

ABSTRACT
NAD+ is an important cofactor for enzymatic oxidation reactions in all living organisms, including (hyper)thermophiles. However, NAD+ is susceptible to thermal degradation at high temperatures. It can thus be expected that (hyper)thermophiles harbor mechanisms that maintain in vivo NAD+ concentrations and possibly remove and/or reuse undesirable degradation products of NAD+. Here we confirmed that at 85°C, thermal degradation of NAD+ results mostly in the generation of nicotinamide and ADP-ribose, the latter known to display toxicity by spontaneously linking to proteins. The hyperthermophilic archaeon Thermococcus kodakarensis possesses a putative ADP-ribose pyrophosphatase (ADPR-PPase) encoded by the TK2284 gene. ADPR-PPase hydrolyzes ADP-ribose to ribose 5-phosphate (R5P) and AMP. The purified recombinant TK2284 protein exhibited activity toward ADP-ribose as well as ADP-glucose. Kinetic analyses revealed a much higher catalytic efficiency toward ADP-ribose, suggesting that ADP-ribose was the physiological substrate. To gain insight into the physiological function of TK2284, a TK2284 gene disruption strain was constructed and examined. Incubation of NAD+ in the cell extract of the mutant strain at 85°C resulted in higher ADP-ribose accumulation and lower AMP production compared with those in experiments with the host strain cell extract. The mutant strain also exhibited lower cell yield and specific growth rates in a synthetic amino acid medium compared with those of the host strain. The results obtained here suggest that the ADPR-PPase in T. kodakarensis is responsible for the cleavage of ADP-ribose to R5P and AMP, providing a means to utilize the otherwise dead-end product of NAD+ breakdown.
IMPORTANCE Hyperthermophilic microorganisms living under high temperature conditions should have mechanisms that deal with the degradation of thermolabile molecules. NAD+ is an important cofactor for enzymatic oxidation reactions and is susceptible to thermal degradation to ADP-ribose and nicotinamide. Here we show that an ADP-ribose pyrophosphatase homolog from the hyperthermophilic archaeon Thermococcus kodakarensis converts the detrimental ADP-ribose to ribose 5-phosphate and AMP, compounds that can be directed to central carbon metabolism. This physiological role for ADP-ribose pyrophosphatases might be universal in hyperthermophiles, as their homologs are widely distributed among both hyperthermophilic bacteria and archaea.
KEYWORDS: ADP-ribose pyrophosphatase, Archaea, NAD+, Thermococcus, hyperthermophiles, thermal degradation
INTRODUCTION
NAD+ is an essential cofactor for enzymatic oxidation reactions in all living organisms. Besides its major role as an electron carrier, NAD+ acts as the adenylate donor in DNA ligase reactions in bacteria (1). NAD+ is also the starting material for ADP-ribosylation of proteins, a type of posttranslational modification found in eukaryotes and a number of bacteria (2, 3). The presence of NAD+-dependent enzymes has been demonstrated in a number of hyperthermophiles (4–8). However, it is also known that NAD+ is readily degraded at high temperature as well as at high pH (9, 10). Therefore, we supposed that hyperthermophiles harbor metabolic features to efficiently (re)generate NAD+ and maintain its in vivo concentration at adequate levels. A system to remove undesirable effects of the degradation products from NAD+ might also be present.
The degradation rate of NAD+ has previously been examined in vitro (9). At 100°C and in 0.1 M Tris buffer (pH 7.60 at 25°C), the half-life (t1/2) of NAD+ was approximately 10 min (rate constant of degradation [kd] = 0.050 · min−1). In the degradation of NAD+, the nicotinamide-ribose linkage is cleaved, resulting in the generation of nicotinamide and ADP-ribose (10). Free ADP-ribose can cause glycation (nonenzymatic glycosidation) of proteins, such as eukaryotic histones (11). Through hydrogen rearrangement, the glycated proteins are converted to ketoamine glycation conjugates. Furthermore, by self-cleavage, the conjugates can also be converted to advanced glycation end products (AGEs) such as Nε-carboxymethyllysine (CML), which can result in cell damage through AGEs acceptors (12), at least in eukaryotes. Therefore, the accumulation of free ADP-ribose can be expected to be detrimental to the cell (13, 14). NADH is also susceptible to thermal damage. At 35°C, pH 6.0, NADH is spontaneously converted to a hydrated form (referred to as NADHX) (15). The produced NADHX is repaired to NADH by two enzymes, NAD(P)HX epimerase and NAD(P)HX dehydratase. Proteins showing similarities to these two enzymes are widely distributed in eukaryotes and bacteria, and the activities of these enzymes have already been examined in several eukaryotes and bacteria. Many archaea possess a gene that is 20 to 30% identical to that of the NAD(P)HX epimerase/dehydratase fusion protein from Escherichia coli, but enzymatic analysis will be required to confirm the function of the gene.
Thermococcus kodakarensis is a hyperthermophilic archaeon with an optimum growth temperature of 85°C (16). A number of enzymes from this organism, such as ketopantoate reductase (6) and glyceraldehyde-3-phosphate dehydrogenase (8), are known to utilize NAD+/NADH. Its genome also harbors a closely related homolog of l-lysine 6-dehydrogenase from Pyrococcus horikoshii (7), which also utilizes NAD+/NADH. Considering the thermolabile properties of NAD+ and the high growth temperature of T. kodakarensis, we took interest in how this organism deals with the degradation of NAD+. In this study, we performed biochemical and genetic analyses on an ADP-ribose pyrophosphatase (ADPR-PPase) homolog in T. kodakarensis. The results strongly suggest that the enzyme is involved in dealing with the thermal degradation of NAD+ by converting one of the degradation products, ADP-ribose, to AMP and ribose 5-phosphate (R5P).
RESULTS
Thermal degradation of NAD+ at 85°C.
As shown in previous reports (9, 10, 17), we confirmed that the thermal degradation products of NAD+ at 85°C in 50 mM Tris-HCl buffer (pH 6.5 at 85°C) were predominantly ADP-ribose and nicotinamide (Fig. 1A and B). We also observed the generation of ADP-ribose when NADH or deamido-NAD+ was incubated at 85°C (data not shown). The degradation rate of NAD+ at 85°C was examined by high-performance liquid chromatography (HPLC) and compared with that of ADP-ribose, one of the main degradation products (Fig. 1C). ADP-ribose was hardly degraded after 60 min, whereas NAD+ degraded to less than 25% of the initial concentration. Using a typical decay equation (dC/dt = −kdC) in which C is the concentration of the compound, t1/2 of NAD+ was calculated as 24.2 min. The results suggest that if enzymes that convert ADP-ribose are not present in T. kodakarensis cells, the relatively thermostable ADP-ribose would accumulate via NAD+ degradation at 85°C, the optimum growth temperature for the organism. As the accumulation of ADP-ribose may have detrimental effects, this raises the possibilities that T. kodakarensis and other hyperthermophiles harbor enzymes to deal with this accumulation and metabolize ADP-ribose.
FIG 1.
