

Abstract
The present study aimed to investigate changes in the expression of interleukin (IL)-1 receptor-associated kinase 4 (IRAK4) and microRNA (miRNA or miR)-93 in mice with cerebral ischemia reperfusion (CIR) injury, as well as the association and regulatory mechanism between IRAK4 and miR-93. The CIR mouse model was constructed and mouse microglia BV2 cells were transfected with miR-93 mimic or miR-93 inhibitor. Quantitative polymerase chain reaction was used to measure the expression of mRNA and miR-93. Western blotting was performed to determine protein expression. Enzyme-linked immunosorbent assays were performed to measure the concentrations pro-inflammatory factors. The expression of miR-93 in CIR mice brains was significantly reduced, while Ago-miR-93 (a type of miRNA analog) increased its expression. Ago-miR-93 alleviated neurological deficits and reduced cerebral infarction volume in the mice. Furthermore, Ago-miR-93 inhibited inflammatory responses following CIR. Ago-miR-93 decreased the rate of cell apoptosis following CIR. In addition, miR-93 downregulated IRAK4 protein expression, but did not alter its mRNA expression levels in BV2 cells. miR-93 expression reduced the expression of pro-inflammatory factors in BV2 cells. Ago-miR-93 inhibited IRAK4 expression in the brain tissues of CIR mice. The present study demonstrated that miR-93 inhibits inflammatory responses and cell apoptosis following CIR by targeting the IRAK4 signaling pathway.
Keywords: cerebral ischemia reperfusion injury, microRNA-93, interleukin-1 receptor-associated kinase 4
Introduction
Cerebral ischemia reperfusion (CIR) injury is a pathological process in which ischemia and hypoxia lead to nerve damage that is further aggravated after recovering blood perfusion in a short term (1). Inflammatory response and apoptosis are the main mechanisms for nerve cell injury after CIR. Therefore, studies on the inhibition of inflammatory response and apoptosis after CIR may be of great clinical significance, and provide novel therapy for the treatment of ischemic cerebrovascular diseases.
Interleukin (IL)-1 receptor-associated kinase 4 (IRAK4) is a key molecule that is found to participate in innate immune response process (2). IRAK4 belongs to IRAK family, which also includes IRAK-1, IRAK-2 and IRAK-M (3). Studies show that IRAK4 connects upstream and downstream signal transductions, induces signal transduction cascades, and plays a role in inflammatory signaling pathways mediated by Toll-like receptors (TLRs)/IL-1 receptor (IL-1R) (4,5). Further studies on the regulatory mechanism and molecular signaling network of IRAK4 may provide effective strategy for the treatment of nerve injury after CIR.
MicroRNA (miRNA or miR) is a small non-coding RNA molecule (containing 18–25 nucleotides) in eukaryotes that inhibits mRNA translation or degrades mRNA by binding with the 3′-untranslated region (UTR) of mRNA molecules transcribed from specific genes (6). In human body, miRNA expression, which is mediated by multiple signaling pathways, tightly regulates physiological and pathological processes in the body (7). Studies demonstrate that miRNAs are closely associated with ischemic cerebrovascular diseases (8,9). Microarray study shows that the levels of miRNAs are altered in the acute phase of cerebral stroke (10). In addition, changes in miRNA levels are also found in brain tissues from mouse model of CIR (11). Importantly, miRNAs also affect the pathological process after CIR by targeting multiple genes (12,13). In the present study, we investigate the regulatory effect of IRAK4/miR-93 on nerve injury and the mechanism under this process using CIR mouse model.
Materials and methods
Cells
Mouse microglia cell line BV2 cells (purchased from Cell Bank, Wuhan University, Wuhan, China) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37°C in an incubator with 5% CO2. Cell growth was monitored every day and medium was changed every two days. When reaching 80–90% confluency, the cells were passaged at a ratio of 1:3 every two or three days.
