

Abstract
Neuromyelitis optica (NMO) is an inflammatory disorder mediated by antibodies to aquaporin-4 (AQP4) with prominent blood-brain barrier (BBB) breakdown in the acute phase of the disease. Anti-AQP4 antibodies are produced mainly in the periphery, yet they target the astrocyte perivascular end feet behind the BBB. We reasoned that an endothelial cell–targeted autoantibody might promote BBB transit of AQP4 antibodies and facilitate NMO attacks. Using monoclonal recombinant antibodies (rAbs) from patients with NMO, we identified two that strongly bound to the brain microvascular endothelial cells (BMECs). Exposure of BMECs to these rAbs resulted in nuclear translocation of nuclear factor κB p65, decreased claudin-5 protein expression, and enhanced transit of macromolecules. Unbiased membrane proteomics identified glucose-regulated protein 78 (GRP78) as the rAb target. Using immobilized GRP78 to deplete GRP78 antibodies from pooled total immunoglobulin G (IgG) of 50 NMO patients (NMO-IgG) reduced the biological effect of NMO-IgG on BMECs. GRP78 was expressed on the surface of murine BMECs in vivo, and repeated administration of a GRP78-specific rAb caused extravasation of serum albumin, IgG, and fibrinogen into mouse brains. Our results identify GRP78 antibodies as a potential component of NMO pathogenesis and GRP78 as a candidate target for promoting central nervous system transit of therapeutic antibodies.
INTRODUCTION
Neuromyelitis optica (NMO) is a severe inflammatory autoimmune disorder of the central nervous system (CNS) that affects both adults and children. NMO predominantly presents with recurrent optic neuritis and transverse myelitis (1, 2). However, lesions may occur throughout the CNS, causing pleiotropic signs and symptoms. NMO was historically considered a variant of multiple sclerosis (MS), but the discovery of an NMO-specific immunoglobulin G (NMO-IgG), which bound in a distinctive perivascular pattern in CNS tissue sections, provided a disease-specific biomarker that unambiguously discriminated NMO from MS (3, 4). Shortly thereafter, it was discovered that NMO-IgG contained autoantibodies specific for aquaporin-4 (AQP4-IgG), the brain’s main water channel protein, primarily expressed on CNS astrocyte end feet (5). Detection of serum AQP4-IgG in a patient’s serum facilitates clinical diagnosis and early treatment of NMO (6, 7). NMO patients often demonstrate serologic and clinical manifestations of systemic autoimmunity (8, 9).
Multiple experimental models demonstrated in vivo and ex vivo that binding of AQP4-IgG to CNS AQP4 initiates complement- and antibody (Ab)–dependent cellular cytotoxicity of target astrocytes (10–12). Additional in vitro studies suggested AQP4-IgG–mediated modulation of both AQP4 localization and expression, as well as perturbation of astrocyte functional phenotypes (13, 14). Patient NMO tissue immunopathology demonstrates that astrocytes are the principal cell target in NMO (15). NMO symptoms emerge abruptly, and symptomatic episodes recur. Anti-inflammatory and Ab-depleting therapies treat symptomatic attacks, whereas therapies targeting B cells are used to reduce attack numbers (16). Although intrathecal plasmablasts can produce AQP4-IgG, most AQP4-IgG–producing cells are in the periphery (10, 17–19). AQP4-IgG can access the CNS at circumventricular organs where the endothelia lack tight junctions (20). It is not yet understood how AQP4-IgG penetrates the blood-brain barrier (BBB) to gain access to other CNS sites. One clue comes from demographics: NMO patients frequently have coexisting autoimmune diseases, such as systemic lupus erythematosus (SLE), in which endothelial cell Abs are common (8, 9, 21, 22). Additionally, intrathecal production of AQP4-IgG is not abundant; AQP4-IgG in cerebrospinal fluid (CSF) generally reflects a high serum level of AQP4-IgG and an impaired blood-CSF barrier (17–19). Accordingly, we hypothesized that a distinct endothelial cell–specific Ab might promote transit of AQP4-IgG across the BBB.
RESULTS
Non-AQP4 Abs in NMO-IgG cause BBB dysfunction
We initially examined whether NMO-IgG could activate BBB endothelial cells in vitro. We incubated IgG pooled from sera of 50 NMO patients, or pooled IgG from 25 healthy controls, with human brain microvascular endothelial cells (BMEC) TY10 cells in monolayer culture and performed immunohistochemical analysis for two markers of cell activation: nuclear factor κB (NF-κB) p65 localization after 1 hour and intracellular adhesion molecule–1 (ICAM-1) expression after 24 hours. Pooled NMO-IgG induced nuclear translocation of NF-κB p65 and up-regulation of ICAM-1 in BMECs. Pooled control-IgG was inert in these assays (Fig. 1, A and B).
Fig. 1. Markers of brain endothelial cell activation after exposure to IgG from pooled serum specimens or individual NMO patients.

(A) Confocal three-dimensional (3D) reconstruction images show immunostaining of BMECs for cytoplasmic versus nuclear NF-κB p65 (top) and ICAM-1 (bottom) after exposure to IgG (400 μg/ml) prepared from pooled NMO patient (50 patients) and control sera. Nuclei are counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Arrowheads indicate representative cells negative for nuclear NF-κB p65, and arrows indicate representative nuclear NF-κB p65–positive cells. Three independent experiments were performed. Scale bar, 50 μm. (B) Quantification of nuclear NF-κB p65–positive BMECs by high-content imaging after exposure to IgG (400 μg/ml) from pooled NMO and control sera. Data normalized to cultures unexposed to human IgG are shown as means ± SEM from three independent experiments with technical triplicates. P values were assessed with one-way analysis of variance (ANOVA), followed by Tukey multiple comparisons test (***P < 0.001). (C) Quantification of the effect of IgG from individual patients (NMO or SLE) and healthy controls on NF-κB p65 nuclear translocation in BMECs. IgG (400 μg/ml) from patients NMO1 and NMO2 as well as patients SLE1, SLE2, and SLE3 shows significantly greater NF-κB p65 nuclear translocation than control-IgG (***P < 0.001, **P < 0.01, *P < 0.05 versus control, one-way ANOVA followed by Sidak multiple comparison test). Data are shown as means ± SEM from four independent experiments with technical triplicates.
