

Abstract
Trastuzumab has been widely applied as a treatment for human epidermal growth factor 2 (HER2)-overexpressing breast cancer. However, the therapeutic efficacy of trastuzumab is limited. Flap endonuclease 1 (FEN1) is a multifunctional endonuclease that has a crucial role in DNA recombination and repair. Inhibition of FEN1 is associated with the reversal of anticancer drug resistance. However, it is unclear whether FEN1 is involved in trastuzumab resistance. In the present study, it was demonstrated that trastuzumab increases the expression of FEN1, and FEN1 knockdown significantly enhanced the sensitivity of BT474 cells to trastuzumab (P<0.05). It was also revealed that trastuzumab induced HER receptor activation, increased binding with FEN1 and estrogen receptor α (ERα), and upregulated ERα-target gene transcription (P<0.05). Upon silencing of FEN1 expression with siRNA, activation of HER receptor and FEN1 binding to ERα were decreased, and trastuzumab-induced ERα target gene upregulation was partially ameliorated (P<0.05). These results suggest that FEN1 may mediate trastuzumab resistance via inducing HER receptor activation and enhancing ERα-target gene transcription. The findings of the present study indicate a novel role of FEN1 in trastuzumab resistance, suggesting that targeting FEN1 may enhance the efficiency of trastuzumab as a treatment for HER2-positive breast cancer.
Keywords: flap endonuclease 1, human epidermal growth factor, trastuzumab, estrogen receptor α
Introduction
Human epidermal growth factor 2 (HER2)-positive breast cancer exhibits aggressive behavior and is regarded as a refractory disease (1). At present, increasing the efficacy of anticancer therapy and developing alternative therapeutic strategies is a considerable challenge. Trastuzumab is a recombination monoclonal antibody that specifically targets the HER2 extracellular domain (2). It has previously been reported that trastuzumab is able to inhibit the activation of ligand-independent HER signaling and block its downstream pathways (1), and in clinical trials it has been reported to markedly improve survival in early and metastatic breast cancers (3,4). However, the therapeutic efficacy of trastuzumab treatment alone is <30% (5), indicating that some underlying mechanisms are not fully understood. When trastuzumab blocks the function of HER2, other members of the HER family are activated to compensate for the loss of HER2 activity via a complex biological network (6). Furthermore, activated HER receptors have been demonstrated to promote trastuzumab resistance by activating shared downstream signaling pathways (1). In the present study, the molecular mechanisms of trastuzumab resistance were investigated with the goal of identifying a crucial factor to predict the efficacy of trastuzumab and to reverse drug resistance.
Flap endonuclease 1 (FEN1) is a crucial enzyme for the maintenance of genomic stability, which functions by processing Okazaki fragment maturation and DNA intermediates during long-patch base excision repair (7,8). Although FEN1 is generally regarded as a tumor suppressor gene, many studies have reported that it is highly expressed in proliferative cancer cells and is essential for cell growth and proliferation in tumor tissues (9–11). Notably, FEN1 expression is significantly upregulated by chemotherapy (5) and other genotoxic stresses, such as DNA-alkylating drugs (12) and radiation treatment (13). Conversely, downregulation of FEN1 enhances cancer cell sensitivity to chemotherapies such as temozolomide, platinum, mitomycin C, and taxol (5,14), which suggests that FEN1 expression is associated with the efficacy of anticancer therapy. However, whether FEN1 mediates resistance to targeted therapy remains unclear.
In the present study, it was demonstrated that trastuzumab increases FEN1 expression, and knockdown of FEN1 increases trastuzumab sensitivity in HER2-overexpressing breast cancer. The results suggest that FEN1 may be a novel target for increasing the anticancer effect of trastuzumab in HER2-overexpressing breast cancer.
Materials and methods
Materials and antibodies
Trastuzumab was obtained from Genentech, Inc., (South San Francisco, CA, USA). Antibody against FEN1 (cat. no. Ab462; 1:1,000) was obtained from Abcam (Cambridge, MA, USA). Antibodies against EGFR (cat. no. 2646; 1:1,000), p-EGFR (Tyr1068; cat. no. 2234; 1:500), phospho-HER2 (Tyr1248; cat. no. 2247S; 1:500), HER3 (cat. no. 4754S; 1:1,000), phospho-HER3 (Tyr1289; cat. no. 2842; 1:250), HER4 (cat. no. 4795; 1:250), phospho-HER4 (Tyr1284; cat. no. 4757S; 1:250), AKT (cat. no. 9272; 1:1,000), p-AKT (Ser473; cat. no. 9271; 1:1,000), ERα (cat. no. 8644S; 1:1,000) and PARP (cat. no. 9542L; 1:1,000) were purchased from Cell Signaling Technology, Inc., (Danvers, MA, USA). HER2 (cat. no. sc-33684; 1:1,000), GAPDH (cat. no. sc-25778; 1:1,000) and secondary goat anti-rabbit IgG-HRP (cat. no. sc-2357; 1:5,000) and goat anti-mouse IgG-HRP (cat. no. sc-516102; 1:5,000) antibodies were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Cell culture and transfection
HER2-positive human breast cancer cell line BT474 cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). According to the literature, the BT474 cell line has naturally high HER2 expression and is regarded as a trastuzumab-sensitive cell line (15). Cells were cultured for 96 h in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (both Invitrogen; Thermo Fisher Scientific, Inc.) in a humidified atmosphere containing 5% CO2 at 37°C. The negative control (NC) and FEN1 siRNA sequences from Guangzhou RiboBio Co., Ltd., (Guangzhou, China) were as follows: NC forward, 5-UUC UCC GAA CGU GUC ACG UTT-3 and reverse, 5-ACG UGA CAC GUU CGG AGA ATT-3; FEN1 forward, 5-GGG UCA AGA GGC UGA GUA AdT dT-3 and reverse, 5-dTd TCC CAG UUC UCC GAC UCA UU-3. The NC or FEN1 siRNA (10 nM) and Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) were diluted in serum-free 1640 medium (Gibco; Thermo Fisher Scientific, Inc.). Following 20 min of incubation at 37°C, the complexes were added to each well of 6-well plates containing serum-free 1640 and cells. Following 72 h of transfection, cells were used in the subsequent experiments.