BV2 cells in log phase were digested and seeded in 6-well plates for further study. When reaching 70% confluency, the cells were transfected with mmu-miR-93 mimics or mmu-miR-93 inhibitor (GenePharma, Shanghai, China) using Lipofectamine® 2000 agent following the manufacturer's manual (Thermo Fisher Scientific, Inc., Waltham, MA, USA). For control, miR-NC was used instead of mmu-miR-93 mimics or mmu-miR-93 inhibitors. In the first vial, 2.5 µl scramble miRNA, mmu-miR-93 mimics, or 2.5 µl mmu-miR-93 inhibitor was mixed with 50 µl Opti Memi medium (Thermo Fisher Scientific, Inc.). In the second vial, 1 µl Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.) was mixed with 50 µl Opti-MEM I medium. After standing still for 5 min, the two vials were combined for another waiting at room temperature for 20 min. Then, the mixtures were added onto cells in respective groups. At 6 h later, the medium was replaced with RPMI-1640 containing 10% fetal bovine serum. At 48 h after transfection, the cells were harvested for further use.
Animals
C57 mice were divided into CIR (control) group (n=6), sham-operation group (n=6), and Ago-miR-93 group (received CIR surgery, n=6). Mice in control group was not treated, sham-operation group received anaesthesia and sham operation, and Ago-miR-93 group was injected with Ago-miR-93 (a kind of miRNA analog) (RiboBio Co., Ltd., Guangzhou, China) via caudal vein. CIR mouse model was constructed using suture embolization method. Anesthesia was induced in spontaneously breathing animals by intraperitoneal injection of ketamine (100 mg/kg body weight) and xylazine (20 mg/kg body weight). During surgery, animals were placed on a heating device to ensure normothermia (37°C). After a midline skin incision in the neck, the left common carotid artery and external carotid artery were isolated and ligated. Following arteriotomy a 4.0 nylon monofilament (Ethilon; Johnson & Johnson, Sint-Stevens-Woluwe, Belgium) with a heat-blunted tip was inserted into the left internal carotid artery and advanced cranially to the origin of the middle cerebral artery until a gentle resistance was felt. The occluding filament was left in situ for 60 min. Then, animals were re-anesthetized and the occluding monofilament was withdrawn to allow for reperfusion. All animal experiments were conducted according to the ethical guidelines of Jining No. 1 People's Hospital. All animal research was approved by Jining No. 1 People's Hospital Committee on Animal Research.
At 24 h after reperfusion, 5 mice in each group were decapitated to obtain brain tissues. The brain was cut evenly into 5 coronal slices, immersed into 2% triphenyl tetrazolium chloride, and incubated at 37°C for 30 min. After staining, the samples were fixed in 4% paraformaldehyde for 24 h before imaging. Image J software (version 1.37c; http://rsb.info.nih.gov/ij/) was used to determine the area and volume of cerebral infarction (red region indicates normal brain tissues, while pale region indicates infarction tissues). Corrected infarction volume percentage = corrected infarction volume/non-ischemic hemisphere volume × 100%. For the scoring of neurologic function loss, the Zea-Longa 5-point scheme was used to evaluate nerve functions of all mice. Normal mice scored 0 point, while higher points represented more severe functional defects. Mice with 0 and 4 points were excluded from the study.
Quantitative polymerase chain reaction (qPCR)
Before total RNA extraction, tissues (100 mg) were ground into powder using liquid nitrogen before addition of 1 ml TRIzol (Thermo Fisher Scientific, Inc.) for lysis, and BV2 cells (2×105) were trypsinized and lysed by 1 ml Trizol (Thermo Fisher Scientific, Waltham, Inc.). After lysis, total RNA was extracted using phenol chloroform method. The purity of RNA was determined by A260/A280 using ultraviolet spectrophotometry (Nanodrop ND1000, Thermo Scientific, Waltham, Inc.). Then, cDNA was obtained by reverse transcription using PrimeScript RT Reagent Kit (Takara Bio, Dalian, China) from 1 µg RNA and stored at −20°C.