Because NMO-IgG contained Abs derived from multiple individuals, it remained uncertain whether NMO-derived Abs from individual patients could mediate these effects or whether complementary effects of multiple Abs were acting in concert to induce inflammatory changes in BMECs. To address this question, we assayed IgGs prepared from four AQP4-IgG–positive NMO cases, four AQP4-IgG–negative SLE cases, and two healthy subjects. IgGs from two of four NMO and three of four SLE patients bound to BMECs and elicited ICAM-1 up-regulation and NF-κB p65 nuclear translocation (Fig. 1C, fig. S1, and table S1), demonstrating that individual patient serum IgG could bind to and activate BBB endothelium. This activity was not only specific for NMO patients but was also present in SLE, consistent with previous reports of antiendothelial cell Abs in SLE sera (22, 23). All NMO patient sera were positive for AQP4-IgG; however, no SLE case or healthy control was AQP4-IgG–positive (fig. S2 and table S1). Therefore, AQP4-IgG was neither necessary nor sufficient for BMEC binding or activation by the IgG preparations.
Recombinant Abs isolated from CSF of NMO patients bind and activate BMECs
It remained possible that NMO-IgG might contain multiple BMEC specificities. To determine whether a monoclonal antibody from the NMO B cell repertoire could bind and activate BMECs, we screened a panel of coded AQP4-reactive and AQP4-nonreactive recombinant antibodies (rAbs) prepared from plasmablasts isolated from the CSF of relapsing NMO (13 rAbs) and one control subject with pathogen-induced CNS Ab production (1 control rAb). Two NMO-rAbs (ON-12-2-46 and ON-07-5-31) strongly bound to BMECs at 50 μg/ml (fig. S3A and table S2). Both rAbs induced up-regulation of ICAM-1 (fig. S3, B and C, and table S2); however, only ON-12-2-46 induced nuclear translocation of NF-κB p65 (Fig. 2, A and B). Neither ON-07-5-31 nor ON-12-2-46 bound to AQP4 (fig. S4 and table S2), consistent with previous experiments demonstrating that the effects of NMO-IgG on BMECs were independent of AQP4 reactivity. Furthermore, immunoblot analysis of endothelial lysates using rAbs ON-12-2-46 and ON-07-5-31 detected a dominant band of identical electrophoretic mobility, suggesting that these two rAbs might target a single protein antigen (see Fig. 4A). Effects on BMEC NF-κB p65 nuclear translocation and ICAM-1 up-regulation were dose-dependent for both pooled NMO-IgG and AQP4-nonreactive NMO-rAb ON-12-2-46 (Fig. 2, C and D, and fig. S3, B and C).
Fig. 2. Effects of an NMO patient-derived rAb on brain endothelial cell activation.

(A) Confocal 3D reconstruction images show immunostaining of BMECs for NF-κB p65 (top) and ICAM-1 (bottom) after exposure to control-rAb or NMO-rAb ON-12-2-46 (50 μg/ml). Nuclei are counterstained with DAPI (blue). Arrowheads indicate representative NF-κB p65 nuclear–negative cells, and arrows indicate representative NF-κB p65-positive cells. Three independent experiments were performed. Scale bar, 50 μm. (B) Quantification of nuclear NF-κB p65–positive BMECs by high-content imaging after exposure to NMO-rAb ON-12-2-46 or isotype-matched control rAbs (50 μg/ml). Data normalized to cultures unexposed to human IgG are shown as means ± SEM from three independent experiments with technical triplicates. P values were assessed with one-way ANOVA followed by Tukey multiple comparisons test (***P < 0.001 and *P < 0.05). (C and D) Quantification of nuclear NF-κB p65–positive BMECs by high-content imaging after exposure to pooled NMO-IgG and pooled control-IgG (C) or NMO-rAb ON-12-2-46 and control-rAb (D). Data normalized to cultures unexposed to human IgG are shown as means ± SEM from three independent experiments with technical triplicates. Statistical significance was assessed with one-way ANOVA followed by Dunnett’s multiple comparisons test [***P < 0.001 and *P < 0.05 versus NMO-IgG (12.5 μg/ml) or NMO-rAb (3.125 μg/ml)].
Fig. 4. Identification of GRP78 as the target antigen of NMO-rAbs ON-12-2-46 and ON-07-5-31.

(A) Proteins isolated from crude membrane lysates of U87MG (glioblastoma) and OL (oligodendroglia) cells were separated by polyacrylamide gel electrophoresis and analyzed by Western blot. Multiple protein bands reacted with the NMO-rAbs. Unlabeled lanes contain molecular weight standards. (B) Schematic detailing the purification scheme (X-linker, cross-linker; DSS, disuccinimidyl suberate). (C) The protein eluted with DTT from rAb cross-linked to the U87MG cells using DTSSP/disuccinimidyl suberate (DSS) was subjected to polyacrylamide gel electrophoresis and immunoblot analysis (lane 1, molecular weight standards; lane 2, protein bound to ON-07-5-31 rAb; lane 3, protein bound to ON-12-2-46 rAb). The blots were probed with the indicated NMO-rAb. (D) Immunofluorescence labeling of U87MG glioblastoma cells with ON-07-5-31 rAb (10 μg/ml) and rabbit anti-GRP78 antisera shows colocalization of the two proteins (merge in yellow). Nuclei are counterstained with DAPI (blue). (E) ON-12-2-46 rAb (20 μg/ml; green channel) and rabbit anti-GRP78 antiserum (red channel) immunoreactivity on U87MG glioblastoma cells after 24 hours of treatment with 0.5 mM thapsigargin (TG) or DMSO (control). The merged image shows colocalization of the two proteins (yellow). (F) Immunoblot analysis of commercial recombinant GRP78 protein purified from transfected human embryonic kidney (HEK) 293 cells (lane 1) or bacteria (lane 2) and probed with rAb or rabbit anti-GRP78 serum demonstrates that NMO-rAbs ON-12-2-46 and ON-07-5-31 recognize GRP78. (G) Binding of ON-12-2-46 rAb (2 μg/ml) (green channel) to the U87MG glioblastoma cells in competition immunofluorescence assay is blocked in the presence of commercial recombinant GRP78 protein (5 μg). DAPI-stained nuclei are depicted in blue.
