

Abstract
Introduction
It is increasingly becoming accepted that inflammation may play an important role in the pathogenesis of Alzheimer’s disease (AD), as several immune-related genes have been associated with AD. Among these is tumor necrosis factor (TNF)-α, a proinflammatory cytokine known to play an important role in autoimmune disorders, including rheumatoid arthritis (RA). Although AD and RA appear to involve similar pathological mechanisms through the production of TNF-α, the relationship between AD and RA remains unknown.
Objective
To determine the relative risk of AD among RA patients and non-RA patients, and whether anti-TNF therapy for RA was associated with a lower risk of AD in RA patients.
Methods
We performed a nested case-control study of more than 8.5 million commercially insured adults (aged ≥18 years) in all 50 US states, Puerto Rico, and US Virgin Islands in the Verisk Health claims database. We derived a sub-cohort of subjects with a diagnosis of RA (controls), or RA and AD (cases), matching cases and controls based on age, sex, exposure assessment period, and methotrexate treatment. We also assessed relative risk of AD following exposure to standard RA therapies, including anti-TNF agents (infliximab, adalimumab, etanercept), methotrexate, prednisone, sulfasalazine, and rituximab. Odds ratios were adjusted for comorbidities, including coronary artery disease, diabetes mellitus, and peripheral vascular disease.
Results
AD was more prevalent (p < 0.0001) among RA patients (0.79 %) than among those without RA (0.11 %). Chronic conditions such as coronary artery disease (odds ratio [OR] 1.48; 95 % confidence interval [CI] 1.04–2.05; p = 0.03), diabetes (OR 1.86; 95 % CI 1.32–2.62; p = 0.0004), and peripheral vascular disease (OR 1.61; 95 % CI 1.06–2.43; p = 0.02) significantly increased the relative risk of AD among RA patients. Exposure to anti-TNF agents as a class, but not other immunosuppressive drugs studied, was associated with lowered risk of AD among RA patients (unadjusted OR 0.44; 95 % CI 0.22–0.87; p = 0.02; adjusted OR 0.45; 95 % CI 0.23–0.90; p = 0.02). Sub-group analysis demonstrated that of the three anti-TNF agents studied, only etanercept (unadjusted OR, 0.33; 95 % CI 0.08–0.94; p = 0.03; adjusted OR 0.30; 95 % CI 0.08–0.89; p = 0.02) was associated with a decreased risk of AD in RA patients.
Conclusion
There is an increased risk of AD in the studied RA population. The relative risk of AD among RA subjects was lowered in those exposed to etanercept. Anti-TNF therapy with etanercept shows promise as a potential treatment for AD.
1 Introduction
Alzheimer’s disease (AD) is the most common form of dementia in the elderly. Its incidence increases with age, and doubles between the ages of 65 and 85 years; it affects 32 % of the US population by the age of 85 years [1, 2]. A number of risk factors have been associated with different forms of dementia, including AD [3]. In addition to the well-known APOE-ε4 gene, genetic studies have identified multiple genes that can increase the risk of AD, many of which are related to immune function [4–6]. The pathological hallmark of AD is the deposition of senile plaques, neurofibrillary tangles, and amyloid angiopathy in the brain [7–9]. The deposition of amyloid beta in senile plaques and the hyper-phosphorylation of the microtubule-associated protein tau in neurofibrillary tangles are pathognomonic for AD, but the molecular mechanisms leading to the development of the symptoms are unknown [10–14]. As a result, there are no curative treatments for AD, and current therapeutic treatments for the disease focus on symptomatic control [13, 14]. While recent molecular and technological advances such as the Alzheimer’s Disease Neuroimaging Initiative have tried to correlate cerebrospinal and imaging findings with different clinical stages, guidelines for the diagnosis of AD have not been adopted in clinical practice owing to a lack of verifiable biomarkers [15–17]. Therefore, a diagnosis of AD remains largely one of exclusion. Among the potential biomarker candidates is tumor necrosis factor (TNF)-α; the level of this proinflammatory cytokine is elevated in the cerebrospinal fluid of AD patients and appears to correlate with disease progression [18–20]. However, the blood levels of TNF in AD patients are inconsistent among different studies and their clinical significance is unknown [20–22].
TNF plays a critical role in rheumatoid arthritis (RA), a systemic inflammatory disease that mainly affects diarthrodial joints, although it can affect any organ system. RA is believed to be driven by TNF, based on clinical evidence and RA patients’ responses to anti-TNF therapies [23–25]. In addition to severe joint damage, a potential complication of the chronic inflammation of RA is secondary amyloidosis as a result of tissue deposition of amyloid protein fibrils (AA) [26, 27]. AA is derived from its circulating precursor known as serum amyloid A, which is an acute-phase protein produced by the liver [26–28]. Although this mechanism appears to parallel the deposition of amyloid beta in AD, the impact of chronic elevation of inflammatory proteins (i.e., TNF-α, AA) in the periphery on the pathogenesis of AD remains under investigation.