To detect 3′-UTR miRNA binding sites of IRAK4 mRNA, TargetScan (http://www.targetscan.org/) was employed. To measure miR-93 expression in tissues and cells, SYBR PrimeScript miRNA RT-PCR kit (Takara Bio) was employed, using U6 as internal reference. The PCR system (25 µl) included 12.5 µl SYBR Premix Ex Taq, 1 µl PCR Forward Primer (miR-93, 5′-AGGCCCAAAGTGCTGTTCGT-3′), 1 µl Uni-miR qPCR Primer, 2 µl template, and 8.5 µl ddH2O. Each sample was tested in triplicate. PCR conditions were: initial denaturation at 95°C for 30 sec; 40 cycles of 95°C for 5 sec and 60°C for 20 sec. The 2−ΔΔCq method was used to calculate the relative expression of miR-93.
To determine IRAK4 (forward, 5′-CAAGTGATGGAGATGACCTCTGCTTAGT-3′; reverse, 5′-TCTAGCAATAACTGAGGTTCACGGTGTT-3′), IL-1β (forward, 5′-AATCTCACAGCACATCAA-3; reverse, 5′-AGCCCATACTTTAGGAAGACA-3′), tumor necrosis factor (TNF)-α (forward, 5′-TTCGAGTGACAAGCCTGTAGC-3′; reverse, 5′-AGATTGACCTCAGCGCTGAGT-3′), BCL-2 (forward, 5′-GAACTGGGGGAGGATTGTGG-3′; reverse, 5′-GCATGCTGGGGCCATATAGT-3′) and Bax (forward, 5′-CTGGATCCAAGACCAGGGTG-3; reverse, 5′-CCTTTCCCCTTCCCCCATTC-3′) mRNA expression in tissues and cells, mRNA SYBR-Green qPCR Kit (Takara Bio) was carried out, using glyceraldehyde-3-phosphate dehydrogenase (forward, 5′-GCAGTGGCAAAGTGGAGATTG-3′; reverse, 5′-TGCAGGATGCATTGCTGACA-3′) as internal reference. The PCR system (20 µl) included 10 µl SYBR EX Taq-Mix, 0.5 µl upstream primer, 0.5 µl downstream primer (Table I), 1 µl cDNA and 8 µl ddH2O. Each sample was tested in triplicate. PCR conditions were: initial denaturation at 95°C for 10 min; 40 cycles of 95°C for 1 min, 60°C for 40 sec, 72°C for 30 sec, and 72°C for 1 min. The 2−ΔΔCq method was used to calculate the relative expression of mRNA.
Table I.
Primer sequences.
| Primer | Sequence (5′-3′) |
|---|---|
| IRAK-4 | |
| Forward | CAAGTGATGGAGATGACCTCTGCTTAGT |
| Reverse | TCTAGCAATAACTGAGGTTCACGGTGTT |
| IL-1β | |
| Forward | AATCTCACAGCACATCAA |
| Reverse | AGCCCATACTTTAGGAAGACA |
| TNF-α | |
| Forward | TTCGAGTGACAAGCCTGTAGC |
| Reverse | AGATTGACCTCAGCGCTGAGT |
| Bax | |
| Forward | CTGGATCCAAGACCAGGGTG |
| Reverse | CCTTTCCCCTTCCCCCATTC |
| GADPH | |
| Forward | GCAGTGGCAAAGTGGAGATTG |
| Reverse | TGCAGGATGCATTGCTGACA |
IRAK-4, interleukin-1 receptor-associated kinase-4; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α; GADPH, glyceraldehyde-3-phosphate dehydrogenase.