To address whether binding of NMO-rAb was restricted to BBB endothelial cells among several sources of microvascular endothelial cells (MECs), we evaluated ICAM-1 up-regulation after binding of ON-12-2-46 to MECs of distinct origins: BMECs (TY10), commercial human umbilical vein endothelial cells (HUVECs), human dermal MECs, and primary human kidney or lung MECs. We observed rAb binding and ICAM-1 up-regulation only in BMECs (fig. S5).
To investigate whether BMEC activation mediated by NMO-rAb ON-12-2-46 was associated with a structural change in tight junctions and enhanced BBB permeability, we evaluated claudin-5 immunoreactivity and the permeability of BMECs to 10-kDa dextran and recombinant human IgG. The area fraction of claudin-5 immunoreactivity in BMECs was significantly decreased (P < 0.001) after exposure to pooled NMO-IgG or rAb ON-12-2-46 but not to control-IgG or control-rAbs (Fig. 3, A and B). The permeability of BMEC monolayers to 10-kDa dextran or recombinant human IgG was significantly increased (P < 0.05 and P < 0.001, respectively) after incubation with rAb ON-12-2-46 but not to control-rAbs (Fig. 3, C and D). These findings indicated that rAb ON-12-2-46 could enhance BBB permeability to small tracers and recombinant human IgG, as well as alter tight junctions.
Fig. 3. Effect of NMO-IgG and NMO-rAb ON-12-2-46 on BMEC function.

(A) Confocal images of claudin-5 (green) and DAPI (blue) in BMECs after exposure to pooled control-IgG (400 μg/ml), pooled NMO-IgG (400 μg/ml), control-rAb (50 μg/ml), or NMO-rAb ON-12-2-46 (50 μg/ml). Scale bar, 50 μm. (B) Claudin-5 area fraction was determined from confocal microscopy images. Data are means ± SEM from three independent experiments with technical triplicates. P values were calculated by one-way ANOVA followed by Sidak multiple comparisons test (***P < 0.001). (C) Effect of NMO-rAb ON-12-2-46 on the permeability of BMECs to 10-kDa dextran. Data are means ± SEM from three independent experiments with technical triplicates. Data are normalized to control-rAb 1. P values were calculated by one-way ANOVA followed by Tukey’s multiple comparisons test (*P < 0.05). (D) Effect of NMO-rAb ON-12-2-46 and control-rAb on the permeability of BMECs to IgG. Data are means ± SEM from three independent experiments with technical triplicates. Data are normalized to control-rAb 1. P values were obtained by Mann-Whitney U test (***P < 0.001).
Brain microvascular endothelial Abs in NMO-IgG target glucose-regulated protein 78
We next used proteomics to identify the antigenic targets of the two NMO-rAbs, ON-12-2-46 and ON-07-5-31, that bound to BMECs. The epitope was preserved after fixation and present on the cell surface: Both rAbs bound to the membrane of multiple cell lines, including fixed U87MG cells and live oligodendrocyte (OL) cells. In addition, both rAbs bound to neuronal and glial cells in mouse brain sections (fig. S6). Western blot analyses of crude membrane fractions from U87MG and OL cells yielded multiple bands of similar molecular weight (Fig. 4A). To reduce background and enhance specificity, we reversibly cross-linked NMO rAbs ON-12-2-46 and ON-7-5-31 to their cell surface antigens on U87MG cells with 3,3′dithiobis (sulfosuccinimidyl propionate) (DTSSP). After solubilization and purification on protein A/G beads, the antigenic targets were released with dithiothreitol (DTT) and analyzed by Western blot. Both ON-12-2-46 and ON-7-5-31 bound to a single ~75-kDa protein (Fig. 4, B and C) that was excised and analyzed by mass spectrometry. The target antigen of both ON-7-5-31 and ON-12-2-46 was identified as glucose-regulated protein 78 (GRP78), also referred to as IgG heavy-chain binding protein (BiP) and heat shock protein A5 (HSPA5); forty-one and 51 tryptic peptides covered 38 and 40% of the GRP78 protein, respectively. Double immunostaining with commercial anti-GRP78 Abs and both rAbs demonstrated colocalization in U87MG cells (Fig. 4, D and E). Preincubating recombinant GRP78 protein with the NMO-rAbs decreased binding to U87MG cells (Fig. 4G). Immunoblot analysis demonstrated that both rAbs reacted with recombinant GRP78 protein (Fig. 4F). Isolation of two rAbs with a single specificity from two different NMO patients suggested that GRP78 may be a CNS endothelial autoantigen pertinent to NMO.
GRP78 is expressed on the plasma membrane of BMECs in vitro and in vivo
In contrast to HUVECs and MECs from the kidney and dermis, we detected abundant cell-cell boundary expression of GRP78 in fixed and live BMECs using commercial GRP78 Abs (fig. S7, A and B). Cell surface GRP78 was also detected on BMECs in vivo by immunostaining nonpermeabilized mouse brain sections with GRP78 rAb ON-12-2-46 in conjunction with endothelial (claudin-5) and astrocyte (AQP4) markers (fig. S7, C and D). Only two commercial GRP78 Abs, however, were capable of inducing nuclear translocation of NF-κB p65 in cultured BMECs (fig. S8), suggesting that BMEC activation is epitope-dependent. This hypothesis is consistent with our finding of differential effects of two GRP78 rAbs, ON-12-2-46 and ON-7-5-31, on inducing NF-κB p65 translocation in BMECs.