In the present study, we test the hypothesis that there is an increased relative risk of AD among RA subjects, and anti-TNF therapy for RA will reduce the risks of AD among these subjects. To test the hypothesis, RA subjects were first evaluated for overall risk of AD among a large general population of 8.5 million adults in a commercial database containing medical and pharmacy claims data. Second, individuals who had a diagnosis of RA were further selected using exclusion criteria, and additionally categorized as with or without AD; nested case-control studies were then performed to examine the association between different comorbidities and different therapeutic treatments for RA on the risk of developing AD.
2 Methods
2.1 Study Population
We analyzed the medical and pharmacy claims data in a large database (Verisk Health, Waltham, MA, USA) obtained from commercially insured adults who were 18 years of age or older between January 2000 and November 2007. This time period is referred to as the “analysis period”, which contains the data analyzed for the present study. The medical claims data contain individuals’ diseases diagnoses in the form of International Classification of Diseases, Ninth Revision (ICD-9) codes, while the pharmacy data contain the names of the medications that individuals were put on. These claims data were based on the diagnoses submitted by qualified medical providers to different insurance carriers after in-or out-patient visits and necessary diagnostic workup. Enrolled individuals were employees and their dependents in the private sector, and were located in all 50 states in the US, Puerto Rico, and the United States Virgin Islands. From this cohort, we derived a sub-cohort of subjects with a diagnosis of RA, based on at least two outpatient claims with the same diagnosis or one inpatient claim as defined by ICD-9 for RA. These individuals must have had no previous identifiable pharmacy or diagnosis claims data for RA at least 6 months prior to the start of the analysis period. A summary of all the ICD-9 codes that were used in our study is shown in supplemental Table 1s. We also identified individuals with RA who had a new diagnosis of AD made at least 120 days after the initial diagnosis of RA to ensure sufficient temporal sequence of RA preceding AD based on claims data. We chose 120 days as it represented a minimal time frame for RA patients to return for follow-up visits (i.e., 90 days) and possible workup for AD (i.e., 30 days). Analysis of de-identified data was exempted from continuing review by the Committee for the Protection of Human Subjects at Dartmouth College and Beth Israel Deaconess Medical Center Committee on Clinical Investigation.
2.2 Exclusion Criteria
All individuals were excluded from our analysis if, (1) they had an identifiable diagnosis of RA prior to the analysis period, (2) they had claims data about RA for 6 months prior to the analysis period, and (3) during any time of the analysis period, they also had a diagnosis of inflammatory bowel disease (Crohn’s disease and ulcerative colitis), psoriatic arthritis, frontotemporal dementia, Lewy body dementia, Parkinson’s disease, stroke, or vascular dementia. An exclusion of individuals with psoriatic arthritis or inflammatory bowel disease eliminated individuals with seronegative spondyloarthroapathy, whereas individuals with stroke likely had vascular dementia. In addition, individuals were excluded if they had a diagnosis of AD made before the index date (i.e., diagnosis of RA) or less than 120 days after the index date (see above). They were also excluded if less than 12 months of data were available for assessment of exposure to different therapeutic agents after the index date.
2.3 Control Sampling and Matching Criteria
We used a nested case-control design, an established approach in pharmacoepidemiological studies [29, 30]. Cases (RA subjects with a diagnosis of AD) and controls (RA subjects without a diagnosis of AD) were matched based on the following matching criteria: age within 2 years, sex, exposure assessment period within 3 months, and methotrexate treatment. A total of 80 % of the cases were matched with ten controls based on all criteria. Because of the multiple matching criteria, each case was matched with up to ten controls to improve the efficiency of analysis [31]. Each control was only allowed to match with one case, and controls could not be used as cases at any time. All cases and controls were required to be in the cohort during the same time period and exposure assessment period.
2.4 Exposure Assessment
The occurrence of AD among RA subjects following exposure to different therapies for RA was examined. The exposures to different treatments of RA examined during the analysis period included methotrexate, prednisone, sulfasalazine, three anti-TNF agents (adalimumab, etanercept, and infliximab), and an anti-CD20 agent (rituximab). These treatments are representative of current standard therapies for RA [32]. The outcomes of the exposure to these agents were defined as occurrence of AD, and were assessed 3 months preceding the diagnosis of AD for cases and the corresponding date for controls.
2.5 Comorbidities
Individuals with claims data in the form of ICD-9 codes for common chronic comorbid conditions, including coronary artery disease, diabetes mellitus, hyperlipidemia, hypertension, and peripheral vascular disease, were identified from the database of Verisk Health. All these comorbid conditions were assessed in both cases and controls throughout the entire analysis period.
