phosphorylation (Bonnen et al. 2013; Gai et al. 2013). FBXL family member proteins, characterized by leucine-rich repeats containing of F-box motifs, interact with SKP1-CUL1-F-box protein (SCF) E3 ubiquitin ligases (Van Rechem et al. 2011). FBXL4 was first identified as a component of the SCF ubiquitin ligase complex via yeast two-hybrid screen (Winston et al. 1999). The SCF complex is active in phosphorylation-dependent ubiquitination. The FBXL4 protein has an N-terminal mitochondrial localization signal and localizes within the intermembrane space of mitochondria (Gai et al. 2013).
A recent review summarized the phenotypic spectrum present in 28 cases with FBXL4 mutations (Huemer et al. 2015). A total of 23 sequence variants in FBXL4, with 14 missense, 7 loss-of-function, and 2 splice site mutations among patients with early-onset mitochondrial encephalomyopathy, were identified. Two additional sequence variants have since been reported (van Rij et al. 2016; Barøy et al. 2016). Patients with nonsense mutations had more severe phenotypes than those with missense changes (Bonnen et al. 2013; Gai et al. 2013). Fibroblasts and myocytes from affected individuals demonstrated decreased mitochondrial DNA (mtDNA) content, loss of the mitochondrial membrane potential, and decreased respiratory chain enzyme activity (Antoun et al. 2015). It is hypothesized that the defect in oxidative phosphorylation is due to the depletion of mtDNA. Expression of wild-type FBXL4 protein in skin fibroblasts from affected patients rescued the defects in mtDNA content and oxidative phosphorylation activity (Antoun et al. 2015).
Here we describe two siblings who both presented in the neonatal period with lactic acidosis, hypotonia, and cerebral atrophy. Unique to these cases, we observed severe depletion of tricarboxylic acid cycle (TCA) intermediates and of the anaplerotic amino acid aspartate. Because aspartate is required to provide nitrogen to form argininosuccinate in the urea cycle, its depletion may be contributing to the severe hyperammonemia observed in these cases. Both biochemically and clinically, the presentation of these siblings with FBXL4 missense variants closely mimicked the findings associated with pyruvate carboxylase deficiency. However, after pyruvate carboxylase deficiency was ruled out by gene sequencing, a muscle biopsy for mitochondrial studies was performed in the older child (sibling 1, S1), which revealed depletion of mtDNA relative to nuclear DNA, consistent with the prior findings of mtDNA depletion in other patients affected by FBXL4 gene mutations.
Case Reports
Sibling 1
Sibling 1 (S1), a male infant, was born at 38 2/7 weeks’ gestation by cesarean section for fetal heart rate decelerations and intrauterine growth restriction (IUGR). At birth he appeared healthy and he was transferred to the newborn nursery. By 18 h of life he displayed temperature instability, deep breathing, and hypoglycemia as low as 1.4 mmol/L (reference interval: 2.2–5.6). Clinical characteristics are summarized in Supplemental Table 1. A venous blood gas at 18 h of life identified pH 7.09, pCO2 16 mmHg, and pO2 92 mmHg. Additional testing revealed a plasma ammonia level of 174 μmol/L (reference interval: 16–47) and plasma lactate level of 27 mmol/L (reference interval: 0.5–2.2). The hyperammonemia was treated with continuous infusions of sodium benzoate and sodium phenyl acetate, while the acidosis was corrected with sodium bicarbonate. Peak plasma ammonia was >362 μmol/L. He also had abnormal coagulation with an international normalized ratio of 2.63 (reference interval: 0.87–1.13), prothrombin time 24.9 s (reference interval: 9.4–11.8), partial thromboplastin time 104.4 s (reference interval: 24–32), and fibrinogen 114 mg/dL (reference interval: 200–400). Specific coagulation factors were not interrogated. Liver function otherwise appeared unaffected as evidenced by normal plasma alanine transaminase activity 14 unit/L (reference interval: 3–54), albumin 3.0 g/dL (reference interval: 2.5–4.0), and total bilirubin 4.0 g/dL (reference interval: 4.2–6.6).