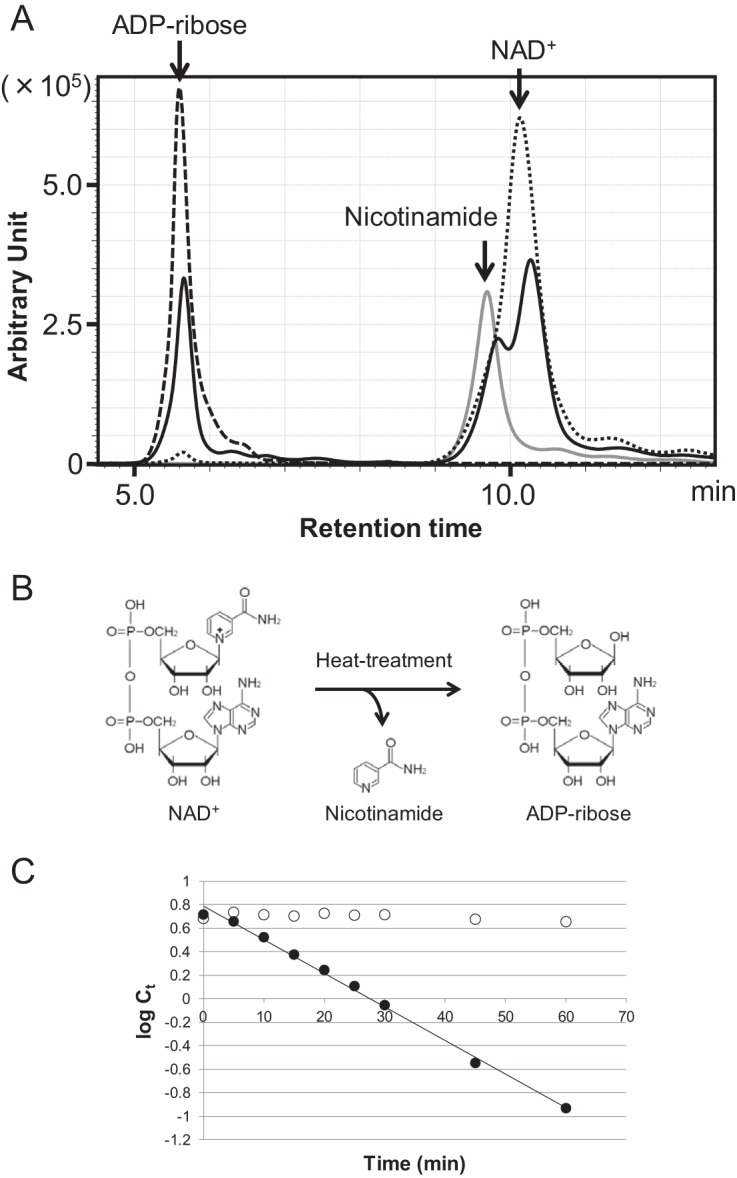
Identification of the thermal degradation products of NAD+ and determination of the thermal degradation rates of NAD+. (A) HPLC analysis was performed to identify the thermal degradation products of NAD+. The solid line shows NAD+ after heat treatment at 85°C for 30 min. The dotted line shows an NAD+ standard, the dashed line shows an ADP-ribose standard, and the gray line shows a nicotinamide standard. All samples were analyzed by monitoring A215. (B) Chemical reaction of the thermal degradation of NAD+. (C) Thermal degradation rate of NAD+ and ADP-ribose at 85°C. Ct indicates concentration of NAD+ or ADP-ribose (millimolar) after heat treatment for t min. log Ct indicates natural logarithm of Ct. Filled circles and open circles indicate NAD+ and ADP-ribose, respectively.
A candidate for an enzyme metabolizing ADP-ribose.
To identify a protein involved in ADP-ribose metabolism, we searched the T. kodakarensis genome for a gene encoding an enzyme that catalyzes the hydrolysis of ADP-ribose. We found a gene, TK2284, which was a homolog of the ADPR-PPase gene from Methanocaldococcus jannaschii (MJ1149, 48% identical) (18). To determine the activity of the protein, the recombinant TK2284 protein produced in E. coli was purified by heat treatment, anion-exchange chromatography, and gel filtration chromatography. The TK2284 protein was purified to apparent homogeneity, judging from the results of SDS-PAGE (Fig. 2A).
FIG 2.
Purified recombinant TK2284 and TK0067 proteins. Purified TK2284 (A) and TK0067 (B) recombinant proteins were analyzed by SDS-PAGE and stained with Coomassie brilliant blue. Four micrograms of protein was applied in each lane.
Examination of hydrolase activity of the TK2284 protein.
In general, ADPR-PPase belongs to the “Nudix” hydrolase family, whose members catalyze the hydrolysis of nucleoside diphosphates (NDPs) linked to a given moiety x. Members of this family from bacteria and eukaryotes display hydrolase activities toward various NDP derivatives, including 8-oxo-2′-deoxyguanosine-5′-triphosphate (8-oxo-dGTP), dATP, NADH, GDP-mannose, diadenosine polyphosphates (ApnA [n = 3 to 6]), ADP-ribose, 5-methyl-dCTP, thiamine pyrophosphate (TPP), diphosphoinositol pentakisphosphate (PP-InsP5), coenzyme A (CoA), UDP-glucose, capped snoRNAs, and capped mRNAs (19). Among archaeal Nudix family enzymes, the MJ1149 protein from M. jannaschii has been identified to exhibit ADPR-PPase activity (18).
Activity of the TK2284 protein was examined toward various NDP derivatives. Ten NDP derivatives, namely, ADP-ribose, ADP-glucose, NAD+, NADH, deamido-NAD+, UDP-glucose, ADP, UDP, CoA, and NADP+, were tested as the substrates at concentrations of 10 mM. Significant amounts of hydrolysis products were detected when ADP-ribose, ADP-glucose, NAD+, NADH, and deamido-NAD+ were incubated with the TK2284 protein (Table 1). However, a detailed HPLC analysis of the products of the reaction with NAD+ indicated that the TK2284 protein did not recognize NAD+ itself, but exhibited activity toward the ADP-ribose generated by thermal degradation of NAD+ during the reaction (data not shown). In the case of NADH and deamido-NAD+ also, the compounds themselves were not the actual substrates, and it was their degradation product, ADP-ribose, that was recognized by TK2284 (data not shown). On the other hand, the results indicated that ADP-glucose itself was hydrolyzed by the TK2284 protein. Therefore, kinetic analyses of hydrolyzing activities toward ADP-ribose and ADP-glucose were carried out (Fig. 3). The hydrolase reactions toward both substrates followed Michaelis-Menten kinetics, with a kcat/Km value for ADP-ribose approximately 53-fold higher than that for ADP-glucose (Table 2). This result suggested that the physiological substrate for TK2284 was ADP-ribose.
TABLE 1.
Hydrolase activity of TK2284 protein toward various substrates
| Substrate | Specific activity (μmol · min−1 · mg−1)a |
|---|---|
| ADP-ribose | 780.8 ± 41.2 |
| ADP-glucose | 34.1 ± 10.8 |
| NAD+ | 245.4 ± 42.0 |
| NADH | 96.8 ± 4.6 |
| Deamido-NAD+ | 38.5 ± 5.3 |
| UDP-glucose | <5.0 |
| ADP | <5.0 |
| UDP | <5.0 |
| CoA | ND |
| NADP+ | ND |
Values include standard deviations when available; ND, not detected.
FIG 3.
