

Abstract
The mitochondrial matrix protein cyclophilin D (CypD) is an essential component of the mitochondrial permeability transition pore (MPTP). Here we characterized the effects of CypD ablation on bioenergetics in the kidney. CypD loss triggers a metabolic shift in Ppif−/− male and female mouse kidneys towards glycolysis and Krebs cycle activity. The shift is accompanied by increased glucose consumption and a transcriptional upregulation of effectors of glucose metabolism in the kidney. These included activation of Akt, AMPK (only in males) and p70S6K kinases. Gender specific differences between the Ppif−/− male and female mouse kidneys were observed including activation of pro-surviving ERK1/2 kinase and inhibited expression of pro-apoptotic and pro-fibrotic JNK and TGFβ1 proteins in Ppif−/− females. They also showed the highest expression of phosphorylated-ERK1/2 and Akt S473 proteins of all four investigated animal groups. Furthermore, Ppif−/− females showed higher lactate concentrations and ATP/ADP-ratios in the kidney than males. These metabolic and transcriptional modifications could provide an additional level of protection to Ppif−/− females. In summary, loss of mitochondrial CypD results in a shift in bioenergetics and in activation of glucose-metabolism regulating Akt/AMPK/p70S6 kinase pathways that is expected to affect the capability of Ppif−/− mice kidneys to react to stimuli and injury.
Introduction
In response to oxidative or other cellular stresses, mitochondrial permeability transition is accompanied by pathological and non-specific mPT pore (mPTP) opening in the inner membrane of mitochondria. mPTP can serve as a target to prevent cell death under pathological conditions such as cardiac and brain ischemia/reperfusion (I/R) injury, myocardial infarction, stroke, and diabetes1–3.
Cyclophilin D (CypD) is encoded by Ppif and is a mitochondrial peptidyl-prolyl cis-trans isomerase that has been shown to regulate the opening of the mPTP4, 5. Genetic deletion of CypD has been shown to reduce I/R injury, presumably by inhibiting the prolonged opening of the mPTP4, 5. Furthermore, the CypD inhibitor cyclosporin A (CsA) mimics the effects of CypD deletion and reduces I/R injury in animal models as well as in humans6–9.
The majority of the studies involving Ppif−/− mice have focused on its role and underlying mechanisms in the setting of myocardial and brain I/R injury. Here, CypD inhibition is beneficial by preventing necrotic cell death5. Recently, CypD deletion has also been shown to reduce the bone loss in aging mice10. On the other hand, Ppif−/− mice are also more susceptible to heart failure initiated by several stimuli, including physiologic exercise-induced hypertrophy11, 12. Mechanistically, in the heart, CypD ablation is associated with elevated levels of mitochondrial matrix Ca2+ that in turn leads to increased glucose oxidation relative to fatty acids. This metabolic switch limits the heart’s ability to adapt during stress11, 13.
In addition to the heart and brain, CypD deletion has also been shown to protect against I/R injury in the kidney14–16. However, much less is known yet about the effects of CypD deletion on the kidney. Ischemic kidney injury is the primary cause of acute kidney injury (AKI) in hospitalized patients17. We hypothesized that CypD deletion will lead to the alteration of cellular metabolism and related signaling pathways including AKT and mitogen-activated protein kinases (MAPK) in the kidney which also act as mediators of ischemic preconditioning18, 19.
Both clinical and experimental observations support the concept that female kidneys are less susceptible to I/R injury20, 21, and that women show a slower progression rate of renal disease22. Thus we also compared the effects of CypD deletion in kidneys from male and female mice.
Materials and Methods
In vivo experiments
Mice were cared for (before and during the experimental procedures) in accordance with the policies of the Institutional Animal Care and Use Committee of the University of Colorado and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All protocols received prior approval from the University of Colorado Institutional Animal Care and Use Committee. Wild-type (WT) B6.129SF2 mice, as well as mice null for Ppif on the same background (Ppif−/−, complete knockout), were purchased from Jackson Laboratory (Bar Harbor, ME) and were used for breeding. Age-matched homozygous KO and WT animals were used for all experiments. Animals from both types were housed in the same cage to control for environmental factors. Twelve week-old male and female mice were sacrificed, and urine (by bladder puncture) and kidney tissues were collected. For kidney collection, the renal artery was clamped, the kidney removed and immediately snap-frozen until further use.
Tissue extraction for Western blot analysis
Frozen kidney tissues were ground to a fine powder using a mortar and pestle and solubilized in lysis buffer containing protease and phosphatase inhibitors (Pierce, Rockford, IL). The extracts were kept frozen at −80 °C for all subsequent analyses. The protein concentrations were determined using the Bradford protein assay kit (BioRad, Hercules, CA).