Because S1 manifested neonatal hypoglycemia with severe and persistent hyperammonemia and metabolic acidosis, characterized by both ketosis and lactic acidosis, his initial metabolic differential diagnosis included the possibilities of pyruvate carboxylase deficiency or an organic acidemia, similarly capable of secondary inhibition of the urea cycle, such as propionic acidemia or methylmalonic acidemia. On his first day at our institution, an emergency urine organic acid profile demonstrated a remarkable, nearly complete absence of TCA cycle intermediates (Fig. 1a). For comparison, a typical urine organic acid profile is shown in Supplemental Fig. 1. Concurrent analysis of plasma amino acids demonstrated elevations of alanine at 1,280 μmol/L (reference interval: 120–499), citrulline at 98 μmol/L (reference interval: 2–50), proline at 916 μmol/L (reference interval: 66–300), and lysine at 593 μmol/L (reference interval: 54–271), whereas the glutamine level was within the reference interval at 716 μmol/L (reference interval: 322–1,084). The amino acid profile also showed a low-normal aspartate level at 3 μmol/L (reference interval: 3–17). These findings were obtained at the same time as the severely elevated plasma lactate level of 27 mmol/L and were consistent with an initial working diagnosis of pyruvate carboxylate (PC) deficiency. Supplementation with citrate, aspartate, biotin, carnitine, and thiamine was commenced in the NICU, and the patient’s biochemical abnormalities stabilized considerably. With this treatment, the depletion of TCA cycle intermediates in the urine was quickly corrected (Fig. 1b). Plasma lactate levels normalized and the hyperammonemia resolved. The use of citrate and aspartate supplementation to promote anaplerosis was based on case reports of its effectiveness in PC deficiency (Mochel et al. 2005). Given the dismal prognosis of neonatal PC deficiency, triheptanoin was also later administered as an anaplerotic substrate for the TCA cycle. However, this was discontinued, since its use appeared to be associated with vomiting.
Fig. 1.
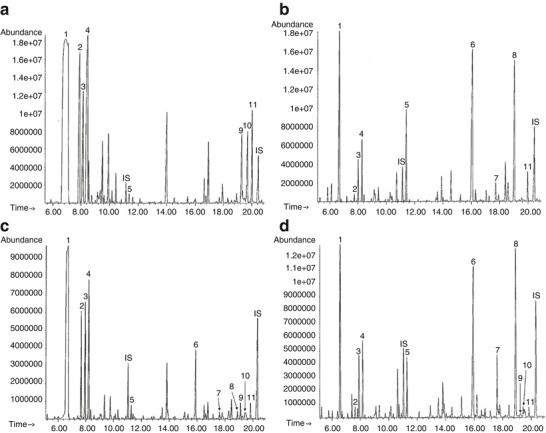
Representative gas chromatography-mass spectrometry (GC-MS) organic acid chromatograms from urine specimens. (a) Urine sample collected from S1 on presentation prior to administering treatment. The tricarboxylic acid cycle intermediates 2-ketoglutarate, aconitate, and citrate were undetectable in the urine on presentation. (b) Urine sample collected from S1 after treatment with aspartate and citrate. (c) Urine sample collected from S2 on presentation prior to administering treatment. (d) Urine sample collected from S2 after treatment with aspartate and citrate. Horizontal axis reflects retention time, and vertical axis reflects total ion abundance. The following peak numbers correspond to compounds detected in both chromatograms: (1) lactate, (2) 2-hydroxybutyrate, (3) pyruvate, (4) 3-hydroxybutyrate, (5) fumarate, (6) 2-ketoglutarate, (7) aconitate, (8) citrate, (9) unsaturated sebacate, (10) sebacate, (11) 4-hydroxyphenyllactate. Internal standards (IS): 2-ketocaproate (approximately 11.0 min) and pentadecanoate (approximately 20.3 min)
Genetic sequencing of the pyruvate carboxylase (PC) gene, as well as 101 nuclear genes associated with mitochondrial disease, was normal. Enzymatic activity assays using patient fibroblasts demonstrated normal activities of PC, pyruvate dehydrogenase, and phosphoenolpyruvate carboxykinase.