2.6 Statistical Analysis
Statistical analyses were performed to the characteristics of the cases and controls and to evaluate the association between different therapeutic agents for RA and the development of AD in the current model of case-control study. The characteristics of the cases and controls were compared using (1) a two-tailed Student’s t test in the case of continuous data and (2) a Chi-square test or Fisher’s exact test for categorical data. Additionally, the Chi-square test was used to compare the relative risk of AD among subjects with and without RA. Because the case and controls are matched from study design, to avoid bias from regular logistic regression, we conducted conditional logistic regression analysis without grouping to determine the association between different therapeutic agents or common comorbidities and the relative risk of AD among RA subjects. Univariate analysis consisted of evaluation of the association between individual therapeutic agents or comorbidities and the relative risk of AD. Multivariate analysis was used to address the association between comorbidities and individual therapeutic agents as well as the interactions among different comorbidities. Each of the binary variables indicating exposure to different therapeutic agents, i.e., adalimumab, etanercept, infliximab, prednisone, rituximab, or sulfasalazine, was used as an independent variable in separate models. A new composite variable was also defined in the presence of any therapeutic agent and its effect was evaluated in a similar manner. The covariates considered were coronary artery disease, diabetes, hyperlipidemia, hypertension, and peripheral vascular disease. Only coronary artery disease, diabetes, and peripheral vascular disease remained significant after the univariate analysis (see Sect. 3.3). These three variables were used in the multivariate analysis to adjust for the effect of the drug on AD. Odds ratios (ORs) were estimated in both univariate and multivariate analyses. Adjusted ORs were obtained to estimate the effects of different therapeutic agents after controlling for covariates that were found to be significant in the univariate analysis. We constructed 95 % confidence intervals (CIs) for the OR. A p value of less than 0.05 was considered statistically significant.
3 Results
3.1 Overall Population
In the first part of the studies, the overall prevalence of RA and AD in the studied population was measured. A total of 8,500,454 adults aged 18 years or older were identified in the database between January 2000 and November 2007 (Table 1). The average age of the studied population was 42.1 years, and the percentages of female (49.6 %) and male (50.4 %) individuals were comparable. We were able to identify 41,109 individuals with a diagnosis of RA during this same period of time. The prevalence of RA found in the studied population (0.48 %) was comparable to that of other studies [33, 34]. In the studied population, 9253 individuals had a diagnosis of AD, which constituted 0.11 % of this population. A total of 325 subjects had a diagnosis of both RA and AD. Therefore, the overall prevalence of AD among RA subjects was 0.79 %, whereas the prevalence of AD among the subjects who did not have RA was 0.11 % (Table 2). This disparity in the prevalence of AD among the RA subjects and non-RA subjects was statistically significant (p < 0.0001). Further analysis of subjects who were equal to or older than 65 years old (Table 3) showed a similar statistically significant increase in the prevalence of AD among RA subjects as compared with non-RA subjects (p < 0.0001). Therefore, our findings are consistent with a previous small-scaled cross-sectional study, which showed that RA subjects were more likely to have cognitive impairment than healthy controls [35].
Table 1.
Baseline characteristics of the population in our database as compared with the U.S. population in 2000a
| Number (%) | US population in 2000a (≥18 years old) | Studied population | Total RAc | Total Alzheimer’s diseasec | RA subjects with Alzheimer’s diseasec |
|---|---|---|---|---|---|
| Total number | 209,130,000 | 8,500,454 | 41,109 | 9253 | 325 |
| Average age (years) | 35.3b | 42.1 | 55.5 | 79.4 | 76.5 |
| Female | 108,133,000 (51.7) | 4,216,225 (49.6) | 29,681 (72.2) | 5714 (61.8) | 229 (70.5) |
| 18–19 years old | 8,178,000 (3.9) | 331,518 (3.9) | 246 (0.6) | 0 (0.0) | 0 (0.0) |
| 20–44 years old | 104,005,000 (49.7) | 4,641,247 (54.6) | 8592 (20.9) | 221 (2.4) | 7 (2.2) |
| 45–64 years old | 61,954,000 (29.6) | 2,949,658 (34.7) | 23,268 (56.6) | 993 (10.7) | 52 (16.0) |
| ≥65 years old | 34,992,000 (16.7) | 578,031 (6.8) | 9003 (21.9) | 8039 (86.9) | 266 (81.8) |
RA rheumatoid arthritis
Only median age is available
Based on our population
Table 2.
Prevalence of AD in the overall population of patients with and without RA
| RA | AD (overall population)
|
Prevalence of AD (%) (95 % CI) |
p value | ||
|---|---|---|---|---|---|
| + | − | Total | |||
| + | 325 | 40,784 | 41,109 | 0.79 (0.71–0.88) | <0.0001 |
| − | 8928 | 8,450,417 | 8,459,345 | 0.11 (0.10–0.11) | |
| Total | 9253 | 8,491,201 | 8,500,454 | ||
AD Alzheimer’s disease, CI confidence interval, RA rheumatoid arthritis
Table 3.