Western blot analysis
For Western blots, tissue extracts were loaded onto Biorad Bis-Tris HCl Criterion gels (various percentages). Proteins were separated using a Biorad Criterion electrophoresis system operating at 120 V and then transferred from the gel to an Immobillon-P membrane (200 mA, Millipore, Billerica, MA). Membranes were incubated with the primary antibody at 4 °C overnight, after blocking with 5% milk/2% BSA in PBS-Tween buffer. Antibodies used in this study included (the majority were raised in a rabbit and to a smaller amount in a mouse): PTEN (phosphatase and tensin homolog); protein phosphatase 2 A (PP2A); AKT unmodified and phosphorylated at Ser473 and Thr308; p70S6K (unmodified and phosphorylated at T389); p38 and p42/44 MAPK (ERK1/2) unmodified and phosphorylated at Thr180/Tyr182 and Thr202/Tyr204, respectively; JNK (unmodified and phosphorylated at Thr183/Tyr185); PAK2 (p21-activated kinase) unmodified and phosphorylated at Ser20; p70 S6 kinase unmodified and phosphorylated at Thr389 and Thr421/Ser424; β-actin (source of all antibodies above Cell Signaling Technology, Danvers, MA); TGFβ1 and TGFβ3 (transforming growth factor, Santa Cruz Biotechnology, Santa Cruz, CA). After membranes were washed three times, the secondary antibody (goat-anti rabbit and horse anti-mouse antibodies conjugated to horseradish peroxidase, Cell Signaling Technology) was added. Membranes were subsequently treated with Pierce SuperSignal West Pico or Femto Solution (Pierce) following the manufacturer’s instructions. A UVP system (BioImaging Systems, Upland, CA) was used to detect the horseradish peroxidase reaction on the membrane. ImageJ software (NIH, Bethesda, MD) was used for quantitative densitometric analysis of select gel band intensities. Densitometry data were normalized to β-actin.
Quantification of high-energy phosphate metabolites in kidney tissues
To assess the effect of CypD ablation on energy metabolism, we determined high-energy phosphate metabolite concentrations in snap-frozen kidneys using a previously described assay23. An average of 100 mg kidney tissue was homogenized in a mortar grinder over liquid nitrogen and extracted with 6 mL ice-cold PCA (12%) as described previously23. The samples were centrifuged, the liquid phase removed and neutralized to a pH of 7.0–7.3 using KOH. To separate from perchlorate salts, the neutralized samples were centrifuged again, and the supernatant lyophilized overnight. Lyophilisates were then re-dissolved in 0.5 ml water and adjusted to pH 6.5. An Agilent series 1100 HPLC (Agilent Technologies, Palo Alto, CA) coupled to an API4000 triple quadrupole mass spectrometer (AB Sciex, Concord, ON) equipped with an electrospray ionization (ESI) source was employed for quantitation of nucleotide mono-, di-, triphosphates, FAD(H2) and NAD(H). Energy charge was calculated as [ATP + (0.5 × ADP)]/(ATP + ADP + AMP).
Quantification of Krebs cycle, and purine degradation metabolites in kidney tissues and urine
Citrate, succinate, glucose, α-ketoglutarate and lactate were quantified in PCA kidney tissue extracts (vide supra) and 1:20-diluted mouse urine samples (diluted with 40 nM NaOH aqueous buffer). The internal standard solution was added at a 25:1 ratio. It was prepared in 40 nM NaOH aqueous buffer and contained 200 µM d6-glucose and d4-succinate.
All samples were analyzed on an Agilent 1100 series HPLC (Agilent Technologies, Palo Alto, CA) interfaced with a positive/negative ESI API4000 tandem mass spectrometer (AB Sciex). Analytes were separated using a 150 × 3 mm Luna HILIC, 3 µm column (Phenomenex, Torrance, CA) at an HPLC solvent flow rate of 450 µL/min. The solvents were 0.1% aqueous formic acid (mobile phase A) and acetonitrile (mobile phase B). The gradient was: 0–1 min 5% acetonitrile, 1.0–3.5 min 5% acetonitrile to 15% acetonitrile, 3.5–4.5 100% acetonitrile. The column was then re-equilibrated to starting conditions (5% acetonitrile) between 4.6 and 5.5 min. The mass spectrometry parameters were (dual polarity: positive: 0–1.79 min, negative: 1.8–5.5 min): ion source gas one: 40, ion source gas two 45, source temperature 500 °C, collision gas 10, curtain gas 20, and ion source voltage of +/−4500V for positive and negative ion modes, respectively.
Kidney histology
For hematoxylin and eosin (H&E) staining, kidney tissue samples were fixed in 10% buffered formaldehyde and embedded in paraffin, incubated for 5 minutes in Harris hematoxylin solution and for 60 seconds in eosin solution. Sections were washed with plain water, differentiated in 1% hydrochloric acid (HCl) + 50% ethanol, and stain intensity was optimized in ammonia water. Finally, sections were rinsed in 70% ethyl alcohol and dehydrated in xylene solution. Semi quantitative scoring system . The slides were scanned using an Aperio Scanscope with Spectrum Software (v. 10.0.1346.1805). The images were then analyzed using Aperio Imagescope software (v. 9.1.19.1574; APERIO Technologies, Vista, CA). Inflammatory infiltration of the kidneys was assessed by measurement of the density of nuclei using color saturation. In addition, 25 high-powered fields were examined in the tubulo-interstitium of the cortex and outer medulla of each section. The kidneys were examined for inflammation, epithelial necrosis, loss of brush border, and tubular dilatation. Twenty five glomeruli were examined for histologic changes and for vascular congestion. Their sizes as well as the average area were determined for each slide. To determine the number of glomeruli, the number of glomeruli within a region of the cortex was counted. The results are reported as the number of glomeruli corrected for the area analyzed.