Brain MRI on day of life six demonstrated diffuse T2 hyperintensity of the supratentorial deep and subcortical white matter within the frontal, parietal, occipital, and temporal lobes (Fig. 2a). The cerebellum and brainstem were hypoplastic (Fig. 2b). Electroencephalogram (EEG) at 2 weeks of life was abnormal due to excessive multifocal sharp waves, delta brushes, and excessive discontinuity in sleep consistent with nonspecific encephalopathy. No epileptic activity was noted.
Fig. 2.

Brain abnormalities in affected siblings on magnetic resonance imaging (MRI). (a) Axial T2-weighted images of S1 at 6 days of life image show diffuse white matter hyperintensity in the periventricular and subcortical white matter. (b) Axial T2-weighted image of S1 at 6 days of age shows enlarged retrocerebellar cerebrospinal fluid (CSF) space and a hypoplastic cerebellar hemisphere. (c) Axial T2-weighted image of S1 at 28 months of age shows marked cerebral atrophy. (d) Fetal MRI scans of S2 at 21+5 (left) and 34+5 weeks’ gestation (right) show enlarged retrocerebellar CSF spaces. (e) Single voxel magnetic resonance spectroscopy of S2 at 5 days old performed at TE 144 ms from the parietal white matter shows an inverted doublet of lactate at 1.33 ppm. (f) Axial T2-weighted image of S2 at 5 days of life shows large areas of extensive intraparenchymal and intraventricular hemorrhage in the bilateral parietal and occipital lobes. Smaller areas of hemorrhage are seen along the expected course of the medullary veins in the frontal white matter (arrows). (g) Axial T2-weighted image of S2 at 5 days of age shows prominence of the retrocerebellar cystic space with a hypoplastic inferior vermis and small, slightly dysmorphic cerebellar hemispheres. (h) Axial T2-weighted image of S2 at 25 months of age shows marked atrophy with ex-vacuo dilatation of the ventricular system and sulcal spaces in the frontal and temporal regions. In addition, sequelae of the prior hemorrhage are seen as porencephalic changes and encephalomalacia in the bilateral (left more than right), parietal, and occipital lobes. Periventricular white matter continues to show abnormal T2 hyperintensity
S1 also had symptomatic persistent pulmonary hypertension (PPHN) requiring inhaled nitric oxide in the first week of life until it resolved. He had cardiomyopathy and progressive dilation of the pulmonary artery with z-score of 5–6, as well as a mildly dilated ascending aorta, without evidence of cerebrovascular dilation on MRI. His hypotonia was persistent, and he had oromotor dysfunction with aspiration of thin liquids. Following NICU discharge, he continued to be treated with biotin, l-aspartic acid, citric acid-potassium citrate and sodium bicarbonate. As an outpatient, his plasma ammonia was usually normal, except during intermittent illnesses. However, his lactic acid levels were chronically elevated (usually between 5 and 10 mmol/L), and his plasma aspartate levels were often at the lower limit of normal despite ongoing enteral aspartate supplementation.
Muscle biopsy at 8 months was notable only for mild excess variation in fiber size diameter affecting type 1 fibers more than type 2 fibers. There were occasional nuclei with prominent regenerative nucleoli on electron microscopy, but findings were otherwise normal. Trichrome staining revealed normal connective tissue and myofibrillar structures; in addition, there were no ragged red fibers. PAS stain revealed a normal content and appearance of glycogen, which digested appropriately with dispase. Oil Red O stain revealed a normal content of lipid. NADH, ATPase, COX, and SDH histochemical stain revealed the normal findings. Mitochondria were normal in number, distribution, and morphology on electron microscopy. Biochemical testing for electronic transport chain complex I–IV activity in skeletal muscle biopsy demonstrated low activity of all complexes, though citrate synthase activity was also low so the corrected enzyme activity was within normal limits.
S1 also had global developmental delay as he began to sit at approximately 1 year of age and independently bear weight on his legs at 2 years. Serial MRI, with the last at 24 months of life, documented progressive cerebral atrophy and cavitation of the basal ganglia without diffusion abnormality (Fig. 2c). The patient had cerebral visual impairment with optic disc atrophy, nystagmus, and mild to moderate bilateral sensorineural hearing loss.
After an initial period of metabolic stability at home, he presented again at 23 months of age with a second episode of hyperammonemia. Subsequently, he continued to have repeated admissions to the ICU for hyperammonemia, some requiring dialysis. He passed away at 40 months while receiving palliative care at home.