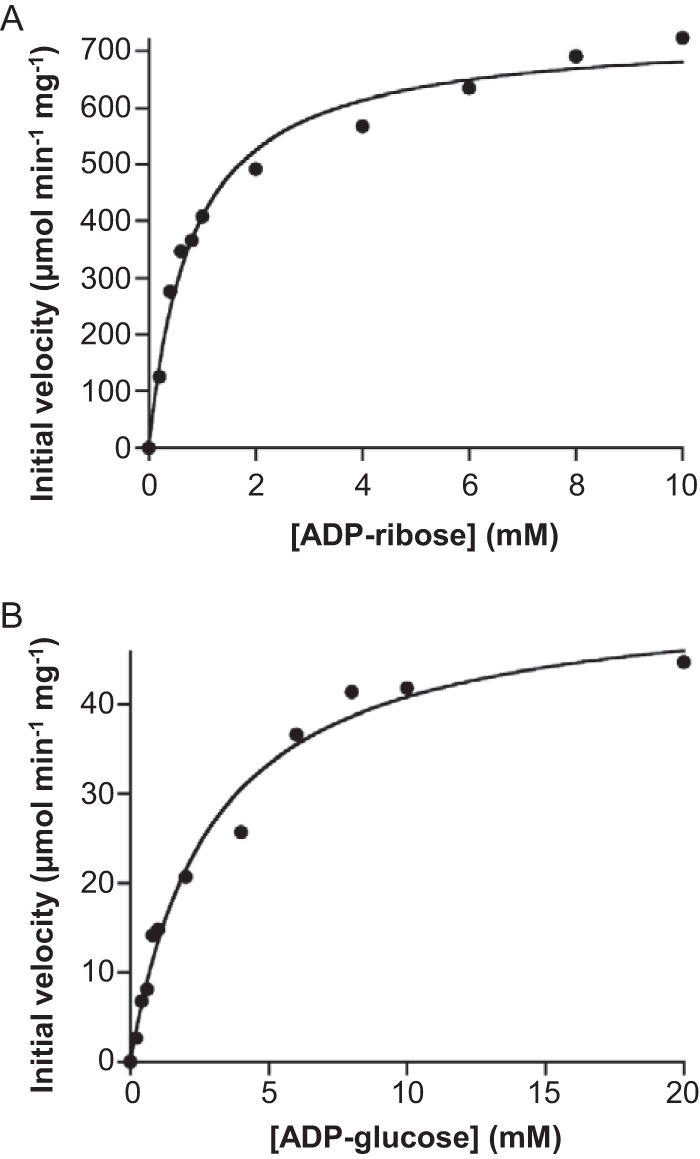
Kinetic analyses of the hydrolase reactions toward ADP-ribose (A) and ADP-glucose (B). Initial velocities were measured in the presence of various concentrations of ADP-ribose and ADP-glucose.
TABLE 2.
Kinetic analyses of the TK2284 reaction toward ADP-ribose and ADP-glucosea
| Substrate | Vmax (μmol · min−1 · mg−1) | kcat (s−1) | Km (mM) | kcat/Km (M−1 · s−1) |
|---|---|---|---|---|
| ADP-ribose | 736 ± 22 | 228 ± 7 | 0.80 ± 0.08 | 2.93 × 105 |
| ADP-glucose | 52 ± 2 | 16 ± 1 | 2.90 ± 0.41 | 5.55 × 103 |
Values include standard errors when appropriate.
Analysis of ADP-ribose hydrolyzing activity in cell extracts of T. kodakarensis cells with and without the TK2284 gene.
To gain insight into the physiological function of the TK2284 gene, we disrupted the gene in T. kodakarensis. The genotype of the transformant was examined by PCR analysis using two primers annealing outside the regions for homologous recombination as well as two within the target gene (Fig. 4A and B). As expected, we could observe a shorter amplified DNA fragment in the transformant using the primers annealing outside the homologous regions and no product using primers within the coding region. The genotype of the transformant was also examined by Southern blotting (Fig. 4A and C). When using a probe that binds to the 3′-flanking region of the TK2284 gene, a signal corresponding to a 2.4-kbp fragment was observed in the genome of the wild-type strain KOD1 and the host strain KU216 (Fig. 4C [a]). On the other hand, a single signal corresponding to the expected shorter fragment (1.9 kbp) was detected in the TK2284 (0.5 kbp) deletion mutant. Using a probe that hybridizes within the TK2284 gene, signals corresponding to a length of 2.4 kbp were observed in the genomes of KOD1 and KU216, while no band was observed in the transformant (Fig. 4C [b]). To confirm the absence of nonhomologous recombination between pUDTK2284 and the chromosome, 12 probes, each 0.5 kbp in length, were constructed (Fig. 4A). Together, the probes cover the entire sequence of the pUDTK2284 vector. When using these probes, 3.0-kbp signals corresponding to fragments including the TK2284 gene along with its flanking regions were observed in KOD1 and KU216 (Fig. 4C [c]). In the case of the transformant, a shorter 2.5-kbp signal was observed, similar to the results shown in Fig. 4C (a). We also observed very weak signals corresponding to fragments of 9.8 kbp (KOD1), 9.2 kbp (KU216), and 9.2 kbp (TK2284 mutant). The signals are expected to correspond to fragments containing the pyrF promoter and/or the pyrF gene, whose sequences are also present in the pUDTK2284 vector. No other signal was observed in the transformant. Based on these results, we could confirm that the genome of the transformant is free of the TK2284 gene and that there is no insertion of any part of the vector via unintended or nonhomologous recombination.
FIG 4.

Genotypic analyses of the ADPR-PPase gene deletion mutant. (A) Schematic drawing of relevant regions of the pUDTK2284 vector and the chromosome in wild-type (KOD1), host (KU216), and the TK2284 disruption strains. Primer sets used in PCR analyses, annealing within the TK2284 coding region (i-dTK2284-F/i-dTK2284-R) and outside the homologous regions (o-dTK2284-F/o-dTK2284-R), are indicated with gray and black arrows, respectively. Probes annealing to regions within the TK2284 gene, the 3′-flanking region of the TK2284 gene, and the entire sequence of pUDTK2284 in Southern blot analysis are indicated with gray, striped, and black bars, respectively. Although there are gaps between bars 1 to 12, the probes actually cover the entire sequence of pUDTK2284 without any gaps. The cleavage sites of the restriction enzymes used in Southern blot analyses are also shown. (B) PCR analyses of the TK2284 gene deletion mutant were performed with primer sets that anneal outside the homologous regions for homologous recombination (a) and within the TK2284 gene (b). (C) Southern blot analyses were performed with probe a binding to the 3′-flanking region of the TK2284 gene (a), probe b binding within the gene (b), and probes 1 to 12 corresponding to the entire sequence of the pUDTK2284 vector (c). The asterisk indicates the signals corresponding to fragments containing the pyrF promoter and/or pyrF gene (9.8 kbp in the wild-type strain and 9.2 kbp in the host strain and the deletion mutant). Ec, cleavage site of EcoRI; Sa, cleavage site of SalI; Sc, cleavage site of ScaI; P, promoter region of the operon including pyrF; M, marker; W, T. kodakarensis wild-type KOD1; H, T. kodakarensis host strain KU216; D, TK2284 gene disruptant.
Cell extracts from the host strain KU216 and the TK2284 gene disruptant were prepared. We investigated whether NAD+ in the cell extracts is degraded to ADP-ribose at high temperature (85°C) and if so, whether the presence of TK2284 had any effect on the generated ADP-ribose. We added NAD+ (2 mM) to the cell extracts (100 μg) of each strain and quantified the NAD+, ADP-ribose, nicotinamide, and AMP present in the reaction mixtures after 30 min of incubation at 85°C. As a result, we observed that NAD+ was degraded to similar degrees in both cell extracts (Fig. 5A and B). We also observed the generation of ADP-ribose and nicotinamide. The concentrations of ADP-ribose were significantly higher in the cell extracts of the TK2284 gene disruptant than in those of the host strain, which may reflect the absence of ADPR-PPase in the TK2284 gene disruptant. This is also supported by the lower concentrations of AMP in the cell extracts of the TK2284 gene disruptant. The result implies that ADP-ribose is also formed in T. kodakarensis cells due to NAD+ degradation at high temperature and that the TK2284 protein hydrolyzes the generated ADP-ribose.