Prevalence of AD in patients aged 65 years and older with and without RA
| RA | AD (≥65 years old)
|
Prevalence of AD (%) (95 % CI) |
p value | ||
|---|---|---|---|---|---|
| + | − | Total | |||
| + | 266 | 8737 | 9003 | 2.95 (2.26–3.33) | <0.0001 |
| − | 7773 | 561,255 | 569,028 | 1.37 (1.34–1.40) | |
| Total | 8039 | 569,992 | 578,031 | ||
AD Alzheimer’s disease, CI confidence interval, RA rheumatoid arthritis
3.2 Subpopulation and Matching
In the second part of the studies, cases were matched with controls following the exclusion criteria to assess comorbidity and outcomes of treatment exposure in the analysis period. For the cohort with both RA and AD, we eliminated those also diagnosed with psoriatic arthritis, ulcerative colitis, or Crohn’s disease, leaving 313 subjects for further analysis (Fig. 1). In addition, 125 RA subjects were excluded because the diagnosis of AD was made either before the diagnosis of RA or within 120 days of the diagnosis of RA. Finally, we excluded 23 subjects who did not have at least 12 months of data available for assessment of treatment after the index date. Of the original group, only 165 RA subjects with a diagnosis of AD (i.e., cases) met all four eligibility criteria for further statistical analysis. These 165 cases were then matched with ten control subjects based on the matching criteria described above. The characteristics of these individuals are shown in Table 4. There were no statistically significant differences between the cases and control subjects for any of the matching criteria.
Fig. 1.
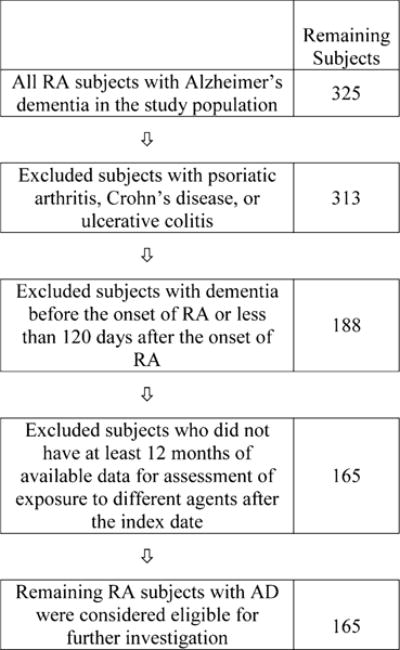
Elimination of RA subjects based on exclusion criteria. AD Alzheimer’s disease, RA rheumatoid arthritis
Table 4.
Characteristics of cases (RA subjects with AD) and matched controls (RA subjects without AD)
| Number (%) | Cases | Matched controls | p values |
|---|---|---|---|
| Total number | 165 | 1383 | – |
| Average age (years) | 76.8 (range 24–98) CI (75.0–78.6) |
75.4 (range 22–98) CI (74.8–76.08) |
0.15a |
| Female | 120 (72.7) CI 65.3–79.5 |
1051 (76.0) CI 73.7–78.2 |
0.36b |
| 18–19 years old | 0 | 0 | – |
| 20–44 years old | 3 (1.8) CI 0.3–5.2 |
28 (2.0) CI 1.4–2.9 |
0.88c |
| 45–64 years old | 22 (13.3) CI 8.6–19.5 |
223 (16.1) CI 14.2–18.2 |
|
| ≥65 years old | 140 (84.8) CI 78.5–90.0 |
1133 (81.9) CI 79.8–83.9 |
AD Alzheimer’s disease, CI confidence interval, RA rheumatoid arthritis
Student’s t test
Chi-square test
Fisher’s exact test
3.3 Comorbidities and the Relative Risk of AD among RA Subjects
We used univariate analysis to estimate the associations of five common chronic conditions with AD for the 165 RA subjects using ORs (Fig. 2). Of the five comorbidities measured, coronary artery disease (OR 1.48; 95 % CI 1.04–2.05; p = 0.03), diabetes (OR 1.86; 95 % CI 1.32–2.62; p = 0.0004), and peripheral vascular disease (OR 1.61; 95 % CI 1.06–2.43; p = 0.02) were found to be correlated significantly with an increased relative risk of AD among RA subjects, based on their p values in a univariate logistic regression model. However, there was little evidence that hyperlipidemia (OR 0.90; 95 % CI 0.64–1.26; p = 0.53) or hypertension (OR 1.15; 95 % CI 0.79–1.66; p = 0.53) was associated with the relative risk of AD among RA patients.
Fig. 2.