Sibling 2
Sibling 2 (S2), a female infant, was born at 38 6/7 weeks’ gestation via vaginal delivery. Given the family history, she had baseline studies taken soon after birth that demonstrated elevated plasma lactic acid level of 13.3 mmol/L (reference interval: 0.5–2.2) and hyperammonemia (plasma ammonia, 134 μmol/L; reference interval: 16–47). Supplementation with citric acid and aspartate was initiated immediately. Extensive metabolic evaluation demonstrated decreased plasma aspartate of 3 μmol/L (reference interval: 3–17) with increased levels of proline at 350 μmol/L (reference interval: 66–300), lysine at 286 μmol/L (reference interval: 54–271), and alanine at 796 μmol/L (reference interval: 120–499), consistent with the pyruvate-carboxylase-deficiency-like syndrome of her brother. Urine organic acids demonstrated significant depletion of TCA cycle intermediates such as citrate (Fig. 1c). After initiation of citric acid at 0.12 mmol/kg/hr and aspartate supplementation of 250 mg every 3 h, hyperammonemia resolved. The urine organic acid profile after supplementation demonstrated the presence of a proportionally abundant citrate peak consistent with exogenous citrate administration (Fig. 1d). No muscle biopsy was conducted given the similarity of her disease phenotype to that of her brother.
Prenatal imaging had raised concerns for enlarged retrocerebellar space and small cerebellum (Fig. 2d). Brain MRI in the first week of life demonstrated extensive hemorrhages within the parenchyma, ventricles, and enlarged extra-axial spaces (Fig. 2f). The vermis and cerebellar hemispheres were hypoplastic (Fig. 2g). Single voxel MR spectroscopy from a voxel placed in the left parietal white matter demonstrated a prominent inverted doublet indicating elevated brain lactate (Fig. 2e).
During her NICU admission, she demonstrated poor oral feeding ability, and even with gastrostomy tube feedings, she had significant daily emesis. She received continuing supplementation with biotin, aspartic acid, and citric acid-potassium citrate. She was discharged home at 3 weeks of age at a weight of 2.66 kg and receiving via gastrostomy tube biotin 20 mg per day, citric acid-sodium citrate 1.5 mEq every 3 h, and l-aspartic acid 250 mg every 3 h. She remained reasonably stable on similar therapy for most of the next 2 years. Like her brother, she had chronic lactic acidosis (usually 5–10 mmol/L), intermittent hyperammonemia, and plasma aspartate levels that remained often close to the lower limit of normal despite her enteral supplements.
Her visual acuity was 20/190 at 12 months and nystagmus was noted. Brain MRI at 24 months demonstrated persistent white matter T2 hyperintensity, progressive predominant frontotemporal atrophy and porencephaly, and encephalomalacia in the parietal and occipital lobes at the sites of prior hemorrhage (Fig. 2h). She had a significant developmental delay and by 23 months of life was unable to sit unsupported.
Between 9 and 23 months of age, S2 had four admissions to the ICU for acidosis with varying degrees of hyperammonemia, from mild to severe. In the most acute episode, at 23 months of age, without a clear inciting illness, she presented with altered mental status, plasma lactic acid 13.4 mmol/L (reference interval: 0.5–2.2), and severe hyperammonemia of 443 μmol/L (reference interval: 12–38), which required emergency hemodialysis for treatment. At 24 months of age, she was hospitalized again with recurrent vomiting, persistent lactic acidosis, and mild hyperammonemia. Lumbar puncture during this final hospitalization revealed a CSF lactic acid level of 10 mmol/L, notably almost twice the level previously obtained in plasma (6 mmol/L). To treat her persistent lactic acidosis, she required continuous infusion with up to 70 mEq/kg/day of sodium bicarbonate. After this 5-week hospitalization, S2 was transitioned to palliative care at home and passed away at 26 months.
Materials and Methods
Patient Enrollment
The proband (S1), both parents, the affected sibling (S2), and an unaffected sibling were enrolled in an IRB-approved study at Boston Children’s Hospital (BCH) (Fig. 3a).
Fig. 3.