FIG 5.
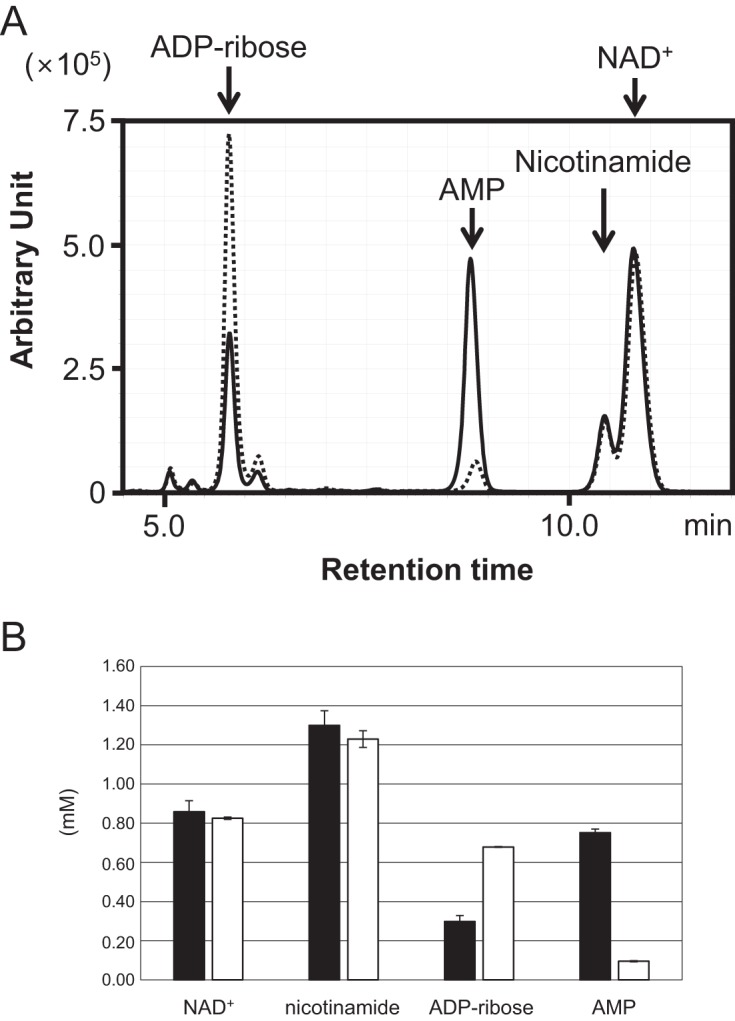
Thermal degradation of NAD+ and enzymatic degradation of ADP-ribose in cell extracts. (A) NAD+ (2 mM) and MgCl2 (10 mM) were added to cell extracts and heat treated at 85°C for 30 min. Solid and dotted lines show products in cell extracts from the KU216 host strain and TK2284 gene disruptant, respectively. Both samples were analyzed by monitoring A254. The experiment was repeated with cell extracts obtained from three independent cultures for each strain, and a representative result is shown. (B) The concentrations of NAD+, nicotinamide, ADP-ribose, and AMP were calculated with the results shown in panel A, and the averages and standard deviations (n = 3) are shown. Filled bars indicate results with the cell extract of the KU216 host strain, and open bars indicate those of the gene disruptant.
Growth properties of T. kodakarensis host strain KU216 and TK2284 gene disruptant.
To examine the growth properties of the host strain KU216 and TK2284 gene disruptant, cells were grown in a synthetic amino acid medium (ASW-AA-S0-Pyr-Ura-W). In this medium, the maximum optical density at 600 nm (OD660) of the TK2284 gene disruptant (0.055) was significantly lower than that of the host strain (0.124) (Fig. 6). In addition to the cell densities, the maximum specific growth rate of the gene disruptant (0.72 · h−1) was lower than that of the host strain (0.92 · h−1). There are two possibilities for the reduced cell yield observed in the TK2284 disruption strain. One is that ADP-ribose, which is normally converted to R5P and AMP, is not hydrolyzed and thus cannot be utilized in the gene disruptant. The other is that ADP-ribose accumulation displays toxicity by spontaneously linking to proteins, which may also affect the specific growth rate.
FIG 6.
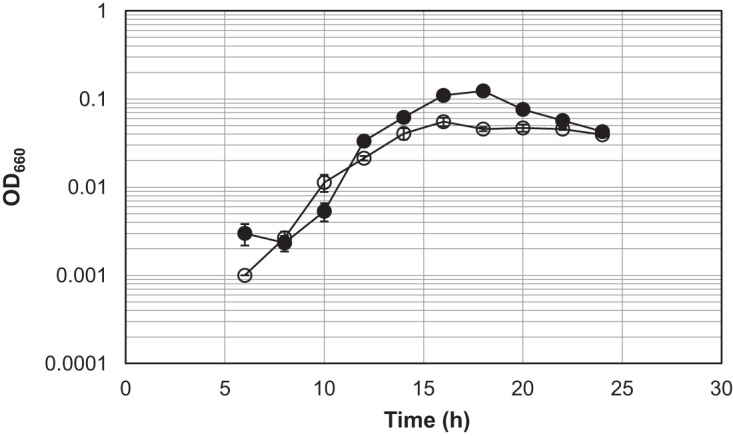
Growth properties of the T. kodakarensis host strain KU216 and TK2284 gene disruptant. Both strains were grown in a synthetic amino acid medium, ASW-AA-S0-Pyr-Ura-W. Filled circles and open circles represent the results from three independent growth experiments with the host strain and the TK2284 gene disruptant, respectively. The vertical axis is represented in logarithmic scale.
Examination of a protein annotated as nicotinamide mononucleotide adenylyltransferase.
We next searched for proteins that might be related to the metabolism involving the TK2284 protein using STRING (http://string-db.org/), which identifies proteins related to the target protein from various aspects. We found that in some bacteria, TK2284 homologs encoding ADPR-PPase are fused to nicotinamide mononucleotide adenylyltransferase (NMNAT) which catalyzes the adenylylation of NMN and functions in the direct salvage of nicotinamide to NAD+. We thus examined the NMNAT homolog in T. kodakarensis, encoded by TK0067.
The recombinant TK0067 protein was produced in E. coli and purified (Fig. 2B). When we measured adenylyltransferase activity, we found that the TK0067 protein displayed robust activity toward both NMN and nicotinic acid mononucleotide (NaMN) (see Fig. S1A in the supplemental material). We also examined activity toward R5P, which shares the R5P moiety with NMN and NaMN. The reaction with R5P and ATP would result in the formation of pyrophosphate and ADP-ribose, the substrate of ADPR-PPase. The TK0067 protein displayed activity toward R5P and generated ADP-ribose, but activity levels were much lower than those observed with NMN and NaMN (Fig. S1B).