Cell viability and colony formation assay
The effect of trastuzumab on cell viability was detected using an MTT assay. Cells were separated into 3 groups: Control, NC siRNA and FEN1 siRNA. BT474 cells were seeded into 96-well plates at a density of 10,000 cells/well following transfection with NC or FEN1 siRNA. 10 µg/ml trastuzumab were added to BT474 cells and incubated for the indicated time points (0, 3, 6, 12, and 24 h) at 37°C. Various concentrations of trastuzumab (0, 0.1, 1 and 10 µg/ml) were added to BT474 cells and incubated for 12 h at 37°C. Following incubation, 25 µl of MTT solution (5 mg/ml; Beyotime Institute of Biotechnology, Haimen, China) was added to each well and cells were incubated for 4 h at 37°C. Cell culture supernatants were carefully removed and 200 µl dimethyl sulfoxide (DMSO) was added. The same volume of DMSO was used as the negative control. Finally, the optical density was measured at a wavelength of 570 nm using a microplate reader (Model 550; Bio-Rad Laboratories, Inc., Hercules, CA, USA). The cell inhibition ratio was calculated using the following formula: Cell inhibition (%)=[1-(optical density of the experimental sample/optical density of the untreated group)]x100.
For colony formation, BT474 cells were separated into 5 groups: untreated, NC siRNA, NC siRNA with trastuzumab, FEN1 siRNA and FEN1 siRNA with trastuzumab. Cells were seeded at 1,000 cells/well in 12-well plates following transfection with NC siRNA or FEN1 siRNA for 48 h. Following transfection, cells were treated with or without 10 µg/ml trastuzumab and incubated for 14 days at 37°C. Finally, cells were stained with Wright Giemsa. The number of colonies was counted and images were captured under a light microscope.
Flow cytometry analysis
BT474 cells were seeded into 6-well plates at a density of 5×105/well and transfected with FEN1 specific or NC siRNA for 72 h, following exposed to 10 µg/ml trastuzumab for 12 h and cultured at 37°C. Wells treated with culture media without trastuzumab served as the untreated control. Cells were harvested, centrifuged at 13,000 × g for 5 min at 4°C and fixed in ice-cold 70% ethanol overnight. Samples were subsequently stained using an Annexin V-fluorescein isothiocyanate/propidium iodide apoptosis detection kit (cat no. BMS500FI-100; Invitrogen; Thermo Fisher Scientific, Inc.) and the number of apoptotic cells was determined by FACSCalibur flow cytometry (BD Biosciences, San Jose, CA, USA), according to the manufacturers protocol. Finally, the results were analyzed with WinMDI v. 2.9 software (The Scripps Research Institute, La Jolla, CA, USA).
Western blotting and immunoprecipitation assay
Total protein was obtained using a cell lysis buffer [1% Triton X-100, 1 mM PMSF, 50 mM Tris-HCl pH 7.4 were purchased from Sigma-Aldrich, Merck KGaA. 10 mM EDTA and 2 µg/ml aprotinin were obtained from Solarbio Science and Technology Co., Ltd. (Beijing, China). 150 mM NaCl, 100 mM NaF and 1 mM Na3VO4 were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China)]. Protein concentration was quantified using the Coomassie brilliant blue method. In brief, 4 µl sample protein or negative control cell lysis buffer were added into 1 ml coomassie brilliant blue solution, samples were added in triplicate at 200 µl/well in 96-well plates. The optical density was measured at a wavelength of 570 nm using a microplate reader (Model 550; Bio-Rad Laboratories).