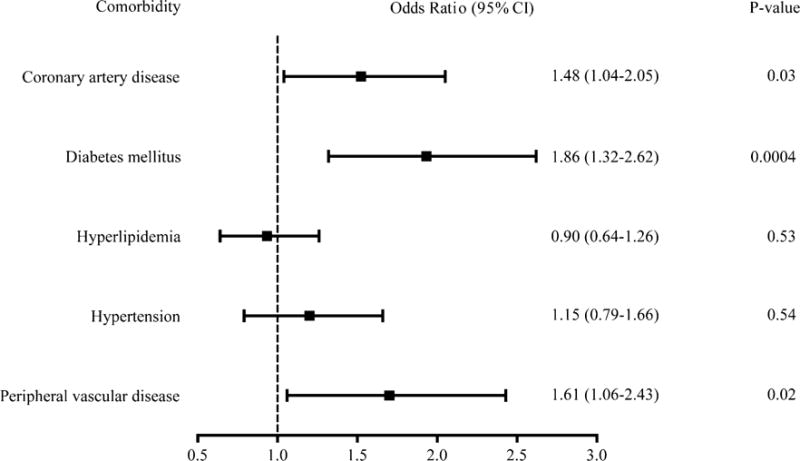
Univariate analysis of the effects of five common chronic medical conditions on the incidence of AD among RA subjects. AD Alzheimer’s disease, CI confidence interval, RA rheumatoid arthritis
3.4 Therapeutic Treatments of RA on AD
The association between different treatments for RA (i.e., prednisone, sulfasalazine, three anti-TNF agents, and rituximab) and the relative risk of AD were examined with or without adjustment for comorbidities (Fig. 3). In both cases, only the anti-TNF agents as a group showed a significant decrease in the relative risk of AD among RA subjects following treatment (unadjusted OR 0.44; 95 % CI 0.22–0.87; p = 0.02; adjusted OR 0.45; 95 % CI 0.23–0.90; p = 0.02). There were no differences in the relative risks of AD in RA patients treated with prednisone, sulfasalazine, and rituximab, either with or without adjustment for comorbidities. Of the three anti-TNF agents studied, only etanercept significantly decreased the relative risk of AD in RA subjects following treatment (unadjusted OR 0.33; 95 % CI 0.08–0.94; p = 0.03; adjusted OR 0.30; 95 % CI 0.08–0.89; p = 0.02). Of the three risk factors (coronary artery disease, diabetes, and peripheral vascular disease), only diabetes was found to be a significant covariate, which suggests that the association between coronary artery disease and peripheral vascular disease and AD are likely dependent on diabetes.
Fig. 3.
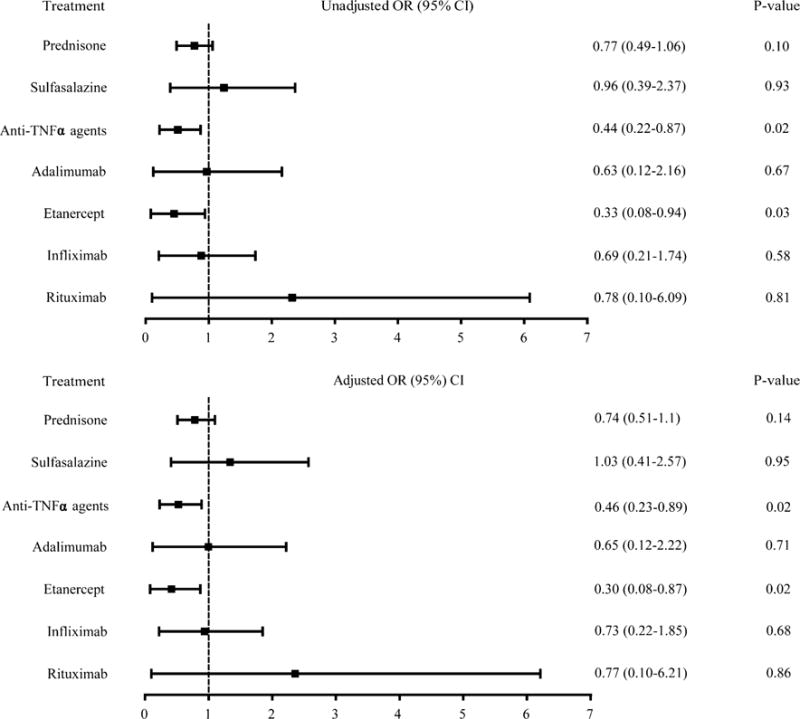
Effects of immunosuppressive treatments for RA on the incidence of AD before adjustment for comorbidities (unadjusted OR) and after adjustment for significant comorbidities (adjusted OR) using univariate analysis and multi-variate analysis, respectively. AD Alzheimer’s disease, CI confidence interval, OR odds ratio, RA rheumatoid arthritis, TNF tumor necrosis factor
4 Discussion
In the present study, we found a significant increase in the overall prevalence of AD among RA subjects as compared with non-RA subjects. Because we studied a commercially insured group, the average member age was younger than the US population based on 2000 census data. Persons aged 20–64 years were somewhat over-represented, and persons aged over 65 years were under-represented in our population. Yet, we found a significant increase in the overall prevalence of AD among RA subjects aged ≥65 years (Table 3). The prevalence of RA among the studied population in the present study was consistent with other studies [33, 34].
Unlike vascular dementia, which is caused by diabetes or peripheral vascular disease, AD-associated vasculopathy is presumably caused by deposition of amyloid protein in the blood vessel wall, i.e., amyloid angiopathy, which is a cardinal feature of AD pathology. Recent studies also demonstrated a previously unknown vasculopathy among subjects with AD: decreased perfusion in different brain areas as compared with control subjects, the exact mechanisms of which remain unknown [36]. Consistent with previous findings, three of the five comorbid conditions examined in the present study, i.e., coronary artery disease, diabetes, and peripheral vascular disease, were shown to correlate with an increase in the relative risk of AD among RA subjects (Fig. 2). In post-mortem analyses, both coronary artery disease and diabetes increased the risk of AD in both non-APOE-ε4 and APOE-ε4 carriers, although the findings were more pronounced in APOE-ε4 carriers [37, 38]. Individuals with diabetes also have less β-amyloid deposition and neurofibrillary tangle formation than those without diabetes [38]. Therefore, further study of the complicated functional relationship between diabetes and AD is warranted.