Statistical analysis
All numerical data are presented as mean ± standard deviation. One-way ANOVA with Tukey’s post hoc test was used to determine differences among groups (either between the WT and Ppif−/− animals within the same gender and between genders). The level of significance was set at p < 0.05 for all tests (SPSS, version 24.0, IBM/SPSS, Armonk, NY).
Results
Kidney histology
Comparison of kidney histologies revealed no visible differences (Fig. 1A), with the only exception that the size (area) of glomeruli were slightly enlarged in Ppif−/− mice as compared to their wild-type counterparts (in females and males, respectively, Fig. 1B). At the same time, the number of glomeruli remained unchanged (Fig. 1C).
Figure 1.

Kidney histologies of WT and Ppif−/− (KO) animals. (A) Evaluation of kidney histologies revealed a (B) larger area of glomeruli in Ppif−/− male and female mice as compared to their WT counterparts while (C) the number of glomeruli per mm2 remained the same. Arrows indicate glomeruli. Size bars indicate 200 µm (10X) and 100 µm (40x). Significance levels are: *p < 0.05 for WT versus Ppif−/− animals.
Glucose metabolism
Metabolite changes in the kidney
Kidneys of male Ppif−/− animals showed a significantly higher energy charge as compared to their wild-type counterparts (Fig. 2A). This augmented energy production was accompanied by increased glucose uptake and increased Krebs cycle activity, as suggested by the significantly higher levels of glucose, citrate and succinate in the kidney tissues of Ppif−/− males (Table 1). Interestingly, female Ppif−/− animals showed only significant increase in succinate when compared to WT females (Table 1). Their expression of glucose transporter Glut1, however, was the highest among all investigated animal groups (Fig. 2B), and so was the concentrations of the glycolysis product lactate (Table 1). Ppif−/− mice of both genders had higher kidney lactate concentrations as compared to their WT counterparts (Table 1).
Figure 2.
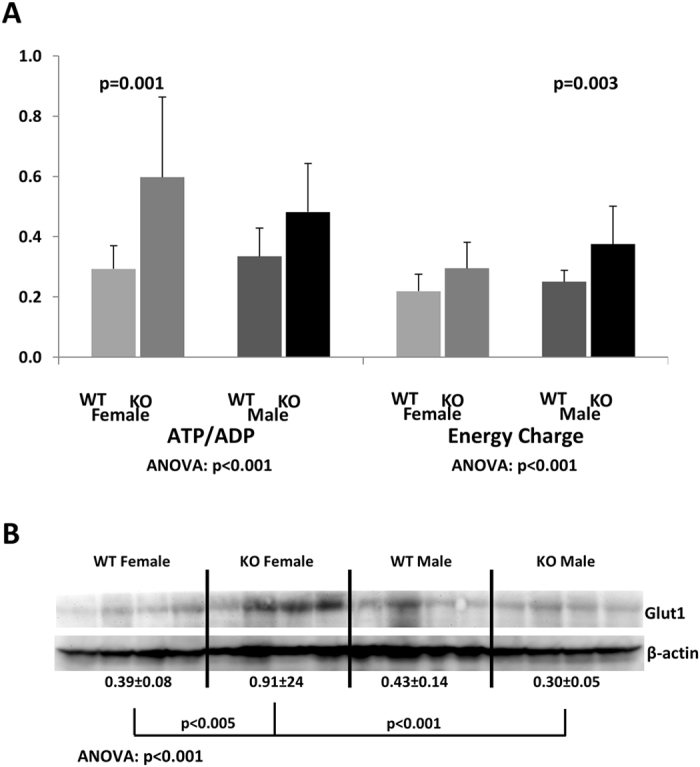
Energy state of the kidney following CypD ablation. (A) Comparison of ATP/ADP-ratios and energy charge between the kidneys of female and male Ppif−/− (KO) and wild-type (WT) mice. Data is presented as average ± SD (n = 9–14) (measured concentrations [µM] were normalized to protein [mg] prior to ratio calculations). (B) Expression of Glut1 glucose transporter. Data was normalized to β-actin and is presented as average ± SD (arbitrary units, n = 4). One-way ANOVA results with Tukey post-hoc significance levels are presented below. Abbreviations: ADP: adenosine diphosphate, ATP: adenosine triphosphate, AMP: adenosine monophosphate, energy charge calculated as [ATP + (0.5 × ADP)]/(ATP + ADP + AMP).
Table 1.
Comparison of metabolite concentrations (glucose metabolism including Krebs cycle and glycolysis) as measured in the kidneys from female and male Ppif−/− (KO) and wild-type (WT) mice.
| Citrate ANOVA: p < 0.001 | α-KGD ANOVA: p = 0.201 | Succinate ANOVA: p = 0.001 | Lactate ANOVA: p = 0.002 | Glucose ANOVA: p = 0.003 | |
|---|---|---|---|---|---|
| WT Female | 105 ± 16 | 48 ± 8.6 | 175 ± 34 | 1823 ± 255 | 1438 ± 481 |
| KO Female | 160 ± 32# | 47 ± 14 | 305 ± 86* | 2612 ± 529** | 2156 ± 455## |
| WT Male | 109 ± 30 | 35 ± 14 | 156 ± 15 | 1890 ± 193 | 1787 ± 680 |
| KO Male | 244 ± 63*** | 31 ± 18 | 401 ± 148*** | 2388 ± 97* | 4498 ± 1766*** |
Data is presented as average ± SD (n = 5–6; in µM/ mg protein). One-way ANOVA significances are presented below. Tukey post hoc significance levels are as follows: *p < 0.05, **p < 0.01, ***p < 0.001 for WT versus Ppif−/− animals (male and female), and #p < 0.05 and ##p < 0.01 for male versus female (Ppif−/− and WT, respectively). Abbreviations: α-KGD: α-ketoglutarate.