For immunoprecipitation, proteins (200 µg of cell lysates) were incubated with 4 µl anti-FEN1 (cat. no. sc-28355; Santa Cruz Biotechnology, 1:100), anti-ERα (cat. no. 8644S; Cell Signaling Technology, 1:100), control immunoglobulin G (cat. no. sc-2025; Santa Cruz Biotechnology, 1:100) mixed with Protein G Agarose beads (GE Healthcare Life Sciences, Little Chalfont, UK) with gentle agitation overnight at 4°C. Immunoprecipitates were washed four times with lysis buffer mixed with SDS-PAGE 5xsample loading buffer (Beyotime Institute of Biotechnology) for western blotting. Proteins (30–40 µg) were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Membranes were blocked in 5% skimmed milk for 1 h at room temperature and incubated with specific antibodies and GAPDH (internal reference) overnight at 4°C. Subsequently, the membranes were incubated with secondary goat anti-rabbit IgG-HRP antibody or goat anti-mouse IgG-HRP antibody for 40 min at room temperature and protein bands were detected with an enhanced chemiluminescence reagent (SuperSignal Western Pico Chemiluminescent Substrate; Pierce, Rockford, IL, USA) and scanned using the Electrophoresis Gel Imaging Analysis System (DNR Bio-Imaging Systems, Jerusalem, Israel).
RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Cells were transfected with FEN1 specific or NC siRNA for 72 h, following exposed to 10 µg/ml trastuzumab for 12 h and cultured at 37°C. Total RNA was isolated with TRIzol reagent (Thermo Fisher Scientific, Inc.), and SuperScript III Reverse Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.) was used for RNA reverse transcription. RT-qPCR was performed with Taq DNA polymerase (cat. no. 10342020; Invitrogen; Thermo Fisher Scientific, Inc.) and a QuantiTect SYBR Green RT-PCR kit (Qiagen SA, Courtaboeuf, France; cat. no. 204443) and measured using the ABI 7500 Real-time PCR detection system (Applied Biosystems; Thermo Fisher Scientific, Inc.). PCR conditions were as follows: 95°C for 30 sec, followed by 45 cycles of 95°C for 5 sec and 58°C for 25 sec. The 2−∆∆Cq method was applied to process the data (16). Relative expression levels of ERα target genes were normalized to the reference gene, 18S. Primer sequences were as follows: FEN1, forward 5-AAG GTC ACT AAG CAG CAC AAT G-3 and reverse 5-GTA GCC GCA GCA TAG ACT TTG-3; trefoil factor 1 (pS2), forward 5-TCC CCT GGT GCT TCT ATC CTA A-3 and reverse 5-ACT AAT CAC CGT GCT GGG GA-3; progesterone receptor (PgR), forward 5-AGC TCA CAG CGT TTC TAT CA-3 and reverse 5-CGG GAC TGG ATA AAT GTA TTC-3; early growth responses protein 3 (EGR3), forward 5-GAG CAG TTT GCT AAA CCA AC-3 and reverse 5-AGA CCG ATG TCC ATT ACA TT-3; and 18S, forward 5-CCC GGG GAG GTA GTG ACG AAA AAT-3 and reverse 5-CGC CCG CCC GCT CCC AAG AT-3.
Statistical analysis
Differences between the two groups were analyzed using Students t-test and are presented as the mean ± standard deviation. SPSS 17.0 computer software (SPSS, Inc., Chicago, IL, USA) was used for statistical analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
Trastuzumab increases FEN1 expression and knockdown of FEN1 enhances trastuzumab sensitivity
To investigate the resistance mechanism of trastuzumab, BT474 cells were exposed to 10 µg/ml trastuzumab for 0 to 24 h. HER2 and AKT were gradually activated from 1 to 12 h, and activation of HER2 and AKT was markedly reduced at 24 h (Fig. 1A). Phosphorylation of HER2 and AKT when cells were treated with 0.1 to 10 µg/ml trastuzumab for 12 h was also investigated. HER2 and AKT were markedly activated following 0.1 to 5 µg/ml trastuzumab treatment; however, the level of phosphorylation was markedly reduced at 10 µg/ml (Fig. 1B). Expression of FEN1 protein was markedly increased in a time- and dose-dependent manner (Fig. 1A and B, respectively). To analyze the effect of FEN1 on trastuzumab sensitivity, the expression of FEN1 was silenced with FEN1-specific siRNA; a histogram indicates that the average gray value of the NC group is 0.799 and the siFEN1 group is 0.234 and therefore, >70% inhibition of FEN1 protein expression was observed following 72 h of transfection (Fig. 1C). Trastuzumab sensitivity of FEN1 cells significantly increased in a time- and dose-dependent manner with transfection compared with NC cells (P<0.05; Fig. 1D and E, respectively). A similar result was also observed in the colony formation assays. Although knockdown of FEN1 did not have a notable inhibitory effect on cell viability (Fig. 1E), the number of colonies in the group treated with trastuzumab and FEN1-knockdown was significantly lower than that in the trastuzumab treatment alone group (P<0.05; Fig. 1F). These results suggest that trastuzumab upregulates the expression of FEN1, and knockdown of FEN1 enhances sensitivity to trastuzumab in BT474 breast cancer cells.
Figure 1.

Trastuzumab upregulates FEN1 expression and FEN1 knockdown sensitizes BT474 breast cancer cells to trastuzumab. (A) BT474 cells were treated with trastuzumab (10 µg/ml) for the indicated time points or (B) incubated for 12 h with increasing concentrations of trastuzumab. Basal and phosphorylation expression levels of HER2, AKT and FEN1 were analyzed by western blotting. (C) BT474 cells were transfected with FEN1-specific siRNAs and the expression levels were measured using western blotting. (D) Cells were transfected with FEN1-specific siRNA followed by treatment with trastuzumab (10 µg/ml) for the indicated time points or (E) incubated for 12 h with different doses of trastuzumab. *P<0.05 vs. NC. Inhibition of cell viability was determined by MTT assay. (F) Transfected cells were treated with or without 10 µg/ml trastuzumab for 14 days. Finally, cells were stained and counted. *P<0.05, data are presented as mean ± standard deviation. FEN1, flap endonuclease 1; HER2, human epidermal growth factor 2; AKT, protein kinase B; siRNA, short interfering RNA; p, phosphorylated; NC, negative control; siFEN1, FEN1-specific siRNA.