Western blotting
Tissues (50 mg) were ground using liquid nitrogen, while BV2 cells (2×105) were trypsinized and collected. Then, precooled Radio-Immunoprecipitation Assay (RIPA) lysis buffer (600 µl; 50 mM Tris-base, 1 mM EDTA, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 1% sodium deoxycholate; Beyotime Institute of Biotechnology, Shanghai, China) was added to the samples. After lysis for 50 min on ice, the mixture was centrifuged at 12,000 × g and 4°C for 5 min. The supernatant was used to determine protein concentration by bicinchoninic acid (BCA) protein concentration determination kit (RTP7102; Real-Times Biotechnology Co., Ltd., Beijing, China). Protein samples (50 µg) were then mixed with equal volume of 2X sodium dodecyl sulfate loading buffer before denaturation in boiling water bath for 5 min. Afterwards, 10 µl (1 µg/µl) samples were subject to sodium dodecyl sulfate-polyacrylamide gel electrophoresis at 100 V. The resolved proteins were transferred to polyvinylidene difluoride membranes on ice (300 mA, 1.5 h) and blocked with 50 g/l skimmed milk at room temperature for 1 h. Then, the membranes were incubated with primary antibodies of IRAK4 (catalogue no. ab13685), BAX (catalogue no. ab182734), IL-1β (catalogue no. ab200478) and TNF-α (catalogue no. ab6671) (all 1:1,000; Abcam, Cambridge, UK) and GAPDH primary antibody (catalogue no. ab9485) (1:2,000; Abcam, Cambridge, UK) at 4°C overnight. After extensive washing with phosphate-buffered saline with Tween-20 for 3 times of 15 min, the membranes were incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (catalogue no. ab6721; 1:1,000; Abcam, Cambridge, UK) for 1 h at room temperature before washing with phosphate-buffered saline with Tween-20 for 3 times of 15 min. Then, the membrane was developed with enhanced chemiluminescence detection kit (Sigma-Aldrich, St. Louis, MO, USA) for imaging. Image lab (Bio-Rad Laboratories, Inc., Hercules, CA, USA) software was used to acquire and analyze images. The contents of target proteins were expressed relative to GAPDH.
Enzyme-linked immunosorbent assay (ELISA)
At 24 h after transfection, the concentrations of IL-1β (product code: MLB00C; R&D Systems, Inc., Minneapolis, MN, USA) and TNF-α (product code: MTA00B; R&D Systems, Inc.) in the supernatant of cells in control, mmu-miR-93 mimics and mmu-miR-93 inhibitor groups were measured using ELISA kits according to the manufacturer's manual (R&D Systems, Inc.). Absorbance at 450 nm was measured using a microplate reader (Bio-Rad Laboratories, Inc.) within 15 min after stopping the reactions.
Statistical analysis
The results were analyzed using SPSS 17.0 statistical software (IBM SPSS, Armonk, NY, USA). Measurement data were expressed as means ± standard deviation. To compare the difference between two groups, t-test was used. P<0.05 indicates statistically significant differences.
Results
The level of miR-93 in CIR mouse brain is significantly reduced, while Ago-miR-93 increases its expression
To measure the expression of miR-93, qRT-PCR was used. The data showed that miR-93 level in brain tissues from CIR mice was significantly lower than that in mice in sham-operation group (P<0.05) (Fig. 1A). In addition, injection of Ago-miR-93 vial caudal vein significantly enhanced the expression of miR-93 compared with CIR group (Fig. 1B). The result suggests that the level of miR-93 in CIR mouse brain is significantly reduced, while Ago-miR-93 increases its expression.
Figure 1.

Effect of Ago-miR-93 injection on the relative expression of miR-93 in mice. (A) miR-93 level in brain tissues from CIR mice compared with that in mice in sham-operation group. (B) Expression of miR-93 in the group injected with Ago-miR-93 vial caudal vein compared with CIR group. qRT-PCR was used to measure the expression of miR-93. qPCR was used to measure the expression of miR-93. *P<0.05 compared with the other group. CIR, cerebral ischemia reperfusion.