In terms of gender differences, higher intracellular kidney glucose and citrate concentrations were detected in Ppif−/− males as compared to corresponding females (Table 1).
Metabolites excretion in the urine
Only urinary citrate concentrations were higher in Ppif−/− males as compared to WT (Table 2). Similarly, in Ppif−/− females, the urinary concentrations of Krebs cycle and glycolysis intermediates and products did not differ as compared to WT females (Table 2).
Table 2.
Comparison of metabolite as measured in the urine from female and male Ppif−/− (KO) and wild-type (WT) mice. Data is presented as average ± SD (n = 7–12 in µM per mM creatinine).
| Citrate ANOVA: p = 0.032 | α-KGD ANOVA: p = 0.053 | Succinate ANOVA: p = 0.496 | Lactate ANOVA: p = 0.002 | Glucose ANOVA: p = 0.058 | |
|---|---|---|---|---|---|
| WT Female | 239 ± 165 | 323 ± 225 | 67 ± 33 | 49 ± 26## | 3373 ± 632 |
| KO Female | 353 ± 184 | 218 ± 128 | 59 ± 34 | 36 ± 9.0 | 4353 ± 2292 |
| WT Male | 144 ± 54 | 111 ± 33 | 42 ± 19 | 19 ± 9.1 | 2864 ± 916 |
| KO Male | 240 ± 105* | 335 ± 222 | 63 ± 37 | 28 ± 13 | 2829 ± 800 |
One-way ANOVA significances are presented below. Tukey post hoc significance levels are as follows: *p < 0.05 for WT versus Ppif−/− animals (male and female), and ##p < 0.01 for male versus female (Ppif−/− and WT, respectively). Abbreviations: α-KGD: α-ketoglutarate.
Glucose Metabolism Regulation: Akt/mTOR Pathway
Activity of Akt kinase that serves as a glucose uptake and metabolism regulator was measured through its phosphorylation at two sites: S473 and T308. A significant increase in the phospho-Akt S473 to Akt ratio was observed in Ppif−/− animals (Fig. 3A), whereas no change in the activation of Akt at the T308 site was noted (Fig. 3B). Although no change in the phospho-Akt T308 to Akt ratio was observed, expression of its downstream target, phospho-p70S6 kinase (expressed as phospho-p70S6K/p70S6K-ratio) was significantly higher in Ppif−/− animals (Fig. 3C). The expression of the energy regulator AMPK was significantly higher in kidneys of malePpif−/− animals (Fig. 3D). Interestingly, these increases of the phosphorylated AktS473, p70S6K and AMPK proteins, were not accompanied by changes in the expression of PPP2A phosphatases (Fig. 3E). Other than noted in heart or lung tissues24, but in alignment with the expression in aortic tissue24, naïve PTEN did not change in the kidneys of WT versus Ppif−/− animals (Fig. 3E).
Figure 3.

Western blot analysis of proteins involved in the regulation of glucose metabolism. (A) Activated Akt (presented as the phospho-Akt S473 to Akt ratio) was significantly higher in kidneys of Ppif−/− than in WT animals. (B) No change in the phospho-Akt T308 levels between Ppif−/− and WT animals was found. (C) The expression of phospho-p70S6 kinase T389 was higher in Ppif−/− animals and (D) so was expression of phospho-AMPK, however significantly only in males. (E) Expression of PP2A and PTEN proteins (with their respective β-actin loading controls) was similar among groups. Data is presented as average ± SD (n = 4). One-way ANOVA significances were as follows: (A) P < 0.001, (B) P < 0.001, (C) P = 0.363 and P < 0.001 for phospho-p70S6K/p70S6K 85 kD (grey bars) and 70 kDa (black bars) forms, respectively, (D) P = 0.006 and (E) P = 0.037 for PPP2CB, P = 0.009 for PP2R2A, and P = 0.281 for PTEN. Significance levels are: *p < 0.05, **p < 0.01 and ***p < 0.001 for WT versus Ppif−/− animals (male and female); #p < 0.05 and ##p < 0.01 for male versus female in WT and Ppif−/− groups.
In regards to gender differences, kidneys of Ppif−/− females contained higher levels of phospho-Akt S473 as compared to Ppif−/− males (Fig. 3A). In addition, both WT and Ppif−/− females expressed higher levels of phospho-Akt T308 as well as phospho-p70S6 kinase when compared to their respective male counterparts (Fig. 3B,D).
Mitogen-activated protein kinases (MAPKs) Pathways: p38, p42/44 (ERK1/2) and JNK
We observed no change in the phosphorylation of either p42/44 or p38 MAPKs in Ppif−/− males, whereas Ppif−/− females showed higher expression of both phosphorylated MAPK proteins (as ratios of phosphorylated to naïve protein forms) as compared to female WT controls (Fig. 4A,B, respectively). No change in the phosphorylation status of JNK MAPK was seen in Ppif−/− versus WT animals (phospho-JNK to JNK, Fig. 4C).