Knockdown of FEN1 increases trastuzumab-induced apoptosis
In the present study, it was further investigated whether FEN1-knockdown-induced enhanced trastuzumab sensitivity had an effect on apoptosis in BT474 cells. It was demonstrated that knockdown of FEN1 led to a marked increase in the number of apoptosis-positive cells, whereas the combination of FEN1 knockdown and trastuzumab lead to higher levels of apoptosis, as estimated by Annexin V staining (Fig. 2A). The level of apoptosis in untreated cells, NC cells, trastuzumab with NC-treated cells, siFEN1-transfected cells, and trastuzumab-treated and siFEN1-transfected cells were 2.93±0.51, 2.77±0.32, 11.63±1.79, 15.90±3.32 and 22.17±0.35%, respectively (Fig. 2B). The percentage of apoptotic cells was significantly higher in the siFEN1 and NC + trastuzumab groups compared with the NC group (P<0.05; Fig. 2B). Furthermore, significantly more apoptotic cells were observed in the siFEN1 + trastuzumab group compared with the NC + trastuzumab group (P<0.05; Fig. 2B). Apoptosis was further investigated by detecting levels of cleaved PARP. Consistent with the above results, the level of cleaved PARP was markedly increased by the combination of trastuzumab and FEN1 knockdown compared with trastuzumab treatment alone (Fig. 2C). These results suggest that FEN1 knockdown enhances trastuzumab-induced apoptosis.
Figure 2.

Knockdown of FEN1 increases trastuzumab-induced apoptosis. BT474 cells were transfected with FEN1-specific prior to treatment with 10 µg/ml trastuzumab for 12 h. NC cells were not treated with trastuzumab. Cells incubated with serum-containing medium served as an untreated control. (A and B) Percentage of apoptosis cells was assessed by flow cytometry analysis. (C) Cells lysates were subjected to detection of PARP expression by western blotting. *P<0.05, data are presented as mean ± standard deviation. FEN1, flap endonuclease 1; PARP, poly ADP-ribose polymerase; siFEN1, FEN1-specific small interfering RNA; NC, negative control.
Knockdown of FEN1 decreases trastuzumab-induced HER receptor activation
To investigate whether FEN1-mediated trastuzumab resistance is associated with HER receptor activation, the HER receptor signaling pathway was analyzed. Phosphorylation levels of all HER receptors' including EGFR, HER2, HER3 and HER4, were enhanced following exposure to 10 µg/ml trastuzumab for 12 h, as was the activation of the downstream AKT pathway. Furthermore, when FEN1 expression in BT474 cells was knocked down, the baseline phosphorylation levels of HER receptors and downstream AKT signaling were markedly reduced, as was the activation of HER receptors and AKT induced by trastuzumab (Fig. 3). These results indicate that knockdown of FEN1 ameliorates the trastuzumab response by decreasing the activation of HER receptors.
Figure 3.
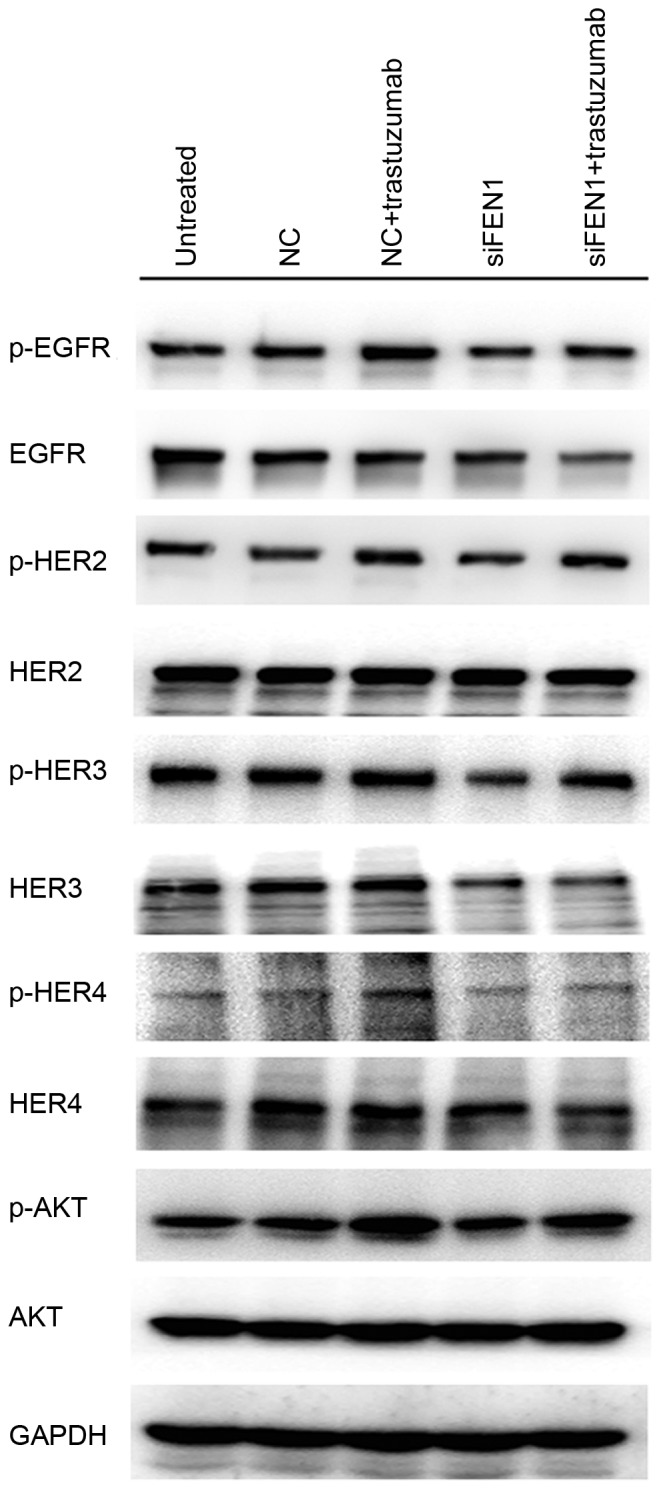