Depletion of GRP78 Abs from NMO-IgG decreases BMEC activation
Although GRP78 rAb ON-12-2-46 was equivalenttopooledNMO-IgGinmediating effects on BMECs in vitro, it remained unclear whether GRP78-specific IgG was the major constituent of pooled NMO-IgG mediatingNF-κBp65nucleartranslocation. Using lysates from cells that overexpressed FLAG-tagged GRP78 or control FLAG-tagged synaptosomal-associated protein 25 (SNAP25), we preexposed pooled NMO-IgG to an excess of recombinant GRP78 protein or SNAP25 to deplete GRP78- or SNAP25-binding Abs. NMO-IgG depleted of GRP78-specific Abs was significantly less effective in promoting NF-κB p65 nuclear translocation in BMECs, but depletion of SNAP25-specific Abs had no effect (Fig. 5).
Fig. 5. Removal of GRP78-specific IgG from pooled NMO-IgG reduces NF-κB p65 nuclear translocation in BMECs.
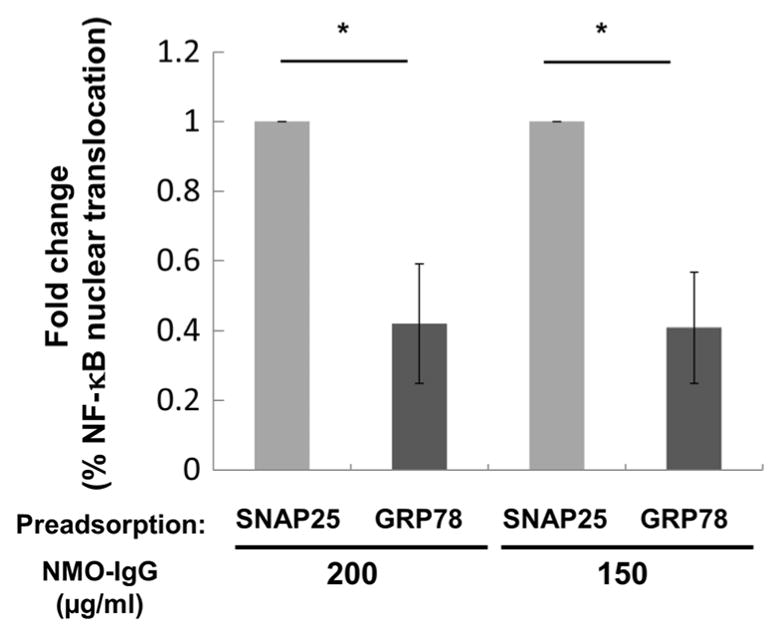
Numbers of cells positive for nuclear NF-κB p65 translocation were compared in cultures exposed to pooled NMO-IgG (200 and 150 μg/ml) immunoadsorbed against GRP78 or control SNAP25 protein. Data are normalized to control SNAP25-absorbed NMO IgG. Data are means ± SEM from five experiments in triplicate. Statistical significance was assessed by paired two-tailed t test (*P < 0.05).
Administration of GRP78-rAb causes localized extravasation of albumin, fibrinogen, and IgG into brain tissue
We next examined whether administration of GRP78 rAb ON-12-2-46 mediated brain vascular permeability in vivo. Either murinized rAb ON-12-2-46 or isotype control rAb was administered to mice (intravenously or intraperitoneally) daily for 7 days. Beginning on day 3, human AQP4-specifc rAb ON-7-5-53 was also administered. On day 9, the animals were sacrificed, and the nonpermeabilized brain tissue was examined for leakage of plasma components (injected human IgG, endogenous fibrinogen, and albumin) into the extravascular space (Fig. 6). Mice treated with isotype control rAb showed no evidence of vascular leakage (Fig. 6, B to D), whereas mice treated with GRP78 rAb ON-12-2-46 (16 to 20 mg/kg) showed discrete foci of leakage of human IgG (Fig. 6, B to D), fibrinogen (Fig. 6C), and albumin (Fig. 6D) that were independent of rAb dosage. In addition, mice treated with GRP78-specific rAb demonstrated abnormally large-diameter blood vessels (Fig. 6E), suggesting either loosened cell-cell endothelial contacts or vasodilatory reaction to GRP78 engagement by rAb.
Fig. 6. In vivo effects of ON-12-2-46 on mouse brain vasculature.

(A) Experimental setup. IV, intravenously; IP, intraperitoneally. 3D images of 20-μm mouse coronal brain sections immunostained for human IgG (rAb ON-7-5-53) and AQP4 (B), human IgG (rAb ON-7-5-53) and mouse fibrinogen (C), or human IgG, mouse albumin, and AQP4 (D) after administration of murinized GRP78-specific NMO rAb ON-12-2-46 or control rAb in combination with human AQP4-specific rAb ON-7-5-53. (E) Measurement of the average diameter of vessels (mean ± SEM) in mice treated with GRP78-specific NMO rAb ON-12-2-46 (six mice) and control rAb (four mice) in two separate experiments. Vessels were quantified from control-rAb–injected (n = 35) and ON-12-2-46–injected (n = 31) 20× high-power fields, and the diameters were measured using AxioVision LE. P values were calculated by Mann-Whitney U test (***P < 0.0001).
DISCUSSION
Ab-based therapeutics have evolved into a major drug development area. Furthermore, proof-of-principle studies in preclinical models have identified numerous CNS drug targets for which Ab-mediated approaches have preclinical therapeutic efficacy. The BBB restricts penetration of large molecules, thus limiting the utility of therapeutic Abs for CNS disease. Typically, only 0.1% of circulating IgG reaches the brain across the intact BBB (23). Transport of therapeutic Ab across the BBB could be enhanced by taking advantage of receptor-mediated transcytosis (RMT) (24, 25). However, the development of RMT-based approaches has been thwarted by both efficacy and safety concerns (26). The discovery of additional mechanisms for manipulating BBB permeability might enhance therapeutic Ab exposure within the CNS parenchyma. Conventional strategies to identify potential BBB transporters have focused mainly on mining databases generated by proteomic or genomic profiling of BBB endothelium or by detecting BBB-crossing Abs using phenotypic in vitro or in vivo screening with phage or yeast display libraries (27).