Sequence analysis reveals compound heterozygosity in affected siblings but not unaffected family members. (a) Pedigree of the families carrying FBXL4 variants. The probands were II:2 (S1) and II:3 (S3). (b) Sequence analysis for nucleotide 1411. Maternally inherited c.1411G>A variant was presented in S1 (II:2) and S2 (II:3) but not the unaffected sibling (II:1). (c) Sequence analysis for nucleotide 1586. Paternally inherited c.1586C>A variant was presented in S1 (II:2) and S2 (II:3) but neither the mother (I:2) nor the unaffected sibling (II:1). (d) Sequence analysis for nucleotide 1790. Maternally inherited c.1790 A>C variant was presented in S1 (II:2), unaffected sibling (II:1), and S2 (II:3) but not the father (I:1). (e) Amino acid sequence alignment at the location of identified variants. Residues highlighted in yellow are the conserved amino acids affected by the genetic variant, and those highlighted in blue are nonconserved amino acids. (f) Predicted protein structure of FBXL4 with sites of identified variants highlighted. Shown in sphere, p. Gln597 is colored in red, p. Ala471 in purple and p. Ala529 in blue. The hypothetical position of a potential binding partner of FBXL4 leucine-rich repeat (LRR) domain is shown in gray
Urine Organic Acid Analysis
Urine organic acids were extracted into ethyl acetate and diethyl ether and converted to trimethylsilyl derivatives prior to analysis by gas-chromatography mass spectrometry using a GC 6890 N/ MS 5975 system with a DB-1 column (Agilent, Santa Clara, CA).
Muscle mtDNA Assay
Total genomic DNA extracted from frozen muscle tissue from S1 was assayed quantitatively for mtDNA and the 18S ribosomal RNA gene using qRT-PCR (MNG Laboratories, Atlanta, Georgia). The ratio of the number of PCR cycles to reach threshold was calculated, with larger values indicating lower numbers of mtDNA.
DNA Preparation, Exome Sequencing, and Data Analysis
Total genomic DNA was extracted from peripheral blood lymphocytes using QIAmp DNA Mini Kit (Qiagen). DNA from the proband, affected sibling, unaffected sibling, and both biological parents was sent for whole exome sequencing (WES) to Axeq Technologies. Samples were prepared as an Illumina sequencing library and enriched for exomic sequences using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using Illumina HiSeq 2000 Sequencers. The reads were mapped to the human genome assembly UCSC hg19 using Burrows-Wheeler Alignment (BWA version 0.5.8). Single nucleotide polymorphisms (SNPs) and small insertions/deletions were called with SAMtools (version 0.1.7). The resulting VCF files were analyzed using a custom-built, rule-based “Variant Explorer” pipeline capable of integrating SNP chip, linkage, sequencing, and functional database information. In this case only the sequencing and database components were used. We selected likely pathogenic variants to include nonsynonymous, splice site, and indel variants with an allele frequency <0.001 in the NHLBI exome variant server database (EVS) (http://evs.gs.washington.edu/EVS/) or <0.01 in 1000 Genomes Project, phase 3, (http://www.1000genomes.org). The variants were also evaluated for frequency by ExAC database (http://www.exac.broadinstitute.org). The pathogenicity of the variants was evaluated in silico using polyphen-2, SIFT, and MutationTaster programs.
Results
Whole Exome Sequencing
Whole exome sequencing was performed on genomic DNA from the proband and both parents. The mean read depth was 100–115×, and >10× coverage of the target region was 98.1–98.7%. Nine hundred nine variants were nonsynonymous, splice site, and indel variants of which 323 satisfied the frequency filtration criteria described above. Of those variants, 34 were de novo dominant or recessive (homozygous or compound heterozygous). Genes carrying those variants were further evaluated for human disease or animal models overlapping with the phenotype. Only one gene, FBXL4, was identified to be significant based on the human phenotype already associated with FBXL4 variants and the protein’s mitochondrial function.
Both S1 and S2 were found to have three variants in FBXL4 (Fig. 3b–d). Two variants in the leucine-rich repeat domains were inherited from the mother: Chr6:99322230 (hg19):NM_012160:exon9: c.1790 A>C:p. Gln597Pro and Chr6:99323582 (hg19):NM_012160:exon8: c.1411G>A:p. Ala471Thr. The paternal variant present in both affected siblings was Chr6:99323407 (hg19):NM_012160:exon8: c.1586C>A:p. Ala529Glu.