We found that RA patients who were on anti-TNF therapy as a group (adalimumab, infliximab, and etanercept) had a lower relative risk of AD, both with and without adjustment for comorbidities (Fig. 3); however, RA patients on other therapies assessed, including prednisone, sulfasalazine, and rituximab had no change in their relative risk of AD. Elevated levels of TNF have previously been shown in the serum and synovial fluid of RA patients, as well as the serum and cerebrospinal fluid of AD subjects [18–22, 39]. Serum levels of TNF in RA do not correlate with disease activities of RA, yet targeted therapy with anti-TNF agents has revolutionized the treatment for RA [40]. The functional role of TNF in AD has not been established; while disease progression in AD can be correlated with or predicted by TNF levels in the cerebrospinal fluid, its relationship with systemic circulation TNF levels remains uncertain [18–22, 41]. Our findings of reduced relative risk of AD among RA subjects following peripherally administered etanercept suggest a critical role for TNF in the development of AD in RA subjects, although at this time the mechanisms remain unclear. Therefore, our findings established parallelism between roles of TNF and anti-TNF therapy with etanercept in both RA and AD.
It is noteworthy that when we analyzed the three anti-TNF agents individually rather than as a class, we found that only etanercept significantly reduced the relative risk of AD among RA subjects (Fig. 3), although the CIs of the three anti-TNF agents did overlap somewhat. It is likely that the overall effect of anti-TNF agents as a group on lowering the risk of AD is driven by etanercept statistically. Another study has also reported improvement in cognitive function and Mini-Mental State Examination in RA subjects following only 6 months of subcutaneous etanercept treatment for RA [42]. It is important to note that while anti-TNF agents including etanercept are too large to cross the blood–brain barrier [43], this does not preclude etanercept from exerting therapeutic benefit in AD, as demonstrated in the present study. The basis for this therapeutic benefit of etanercept and a lack of significance of adalimumab and infliximab on the relative risk of AD among RA patients is unknown, as all three agents have similar molecular weights (adalimumab MW 148 kDa, etanercept MW 150 kDa, and infliximab MW 149 kDa), but a few points are worth mentioning. First, it is known that the three anti-TNF agents exhibit differential therapeutic efficacies in other diseases, such as Crohn’s disease or Wegener’s granulomatosis [44–47]. Second, two studies have suggested the disparity in therapeutic efficacies among the three anti-TNF agents is owing to the difference in the immunologic responses elicited by the monoclonal antibodies against TNF (adalimumab and infliximab) and engineered human type II TNF receptor (etanercept) [47, 48]. Third, it has been shown that etanercept is more likely to bind the active form of TNF-α, while infliximab binds both the active and inactive forms [49]. Additionally, because etanercept is an engineered version of TNF receptor 2, it also binds TNF-β [49]. It is conceivable that downregulation of both TNF-α and -β is required to decrease the risk of AD in RA patients; however, further investigation is required to confirm this. Therefore, variable therapeutic benefit among anti-TNF agents is not unprecedented, and there is a possible physiologic explanation for the phenomenon. Further studies directly comparing the efficacy of each agent in treating or preventing AD are required to determine the difference in the clinical benefits among these three agents.
The benefit of anti-TNF agents on AD relative risk was not impacted by adjustment for the co-morbid associations of coronary artery disease, diabetes, and peripheral vascular disease (Fig. 3). Therefore, the association between anti-TNF agents and lowered relative risk of AD among RA subjects are independent of the vasculopathy associated with these significant comorbidities. However, using multivariate analysis, only diabetes was seen to affect the relative risk of AD, suggesting that the effect of diabetes is independent of coronary artery disease and peripheral vascular disease.
A definitive diagnosis of AD is based on pathological findings in the brain, which renders the study of molecular mechanisms of AD in humans difficult. Furthermore, studies of AD are often controversial and difficult to interpret owing to small sample sizes. For example, a recent pilot study showed that subcutaneous etanercept was well tolerated in patients with established AD [50]. The authors noted a trend toward improved cognitive function that, while not statistically significant owing to the small sample size (n = 41) and acuteness of the treatment period (24 weeks), offered encouraging results, particularly for a study designed to determine the safety of etanercept in AD patients [50]. In the present study, we showed that administration of subcutaneous etanercept for RA lowers the long-term risk of AD in RA subjects. Taken together, these two studies support a role for TNF-α in the pathogenesis of AD, and suggest that anti-TNF therapy may be a viable candidate to modify the risk or course of AD.
A nested case-control study using a database containing more than eight million subjects allowed us to examine the associations between rare events such as exposure to different treatments in RA and AD in a statistically meaningful and cost-effective manner. In addition, our studies eliminated recall bias and temporal ambiguity by virtue of the study design. Like all nested case-control studies, the major limitation of the present study is that not all pertinent risk factors were recorded in our database. In addition, because many healthcare providers were involved in patient care, risk factors and outcomes will probably not have been recorded or measured with the same accuracy and consistency throughout. Last, the age of the current data set is also a limiting factor as more biologic agents have been added to the treatment of RA recently.