The present study followed a strategy to identify naturally occurring autoantibodies that modulate BBB permeability. We first identified two BBB-reactive Abs cloned from CSF plasmablasts recovered from NMO patients. This IgG-mediated CNS disease involves BBB transit of pathogenic AQP4-specific IgG that is synthesized in peripheral tissues. Characterizing the functional effect of these rAbs on BMECs concurrent with proteomic identification of the antigen as GRP78 allowed validation of GRP78 as a mediator of BBB permeability.
BBB breakdown and diffusion of astrocyte-specific AQP4-IgG into the CNS are critical for NMO pathogenesis (2, 28). Increasing evidence suggests that AQP4-specific IgG in serum is necessary but not sufficient to initiate the inflammatory cascade of NMO (29). Recent studies have demonstrated that NMO patients’ sera directly increase BBB endothelial permeability and reduce tight junction proteins in the BBB endothelium (9, 30). Here, we demonstrate that a non–AQP4-specific rAb IgG derived from NMO patients activates BMECs via canonical NF-κB signaling, associated with increased macromolecular permeability and decreased claudin-5 expression. Proteomic analysis identified GRP78 as the target antigen of this NMO-rAb of previously unknown specificity. Moreover, depleting GRP78-specific IgG from NMO-IgG by immunoadsorption significantly and substantially decreased NF-κB translocation in BMECs. Together, our results suggest that in at least some patients, autoantibodies specific for GRP78 could contribute to NMO attacks.
GRP78/BiP/HSPA5 is a stress protein of the heat shock protein 70 family, strongly expressed in all CNS cells as an endoplasmic reticulum (ER) chaperone (31), accounting for rAb ON-12-2-46 immunoreactivity throughout brain tissue sections, as well as CNS-derived cell lines. Under several ER stress conditions, including deprivation of amino acids or oxygen, and glucose starvation, GRP78 up-regulation prevents the accumulation of unfolded protein and protects cells from apoptosis (32). In addition to its role as an ER-localized chaperone, GRP78 can be translocated to other cellular locations, including the cytosol, mitochondria, nucleus, and plasma membrane (33). GRP78 on the cell surface is involved in signal transduction. By recognizing extracellular ligands, such as human tumor cell DnaJ-like protein 1 (HTJ-1) and α2-macroglobulin, GRP78 regulates proliferation and viability by activating phosphatidylinositol 3-kinase/AKT or NF-κB p65 signaling pathways (34). NF-κB signaling downstream of GRP78 has been studied extensively in cancer cells, including those derived from the breast, ovary, lung, prostate, and glial tumors (35). Because plasma membranes of malignant cells, but not nontumor cells, express high levels of cell surface GRP78 in vivo, GRP78 has been proposed as a cancer therapeutic target (36). Cell surface expression of GRP78 in human lung MECs or HUVECs has been described, but no consistent relationship to endothelial integrity has been reported (37, 38).
We now report a previously unappreciated role for GRP78 at the BBB. Our present study demonstrates that GRP78 is localized on the cell surface of BMECs in vitro and in vivo, and exposure to GRP78-specific rAb results in leakage of endogenous proteins into the brain extracellular space. GRP78-specific NMO-rAb and some commercial GRP78 Abs mediate canonical NF-κB signaling with or without ICAM-1 induction in cultured BMECs. Ab binding to cell surface protein may initiate signal transduction, internalize target protein, interfere with membrane-binding partners, or modulate extracellular ligand binding. In addition, ICAM-1 induction may facilitate the binding of activated immune cells (39). In endothelial cells, ICAM-1 transcription is induced by both NF-κB and cyclic adenosine monophosphate response element–binding protein (CREB) signaling (40), indicating that NF-κB signaling may play a paramount role in mediating the effects of GRP78 autoantibodies at the BBB. The large-diameter cerebral blood vessels noted after GRP78-specific rAb administration may be an indirect indication of vascular instability as similar findings are reported in pericyte-deficient mice with permeable BBBs (41, 42).
GRP78 autoantibodies have been found in patients with cancer and autoimmune disease. GRP78-IgG has been detected in low titer in sera of patients with carcinomas of prostate, colorectum, stomach, and liver and noted as a potential biomarker for early cancer detection and metastatic progression (43–45). GRP78 is present at high levels in the synovial fluids of patients with rheumatoid arthritis (RA) and in sera of 60% of RA patients (46). Although BBB integrity is impaired in an animal model of RA (47), CNS involvement is rare in RA patients. We would anticipate a pathogenic role for GRP78-IgG in CNS dysfunction only when accompanied by a CNS-specific autoantibody.
SLE is a chronic multisystem autoimmune disorder, which sometimes manifests with neuropsychiatric symptoms. Numerous mechanisms have been proposed to explain the etiology of neuropsychiatric SLE (48). A compromised BBB and antiendothelial cell Abs have been reported in patients with SLE (22, 23). Our data showing that IgG from three of four SLE patients elicits the nuclear translocation of NF-κB p65 in BMECs suggest the hypothesis that GRP78 may be a target of anti-endothelial cell Abs in SLE.
The present study carries limitations. It remains unknown how frequently GRP78 Abs are present in NMO patients as compared with suitable controls. If GRP78 Abs are detected, then their relation to disease activity remains a crucial question. AQP4 Abs have been reported (20) to cause initial symptoms by engaging targets in circumventricular organs (lacking BBB properties) so that our findings do not suggest a universal model by which AQP4 Abs access CNS targets.