Knockdown of FEN1 decreases trastuzumab-induced HER receptor activation. BT474 cells were transfected with FEN1-specific siRNA prior to treatment with or without 10 µg/ml trastuzumab for 12 h. Cells incubated with serum-containing medium served as untreated controls. Cell lysates were collected to determine phosphorylation levels of HER receptors and AKT expression. FEN1, flap endonuclease 1; HER, human epidermal growth factor; siRNA, short interfering RNA; AKT, protein kinase B; NC, negative control; p, phosphorylated; EGFR, epidermal growth factor receptor; siFEN1, FEN1-specific siRNA.
Trastuzumab increases binding of FEN1 with ERα and FEN1 knockdown reduces trastuzumab-induced ERα-target gene transcription
Previous reports have demonstrated that ERα is upregulated in patients overexpressing HER2 who do not benefit from trastuzumab treatment, which suggests an association between ERα and trastuzumab resistance (17,18). Furthermore, it has been reported that FEN1 is able to interact with multiple ERα domains and modulate ERα-mediated transcription in breast cancer cells (19). To elucidate whether trastuzumab increases the interaction between FEN1 and ERα, an immunoprecipitation assay was performed. The results indicated that the expression of FEN1 was upregulated following trastuzumab stimulation, whereas ERα expression was not. The binding of FEN1 and ERα was increased following trastuzumab treatment, and similar results were observed by immunoprecipitation with an anti-FEN1 antibody. The expression of FEN1 was upregulated, and binding of FEN1 with ERα was markedly increased following exposure to trastuzumab (Fig. 4A). Relative mRNA expression levels of ERα target genes were investigated using RT-qPCR (20) and the levels of pS2, PgR and EGR3 were increased in trastuzumab-treated cells by 6.78-, 3.54- and 2.2-fold compared with NC cells, respectively (all P<0.05; Fig. 4B). However, when BT474 cells were transfected with FEN1-specific siRNA, a significant decrease was observed in the basal levels of pS2, PgR and EGR3 mRNA to approximately 0.26-, 0.31- and 0.34-fold, respectively compared with the Trastuzumab-treated NC cells (P<0.05; Fig. 4B). Furthermore the combination of trastuzumab and FEN1 knockdown significantly reversed trastuzumab-induced ERα target gene upregulation (P<0.05; Fig. 4B). These results indicate that FEN1 induces trastuzumab resistance via increasing interactions with ERα and promoting ERα-mediated gene transcription.
Figure 4.

Trastuzumab increases binding of FEN1 with ERα and knockdown of FEN1 decreases trastuzumab-induced ERα target gene expression. (A) IP analysis was performed to evaluate the interaction between FEN1 and ERα following treatment with 10 µg/ml trastuzumab for 12 h. (B) Reverse transcription-quantitative polymerase chain reaction was performed to measure the expression levels of FEN1, pS2, PgR and EGR3. *P<0.05, data are presented as mean ± standard deviation. FEN1, flap endonuclease 1; ERα, estrogen receptor α; pS2, trefoil factor 1; PgR, progesterone receptor; EGR3, early growth response factor 3; IP, immunoprecipitation; IgG, immunoglobulin G; NC, negative control; siFEN1, FEN1-specifice short interfering RNA.