DISCUSSION
In high-temperature environments, such as 85°C where hyperthermophiles are known to thrive, NAD+ is degraded into ADP-ribose and nicotinamide. This presents a nonenzymatic mechanism of ADP-ribose generation, in addition to the enzymatic mechanisms previously described in (mesophilic) eukaryotes and bacteria. This study suggests that the hyperthermophilic archaeon T. kodakarensis possesses a system to metabolize the potentially detrimental ADP-ribose derived from thermal degradation of NAD+ with the TK2284-encoded ADPR-PPase. TK2284 homologs are widely distributed in hyperthermophilic archaea and bacteria, suggesting that most hyperthermophiles harbor the ADP-ribose-degrading system clarified in this study. We would like to note that the distribution of TK2284 homologs is not at all biased toward (hyper)thermophiles. These homologs are also widely distributed in mesophilic organisms. Whether they share roles similar to those in (hyper)thermophiles must await further analysis, but the results of this study, emphasized by the lower cell yield and specific growth rate of the TK2284 disruption strain observed in growth experiments, clearly suggest the need for this enzyme in dealing with the thermal degradation of NAD+ in (hyper)thermophiles. The degradation product of ADP-ribose, R5P, also possesses the C1 semialdehyde that can react with proteins. However, R5P can be readily converted to other metabolites, which might prevent any detrimental effects of this compound.
In the thermoacidophilic archaeon Sulfolobus solfataricus MT-4 (DSM5833), there are several reports that suggest the presence of an ADP-ribosylation system (20–26). These reports detected the ADP-ribosyl reaction in this organism, and an enzyme that exhibited poly(ADP-ribose) polymerase (PARP) activity was identified by purifying the protein from the cell extract of S. solfataricus strain MT-4 using an anti-calf thymus PARP antibody. Partial amino acid sequences of a number of peptide fragments of the candidate PARP protein were clarified by Edman degradation. A sequence (231 residues) containing the majority of these sequences is present in the protein database. When we performed a BLAST search using this sequence, homologs of this protein were not found in other archaeal species, including the S. solfataricus P2 strain. Furthermore, proteins displaying homology with PARP and mono(ADP)-ribosyltransferase (MART) from eukaryotes and bacteria are not found in archaeal genomes. These findings suggest that the classical ADP-ribosylation system and the system in S. solfataricus strain MT-4 are not widely distributed in the Archaea. In addition, genes that display similarity with the bacterial or eukaryotic NAD+ glycohydrolase (NADase) are not found in the archaeal genomes. Therefore, the role of ADPR-PPase in most thermophilic archaea may be in metabolism to deal with the thermal degradation of NAD+. By examining the NMNAT homolog (TK0067), we also observed that ADP-ribose could be generated by a side reaction of the NMNAT of T. kodakarensis. As the activity levels were low, we presume that the contributions of this side reaction to ADP-ribose formation in the cell are small and that the main source of ADP-ribose in this organism is the thermal degradation of NAD+. Regardless of the source, ADPR-PPase in T. kodakarensis would be able to salvage the generated ADP-ribose and provide the useful metabolites AMP and R5P.
Many bacteria that harbor NMNAT/ADPR-PPase fusion genes also possess a NaMN adenylyltransferase (NaMNAT). NaMNAT and NMNAT catalyze similar reactions and are involved in the de novo and salvage pathways for NAD+ biosynthesis, respectively. Interestingly, archaeal genomes harbor an NMNAT gene homolog but do not possess the homolog for the NaMNAT gene. As archaea also harbor a gene corresponding to NAD+ synthetase, it has been presumed that the archaeal NMNAT homologs exhibit NaMNAT activity in addition to NMNAT activity, as in the case of the bacterial enzymes from Synechocystis sp. strain PCC 6803 or Francisella tularensis (27). Our biochemical examination of the TK0067 protein demonstrates that this is actually the case for the T. kodakarensis enzyme, as with the protein from S. solfataricus (28).
The presence of a standalone NaMNAT in bacteria that harbor a fusion of ADPR-PPase to NMNAT suggests that ADPR-PPase is involved in the regeneration of NAD+ from ADP-ribose in these bacteria (27, 29). On the other hand, a fusion protein of NMN deamidase and ADPR-PPase is present in Agrobacterium tumefaciens, although the A. tumefaciens ADPR-PPase belongs to clusters of orthologous groups (COG) that differ from that of TK2284 (30). As NMN deamidase is also known to function in NAD+ regeneration, this finding further supports that ADPR-PPase functions in a salvage pathway to supply NAD+.
The occurrence of the protein fusions ADPR-PPase/NMNAT and ADPR-PPase/NMN deamidase and our data imply that TK2284 and TK0067 are involved in NAD+ regeneration in T. kodakarensis. A de novo pathway and a predicted salvage pathway from ADP-ribose to NAD+ in T. kodakarensis are shown in Fig. 7A and B, respectively. NAD+ is degraded into ADP-ribose and nicotinamide at high temperature. ADP-ribose is then hydrolyzed to R5P and AMP by ADPR-PPase (TK2284). R5P is converted to phosphoribosyl pyrophosphate (PRPP) by PRPP synthetase (TK2235). The other major thermal degradation product of NAD+, nicotinamide, is converted to nicotinic acid by nicotinamidase (TK1650). Subsequently, NaMN can be synthesized from PRPP and nicotinic acid by nicotinic acid phosphoribosyltransferase (TK1676). With ATP, NaMN, which is also utilized in NAD+ de novo synthesis, is converted to deamido-NAD+ by NaMNAT (TK0067). Finally, NAD+ is resynthesized by NAD+ synthetase (TK1798). It should be noted that TK0067 also catalyzed the reaction converting NMN and ATP to NAD+. Thus, there is a possibility that this organism possesses an alternative route for NAD+ regeneration in which nicotinamide, instead of nicotinic acid, and PRPP are converted to NMN instead of NaMN by TK1676, and then NMN with ATP is converted to NAD+ by TK0067.
FIG 7.

Predicted NAD+ biosynthesis and salvage pathways in T. kodakarensis. (A) De novo NAD+ biosynthesis pathway from aspartate. (B) Salvage pathway for NAD+ regeneration predicted from genomic information and the results obtained in this study and elsewhere (34–38). Bold arrows represent reactions examined in this study and the gray arrow indicates the side reaction of the TK0067 protein. Solid arrows show generally predicted pathways in Archaea and the dotted arrow indicates the reaction proposed in Discussion. Proteins examined in this study are in bold boxes, proteins that have been examined in other archaea are in solid boxes, and proteins only predicted by amino acid sequence and/or structure are in dashed boxes. DHAP, dihydroxyacetone phosphate; deamido-NAD+, nicotinic acid adenine dinucleotide; PRPP, phosphoribosyl pyrophosphate.
MATERIALS AND METHODS
Identification of the thermal degradation products of NAD+.
NAD+ (2 mM) in 50 mM Tris-HCl buffer (pH 6.5) was incubated at 85°C for 30 min. After cooling on ice for 10 min, the mixture was examined by HPLC. HPLC was performed with a Cosmosil 5-μm C18 (5C18)-PAQ column (Nacalai Tesque, Kyoto, Japan) using 50 mM NaH2PO4 (pH 4.7) as the mobile phase with a column temperature of 40°C. Absorbance at 215 nm (A215) was monitored for detection.
Determining the degradation rates of NAD+ and ADP-ribose at 85°C.
To examine the degradation rates of NAD+ and ADP-ribose, 2 mM NAD+ and ADP-ribose in 50 mM Tris-HCl buffer (pH 6.5) were incubated at 85°C for 5, 10, 15, 20, 25, 30, 45, and 60 min. Samples were applied to an NH2P-50 4E column and analyzed by HPLC using 300 mM NaH2PO4 (pH 4.4) as the mobile phase. The column temperature was set at 40°C, and A254 was monitored for detection. NAD+ and ADP-ribose were quantified by comparing peak areas with a standard curves obtained with 0.5 to 2.5 mM NAD+ and ADP-ribose, respectively.