In summary, we identified GRP78 as a target of antiendothelial cell Abs in NMO patients. Our animal studies indicate that these antiendothelial cell Abs also compromise BBB barrier functions. Therefore, GRP78-IgG may provide a tool that can be exploited to promote BBB transit of large-molecule therapies for CNS diseases.
MATERIALS AND METHODS
Study design
This is an experimental laboratory study designed to identify NMO serum IgG that binds to human BMECs and permeabilizes the BBB for entry of pathogenic AQP4 autoantibodies. This study used human sera and monoclonal rAbs. Human sera were obtained from NMO, SLE, and healthy control patients, and human rAbs were derived from expanded CSF plasmablasts recovered from NMO patients and patients with inflammatory CNS disorders. Initially, we examined serum IgG obtained from NMO patients, SLE patients, and healthy controls for autoantibodies that bind and activate BMECs in vitro. Subsequently, human rAbs derived from NMO CSF plasmablasts and inflammatory controls were evaluated to identify monoclonal NMO rAbs with identical properties. Serum NMO-IgG and rAbs were then tested for functional effects on BMECs in vitro: reduction in tight junction expression and permeabilization to low–molecular weight dextran and divalent IgG. Using chemical cross-linking, immunoprecipitation, and mass spectrometry, we identified the target antigen GRP78. Adsorption of NMO-IgG against target antigen confirmed that GRP78 autoantibodies were the major Ab species in NMO patient sera that bind and activate BMECs. To further understand the target specificity to brain microvasculature, we evaluated GRP78 autoantibody binding on multiple endothelial cell lines in vitro and brain microvasculature in vivo. Last, the effect of GRP78 autoantibodies on brain microvessel permeability in vivo was evaluated after chronic administration of monoclonal anti-GRP78 rAb. Primary data are located in table S3.
Patient samples
We prepared NMO-IgG from waste serum specimens (pooled from 50 NMO patients with high AQP4-IgG titers) and control-IgG from pooled healthy individuals’ serum specimens (Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN). Additional IgG was prepared from individual cases (four NMO patients, four SLE patients, and two healthy volunteers; Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN). IgG was purified by adsorption to protein G and, for functional assays, was dialyzed against physiologic saline.
Construction, expression, and purification of rAbs
Monoclonal rAbs were generated from CSF plasmablasts of seven acutely relapsing NMO patients who were AQP4-IgG–seropositive or AQP4-IgG–seronegative and two control patients with idiopathic meningitis. None of the NMO or control patients had clinical evidence of cancer; testing for paraneoplastic Abs was not performed. CD19+CD138+ plasmablasts were isolated from CSF by single-cell fluorescence-activated cell sorting, and variable region heavy- and light-chain sequences were recovered by reverse transcriptase polymerase chain reaction and DNA sequencing (10). rAbs were produced in HEK293 cells (Invitrogen), as described previously (10); AQP4-reactive and nonreactive rAbs were identified using a quantitative immunofluorescence live cell binding assay (18). As isotype-matched controls, we used a human IgG1 rAb generated from a patient with chronic meningitis (IC05-2-2) (49) and a measles virus–specific rAb (2B4) (50). For in vivo administration, human-mouse chimeric (murinized) rAbs were generated by replacing the human IgG1 Fc region of the human rAbs with a mouse IgG2a Fc region.
Cell culture and treatment
Adult human BMECs immortalized with temperature-sensitive SV40-LTA (51) were grown in MCDB 131 medium (Sigma-Aldrich) supplemented with EGM-2 SingleQuot Kit Supplements and Growth Factors (Lonza) and 20% fetal bovine serum (FBS). HUVECs and dermal MECs were purchased from Lonza. Human primary MECs from kidney or lung were isolated, as previously described (52). Human astrocytes and transfected astrocytes expressing AQP4 M23 were grown in Astrocyte Medium (ScienCell) containing 2% heat-inactivated FBS, astrocyte growth supplement, and penicillin/streptomycin solution (ScienCell), as previously reported (53). Only transfected astrocytes expressing AQP4 M23 expressed cell surface AQP4, but transfected astrocytes expressing AQP4 M23 and human astrocytes both expressed glial fibrillary acidic protein (53). Cells were cultured at 33°C in a humidified atmosphere of 5% carbon dioxide/95% air. Analyses on BMECs were performed 2 days after shifting the temperature to 37°C. IgG (polyclonal human IgGs, 400 μg/ml; rAbs, 50 μg/ml) was added 24 hours after substituting serum-free MCDB 131 medium for 1 hour for NF-κB p65 analyses and for 24 hours for ICAM-1 and claudin-5 analyses. For the pilot study, we used 14 rAbs, 13 from the CSF of NMO patients, and 1 from the brain of a patient with measles encephalitis and randomly assigned “A to N” to ensure blinded investigation. Cells treated with tumor necrosis factor–α (TNF-α) (10 U/ml) or TNF-α (10 U/ml)/interferon-γ (5 U/ml) served as positive controls for NF-κB p65, ICAM-1, and claudin-5 expression.
Abs for immunohistochemistry
Primary Abs were NF-κB p65 rabbit monoclonal antibody (mAb) (1:400; Cell Signaling Technology, #8242), ICAM-1 mouse mAb (1:400; Santa Cruz Biotechnology, sc-18908), claudin-5 rabbit mAb (1:1000; Abcam, ab53765) or mouse mAb (1:50; Life Technologies, #187364), GRP78 mouse mAb (1:100; Santa Cruz Biotechnology, sc-376768) or rabbit polyclonal (1:100; Santa Cruz Biotechnology, sc-13968), albumin goat polyclonal (1:60; Abcam, ab19194), rabbit polyclonal fluorescein isothiocyanate (FITC)–fibrinogen (1:60; Dako, 2022-01), AQP4 rabbit polyclonal (1:200; Santa Cruz Biotechnology, sc-20812), rabbit polyclonal VE-cadherin Ab (1:1000; Abcam, ab33168), and PECAM mouse mAb (1:100; R&D Systems, BBA7). Secondary Abs were Alexa Fluor 488 goat anti-rabbit IgG, Alexa Fluor 488 goat anti-human IgG, Alexa Fluor 594 goat anti-rabbit IgG, Alexa Fluor 594 goat anti-mouse IgG, Alexa Fluor 594 donkey anti-goat, Alexa Fluor 488 donkey anti-human, or Alexa Fluor 647 donkey anti-rabbit (1:400; Life Technologies). Human IgG pools, individual patient IgGs, and rAbs also were used as primary Abs to determine the pattern of IgG binding to cells.