Ago-miR-93 alleviates neurological deficits in mice
To evaluate neurological deficits in mice, Zea-Longa 5-point scheme was used. Mice in sham-operation group showed no nervous functional defects. At 24, 48, and 72 h after reperfusion, the CIR mice had scores of 2.12±0.31, 2.32±0.20 and 2.15±0.21, respectively. After injection of Ago-miR-93, the mice scored 1.95±0.28, 1.8±0.2 and 1.47±0.27 at 24, 48 and 72 h, respectively. Of note, the scores in Ago-miR-93 at each time point were significantly lower than those in CIR group at each time point (P<0.05) (Fig. 2). The result indicates that Ago-miR-93 alleviates nervous functional defects in mice.
Figure 2.

Effect of Ago-miR-93 on neurological deficits of mice with CIR. Neurological deficit scores were determined according to Zea-Longa 5-point scheme. *P<0.05 compared with CIR group. CIR, cerebral ischemia reperfusion.
Ago-miR-93 reduces cerebral infarction volume in mice
To examine the effect of Ago-miR-93 on cerebral infarction, brain tissues were stained using triphenyl tetrazolium chloride. Mice in sham-operation group had no stained area, but those in CIR group had pale infarction area. In addition, the volume of infarction in Ago-miR-93 group (16±3.15 mm3) was significantly lower than that in CIR group (37±5.42 mm3) (P<0.05) (Fig. 3). The result suggests that Ago-miR-93 reduces cerebral infarction volume in mice.
Figure 3.
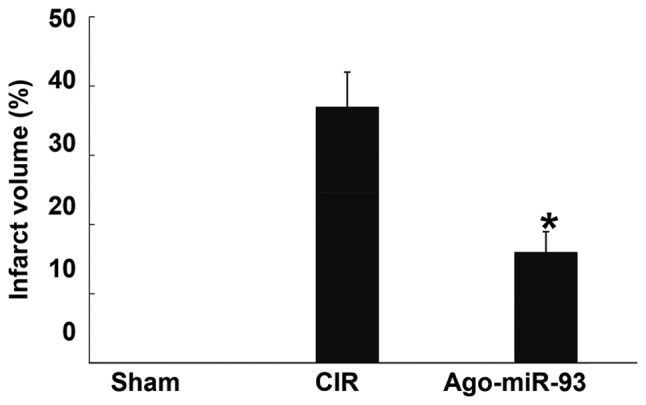
Effect of Ago-miR-93 on cerebral infarction in mice with CIR. Brain tissues were stained using triphenyl tetrazolium chloride and the volume of infarction was measured. *P<0.05 compared with CIR group. CIR, cerebral ischemia reperfusion.
Ago-miR-93 inhibits inflammatory responses after CIR
To detect inflammatory responses after CIR, the levels of IL-1β and TNF-α proteins and mRNA were determined using western blotting and qRT-PCR, respectively. Western blots showed that the contents of IL-1β and TNF-α proteins in Ago-miR-93 group were significantly lower than those in CIR group (P<0.05) (Fig. 4A and B). Similarly, qPCR data showed that IL-1β and TNF-α mRNA expression in Ago-miR-93 group were significantly lower than those in CIR group (P<0.05) (Fig. 4C). These results suggest that Ago-miR-93 inhibits inflammatory responses after CIR.
Figure 4.

Effect of Ago-miR-93 on inflammatory responses after CIR. The levels of (A and B) IL-1β and TNF-α proteins and (C) mRNA were determined using western blotting and qPCR, respectively. *P<0.05 compared with CIR group. CIR, cerebral ischemia reperfusion..
Ago-miR-93 decreases the degree of cell apoptosis after CIR
To detect cell apoptosis after CIR, the ratio of Bax/Bcl-2 was measured using Western blotting and qPCR. The data showed that the ratio of Bax/Bcl-2 protein and mRNA in Ago-miR-93 group were both significantly decreased compared with those in CIR group (P<0.05) (Fig. 5A-C). The result indicates that Ago-miR-93 decreases the degree of cell apoptosis after CIR.