Strains and culture conditions.
The cultivation of T. kodakarensis KOD1, KU216 (ΔpyrF) (31), and derivative strains was performed under anaerobic conditions at 85°C in nutrient-rich medium (artificial seawater [ASW]-yeast extract-tryptone [YT]-sodium pyruvate [Pyr] or ASW-YT-elemental sulfur [S0]) or a synthetic medium (ASW-amino acid mixture [AA]-S0). The nutrient-rich media were composed of 0.8× ASW, 5.0 g · liter−1 yeast extract, 5.0 g · liter−1 tryptone, and 0.8 mg · liter−1 resazurin supplemented with 5.0 g · liter−1 Pyr (for ASW-YT-Pyr) or 2.0 g · liter−1 elemental sulfur (for ASW-YT-S0). ASW-AA-S0 consisted of 0.8× ASW, a mixture of 20 amino acids, modified Wolfe's trace minerals, a vitamin mixture, and 2.0 g · liter−1 elemental sulfur (31, 32). Prior to inoculation, Na2S·9H2O was added to these media until they became colorless (12.5 mg · liter−1). In the case of plate cultures used to isolate transformants, elemental sulfur and Na2S·9H2O in ASW-AA-S0 were replaced with 2 ml · liter−1 of a polysulfide solution (10 g of Na2S·9H2O and 3 g sulfur flowers in 15 ml of H2O), and 10 g · liter−1 of Gelrite was added to solidify the medium. E. coli strain DH5α used for plasmid construction was cultivated at 37°C in Luria-Bertani (LB) medium containing ampicillin (100 mg · liter−1). Unless mentioned otherwise, all chemicals were purchased from Wako Pure Chemicals (Osaka, Japan) or Nacalai Tesque.
Overexpression of the TK2284 and TK0067 genes and purification of the recombinant proteins.
To construct TK2284 and TK0067 gene expression vectors, the coding regions of both genes were amplified from T. kodakarensis genomic DNA by PCR using primer sets (eTK2284-F/eTK2284-R and eTK0067-F/eTK006-R) with NdeI and BamHI restriction sites (see Table S1 in the supplemental material for all primer sequences) and were inserted into pET-21a(+) (EMD Millipore, Billerica, MA) at the NdeI-BamHI sites. After confirming the absence of unintended mutations, the resulting expression vectors, pETTK2284 and pETTK0067, were introduced into E. coli strain BL21-CodonPlus (DE3)-RIL. Transformants were cultivated until the optical density at 660 nm reached 0.4 to 0.6 and were supplemented with isopropyl-1-thio-β-d-galactopyranoside at a final concentration of 0.1 mM to induce protein expression. After a further 4 h of culture, cells were harvested, resuspended in 50 mM Tris-HCl buffer (pH 7.5), and disrupted by sonication. After centrifugation (5,000 × g at 4°C for 10 min), the soluble cell extracts were incubated at 85°C for 20 min (TK2284) or 10 min (TK0067) to remove thermolabile proteins. After centrifugation (5,000 × g at 4°C for 10 min), the supernatants were applied to anion-exchange chromatography, Resource Q (GE Healthcare, Chicago, IL), and proteins were eluted with a linear gradient of NaCl (0 to 1.0 M) in 50 mM Tris-HCl (pH 7.5) at a flow rate of 2.0 ml · min−1. As for the TK2284 protein, after concentrating relevant fractions using an Amicon Ultra centrifugal filter unit (molecular weight cutoff [MWCO], 10,000; EMD Millipore), the sample was separated with a Superdex 200 10/300 gel filtration column (GE Healthcare) with a mobile phase of 50 mM Tris-HCl (pH 7.5) with 150 mM NaCl at a flow rate of 0.8 ml · min−1. Protein concentrations were determined with the protein assay system (Bio-Rad, Hercules, CA) using bovine serum albumin as a standard.
Measurement of activity toward NDP and NDP derivatives.
Hydrolase activities, including ADP-ribose pyrophosphatase activity, were measured in a 100-μl reaction mixture containing 50 mM Tris-HCl (pH 6.5), 50 mM MgCl2, 0.1 to 20 mM substrates, and 0.02 to 1 μg of the TK2284 recombinant protein. After preincubating at 85°C for 1 min, the reactions were initiated by adding each substrate. The hydrolase reactions were carried out at 85°C for 1, 3, and 5 min and were terminated by cooling on ice for 10 min. After removing enzymes with an Amicon Ultra centrifugal filter unit (MWCO, 10,000), AMP, UMP, or nicotinamide mononucleotide generation was quantified by HPLC with a 5C18-PAQ column using 50 mM NaH2PO4 (pH 4.7) as the mobile phase. Column temperatures were set at 40°C, and products were detected by monitoring A254.
Construction of TK2284 gene disruptant of T. kodakarensis.
For constructing the TK2284 gene disruption vector, the TK2284 gene with its 5′- and 3′-flanking regions was amplified by PCR with primers dTK2284-F and dTK2284-R and inserted into plasmid pUD3, which harbors a pyrF gene, at the HincII site. The TK2284 gene was removed by inverse PCR with primers inv-dTK2284-F and inv-dTK2284-R, and the amplified DNA fragment was self-ligated resulting in the TK2284 disruption vector, pUDTK2284. To construct the TK2284 deletion mutant (ΔTK2284), T. kodakarensis KU216 cells were grown in ASW-YT-S0 for 12 h, harvested, resuspended in 200 μl of 0.8× ASW, and kept on ice for 30 min. pUDTK2284 (3 μg) was added to the cells and the mixture was kept on ice for 1 h. After a heat shock at 85°C for 45 s, the mixture was kept on ice for 10 min. Cells were inoculated in ASW-AA-S0 liquid medium and incubated at 85°C for 2 days twice to enrich for transformants harboring the pyrF gene due to pop-in recombination. Cells were then grown at 85°C for 3 to 5 days on ASW-AA solid medium with 0.75% 5-fluoroorotic acid (FOA) and 10 μg · ml−1 of uracil to select for transformants without the pyrF gene. Genotypes of the transformants were examined by PCR with primer sets that anneal within the gene (i-dTK2284-F/i-dTK2284-R) or outside the homologous regions for homologous recombination (o-dTK2284-F/o-dTK2284-R) and were confirmed by DNA sequencing analysis.
Southern blot analysis was carried out with different probes. For examining the presence or absence of the TK2284 coding region in the original locus, two probes were used. Probe a, corresponding to the 3′-flanking region of TK2284, was amplified with the primer set so-dTK2284-F/so-dTK2284-R. Probe b anneals to a region within the TK2284 coding region and was amplified with i-dTK2284-F/i-dTK2284-R. EcoRI was used to digest the genomic DNA. To confirm the absence of nonhomologous recombination between pUDTK2284 and the chromosome, 12 probes (probe 1 to probe 12) were prepared with the following primer sets: s-dTK2284-1-f/s-dTK2284-1-r, s-dTK2284-2-f/s-dTK2284-2-r, s-dTK2284-3-f/s-dTK2284-3-r, s-dTK2284-4-f/s-dTK2284-4-r, s-dTK2284-5-f/s-dTK2284-5-r, s-dTK2284-6-f/s-dTK2284-6-r, s-dTK2284-7-f/s-dTK2284-7-r, s-dTK2284-8-f/s-dTK2284-8-r, s-dTK2284-9-f/s-dTK2284-9-r, s-dTK2284-10-f/s-dTK2284-10-r, s-dTK2284-11-f/s-dTK2284-11-r, and s-dTK2284-12-f/s-dTK2284-12-r. These probes are approximately 0.5 kbp in length and together cover the entire sequence of the disruption vector, pUDTK2284 (see below). In this case, genomic DNA was digested with SalI and ScaI. All primer sequences are indicated in Table S1. Probes were labeled by DIG High Prime DNA labeling and detection starter kit ΙΙ (Roche, Basel, Switzerland).