Discussion
FEN1, a structure-specific endonuclease, maintains genomic integrity by regulating multiple DNA metabolic pathways (7,21). Functional deficiency or mutations of FEN1 that result in nuclease activity deficiency lead to genomic instability and a predisposition to cancer (22,23). Previous reports have demonstrated that FEN1 is dysregulated in breast cancer and multiple other cancerous tissues (23–25). High expression of FEN1 is associated with clinicopathological significance and poor survival (26), suggesting that FEN1 has an important role in the development of breast cancer. It has previously been demonstrated that anticancer therapies are able to induce FEN1 expression (5), and inhibition of FEN1 combined with DNA injury agents results in increased inhibition of proliferation via endogenous DNA damage (12,27–29). In the present study, it was demonstrated that trastuzumab promoted the activation of HER2 and AKT, and concurrently upregulated FEN1 protein expression in BT474 breast cancer cells. FEN1 upregulation triggered by trastuzumab contributed to trastuzumab resistance, indicating that inhibition of FEN1 may be a novel and promising therapeutic strategy to enhance the anticancer effects of trastuzumab.
In HER2-positive breast cancer, the incorporation of trastuzumab in treatment regimens has been reported to improve the clinical outcome (30–33). Regardless, some patients also experience intrinsic and acquired resistance, eventually leading to disease progression (2). Several mechanisms of trastuzumab resistance have been elucidated, such as intrinsic alteration in HER2-like truncated HER2 receptors (34), heterodimer formation between other receptor tyrosine kinases (RTKs) and HER2 (35–37), and constitutive activation of the phosphoinositide 3-kinase (PI3K)/AKT pathway or loss of phosphatase and tensin homolog function (38). Studies have suggested that dual HER2 blockade with trastuzumab and pertuzumab may be significantly associated with clinical outcomes (39,40). The combination of other RTK inhibitors or the PI3K/AKT pathway with trastuzumab partially reversed trastuzumab resistance by acting on bypass-activation or compensation signals (41,42). In the present study, a novel mechanism of FEN1 involving trastuzumab resistance was elucidated, which is completely different from the above mechanisms. The results suggest that knockdown of FEN1 increases sensitivity to trastuzumab in a time- and dose-dependent manner and further enhances trastuzumab-induced inhibition of cell viability and apoptosis in BT474 cells; this suggests that such dual inhibition produces superior effects, and that FEN1 may have a critical role in enhancing sensitivity to trastuzumab in HER2-overexpressing breast cancer cells.
Previous findings have indicated that HER receptor activation induced by trastuzumab via stimulating ligand release and activating survival pathways was responsible for trastuzumab resistance (43,44). Disruption of HER2 activity with inhibitors has been demonstrated to resensitize cancer cells to trastuzumab (44). These reports suggested that HER receptors and downstream signaling activation contribute to trastuzumab resistance. In the present study, knockdown of FEN1 decreased the activation of HER receptors and the downstream activity of AKT, counteracting the effects of trastuzumab in maintaining HER receptor activation. The mechanisms of FEN1-related HER receptor activation remain to be elucidated; therefore, the current study hypothesizes that, one of the possible mechanisms is that FEN1 may participate in HER receptor-mediated DNA synthesis, and another is that FEN1 may stimulate bypass-activation and homo- or heterodimer formation. The association between FEN1 and the activation and dimerization status of HER receptor requires further investigation.
FEN1 interacts with various types of proteins to act through different pathways (7). A previous report indicated that, in breast cancer cells, FEN1 interacts with multiple ERα domains and serves as an effective modulator of ERα-mediated estrogen-responsive gene transcription (19). ERα binds directly to the estrogen response element and regulates the transcription of target genes that control cell proliferation, survival and sensitivity to anticancer drugs (45). A previous report demonstrated that estrogen-responsive genes were highly expressed in trastuzumab-resistant breast cancer cells, and the combination of tamoxifen and trastuzumab reversed trastuzumab resistance by abrogating ERα activation and ERα target gene transcription (46). It remains necessary to elucidate whether trastuzumab treatment increases interactions between FEN1 and ERα to further influence estrogen-responsive gene expression. The results of the present study demonstrated that an increase binding with FEN1 and ERα occurred concomitantly with an increase in FEN1 expression, suggesting a correlation between FEN1 and ERα in trastuzumab sensitivity. It has been reported that pS2, PgR and EGR3 are able to be transcribed by ERα (20). The present findings also showed that the mRNA levels of ERα target genes were increased in trastuzumab-treated cells and were partially restored by knockdown of FEN1. Considering that trastuzumab increases interactions between FEN1 and ERα, FEN1 may serve as a crucial modulator for counteracting trastuzumab treatment.
In conclusion, the results of the present study indicate that trastuzumab upregulates FEN1 expression while concurrently increasing binding of FEN1 and ERα. Inhibition of HER receptor activation and ERα target gene transcription by FEN1 knockdown may enhance the trastuzumab response. These findings indicate that targeting FEN1 may increase trastuzumab sensitivity, and suggest that FEN1 has the potential to be a useful preclinical biomarker in the treatment of HER2-positive breast cancer.
Acknowledgements
The present study was supported by the National Science and Technology Major Project of the Ministry of Science and Technology of China (grant no. 2013JX09303002), the Science and Technology Plan Project of Liaoning Province (grant nos. 2014225013 and 2015020458), the General Project of Liaoning Province Department of Education (grant no. L2015588) and the National Natural Science Foundation of China (grant no. 81172535).