Figure 5.

Effect of Ago-miR-93 on cell apoptosis after CIR. The expressions of Bax and Bcl-2 (A and B) protein and (C) mRNA were measured using western blotting and qPCR, respectively. *P<0.05 compared with CIR group. CIR, cerebral ischemia reperfusion.
miR-93 downregulates IRAK4 protein expression, but does not alter its mRNA levels in BV2 cells
Using bioinformatics tools (microRNA.org), the target genes of mmu-miR-93 were predicted, and the data showed that miR-93 could probably target and regulate IRAK4, a key factor in TLRs/IL-1R-mediated inflammatory signaling pathway (Fig. 6A). To test this idea, we transfected BV2 cells with mmu-miR-93 mimics or mmu-miR-93 inhibitor, and tested the expression of IRAK4 using Western blotting and qPCR. qPCR showed that miR-93 expression in cells transfected with mmu-miR-93 mimics was significantly elevated compared with scramble miRNA transfected cells (NC group) (P<0.05) (Fig. 6B), while miR-93 expression in cells transfected with mmu-miR-93 inhibitor was significantly decreased compared with NC group (P<0.05) (Fig. 6C). Western blotting data showed that the level of IRAK4 in cells transfected with mmu-miR-93 mimics was significantly lower than that in NC group (P<0.05) (Fig. 6D). By contrast, the level of IRAK4 in cells transfected with mmu-miR-93 inhibitor was significantly higher than that in NC group (P<0.05) (Fig. 6E). Interestingly, qRT-PCR data showed that IRAK4 mRNA levels in cells transfected with mmu-miR-93 mimics or mmu-miR-93 inhibitor were not significantly different from that in NC group (P>0.05) (Fig. 6F and G). These results suggest that miR-93 down-regulates IRAK4 protein expression, but does not alter its mRNA levels in BV2 cells.
Figure 6.

Effect of miR-93 on IRAK4 expression in BV2 cells. (A) Predicted interactions between miR-93 and IRAK4 using bioinformatics. (B-C) Relative miR-93 expression in BV2 cells transfected with (B) mmu-miR-93 mimics or (C) mmu-miR-93 inhibitor. (D and E) Relative expression of IRAK4 protein in BV2 cells transfected with (D) mmu-miR-93 mimics or (E) mmu-miR-93 inhibitor. (F and G) Relative expression of IRAK4 mRNA in BV2 cells transfected with (F) mmu-miR-93 mimics or (G) mmu-miR-93 inhibitor. *P<0.05 compared with NC group. IRAK 4, interleukin-1 receptor-associated kinase 4; NC, negative control.
miR-93 expression lowers the expression of pro-inflammatory factors in BV2 cells
To test the effect of miR-93 on the levels of pro-inflammatory factors secreted by BV2 cells, ELISA was performed. The data showed that cells transfected with mmu-miR-93 mimics had significantly reduced levels of IL-1β and TNF-α compared with NC group (P<0.05), while those transfected with mmu-miR-93 inhibitor had significantly elevated IL-1β and TNF-α levels compared with NC group (P<0.05) (Fig. 7). The result indicates that miR-93 expression lowers the expression of pro-inflammatory factors in BV2 cells.
Figure 7.

Effect of miR-93 on the levels of pro-inflammatory factors secreted by BV2 cells. (A and B) Levels of IL-1β secreted by cells transfected with (A) mmu-miR-93 mimics or (B) mmu-miR-93 inhibitor. (C and D) Levels of TNF-α secreted by cells transfected with (C) mmu-miR-93 mimics or (D) mmu-miR-93 inhibitor. ELISA was used to measure the levels of IL-1β and TNF-α. *P<0.05 compared with NC group. NC, negative control; IL, interleukin; TNF, tumour necrosis factor.