ADP-ribose pyrophosphatase activity in cell extracts.
T. kodakarensis KOD1 cells, after cultivating in ASW-YT-Pyr for 16 h, were harvested by centrifugation (20,400 × g at 4°C for 15 min) and washed with an approximately 1/15 volume of 0.8× ASW. After centrifugation (5,800 × g at 4°C for 15 min), cells were lysed in an approximately 1/15 volume of 50 mM Tris-HCl (pH 7.5) containing 0.1% Triton X-100. After mixing with a vortex mixer, the supernatant after centrifugation (20,400 × g at 4°C for 30 min) was used as the cell extract. To analyze ADP-ribose pyrophosphatase activity, ADP-ribose and AMP accumulation levels were measured with methods described above after the 100-μg cell extract, 10 mM MgCl2, and 2 mM NAD+ mixture was heat treated in 50 mM Tris-HCl (pH 6.5) at 85°C for 30 min.
Growth experiments of T. kodakarensis host strain KU216 and the TK2284 gene disruptant.
Growth characteristics of the host strain KU216 and the TK2284 gene disruptant were examined in ASW-AA-S0 medium supplemented with 2.5 g · liter−1 sodium pyruvate, 10 μg · ml−1 uracil, and 10 μM tungsten (ASW-AA-S0-Pyr-Ura-W). Each strain was grown in ASW-YT-S0 medium for 12 to 16 h and inoculated in ASW-AA-S0-Pyr-Ura-W. Growth at 85°C was examined by monitoring the OD660.
Examination of adenylyltransferase activity.
Adenylyltransferase activity was measured in 100-μl reaction mixtures containing 50 mM Tris-HCl (pH 6.5), 50 mM MgCl2, 2 mM substrate (nicotinamide mononucleotide, nicotinic acid mononucleotide, or R5P), and 1 or 10 μg of purified TK0067 recombinant protein, as described previously (33) with slight modifications. After preincubating at 85°C for 1 min, the reactions were initiated by the addition of substrates. The reactions were performed at 85°C for 10 min and were terminated by cooling on ice for 10 min. After removing the enzyme with an Amicon Ultra centrifugal filter unit (MWCO, 10,000), the reaction products (NAD+, deamido-NAD+, or ADP-ribose) were quantified with HPLC. A 5C18-PAQ column was applied and 50 mM NaH2PO4 (pH 4.7) was used as the mobile phase. Column temperatures were set at 40°C, and the products were quantified by monitoring A254.
Supplementary Material
ACKNOWLEDGMENTS
This study was partially funded by the Core Research for Evolutional Science and Technology program of the Japan Science and Technology Agency (JST) to H.A. within the research area Creation of Basic Technology for Improved Bioenergy Production through Functional Analysis and Regulation of Algae and Other Aquatic Microorganisms.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00162-17.
REFERENCES
- 1.Dwivedi N, Dube D, Pandey J, Singh B, Kukshal V, Ramachandran R, Tripathi RP. 2008. NAD+-dependent DNA ligase: a novel target waiting for the right inhibitor. Med Res Rev 28:545–568. doi: 10.1002/med.20114. [DOI] [PubMed] [Google Scholar]
- 2.Hassa PO, Haenni SS, Elser M, Hottiger MO. 2006. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev 70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hassa PO, Hottiger MO. 2008. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci 13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 4.Song SH, Ahluwalia N, Leduc Y, Delbaere LT, Vieille C. 2008. Thermotoga maritima TM0298 is a highly thermostable mannitol dehydrogenase. Appl Microbiol Biotechnol 81:485–495. doi: 10.1007/s00253-008-1633-9. [DOI] [PubMed] [Google Scholar]
- 5.Kallnik V, Schulz C, Schweiger P, Deppenmeier U. 2011. Properties of recombinant Strep-tagged and untagged hyperthermophilic d-arabitol dehydrogenase from Thermotoga maritima. Appl Microbiol Biotechnol 90:1285–1293. doi: 10.1007/s00253-011-3187-5. [DOI] [PubMed] [Google Scholar]
- 6.Tomita H, Imanaka T, Atomi H. 2013. Identification and characterization of an archaeal ketopantoate reductase and its involvement in regulation of coenzyme A biosynthesis. Mol Microbiol 90:307–321. doi: 10.1111/mmi.12363. [DOI] [PubMed] [Google Scholar]
- 7.Yoneda K, Fukuda J, Sakuraba H, Ohshima T. 2010. First crystal structure of l-lysine 6-dehydrogenase as an NAD-dependent amine dehydrogenase. J Biol Chem 285:8444–8453. doi: 10.1074/jbc.M109.084384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia B, Linh le T, Lee S, Pham BP, Liu J, Pan H, Zhang S, Cheong GW. 2011. Biochemical characterization of glyceraldehyde-3-phosphate dehydrogenase from Thermococcus kodakarensis KOD1. Extremophiles 15:337–346. doi: 10.1007/s00792-011-0365-4. [DOI] [PubMed] [Google Scholar]
- 9.Anderson BM, Anderson CD. 1963. The effect of buffers on nicotinamide adenine dinucleotide hydrolysis. J Biol Chem 238:1475–1478. [PubMed] [Google Scholar]
- 10.Kaplan NO, Colowick SP, Barnes CC. 1951. Effect of alkali on diphosphopyridine nucleotide. J Biol Chem 191:461–472. [PubMed] [Google Scholar]
- 11.Jacobson EL, Cervantes-Laurean D, Jacobson MK. 1994. Glycation of proteins by ADP-ribose. Mol Cell Biochem 138:207–212. doi: 10.1007/BF00928463. [DOI] [PubMed] [Google Scholar]
- 12.Cipollone F, Iezzi A, Fazia M, Zucchelli M, Pini B, Cuccurullo C, De Cesare D, De Blasis G, Muraro R, Bei R, Chiarelli F, Schmidt AM, Cuccurullo F, Mezzetti A. 2003. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation 108:1070–1077. doi: 10.1161/01.CIR.0000086014.80477.0D. [DOI] [PubMed] [Google Scholar]
- 13.Cervantes-Laurean D, Jacobson EL, Jacobson MK. 1996. Glycation and glycoxidation of histones by ADP-ribose. J Biol Chem 271:10461–10469. doi: 10.1074/jbc.271.18.10461. [DOI] [PubMed] [Google Scholar]
- 14.Sell DR, Monnier VM. 1989. Structure elucidation of a senescence cross-link from human extracellular matrix. Implication of pentoses in the aging process. J Biol Chem 264:21597–21602. [PubMed] [Google Scholar]