The unaffected sibling carried both the maternal variants but not the paternal variant. In the ExAC database, the variant p. Gln597Pro is present in heterozygous state in two controls (allele frequency of 0.000016), while the other two variants (p. Ala471Thr and p. Ala529Glu) are absent. Both p. Ala471Thr and p. A529E variants have not been previously seen in patients with FBXL4-related disease. All the three variants were predicted to be pathogenic by MutationTaster, while the polyphen-2 and SIFT software programs deemed the Gln597Pro and Ala529Glu variants pathogenic and Ala471Thr benign. All three amino acid residues are very well conserved among the vertebrates (Fig. 3e).
FBXL4 belongs to the F-box protein family, containing an approximately 40-amino acid motif, the F-box in the N-terminus. The C-terminus of FBXL4 contains at least nine tandem leucine-rich repeats (LRR), which is similar to the LRR domain of S-phase kinase-associated protein 2 (SCF Skp2) with more than 40% similarity, suggesting the overall structure of FBXL4 LRR domain is similar to that of Skp2 LRR domain. Based on the structure of Skp2 LRR domain (PDB code: 2ASS), we built a structural model of FBXL4 LRR domain with the highlights of three altered residues using PyMOL (The PyMOL Molecular Graphics System) (Fig. 3f). In the model, a Skp2 LRR binding partner, cyclin-dependent kinases regulatory subunit 1 (CKS1), is shown in gray to suggest the hypothetical position of binding partners of FBXL4 LRR domain. Colored in red, Gln597 is located at the loop near the potential binding partner. A mutation at residue 597 likely affects the binding of FBXL4 to its binding partners. Residues Ala471 in purple and Ala529 in blue are at the back of the LRR domain. Mutations at these two residues might change the structural stability or orientation of the leucine-rich repeats, which can subsequently disrupt the function of FBXL4.
Mitochondrial DNA Depletion Assay
Clinical diagnostic testing of the frozen muscle tissue for mtDNA depletion (MNG Laboratories, Atlanta, GA) revealed a statistically significant depletion of mtDNA relative to nuclear DNA, with a prolonged quantitative PCR mtDNA/18S DNA amplification time ratio of 0.8 (mean 0.68; 95th percentile 0.75), a test in which the higher ratio indicates decreased mtDNA copy number.
Discussion
Previous studies have identified 25 mutations in FBXL4 that were associated with mitochondrial diseases (Bonnen et al. 2013; Gai et al. 2013; Huemer et al. 2015; van Rij et al. 2016; Barøy et al. 2016). Common clinical phenotypes include hypotonia, cerebral atrophy, developmental delay, and episodic lactic acidemia with or without hyperammonemia. Here we report a case of two siblings who shared three genetic variants in FBXL4 and presented with hyperammonemia, depletion of TCA intermediates in urine, low plasma aspartate levels, poor feeding, and frequent vomiting in addition to the shared characteristics mentioned above. We hypothesize that the depletion of TCA cycle intermediates contributed our patients’ frequently low aspartate levels, which then led to secondary urea cycle dysfunction and resulted in hyperammonemia. Given the known role of FBXL4 in protein ubiquitination, it is also possible that FBXL4 participates directly in ubiquitination of proteins related to the urea cycle.
One of the FBXL4 variants (p. Gln597Pro) found in this sibling pair has been previously identified in a 9-year-old girl from a previous case series (Gai et al. 2013). Interestingly, she presented with relatively mild disease and was alive at 8 years of age. She also carried a deleterious frame-shift mutation in FBXL4, so the Gln597Pro mutation is likely to have some residual function given her relatively long survival. Though there are relatively few cases reported from which to draw genotype-phenotype correlations, it seems that the presence of three FBXL4 variants in our patients increased their disease severity.
Patients with FBXL4 mutations have been found to have additional clinical features not commonly associated with mitochondrial encephalomyopathy such as IUGR and microcephaly. In the recent review of mitochondrial encephalomyopathy due to FBXL4 mutation, the mean age at presentation was 115 days, and 11 of 18 patients had survived to a median follow-up time of 46 months (Huemer et al. 2015). The phenotypes of the siblings described here represent a more extreme presentation of the FBXL4 disease spectrum as evidenced by the early presentation, short lifespan, and presence of other significant metabolic derangement in the form of severe hyperammonemia.