Figure 4.

Western blot analysis of MAPK proteins. (A) Increased phosphorylation of p42/44 (expressed as phospho-p42/44 at Thr202/Tyr204 to p42/44-ratio) and (B) p38 (expressed as phospho-p38 at Thr180/Tyr182 to p38-ratio) proteins was evident in the kidneys of Ppif−/− females. (C) However, a reduction of JNK phosphorylation (phospho-JNK at Thr183/Tyr185 to JNK) was evident in both WT and Ppif−/− females as compared to their respective males (grey bars: band at 54 kDa, black bars: band at 46 kDa). Data is presented as average ± SD (n = 4). One-way ANOVA significances were as follows: (A) P = 0.005, (B) P = 0.035 and (C) P = 0.156 and P = 0.005 for pJNK/JNK 54 kD and 46 kDa forms, respectively. Significance levels are: *p < 0.05 and **p < 0.01 for WT versus Ppif−/− animals (male and female); #p < 0.05 and ##p < 0.01 for male versus female in WT and Ppif−/− groups.
Kidneys of Ppif−/− females contained significantly higher levels of phospho- p42/44 MAPK, but lower levels of phospho-JNK MAPK as compared to Ppif−/− males (Fig. 4A,C).
TGF-β signaling
As an interaction between high glucose concentration and the synthesis of TGFβ in renal cells25, 26 had been reported24, 25, we examined the expression of this growth factor in kidneys of Ppif−/− animals. Expression of TGFβ1 and TGFβ3, two proteins within the TGFβ-family associated with the development of renal fibrosis27, 28, remained unchanged between the Ppif−/− and WT animals (Fig. 5A). However, Ppif−/− females showed significantly lower expression of TGFβ1 as compared to the corresponding males (Fig. 5A). This observation seems interesting when considering the often slower rate of progression of renal disease in women22.
Figure 5.

Western blot analysis of TGFβ signaling-associated proteins. (A) TGFβ1 was significantly higher in Ppif−/− males as compared to females, while no change in TGFβ3 expression was noted among the groups. (B) No change in the activation of PAK2 (as pPAK2 Ser20) was observed. Data is presented as average ± SD (n = 4). One-way ANOVA significances were as follows: (A) P = 0.004 and P = 0.469 for TGFβ1 and TGFβ3, respectively, and (B) P = 0.164 for pPAK Ser20/PAK2, respectively. Significance levels are: ##p < 0.01 for male versus female in WT and Ppif−/− groups.
In addition to TGFβ, we examined the expression and activation status of the PAK2 kinase that are also involved in the development and progression of renal fibrosis. No change in the expression of unmodified or phosphorylated PAK2 protein was observed (Fig. 5B).
Discussion
As shown in the present study, ablation of CypD is associated with reorganization of energy pathways in the kidney towards a state of higher glucose utilization.
The kidney contributes to glucose homeostasis through processes of gluconeogenesis, glucose filtration, glucose reabsorption, and glucose consumption. Krebs cycle intermediates are important substrates for renal metabolism as they account for 10–15% of oxidative metabolism in the kidney29. Interestingly, in vitro studies have shown that renal cells can be salvaged from hypoxia-reoxygenation-induced mitochondrial injury by the supplementation of Krebs cycle intermediates which mediate ATP production and prevent an energy deficit30–32. Furthermore, while the renal cortex uses fatty acid oxidation as the main source of energy, the renal medulla is mainly producing and taking up lactate33.
Morphologically, kidneys lacking CypD did not look different from those containing CypD with the only exception that CypD ablation led to enlargement of glomeruli. Metabolically, kidneys from CypD knockout mice displayed higher glucose consumption, Krebs cycle and glycolytical activity and ATP production than kidneys from WT mice. In Ppif−/− males, increased ATP levels were accompanied by a higher energy charge, whereas in Ppif−/− females concentration of AMP also increased resulting in an unchanged energy charge.
Consistent with our data, previous metabolic profiling studies suggested a switch away from fatty acid oxidation towards glycolytic and Krebs cycle metabolism in hearts, brains and livers of Ppif−/− animals12, 13, 34. In the mouse heart, genetic deletion of CypD correlated with elevated levels of mitochondrial Ca2+ and led to alteration in branched chain amino acid metabolism, pyruvate metabolism and the Krebs cycle13. However, CypD deletion did not reduce the activity of proteins involved in cellular respiration13, 35. Yet, in primary hepatocytes and embryonic fibroblasts, CypD loss triggered a global compensatory shift towards glycolysis, with increased glucose consumption and higher ATP production34. The same metabolic switch was seen in our study.
We also observed an activation of AMPK, a crucial cellular energy sensor, in male CypD-deleted mouse kidneys. In females, the degree of activation was not as pronounced and did not reach statistical significance, as was the case with the energy charge. Once activated by impaired energy metabolism, AMPK promotes ATP production by increasing the activity or expression of proteins involved in catabolism while conserving ATP by switching off biosynthetic pathways36. This could mean that the CypD-deleted mouse kidney is reacting to an accumulation of mitochondrial Ca2+ through a lower cell energy demand and redistribution of metabolic pathways, such as a reduction in fatty acid β-oxidation13, 34, 37, rather than stimulation of mitochondrial respiration38, 39. The question if this state of metabolic alteration is ultimately causing mitochondrial stress while decreasing the kidney’s metabolic reserves and rendering it more sensitive to decompensation even under subtle stress conditions, such as described for Ppif−/− heart12, remains to be investigated.