- 15.Marbaix AY, Noel G, Detroux AM, Vertommen D, Van Schaftingen E, Linster CL. 2011. Extremely conserved ATP- or ADP-dependent enzymatic system for nicotinamide nucleotide repair. J Biol Chem 286:41246–41252. doi: 10.1074/jbc.C111.310847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atomi H, Fukui T, Kanai T, Morikawa M, Imanaka T. 2004. Description of Thermococcus kodakaraensis sp. nov., a well studied hyperthermophilic archaeon previously reported as Pyrococcus sp. KOD1. Archaea 1:263–267. doi: 10.1155/2004/204953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honda K, Hara N, Cheng M, Nakamura A, Mandai K, Okano K, Ohtake H. 2016. In vitro metabolic engineering for the salvage synthesis of NAD+. Metab Eng 35:114–120. doi: 10.1016/j.ymben.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Sheikh S, O'Handley SF, Dunn CA, Bessman MJ. 1998. Identification and characterization of the Nudix hydrolase from the archaeon, Methanococcus jannaschii, as a highly specific ADP-ribose pyrophosphatase. J Biol Chem 273:20924–20928. doi: 10.1074/jbc.273.33.20924. [DOI] [PubMed] [Google Scholar]
- 19.McLennan AG. 2006. The Nudix hydrolase superfamily. Cell Mol Life Sci 63:123–143. doi: 10.1007/s00018-005-5386-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faraone-Mennella MR, Piccialli G, De Luca P, Castellano S, Giordano A, Rigano D, De Napoli L, Farina B. 2002. Interaction of the ADP-ribosylating enzyme from the hyperthermophilic archaeon S. solfataricus with DNA and ss-oligo deoxy ribonucleotides. J Cell Biochem 85:146–157. doi: 10.1002/jcb.10107. [DOI] [PubMed] [Google Scholar]
- 21.Faraone-Mennella MR, Farina B. 1995. In the thermophilic archaeon Sulfolobus solfataricus a DNA-binding protein is in vitro (Adpribosyl)ated. Biochem Biophys Res Commun 208:55–62. doi: 10.1006/bbrc.1995.1304. [DOI] [PubMed] [Google Scholar]
- 22.Faraone-Mennella MR, Gambacorta A, Nicolaus B, Farina B. 1996. Immunochemical detection of ADP-ribosylating enzymes in the archaeon Sulfolobus solfataricus. FEBS Lett 378:199–201. doi: 10.1016/0014-5793(95)01455-1. [DOI] [PubMed] [Google Scholar]
- 23.Faraone-Mennella MR, De Luca P, Giordano A, Gambacorta A, Nicolaus B, Farina B. 2002. High stability binding of poly(ADPribose) polymerase-like thermozyme from S. solfataricus with circular DNA. J Cell Biochem 85:158–166. doi: 10.1002/jcb.10108. [DOI] [PubMed] [Google Scholar]
- 24.Faraone-Mennella MR, De Lucia F, De Maio A, Gambacorta A, Quesada P, De Rosa M, Nicolaus B, Farina B. 1995. ADP-ribosylation reactions in Sulfolobus solfataricus, a thermoacidophilic archaeon. Biochim Biophys Acta 1246:151–159. doi: 10.1016/0167-4838(94)00169-H. [DOI] [PubMed] [Google Scholar]
- 25.Castellano S, Farina B, Faraone-Mennella MR. 2009. The ADP-ribosylation of Sulfolobus solfataricus Sso7 modulates protein/DNA interactions in vitro. FEBS Lett 583:1154–1158. doi: 10.1016/j.febslet.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Di Maro A, De Maio A, Castellano S, Parente A, Farina B, Faraone-Mennella MR. 2009. The ADP-ribosylating thermozyme from Sulfolobus solfataricus is a DING protein. Biol Chem 390:27–30. doi: 10.1515/BC.2009.006. [DOI] [PubMed] [Google Scholar]
- 27.Huang N, Sorci L, Zhang X, Brautigam CA, Li X, Raffaelli N, Magni G, Grishin NV, Osterman AL, Zhang H. 2008. Bifunctional NMN adenylyltransferase/ADP-ribose pyrophosphatase: structure and function in bacterial NAD metabolism. Structure 16:196–209. doi: 10.1016/j.str.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raffaelli N, Amici A, Emanuelli M, Ruggieri S, Magni G. 1994. Pyridine dinucleotide biosynthesis in archaebacteria: presence of NMN adenylyltransferase in Sulfolobus solfataricus. FEBS Lett 355:233–236. doi: 10.1016/0014-5793(94)01195-8. [DOI] [PubMed] [Google Scholar]
- 29.Gerdes SY, Kurnasov OV, Shatalin K, Polanuyer B, Sloutsky R, Vonstein V, Overbeek R, Osterman AL. 2006. Comparative genomics of NAD biosynthesis in cyanobacteria. J Bacteriol 188:3012–3023. doi: 10.1128/JB.188.8.3012-3023.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cialabrini L, Ruggieri S, Kazanov MD, Sorci L, Mazzola F, Orsomando G, Osterman AL, Raffaelli N. 2013. Genomics-guided analysis of NAD recycling yields functional elucidation of COG1058 as a new family of pyrophosphatases. PLoS One 8:e65595. doi: 10.1371/journal.pone.0065595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato T, Fukui T, Atomi H, Imanaka T. 2005. Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl Environ Microbiol 71:3889–3899. doi: 10.1128/AEM.71.7.3889-3899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robb FT, Place AR. 1995. Media for thermophiles, p 167–168. In Robb FT, Place AR, Sowers AR, Schreier HJ, DasSarma S, Fleischmann EM (ed), Archea: a laboratory manual. Thermophiles. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 33.Emanuelli M, Amici A, Carnevali F, Pierella F, Raffaelli N, Magni G. 2003. Identification and characterization of a second NMN adenylyltransferase gene in Saccharomyces cerevisiae. Protein Expr Purif 27:357–364. doi: 10.1016/S1046-5928(02)00645-9. [DOI] [PubMed] [Google Scholar]
- 34.Kadziola A, Jepsen CH, Johansson E, McGuire J, Larsen S, Hove-Jensen B. 2005. Novel class III phosphoribosyl diphosphate synthase: structure and properties of the tetrameric, phosphate-activated, non-allosterically inhibited enzyme from Methanocaldococcus jannaschii. J Mol Biol 354:815–828. doi: 10.1016/j.jmb.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 35.Stekhanova TN, Bezsudnova EY, Mardanov AV, Osipov EM, Ravin NV, Skryabin KG, Popov VO. 2014. Nicotinamidase from the thermophilic archaeon Acidilobus saccharovorans: structural and functional characteristics. Biochemistry (Mosc) 79:54–61. doi: 10.1134/S0006297914010088. [DOI] [PubMed] [Google Scholar]
- 36.Sakuraba H, Satomura T, Kawakami R, Yamamoto S, Kawarabayasi Y, Kikuchi H, Ohshima T. 2002. l-Aspartate oxidase is present in the anaerobic hyperthermophilic archaeon Pyrococcus horikoshii OT-3: characteristics and role in the de novo biosynthesis of nicotinamide adenine dinucleotide proposed by genome sequencing. Extremophiles 6:275–281. doi: 10.1007/s00792-001-0254-3. [DOI] [PubMed] [Google Scholar]
- 37.De Ingeniis J, Kazanov MD, Shatalin K, Gelfand MS, Osterman AL, Sorci L. 2012. Glutamine versus ammonia utilization in the NAD synthetase family. PLoS One 7:e39115. doi: 10.1371/journal.pone.0039115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soriano EV, Zhang Y, Colabroy KL, Sanders JM, Settembre EC, Dorrestein PC, Begley TP, Ealick SE. 2013. Active-site models for complexes of quinolinate synthase with substrates and intermediates. Acta Crystallogr D Biol Crystallogr 69:1685–1696. doi: 10.1107/S090744491301247X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.