Ago-miR-93 inhibits IRAK4 expression in brain tissues of CIR mice
To investigate the effect of Ago-miR-93 on IRAK4 expression in brain tissues of CIR mice, Western blotting was carried out. The data showed that the expression of IRAK4 protein in mice injected with Ago-miR-93 was significantly lower than that in CIR group (P<0.05) (Fig. 8). The result suggests that Ago-miR-93 inhibits IRAK4 expression in brain tissues of CIR mice.
Figure 8.

Effect of Ago-miR-93 on IRAK4 expression in brain tissues of CIR mice. Western blotting was used to measure IRAK4 protein expression. (A) Western blots of IRAK4 and GADPH. (B) Quantification of relative intensity of IRAK4 against GADPH. *P<0.05 compared with CIR group. CIR, cerebral ischemia reperfusion. IRAK 4, interleukin-1 receptor-associated kinase.
Discussion
TLR/IL-1R family plays a central regulatory role in immune responses induced by various infectious diseases, and IRAK4 is a key factor in TLR/IL-1R family common signal transduction system (14). TIR family receptors are activated at the start of inflammation, facilitate the binding of IRAK4 and IRAK1 in the receptors, and participate in the formation of receptor complex after binding with Toll-interacting protein (Tollip), myeloid differentiation factor 88 (MyD88) and tumor necrosis factor-associated factor 6 (TRAF6). Then, IRAK-1 is phosphorylated by IRAK4, and the autophosphorylation of IRAK-1 is initiated, finally leading to the hyperphosphorylation status of IRAK-1. After the configuration of IRAK-1 is changed, it dissociates from MyD88 due to reduced affinity, and forms a complex with TRAF6, leading to the oligomerization of TRAF6 and the activation of NF-κB. As a consequence, activated NF-κB stimulates the activation of pro-inflammatory factors IL-1β, IL-6 and TNF-α, and induces downstream cascade inflammatory responses, leading to inflammatory injuries to tissues (15). It is reported that cytokines like NF-κB cannot be activated by lipopolysaccharide or IL-1 in mice or cells lacking IRAK4, and IRAK4-/- mice have high tolerance against lipopolysaccharide (16). These findings show that IRAK4 is of great importance in TLR/IL-1R signal transduction.
Microglia cells are the main immune cells in the central nervous system. In vitro study shows that activated microglia cells stimulated by lipopolysaccharide or interferon produce multiple immune effector molecules, such as IL-1β, IL-18, TNF-α, active nitrogen, chemokines, and secreted proteins (17). Abnormal expression of miRNA is closely related to the occurrence and development of CIR injury (8,9).
Compared with normal mice, the expression of miR-93 in cerebral tissues from CIR mice was reduced, suggesting that the reduction in miR-93 expression is related to the occurrence and development of CIR injury. Mouse model with overexpression of miR-93 was constructed by injection of chemically synthesized Ago-miR-93. Our results show that neurological deficits and cerebral infarction areas of mice in Ago-miR-93 group were lower than those in CIR group and sham-operation group, suggesting that overexpression of miR-93 alleviates CIR injury symptoms. In the meantime, Western blotting data demonstrate that the expression of pro-inflammatory factors (IL-1β and TNF-α) and the ratio of Bax/Bcl-2 in Ago-miR-93 group is also lower than that in CIR group, indicating that miR-93 participates in CIR injury process by regulating the expression of inflammatory factors and apoptotic proteins.