The alteration of kidney metabolism was associated with alterations in cellular signaling pathways. An upregulation of kinase cascades associated with insulin signaling including phosphorylated AMPK, Akt and mTOR was observed in kidneys of Ppif−/− mice. Previous studies had shown that Ppif−/− animals experience the expansion of insulin-producing β-cells and mild hyperinsulinemia34. Increased secretion of insulin could be the driving force behind the increase in Akt/mTOR and glycolytic activity, since the kidney is known to respond to insulin signaling40, 41. Specifically, while no change in the phosphorylation of Akt at T308 occurred, kidneys of Ppif−/− mice had higher levels of Akt phosphorylated at S473. Downstream, these two sites have different targets: while Akt phosphorylated at S473 is a regulating member of the glucose and lipid metabolism42, 43, Akt phosphorylated at T308 primarily leads to the activation of mTORC1, p70S6K, and protein synthesis44, 45. Therefore, the observed activation of AktS473 could be responsible for the observed glucose metabolism shift and increase in glycolysis42, 43. However, notwithstanding a lack of alteration in AktT308, an activation of p70S6 kinase in the kidneys of Ppif−/− mice was seen. This kinase has been shown to protect against I/R injury and to promote cell survival and protein production46.
The activation of Akt is tied to MAPK proteins including p38, p42/44 (ERK1/2) and JNK47, 48. These MAPKs are regulated by stress or injury not only in the kidney but also other organs. ERK promotes cell survival while JNK and p38 lead to cell death. Some studies postulate that it may be the balance between the two (ERK versus JNK) that determines the fate of the cell49. Interestingly, while we did not observe a change in either of these MAPKs in kidneys of Ppif−/− males, Ppif−/− females expressed higher levels of phosphorylated ERK1/2 and lower level of activated JNK kinases as compared to their wild type counterparts. Ppif−/− females had the highest expression of phosphorylated ERK1/2 and Akt S473 proteins among all four investigated animal groups. Furthermore, expression of the glucose transporter Glut1 was the highest in the female Ppif−/− kidney, which metabolically also showed the highest lactate concentration. Based on these gender differences, it may be speculated that the CypD ablation is augmenting the protective effects of estrogen in the female kidney that is known to block inflammatory and apoptotic activities50–53.
With the close interaction between energy demand, supply and activity of protein kinases including Akt, AMPK and mTORC1/mTORC244, the observed switch in energy metabolism following CypD ablation is not surprising. The reorganization of this central controlling hub that regulates cellular functions may also be responsible for the protective effects CypD ablation has during I/R injury or stroke5, 12, 54–56.
In conclusion, it is reasonable to expect that the reorganization of cell signaling pathways and resulting metabolic changes following the loss of CypD as found in the present study (Fig. 6) leads to changes in the response of kidneys of Ppif−/− mice to stimuli and injury and that such responses may differ between genders.
Figure 6.
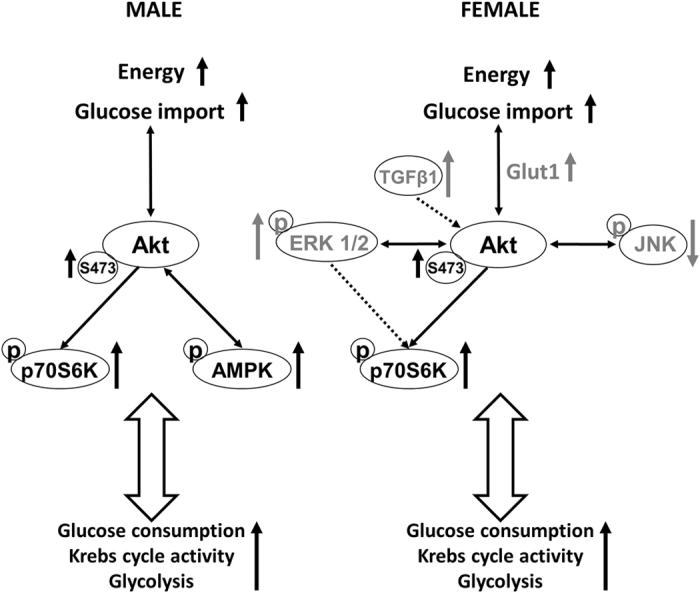
Summary of changes in cell signaling pathways in mouse kidneys as a result of CypD ablation. Increased expressions of phospho-AKT S473 and phospho-p70S6K and pAMPK (in males only) were observed in Ppif−/− mice. Additionally, Ppif−/− females (presented on the right side of the image) showed a reduced expression of TGFβ1 as well as higher expression of phosphorylated ERK1/2 and p38 proteins. ↑: increase compared to WT controls, ↓: decrease compared to WT controls. Please note that a direct link between the phosphorylation of Akt and phosphorylation of MAP kinases p38 and ERK1/2 is not fully confirmed in the here used mouse model. Please also note that, since our experiments were performed in the whole kidney, the exact contribution of each subcellular fraction to the observed changes requires further investigation.