Glossary
Abbreviations
- FEN1
flap endonuclease 1
- HER2
human epidermal growth factor 2
- ERα
estrogen receptor α
- PARP
poly ADP-ribose polymerase
References
- 1.Hynes NE, Lane HA. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1667. [DOI] [PubMed] [Google Scholar]
- 2.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 3.Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365:1273–1283. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez EA, Romond EH, Suman VJ, Jeong JH, Sledge G, Geyer CJ, Martino S, Rastogi P, Gralow J, Swain SM, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: Planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32:3744–3752. doi: 10.1200/JCO.2014.55.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie C, Wang K, Chen D. Flap endonuclease 1 silencing is associated with increasing the cisplatin sensitivity of SGC-7901 gastric cancer cells. Mol Med Rep. 2016;13:386–392. doi: 10.3892/mmr.2015.4567. [DOI] [PubMed] [Google Scholar]
- 6.Narayan M, Wilken JA, Harris LN, Baron AT, Kimbler KD, Maihle NJ. Trastuzumab-induced HER reprogramming in ‘resistant’ breast carcinoma cells. Cancer Res. 2009;69:2191–2194. doi: 10.1158/0008-5472.CAN-08-1056. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: A central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 8.Shen B, Singh P, Liu R, Qiu J, Zheng L, Finger LD, Alas S. Multiple but dissectible functions of FEN-1 nucleases in nucleic acid processing, genome stability and diseases. Bioessays. 2005;27:717–729. doi: 10.1002/bies.20255. [DOI] [PubMed] [Google Scholar]
- 9.Kim IS, Lee MY, Lee IH, Shin SL, Lee SY. Gene expression of flap endonuclease-1 during cell proliferation and differentiation. Biochim Biophys Acta. 2000;1496:333–340. doi: 10.1016/S0167-4889(00)00029-X. [DOI] [PubMed] [Google Scholar]
- 10.Wang K, Xie C, Chen D. Flap endonuclease 1 is a promising candidate biomarker in gastric cancer and is involved in cell proliferation and apoptosis. Int J Mol Med. 2014;33:1268–1274. doi: 10.3892/ijmm.2014.1682. [DOI] [PubMed] [Google Scholar]
- 11.Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, Kernstine KH, Lin D, Shen B. Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol Cancer Res. 2008;6:1710–1717. doi: 10.1158/1541-7786.MCR-08-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikolova T, Christmann M, Kaina B. FEN1 is overexpressed in testis, lung and brain tumors. Anticancer Res. 2009;29:2453–2459. [PubMed] [Google Scholar]
- 13.Christmann M, Tomicic MT, Origer J, Kaina B. Fen1 is induced p53 dependently and involved in the recovery from UV-light-induced replication inhibition. Oncogene. 2005;24:8304–8313. doi: 10.1038/sj.onc.1208994. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Zhou L, Li Z, Zhang T, Liu W, Liu Z, Yuan YC, Su F, Xu L, Wang Y, et al. YY1 suppresses FEN1 over-expression and drug resistance in breast cancer. BMC Cancer. 2015;15:50. doi: 10.1186/s12885-015-1043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, Botero MF, Healy NA, Hilsenbeck SG, Phillips GL, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers-role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011;13:R121. doi: 10.1186/bcr3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Park YH, Shin HT, Jung HH, Choi YL, Ahn T, Park K, Lee A, Do IG, Kim JY, Ahn JS, et al. Role of HER2 mutations in refractory metastatic breast cancers: Targeted sequencing results in patients with refractory breast cancer. Oncotarget. 2015;6:32027–32038. doi: 10.18632/oncotarget.5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z, Barnes CJ, Kumar R. Human epidermal growth factor receptor 2 status modulates subcellular localization of and interaction with estrogen receptor alpha in breast cancer cells. Clin Cancer Res. 2004;10:3621–3628. doi: 10.1158/1078-0432.CCR-0740-3. [DOI] [PubMed] [Google Scholar]
- 19.Schultz-Norton JR, Walt KA, Ziegler YS, McLeod IX, Yates JR, Raetzman LT, Nardulli AM. The deoxyribonucleic acid repair protein flap endonuclease-1 modulates estrogen-responsive gene expression. Mol Endocrinol. 2007;21:1569–1580. doi: 10.1210/me.2006-0519. [DOI] [PubMed] [Google Scholar]
- 20.Fujiki N, Konno H, Kaneko Y, Gohno T, Hanamura T, Imami K, Ishihama Y, Nakanishi K, Niwa T, Seino Y, et al. Estrogen response element-GFP (ERE-GFP) introduced MCF-7 cells demonstrated the coexistence of multiple estrogen-deprivation resistant mechanisms. J Steroid Biochem Mol Biol. 2014;139:61–72. doi: 10.1016/j.jsbmb.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Lieber MR. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays. 1997;19:233–240. doi: 10.1002/bies.950190309. [DOI] [PubMed] [Google Scholar]
- 22.Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, Tsark W, Huang Q, Kernstine K, Zhang X, et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat Med. 2007;13:812–819. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- 23.Yang M, Guo H, Wu C, He Y, Yu D, Zhou L, Wang F, Xu J, Tan W, Wang G, et al. Functional FEN1 polymorphisms are associated with DNA damage levels and lung cancer risk. Hum Mutat. 2009;30:1320–1328. doi: 10.1002/humu.21060. [DOI] [PubMed] [Google Scholar]