Cell lines and immunohistochemistry
BMECs were fixed with either 4% paraformaldehyde (PFA) (NF-κB p65, GRP78, ICAM-1, and VE-cadherin staining) or 100% ethanol (claudin-5 staining), washed, and then permeabilized with 0.3% Triton X-100 (NF-κB p65, GRP78, claudin-5, and VE-cadherin). After blocking overnight in 5% goat serum in phosphate-buffered saline (PBS) (ICAM-1) or 5% goat serum/0.3% Triton X-100 in PBS (NF-κB p65, claudin-5, GRP78, and VE-cadherin), primary Abs were added (for 2 hours at room temperature) and then species-specific Alexa Fluor secondary Abs (1:400). To test the binding of NMO-IgG/NMO-rAb to cells, we incubated fixed BMECs with either pooled IgG or a rAb (1 or 50 μg/ml) as primary Ab (1 hour, room temperature). Live human astrocytes or transfected astrocytes expressing AQP4 M23 cells were incubated (for 1 hour at 37°C) with rAb (20 μg/ml) as primary Ab, then fixed with 4% PFA, and incubated with goat anti-human IgG Alexa Fluor 488 as secondary Ab (1 hour). Slides were mounted with ProLong Gold antifade reagent (Life Technologies). Nuclei were stained with DAPI or Hoechst 33342 (Life Technologies). Images were captured on an LSM780 confocal microscope operated by ZEN software version 2010 (Carl Zeiss) or a Leica DM2500 microscope (Leica Microsystems) using 20× air, 40× oil, or 60× oil objectives (digital zoom factor, 2× to 4×). All image parameters remained constant (pinhole, power, and gain) to capture images from independent experiments in a comparable manner. Noise was removed after deleting nonspecific signal, and binary images were created automatically. The area fraction (percentage of claudin-5–positive area per total window area) was calculated using ImageJ software [National Institutes of Health (NIH)].
The U87MG glioblastoma (U87MG) and human OL cell lines (54) were cultured on poly-L-ornithine–coated glass coverslips for 24 to 48 hours in minimum essential medium alpha or Dulbecco’s modified Eagle’s medium with GlutaMAX containing 10% fetal calf serum, 1 mM sodium pyruvate, and 1× nonessential amino acids, fixed with 4% PFA and stored at −20°C until staining. For some experiments, U87MG cells were treated with 1 mM thapsigargin (Sigma-Aldrich) overnight in the stated medium. For immunostaining, cells were rehydrated in PBS and blocked for an hour in PBS containing 4% normal goat sera with 0.3% Triton X-100. rAbs (10 to 20 μg/ml) were applied for 16 hours at 4°C in blocking buffer, washed four times in PBS, incubated for 2 hours with goat anti-human Alexa Fluor 488 (1:500) in blocking buffer, and washed in PBS. Coverslips were mounted using ProLong Gold plus DAPI.
Animal injections, tissue preparation, and immunohistochemistry
All animal protocols and procedures were approved by the Animal Use and Care Committee of the University of Colorado Denver and conform to the NIH guidelines. Adult C57BL/6 mice were administered with murinized NMO rAb ON-12-2-46 or isotype control rAb (2B4 or ICO-5-2-2) in combination with NMO AQP4-specific rAb ON-7-5-53 to induce BBB permeability. Two experimental paradigms were used: (i) Murinized ON-12-2-46 (16 mg/kg) or isotype control rAb was delivered daily for 7 days (intravenously on days 1, 3, 5, and 7 and intraperitoneally on days 2, 4, and 6) in conjunction with NMO human rAb ON-7-5-53 (5 mg/kg) (intravenously on days 3, 5, and 7) and (ii) murinized ON-12-2-46 (20 mg/kg) or isotype control rAb was delivered daily for 7 days (intravenously on days 1, 3, and 5 and intraperitoneally on days 2, 4, 6, and 7) in conjunction with an escalating dose of NMO human rAb ON-7-5-53 (4 to 10 mg/kg) on days 3 to 7 (delivered intravenously or intraperitoneally in combination with the murinized rAb). For each paradigm, two animals were treated with isotype control rAb, and three animals were treated with NMO rAb ON-12-2-46. Two days after the last injection, the animals were deeply anesthetized with ketamine/xylazine and perfused via cardiac puncture with 3 mM EDTA in PBS, followed by 4% PFA in PBS.
Brains were removed, postfixed overnight in 4% PFA, and cryoprotected overnight in 20% sucrose and then overnight in 30% sucrose, all at 4°C. Cryostat sections (20 μm) of tissue embedded in optimal cutting temperature freezing medium were collected on Superfrost Plus microscope slides (Fisher Scientific) and then stored at −80°C. Before immunostaining, sections were thawed for 10 min, heated at 37°C for 30 min, and blocked for 1 hour in PBS containing 4% normal goat serum (NGS) in the presence (permeabilized) or absence (non-permeabilized) of 0.3% Triton X-100. Patient-derived rAbs were used at 10 to 20 μg/ml, and commercial Abs were used at the concentrations noted above. Primary Abs were applied to mouse tissues for 16 hours at 4°C in PBS containing 4% NGS ± 0.3% Triton X-100. Sections were washed with PBS (three times for 3 min), and Alexa Fluor 488 anti-human IgG (1:500 in PBS containing 4% NGS and 0.3% Triton X-100; Life Technologies) was applied. After 2 hours at room temperature and three 3-min washes in PBS, the slides were mounted using ProLong Gold (Life Technologies) containing DAPI. In some experiments, brain sections were first blocked and incubated with primary Ab for human IgG, albumin, or fibrinogen under nonpermeabilized conditions, then fixed in 4% PFA for 10 min, washed three times in PBS, permeabilized, and reblocked in the presence of Triton X-100. Sections were then incubated with primary Ab against rat anti-AQP4 (astrocytes) or claudin-5 (BMECs), washed, and further visualized with the appropriate secondary fluorophore–conjugated Ab, as described above. Images were visualized on a Nikon E800 fluorescence microscope using a 40× or 60× objective lens and processed using AxioVision 4.8 software. Confocal images were acquired on an Olympus Fluoview 1000 and processed using FV10-ASW software (Olympus).