Using bioinformatics tools, we identify that IRAK4 has regulatory relationship with miR-93. Recently, it is discovered that miR-93 inhibits inflammatory cytokine secretion from macrophages stimulated by lipopolysaccharide by targeting IRAK4 (18). Our data in the present study show that miR-93 expression is negatively correlated with IRAK4 protein expression in BV2 cells. These findings provide evidence that miR-93 plays a role by regulating IRAK4 expression. However, we also find that despite IRAK4 protein expression in BV2 cells is negatively correlated with miR-93, IRAK4 mRNA expression is not correlated with miR-93. ELISA results show that the levels of IL-1β and TNF-α secreted by cells with overexpression of miR-93 are lower than NC group of cells, while the levels of IL-1β and TNF-α secreted by cells with inhibited expression of miR-93 are higher than NC group of cells. Further analysis of IRAK4 protein expression in cerebral tissues shows that IRAK4 protein expression in mice in Ago-miR-93 group is decreased compared with CIR group. The result indicates that miR-93 exerts its anti-inflammatory effect by targeting IRAK4 in CIR injury. However, direct binding between miR-93 and 3′-untranslated region of IRAK4 still needs to be confirmed in future studies. In conclusion, elevated miR-93 expression after CIR injury is correlated with reduced expression of IRAK4, and enhanced expression of pro-inflammatory factors and pro-apoptotic protein. The present study elucidates the mechanism by which miR-93 regulates CIR injury via IRAK4.
Acknowledgements
We would like to thank Dr Zhongrui Yan for his valuable help in completing this study.
References
- 1.Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: The role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43:1382–1397. doi: 10.1227/00006123-199812000-00076. [DOI] [PubMed] [Google Scholar]
- 2.Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: A novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci USA. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 4.Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, Vandenburg Y, Xiao H, Qian W, Hamilton T, et al. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J Exp Med. 2007;204:1025–1036. doi: 10.1084/jem.20061825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeuchi O, Akira S. Toll-like receptors; their physiological role and signal transduction system. Int Immunopharmacol. 2001;1:625–635. doi: 10.1016/S1567-5769(01)00010-8. [DOI] [PubMed] [Google Scholar]
- 6.Pasquinelli AE. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13:271–282. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- 7.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 8.Gan CS, Wang CW, Tan KS. Circulatory microRNA-145 expression is increased in cerebral ischemia. Genet Mol Res. 2012;11:147–152. doi: 10.4238/2012.January.27.1. [DOI] [PubMed] [Google Scholar]
- 9.Tsai PC, Liao YC, Wang YS, Lin HF, Lin RT, Juo SH. Serum microRNA-21 and microRNA-221 as potential biomarkers for cerebrovascular disease. J Vasc Res. 2013;50:346–354. doi: 10.1159/000351767. [DOI] [PubMed] [Google Scholar]
- 10.Tan KS, Armugam A, Sepramaniam S, Lim KY, Setyowati KD, Wang CW, Jeyaseelan K. Expression profile of MicroRNAs in young stroke patients. PLoS One. 2009;4:e7689. doi: 10.1371/journal.pone.0007689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lei P, Li Y, Chen X, Yang S, Zhang J. Microarray based analysis of microRNA expression in rat cerebral cortex after traumatic brain injury. Brain Res. 2009;1284:191–201. doi: 10.1016/j.brainres.2009.05.074. [DOI] [PubMed] [Google Scholar]
- 12.Zhao H, Wang J, Gao L, Wang R, Liu X, Gao Z, Tao Z, Xu C, Song J, Ji X, Luo Y. MiRNA-424 protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke. 2013;44:1706–1713. doi: 10.1161/STROKEAHA.111.000504. [DOI] [PubMed] [Google Scholar]
- 13.Ouyang YB, Lu Y, Yue S, Xu LJ, Xiong XX, White RE, Sun X, Giffard RG. miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol Dis. 2012;45:555–563. doi: 10.1016/j.nbd.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loiarro M, Ruggiero V, Sette C. Targeting TLR/IL-1R signalling in human diseases. Mediators Inflamm. 2010;2010:674363. doi: 10.1155/2010/674363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 17.Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y, Jin H, Yang X, Wang L, Su L, Liu K, Gu Q, Xu X. MicroRNA-93 inhibits inflammatory cytokine production in LPS-stimulated murine macrophages by targeting IRAK4. FEBS Lett. 2014;588:1692–1698. doi: 10.1016/j.febslet.2014.03.013. [DOI] [PubMed] [Google Scholar]