5 Conclusions
AD is a common neurodegenerative disease with no known cure or disease-modifying therapies. Our findings in the present study support a growing consensus that inflammation likely plays an important role in the pathogenesis of AD. Specifically, we have shown that the relative risk of AD is higher among patients with RA, a common autoimmune disease, and that anti-TNF therapy for RA lowers the risk of AD among these patients. Our data support a role for TNF-α in AD, though further research will be required to establish its role in the molecular pathogenesis of AD. Our data suggest that etanercept is a promising candidate for the treatment of AD in the future.
Supplementary Material
Key Points.
Patients with rheumatoid arthritis have an increased risk for Alzheimer’s disease (AD).
Treatment with subcutaneous etanercept, but not other immunosuppressive agents studied (including other anti-tumor necrosis factor [TNF] therapies), mitigated this increased risk.
Our findings support a role for TNF-α in the pathogenesis of AD, and provide evidence that anti-TNF therapies could potentially serve as disease-modifying drugs for the treatment of AD.
Acknowledgments
Funding: This work was partially supported by Grant No. UL1 RR025758–Harvard Clinical and Translational Science Center, from the National Center for Research Resources for Shiva Gautam.
Footnotes
Electronic supplementary material: The online version of this article (doi:10.1007/s40263-016-0374-z) contains supplementary material, which is available to authorized users.
Conflict of interest: Richard C. Chou, Michael Kane, Sanjay Ghimire, and Jiang Gui have no conflicts of interest to declare. Shiva Gautam was partially supported by the aforementioned grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. The funder had no role in the study design, data collection and analysis, decision to publish, or preparation of the article.
Ethical approval: Analysis of de-identified data was exempted from continuing review by the Committee for the Protection of Human Subjects at Dartmouth College and Beth Israel Deaconess Medical Center Committee on Clinical Investigation.
References
- 1.Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015;11(3):332–84. doi: 10.1016/j.jalz.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology. 2013;80(19):1778–83. doi: 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qiu C, De Ronchi D, Fratiglioni L. The epidemiology of the dementias: an update. Curr Opin Psychiatry. 2007;20(4):380–5. doi: 10.1097/YCO.0b013e32816ebc7b. [DOI] [PubMed] [Google Scholar]
- 4.Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8) doi: 10.1101/cshperspect.a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–72. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- 7.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8(6):429–31. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 8.Beljahow S. Pathological changes in the brain in dementia senilis. J Ment Sci. 1889;35:261–2. [Google Scholar]
- 9.Simchowicz T. Sur la signification des plaques seniles et sur la formule senile de l‘ecorce cerebrale. Rev Neurol. 1924;31:221–7. [Google Scholar]
- 10.Gu L, Guo Z. Alzheimer’s Abeta42 and Abeta40 peptides form interlaced amyloid fibrils. J Neurochem. 2013;126(3):305–11. doi: 10.1111/jnc.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr, Younkin LH, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain: biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43) J Biol Chem. 1995;270(13):7013–6. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 12.Younkin SG. Evidence that A beta 42 is the real culprit in Alzheimer’s disease. Ann Neurol. 1995;37(3):287–8. doi: 10.1002/ana.410370303. [DOI] [PubMed] [Google Scholar]
- 13.Buckholtz NS. Perspective: in search of biomarkers. Nature. 2011;475(7355):S8. doi: 10.1038/475S8a. [DOI] [PubMed] [Google Scholar]
- 14.Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet. 2016 doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- 15.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarkowski E, Blennow K, Wallin A, Tarkowski A. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol. 1999;19(4):223–30. doi: 10.1023/a:1020568013953. [DOI] [PubMed] [Google Scholar]
- 19.Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Pychiatry. 2003;74(9):1200–5. doi: 10.1136/jnnp.74.9.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brosseron F, Krauthausen M, Kummer M, Heneka MT. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol Neurobiol. 2014;50(2):534–44. doi: 10.1007/s12035-014-8657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13(11):1359–62. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 22.Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–74. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118(11):3537–45. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. New Engl J Med. 2011;365(23):2205–19. doi: 10.1056/NEJMra100496510.7748/phc2011.11.21.9.29.c8797. [DOI] [PubMed] [Google Scholar]
- 25.McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis: shaping the immunological landscape. Nat Rev Rheumatol. 2016;12(1):63–8. doi: 10.1038/nrrheum.2015.171. [DOI] [PubMed] [Google Scholar]