The early treatment of our second patient’s neonatal hyperammonemia with supplemental citrate and aspartate appears to have been effective, such that she did not require treatment with sodium phenyl acetate and sodium benzoate during the neonatal period, as was needed by her older sibling. Its successful use here suggests possible consideration of the same treatment for other patients with FBXL4 deficiency. Elevations of TCA cycle intermediates are generally observed in patients with mitochondrial disease, so the deficiency in TCA cycle intermediates in these patients may suggest a more direct role for FBXL4 function in the TCA cycle. This may be why our patients responded well to supplementation of TCA cycle intermediates.
Other genes have been associated with syndromes of early-onset mitochondrial encephalopathy with hyperammonemia. Recently, a family with multiple members presenting with lethargy, lactic acidosis, and hyperammonemia were found to have a deficiency of carbonic anhydrase VA (van Karnebeek et al. 2014). The hyperammonemia resolved with supplementation of carglumic acid, indicating that there were effects on both oxidative phosphorylation and urea cycle metabolism as in the cases presented here. However, the mechanism of hyperammonemia in that case is presumed to be due to decreased intracellular bicarbonate inhibiting urea cycle initiation. In these siblings with genetic variants in FBXL4, ongoing supplementation with citric acid and aspartate leads to temporary resolution of lactic acid and hyperammonemia. We hypothesize that this contributed to resolving both abnormalities because aspartate is required to provide nitrogen to form argininosuccinate in the urea cycle and therefore its depletion may be contributing to the hyperammonemia. Identification of additional patients with deleterious FBXL4 variants will help associate specific mutations with the range of disease phenotypes.
Conclusion
In summary, we describe two siblings with three missense variants in FBXL4 presenting with lactic acidosis and hyperammonemia. This case report expands the phenotype of FBXL4-associated mitochondrial encephalomyopathy to include early, severe hyperammonemia in the setting of TCA cycle disruption. This finding points to an expanded role for FBXL4 beyond oxidative phosphorylation. Further studies into the role of FBLX4 in TCA and urea cycle chemistry could help to guide directed therapy for patients with FBXL4 mutations, given the improvement noted with supplementation of citric acid and aspartate.
Electronic Supplementary Material
{kind=link}
Representative gas chromatography-mass spectrometry (GC-MS) urine organic acid chromatogram of a healthy child in the first week of life. Peaks representing tricarboxylic acid cycle intermediates including fumarate, 2-ketoglutarate, aconitate, and citrate are often elevated in this age group and are among the most abundant peaks in the chromatogram. The Internal standards (IS) 2-ketocaproate (approximately 11 min) and pentadecanoate (approximately 20 min) are also shown for comparison of scale (TIFF 2467 kb)
Acknowledgments
The authors will like to thank the family for their support and enrollment. This work was made possible by grants from the following institutions: PBA was supported by 1R01AR068429-01 from the National Institute of Arthritis and Musculoskeletal and Skeletal Diseases (NIAMS) of National Institute of Health (NIH) and U19HD077671 from NICHD/NHGRI/NIH. SUM was supported by T32 HD7466-19 awarded to the Newborn Medicine Division at BCH. The Manton Center for Orphan Disease Research Gene Discovery Core, Boston Children’s Hospital, also supported the work. Sanger sequencing was performed by the Molecular Genetics Core Facility of the IDDRC at Boston Children’s Hospital, supported by National Institutes of Health grant P30 HD18655.
Synopsis
FBXL4 variants are associated with mitochondrial encephalopathy, and this report expands upon the known phenotype by describing a sibling pair with early-onset mitochondrial encephalopathy complicated by hyperammonemia associated with low-normal aspartate levels.
Compliance with Ethics Guidelines
Sarah Morton, Edward Neilan, Roy Peake, Jiahai Shi, Klaus Schmitz-Abe, Meghan Towne, Kyriacos Markianos, Sanjay Prabhu, and Pankaj Agrawal declare that they have no conflict of interest.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the parents of the children included in the study.