Author Contributions
Je. K. and U.C. have designed the work, A.P. and Je. K. have performed the animal studies, J.T. has performed the histology evaluations, has all authors (Je. K., A.P., J.T., Jo. K., U.C.) have been involved in data collection and data analysis and interpretation; all authors have helped with the drafting of the article and its critical revision.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/S0014-5793(01)02316-X. [DOI] [PubMed] [Google Scholar]
- 2.Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis: an international journal on programmed cell death. 2007;12:835–840. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- 3.Brenner C. & Moulin, M. Physiological roles of the permeability transition pore. Circulation research. 2012;111:1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 5.Baines CP, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 6.Piot C, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. The New England journal of medicine. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 7.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. Journal of molecular and cellular cardiology. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 8.Gomez L, et al. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. American journal of physiology. Heart and circulatory physiology. 2007;293:H1654–1661. doi: 10.1152/ajpheart.01378.2006. [DOI] [PubMed] [Google Scholar]
- 9.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovascular research. 2003;60:617–625. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 10.Shum LC, et al. Cyclophilin D Knock-Out Mice Show Enhanced Resistance to Osteoporosis and to Metabolic Changes Observed in Aging Bone. PloS one. 2016;11 doi: 10.1371/journal.pone.0155709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circulation journal: official journal of the Japanese Circulation Society. 2013;77:1111–1122. doi: 10.1253/circj.CJ-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elrod JW, et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2 + ) exchange, metabolic flexibility, and propensity for heart failure in mice. The Journal of clinical investigation. 2010;120:3680–3687. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menazza S, et al. CypD(−/−) hearts have altered levels of proteins involved in Krebs cycle, branch chain amino acid degradation and pyruvate metabolism. Journal of molecular and cellular cardiology. 2013;56:81–90. doi: 10.1016/j.yjmcc.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol. 2009;297:F749–759. doi: 10.1152/ajprenal.00239.2009. [DOI] [PubMed] [Google Scholar]
- 15.Hu W, et al. Knockdown of Cyclophilin D Gene by RNAi Protects Rat from Ischemia/ Reperfusion-Induced Renal Injury. Kidney & blood pressure research. 2010;33:193–199. doi: 10.1159/000316704. [DOI] [PubMed] [Google Scholar]
- 16.Park JS, Pasupulati R, Feldkamp T, Roeser NF, Weinberg JM. Cyclophilin D and the mitochondrial permeability transition in kidney proximal tubules after hypoxic and ischemic injury. Am J Physiol Renal Physiol. 2011;301:F134–150. doi: 10.1152/ajprenal.00033.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singbartl K, Kellum JA. AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012;81:819–825. doi: 10.1038/ki.2011.339. [DOI] [PubMed] [Google Scholar]
- 18.Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart failure reviews. 2007;12:181–188. doi: 10.1007/s10741-007-9025-2. [DOI] [PubMed] [Google Scholar]
- 19.Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovascular drugs and therapy/sponsored by the International Society of Cardiovascular Pharmacotherapy. 2010;24:225–234. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kher A, et al. Cellular and molecular mechanisms of sex differences in renal ischemia-reperfusion injury. Cardiovascular research. 2005;67:594–603. doi: 10.1016/j.cardiores.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Wei Q, Wang MH, Dong Z. Differential gender differences in ischemic and nephrotoxic acute renal failure. Am J Nephrol. 2005;25:491–499. doi: 10.1159/000088171. [DOI] [PubMed] [Google Scholar]
- 22.Neugarten J, Golestaneh L. Gender and the prevalence and progression of renal disease. Advances in chronic kidney disease. 2013;20:390–395. doi: 10.1053/j.ackd.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Klawitter J, Schmitz V, Klawitter J, Leibfritz D, Christians U. Development and validation of an assay for the quantification of 11 nucleotides using LC/LC-electrospray ionization-MS. Anal Biochem. 2007;365:230–239. doi: 10.1016/j.ab.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Marcu R, et al. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circ Res. 2015;116:1336–1345. doi: 10.1161/CIRCRESAHA.116.304881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lan HY, Chung AC. Transforming growth factor-beta and Smads. Contrib Nephrol. 2011;170:75–82. doi: 10.1159/000324949. [DOI] [PubMed] [Google Scholar]
- 26.Diamond-Stanic MK, You YH, Sharma K. Sugar, sex, and TGF-beta in diabetic nephropathy. Semin Nephrol. 2012;32:261–268. doi: 10.1016/j.semnephrol.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gewin L, Zent R. How does TGF-beta mediate tubulointerstitial fibrosis? Semin Nephrol. 2012;32:228–235. doi: 10.1016/j.semnephrol.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moses HL, Yang EY, Pietenpol JA. TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell. 1990;63:245–247. doi: 10.1016/0092-8674(90)90155-8. [DOI] [PubMed] [Google Scholar]
- 29.Simpson DP. Citrate excretion: a window on renal metabolism. The American journal of physiology. 1983;244:F223–234. doi: 10.1152/ajprenal.1983.244.3.F223. [DOI] [PubMed] [Google Scholar]