- 26.Cunnane G. Amyloid precursors and amyloidosis in inflammatory arthritis. Curr Opin Rheumatol. 2001;13(1):67–73. doi: 10.1097/00002281-200101000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Westermark GT, Fandrich M, Westermark P. AA amyloidosis: pathogenesis and targeted therapy. Ann Rev Pathol. 2015;10:321–44. doi: 10.1146/annurev-pathol-020712-163913. [DOI] [PubMed] [Google Scholar]
- 28.Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337(13):898–909. doi: 10.1056/NEJM199709253371306. [DOI] [PubMed] [Google Scholar]
- 29.Corrao G, Zambon A, Faini S, Bagnardi V, Leoni O, Suissa S. Short-acting inhaled beta-2-agonists increased the mortality from chronic obstructive pulmonary disease in observational designs. J Clin Epidemiol. 2005;58(1):92–7. doi: 10.1016/j.jclinepi.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Bernatsky S, Hudson M, Suissa S. Anti-rheumatic drug use and risk of serious infections in rheumatoid arthritis. Rheumatology (Oxford) 2007;46(7):1157–60. doi: 10.1093/rheumatology/kem076. [DOI] [PubMed] [Google Scholar]
- 31.Rose S, Laan MJ. Why match? Investigating matched case-control study designs with causal effect estimation. Int J Biostat. 2009;5(1) doi: 10.2202/1557-4679.1127. Article 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McInnes IB, O’Dell JR. State-of-the-art: rheumatoid arthritis. Ann Rheum Dis. 2010;69(11):1898–906. doi: 10.1136/ard.2010.134684. [DOI] [PubMed] [Google Scholar]
- 33.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: part I. Arthritis Rheum. 2008;58(1):15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 34.Tobon GJ, Youinou P, Saraux A. The environment, geo-epidemiology, and autoimmune disease: eheumatoid arthritis. J Autoimmun. 2010;35(1):10–4. doi: 10.1016/j.jaut.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 35.Appenzeller S, Bertolo MB, Costallat LT. Cognitive impairment in rheumatoid arthritis. Methods Find Exp Clin Pharmacol. 2004;26(5):339–43. doi: 10.1358/mf.2004.26.5.831324. [DOI] [PubMed] [Google Scholar]
- 36.Mattsson N, Tosun D, Insel PS, Simonson A, Jack CR, Jr, Beckett LA, et al. Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain. 2014;137(Pt 5):1550–61. doi: 10.1093/brain/awu043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beeri MS, Rapp M, Silverman JM, Schmeidler J, Grossman HT, Fallon JT, et al. Coronary artery disease is associated with Alzheimer disease neuropathology in APOE4 carriers. Neurology. 2006;66(9):1399–404. doi: 10.1212/01.wnl.0000210447.19748.0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, et al. Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology. 2010;75(13):1195–202. doi: 10.1212/WNL.0b013e3181f4d7f8. [DOI] [PubMed] [Google Scholar]
- 39.Tetta C, Camussi G, Modena V, Di Vittorio C, Baglioni C. Tumour necrosis factor in serum and synovial fluid of patients with active and severe rheumatoid arthritis. Ann Rheum Dis. 1990;49(9):665–7. doi: 10.1136/ard.49.9.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Centola M, Cavet G, Shen Y, Ramanujan S, Knowlton N, Swan KA, et al. Development of a multi-biomarker disease activity test for rheumatoid arthritis. PLoS One. 2013;8(4):e60635. doi: 10.1371/journal.pone.0060635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tedde A, Putignano AL, Nacmias B, Bagnoli S, Cellini E, Sorbi S. Lack of association between TNF-alpha polymorphisms and Alzheimer’s disease in an Italian cohort. Neurosci Lett. 2008;446(2–3):139–42. doi: 10.1016/j.neulet.2008.09.044. [DOI] [PubMed] [Google Scholar]
- 42.Chen YM, Chen HH, Lan JL, Chen DY. Improvement of cognition, a potential benefit of anti-TNF therapy in elderly patients with rheumatoid arthritis. Jt Bone Spine. 2010;77(4):366–7. doi: 10.1016/j.jbspin.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 43.Tweedie D, Sambamurti K, Greig NH. TNF-alpha inhibition as a treatment strategy for neurodegenerative disorders: new drug candidates and targets. Curr Alzheimer Res. 2007;4(4):378–85. doi: 10.2174/156720507781788873. [DOI] [PubMed] [Google Scholar]
- 44.Mukhtyar C, Luqmani R. Current state of tumour necrosis factor {alpha} blockade in Wegener’s granulomatosis. Ann Rheum Dis. 2005;64(Suppl 4):iv31–6. doi: 10.1136/ard.2005.042416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease: Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337(15):1029–35. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 46.Wegener’s Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med. 2005;352(4):351–61. doi: 10.1056/NEJMoa041884. [DOI] [PubMed] [Google Scholar]
- 47.Baert FJ, D’Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, et al. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology. 1999;116(1):22–8. doi: 10.1016/s0016-5085(99)70224-6. [DOI] [PubMed] [Google Scholar]
- 48.Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009;131(2):308–16. doi: 10.1016/j.clim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 49.Mpofu S, Fatima F, Moots RJ. Anti-TNF-alpha therapies: they are all the same (aren’t they?) Rheumatology (Oxford) 2005;44(3):271–3. doi: 10.1093/rheumatology/keh483. [DOI] [PubMed] [Google Scholar]
- 50.Butchart J, Brook L, Hopkins V, Teeling J, Puntener U, Culliford D, et al. Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology. 2015;84(21):2161–8. doi: 10.1212/WNL.0000000000001617. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.