Author Contributions: Drs. Morton, Neilan, and Agrawal had full access to all of the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Morton, Neilan, Agrawal. Acquisition of Data: Morton, Neilan, Towne, Agrawal. Analysis and interpretation of data: Morton, Neilan, Peake, Shi, Schmitz-Abe, Markianos, Agrawal. Drafting of the manuscript: Morton, Neilan, Agrawal. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: Schmitz-Abe, Markianos. Obtained funding: Agrawal. Administrative, technical, or material support: Towne, Agrawal. Study supervision: Morton, Neilan, Agrawal.
Footnotes
Authors Sarah U. Morton and Edward G. Neilan equally contributed to the manuscript.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2016_17) contains supplementary material, which is available to authorized users.
Contributor Information
Pankaj B. Agrawal, Email: pagrawal@enders.tch.harvard.edu
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Antoun G, McBride S, Vanstone JR, et al. Detailed biochemical and bioenergetic characterization of FBXL4-related encephalomyopathic mitochondrial DNA depletion. JIMD Rep. 2015 doi: 10.1007/8904_2015_491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barøy T, Pedurupillay CRJ, Bliksrud YT, et al. A novel mutation in FBXL4 in a Norwegian child with encephalomyopathic mitochondrial DNA depletion syndrome 13. Eur J Med Genet. 2016;59:342–346. doi: 10.1016/j.ejmg.2016.05.005. [DOI] [PubMed] [Google Scholar]
- Bonnen PE, Yarham JW, Besse A, et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet. 2013;93:471–481. doi: 10.1016/j.ajhg.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Seitz A, Kölker S, Hoffmann GF. Recurrent stroke-like episodes in FBXL4-associated early-onset mitochondrial encephalomyopathy. Pediatr Neurol. 2015;53:549–550. doi: 10.1016/j.pediatrneurol.2015.08.018. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013;10:186–198. doi: 10.1007/s13311-013-0177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai X, Ghezzi D, Johnson MA, et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet. 2013;93:482–495. doi: 10.1016/j.ajhg.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huemer M, Karall D, Schossig A, Abdenur JE. Clinical, morphological, biochemical, imaging and outcome parameters in 21 individuals with mitochondrial maintenance defect related to FBXL4 mutations. J Inherit Metab Dis. 2015;38(5):905–914. doi: 10.1007/s10545-015-9836-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochel F, DeLonlay P, Touati G, et al. Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy. Mol Genet Metab. 2005;84:305–312. doi: 10.1016/j.ymgme.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Tang S, Wang J, Lee N-C, et al. Mitochondrial DNA polymerase gamma mutations: an ever expanding molecular and clinical spectrum. J Med Genet. 2011;48:669–681. doi: 10.1136/jmedgenet-2011-100222. [DOI] [PubMed] [Google Scholar]
- van Karnebeek CD, Sly WS, Ross CJ, et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am J Hum Genet. 2014;94:453–461. doi: 10.1016/j.ajhg.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rechem C, Black JC, Abbas T, et al. The SKP1-Cul1-F-box and leucine-rich repeat protein 4 (SCF-FbxL4) ubiquitin ligase regulates lysine demethylase 4 A (KDM4A)/Jumonji domain-containing 2 A (JMJD2A) protein. J Biol Chem. 2011;286:30462–30470. doi: 10.1074/jbc.M111.273508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rij MC, Jansen FAR, Hellebrekers DMEI, et al. Polyhydramnios and cerebellar atrophy: a prenatal presentation of mitochondrial encephalomyopathy caused by mutations in the FBXL4 gene. Clin Case Rep. 2016;4:425–428. doi: 10.1002/ccr3.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston JT, Koepp DM, Zhu C, et al. A family of mammalian F-box proteins. Curr Biol. 1999;9:1180–1182. doi: 10.1016/S0960-9822(00)80021-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative gas chromatography-mass spectrometry (GC-MS) urine organic acid chromatogram of a healthy child in the first week of life. Peaks representing tricarboxylic acid cycle intermediates including fumarate, 2-ketoglutarate, aconitate, and citrate are often elevated in this age group and are among the most abundant peaks in the chromatogram. The Internal standards (IS) 2-ketocaproate (approximately 11 min) and pentadecanoate (approximately 20 min) are also shown for comparison of scale (TIFF 2467 kb)