- 30.Weinberg JM, Venkatachalam MA, Roeser NF, Nissim I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci USA. 2000;97:2826–2831. doi: 10.1073/pnas.97.6.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldkamp T, Kribben A, Roeser NF, Ostrowski T, Weinberg JM. Alleviation of fatty acid and hypoxia-reoxygenation-induced proximal tubule deenergization by ADP/ATP carrier inhibition and glutamate. Am J Physiol Renal Physiol. 2007;292:F1606–1616. doi: 10.1152/ajprenal.00476.2006. [DOI] [PubMed] [Google Scholar]
- 32.Feldkamp T, et al. Evidence for involvement of nonesterified fatty acid-induced protonophoric uncoupling during mitochondrial dysfunction caused by hypoxia and reoxygenation. Nephrol Dial Transplant. 2009;24:43–51. doi: 10.1093/ndt/gfn436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabetic medicine: a journal of the British Diabetic Association. 2010;27:136–142. doi: 10.1111/j.1464-5491.2009.02894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tavecchio M, Lisanti S, Bennett MJ, Languino LR, Altieri DC. Deletion of cyclophilin D impairs beta-oxidation and promotes glucose metabolism. Scientific reports. 2015;5 doi: 10.1038/srep15981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez-Aguilar M, Baines CP. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochimica et biophysica acta. 2015;1850:2041–2047. doi: 10.1016/j.bbagen.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews. Molecular cell biology. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen TT, et al. Cyclophilin D modulates mitochondrial acetylome. Circulation research. 2013;113:1308–1319. doi: 10.1161/CIRCRESAHA.113.301867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jennings RB, Murry CE, Reimer KA. Energy metabolism in preconditioned and control myocardium: effect of total ischemia. Journal of molecular and cellular cardiology. 1991;23:1449–1458. doi: 10.1016/0022-2828(91)90190-W. [DOI] [PubMed] [Google Scholar]
- 39.Nadtochiy SM, et al. Metabolomic profiling of the heart during acute ischemic preconditioning reveals a role for SIRT1 in rapid cardioprotective metabolic adaptation. Journal of molecular and cellular cardiology. 2015;88:64–72. doi: 10.1016/j.yjmcc.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hale LJ, Coward RJ. Insulin signalling to the kidney in health and disease. Clinical science. 2013;124:351–370. doi: 10.1042/CS20120378. [DOI] [PubMed] [Google Scholar]
- 41.Kuczkowski, A. & Brinkkoetter, P. T. Metabolism and homeostasis in the kidney: metabolic regulation through insulin signaling in the kidney. Cell and tissue research, doi:10.1007/s00441-017-2619-7 (2017). [DOI] [PubMed]
- 42.Hagiwara A, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell metabolism. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 43.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Seminars in cancer biology. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vadlakonda L, Dash A, Pasupuleti M, Anil Kumar K, Reddanna P. The Paradox of Akt-mTOR Interactions. Frontiers in oncology. 2013;3 doi: 10.3389/fonc.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plas DR, Thompson CB. Akt-dependent transformation: there is more to growth than just surviving. Oncogene. 2005;24:7435–7442. doi: 10.1038/sj.onc.1209097. [DOI] [PubMed] [Google Scholar]
- 46.Reinhard C, Thomas G, Kozma SC. A single gene encodes two isoforms of the p70 S6 kinase: activation upon mitogenic stimulation. Proc Natl Acad Sci USA. 1992;89:4052–4056. doi: 10.1073/pnas.89.9.4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rane MJ, et al. Interplay between Akt and p38 MAPK pathways in the regulation of renal tubular cell apoptosis associated with diabetic nephropathy. Am J Physiol Renal Physiol. 2010;298:F49–61. doi: 10.1152/ajprenal.00032.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwon DS, et al. Signal transduction of MEK/ERK and PI3K/Akt activation by hypoxia/reoxygenation in renal epithelial cells. European journal of cell biology. 2006;85:1189–1199. doi: 10.1016/j.ejcb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 49.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 50.Srivastava S, et al. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. The Journal of clinical investigation. 1999;104:503–513. doi: 10.1172/JCI7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu J, Eto M, Akishita M, Okabe T, Ouchi Y. A selective estrogen receptor modulator inhibits TNF-alpha-induced apoptosis by activating ERK1/2 signaling pathway in vascular endothelial cells. Vascular pharmacology. 2009;51:21–28. doi: 10.1016/j.vph.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 52.Baylis C. Sexual dimorphism in the aging kidney: differences in the nitric oxide system. Nature reviews. Nephrology. 2009;5:384–396. doi: 10.1038/nrneph.2009.90. [DOI] [PubMed] [Google Scholar]
- 53.Silbiger SR, Neugarten J. The impact of gender on the progression of chronic renal disease. American journal of kidney diseases: the official journal of the National Kidney Foundation. 1995;25:515–533. doi: 10.1016/0272-6386(95)90119-1. [DOI] [PubMed] [Google Scholar]
- 54.Ban K, Kozar RA. Protective role of p70S6K in intestinal ischemia/reperfusion injury in mice. PloS one. 2012;7 doi: 10.1371/journal.pone.0041584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tavecchio M, et al. Cyclophilin D extramitochondrial signaling controls cell cycle progression and chemokine-directed cell motility. J Biol Chem. 2013;288:5553–5561. doi: 10.1074/jbc.M112.433045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim SY, et al. Mitochondrial cyclophilin-D as a potential therapeutic target for post-myocardial infarction heart failure. Journal of cellular and molecular medicine. 2011;15:2443–2451. doi: 10.1111/j.1582-4934.2010.01235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]