- 24.Lv Z, Liu W, Li D, Liu L, Wei J, Zhang J, Ge Y, Wang Z, Chen H, Zhou C, et al. Association of functional FEN1 genetic variants and haplotypes and breast cancer risk. Gene. 2014;538:42–45. doi: 10.1016/j.gene.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 25.Corral R, Lewinger JP, Joshi AD, Levine AJ, Vandenberg DJ, Haile RW, Stern MC. Genetic variation in the base excision repair pathway, environmental risk factors, and colorectal adenoma risk. PLoS One. 2013;8:e71211. doi: 10.1371/journal.pone.0071211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdel-Fatah TM, Russell R, Albarakati N, Maloney DJ, Dorjsuren D, Rueda OM, Moseley P, Mohan V, Sun H, Abbotts R, et al. Genomic and protein expression analysis reveals flap endonuclease 1 (FEN1) as a key biomarker in breast and ovarian cancer. Mol Oncol. 2014;8:1326–1338. doi: 10.1016/j.molonc.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panda H, Jaiswal AS, Corsino PE, Armas ML, Law BK, Narayan S. Amino acid Asp181 of 5-flap endonuclease 1 is a useful target for chemotherapeutic development. Biochemistry. 2009;48:9952–9958. doi: 10.1021/bi9010754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tumey LN, Huck B, Gleason E, Wang J, Silver D, Brunden K, Boozer S, Rundlett S, Sherf B, Murphy S, et al. The identification and optimization of 2,4-diketobutyric acids as flap endonuclease 1 inhibitors. Bioorg Med Chem Lett. 2004;14:4915–4918. doi: 10.1016/j.bmcl.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 29.van Pel DM, Barrett IJ, Shimizu Y, Sajesh BV, Guppy BJ, Pfeifer T, McManus KJ, Hieter P. An evolutionarily conserved synthetic lethal interaction network identifies FEN1 as a broad-spectrum target for anticancer therapeutic development. PLoS Genet. 2013;9:e1003254. doi: 10.1371/journal.pgen.1003254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 31.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga J, Bell R, Jackisch C, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 32.Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 33.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 34.Mukohara T. Mechanisms of resistance to anti-human epidermal growth factor receptor 2 agents in breast cancer. Cancer Sci. 2011;102:1–8. doi: 10.1111/j.1349-7006.2010.01711.x. [DOI] [PubMed] [Google Scholar]
- 35.DiGiovanna MP, Stern DF, Edgerton SM, Whalen SG, Moore D, II, Thor AD. Relationship of epidermal growth factor receptor expression to ErbB-2 signaling activity and prognosis in breast cancer patients. J Clin Oncol. 2005;23:1152–1160. doi: 10.1200/JCO.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 36.Nieto Y, Nawaz F, Jones RB, Shpall EJ, Nawaz S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J Clin Oncol. 2007;25:4405–4413. doi: 10.1200/JCO.2006.09.8822. [DOI] [PubMed] [Google Scholar]
- 37.Gallardo A, Lerma E, Escuin D, Tibau A, Muñoz J, Ojeda B, Barnadas A, Adrover E, Sánchez-Tejada L, Giner D, et al. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br J Cancer. 2012;106:1367–1373. doi: 10.1038/bjc.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engelman JA. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 39.Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, Bianchi G, Cortes J, McNally VA, Ross GA, et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. 2010;28:1138–1144. doi: 10.1200/JCO.2009.24.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cortés J, Fumoleau P, Bianchi GV, Petrella TM, Gelmon K, Pivot X, Verma S, Albanell J, Conte P, Lluch A, et al. Pertuzumab monotherapy after trastuzumab-based treatment and subsequent reintroduction of trastuzumab: Activity and tolerability in patients with advanced human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2012;30:1594–1600. doi: 10.1200/JCO.2011.37.4207. [DOI] [PubMed] [Google Scholar]
- 41.Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res. 2001;61:8887–8895. [PubMed] [Google Scholar]
- 42.Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL. Anti-tumor activity of GW572016: A dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21:6255–6263. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- 43.Feldinger K, Generali D, Kramer-Marek G, Gijsen M, Ng TB, Wong JH, Strina C, Cappelletti M, Andreis D, Li JL, et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget. 2014;5:6633–6646. doi: 10.18632/oncotarget.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kong A, Calleja V, Leboucher P, Harris A, Parker PJ, Larijani B. HER2 oncogenic function escapes EGFR tyrosine kinase inhibitors via activation of alternative HER receptors in breast cancer cells. PLoS One. 2008;3:e2881. doi: 10.1371/journal.pone.0002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashita H, Nishio M, Kobayashi S, Ando Y, Sugiura H, Zhang Z, Hamaguchi M, Mita K, Fujii Y, Iwase H. Phosphorylation of estrogen receptor alpha serine 167 is predictive of response to endocrine therapy and increases postrelapse survival in metastatic breast cancer. Breast Cancer Res. 2005;7:R753–R764. doi: 10.1186/bcr1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen B, Wang Y, Kane SE, Chen S. Improvement of sensitivity to tamoxifen in estrogen receptor-positive and Herceptin-resistant breast cancer cells. J Mol Endocrinol. 2008;41:367–377. doi: 10.1677/JME-08-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]