High-content and high-throughput imaging assay using the Operetta imager and Harmony software
Cells (10,000) were plated per well of a CellCarrier 96-well collagen-coated plate (PerkinElmer), followed by immunostaining for NF-κB p65 or ICAM-1. Plates were scanned, and images were collected with Operetta HTS imaging system (PerkinElmer) at ×20 magnification with 13 fields of view per well, equivalent to between 800 and 1000 cell events. Images were then analyzed with Harmony software (PerkinElmer). Data are means of triplicates.
Live cell imaging
Cells grown on 3.5-cm glass bottom dishes (MatTek dish) were labeled 1 hour using anti-GRP78 IgG (5 μg/ml) (Santa Cruz Biotechnology, sc-376768; Mix-n-Stain CF 568A Ab labeling kits, Sigma-Aldrich, MX568S100). After a 10-min exposure of CellMask Deep Red Plasma membrane Stain (Life Technology, C10046) and Hoechst 33342 (Life Technologies, H3570), live cell images were captured.
Cell line homogenates and Western blot
U87MG and OL cells were harvested at 4°C by scraping in PBS, collected by centrifugation, homogenized in tris-HCl buffer [0.1 M tris (pH 8.0), with 1% Triton X-100] containing protease inhibitors (cOmplete tablets, Roche Diagnostics), and then centrifuged for 30 min at 14,000g. Supernatants were collected and stored at −80°C. Total protein concentrations were determined by bicinchoninic acid assay kit (Pierce Biotechnology).
Forty micrograms of total protein from U87MG and OL cell homo-genates was resolved using 4 to 15% gradient SDS–polyacrylamide gel electrophoresis (Bio-Rad Laboratories). The proteins from the gels were transferred to polyvinylidene difluoride (PVDF) membrane (Millipore) using a semidry transfer system (Bio-Rad Laboratories). The PVDF membranes were blocked with 1× casein solution (Vector Laboratories) in 1× tris-buffered saline (TBS) (Bio-Rad Laboratories) and incubated with human rAbs (10 to 20 μg/ml) in casein block overnight at 4°C. After washing three times with TBST (tris-buffered saline containing 0.02% Tween 20), the membranes were incubated with horseradish peroxidase–conjugated goat anti-human IgG, 25°C for 2 hours. After three washes with TBST, the blot was transferred to PBS. Protein bands were visualized with chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Thermo Fisher Scientific) and analyzed with an Alpha Innotech FluorChem Q. For Western blot verification, purified GRP78 protein [from OriGene (100 ng per well) and Novus Biologicals (0.5 μg per well)] was subjected to SDS–polyacrylamide gel electrophoresis, transferred to PVDF, incubated with 0.5% glutaraldehyde in PBS (15 min), with 0.25 M tris (pH 7.5) in PBS (15 min), washed three times in TBS (10 min), blocked, and then processed as described above.
Cross-linking and mass spectrometry of non-AQP4 antigen
Protein A/G beads, DSS, and DTSSP were obtained from Thermo Fisher Scientific. U87MG cells were incubated with AQP4-nonreactive rAbs (20 μg/ml) in 1× Hepes-buffered saline (HBS; Sigma-Aldrich) containing 0.4 M sucrose, 2 mM MgCl2, and CaCl2 at room temperature for an hour with intermittent rocking. After washing twice with the same solution, the cells were incubated with 12.5 mg of DTSSP in the same buffer for 30 min. Tris-HCl (pH 7.5) was added to 25 mM and incubated for 15 min at room temperature. Cells were lysed in HBS containing 0.32 M sucrose, 1 mM EDTA, and 0.1% Triton X-100 with protease inhibitors. Lysates were incubated with Protein A/G beads overnight with end-over-end mixing and then washed four times with 1× HBS. DSS (1 mg) was added and incubated with end-over-end mixing for 30 min at room temperature. Tris-HCl (pH 7.5) was added to 25 mM and incubated at room temperature for 15 min. Protein A/G beads were washed twice in 1× HBS, and the antigen was eluted with 50 mM DTT in TBS [10 mM tris (pH 7.5) and 150 mM NaCl] and analyzed by SDS–polyacrylamide gel electrophoresis, with and without Western blot. Gel slices corresponding to the Western blot (75-kDa band) were excised. Sample preparation was performed at the Mass Spectrometry Research Center/Proteomics Core at the University of Colorado Cancer Center as follows. Excised gel pieces were destained in ammonium bicarbonate/50% acetonitrile (ACN) and dehydrated in 100% ACN. Disulfide bonds were reduced by dithiothreitol, and cysteine residues were alkylated with iodoacetamide. Proteins were digested with trypsin. After digestion, the tryptic mixtures were extracted in 1% formic acid/50% ACN. Samples were analyzed on a linear trap quadrupole Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) coupled to an Eksigent NanoLC-2D system through a nanoelectrospray liquid chromatography–mass spectrometry interface using a 90-min gradient from 6 to 40% ACN. Peptide fragmentation was performed in a higher-energy collisional dissociation cell with normalized collision energy of 40%, and tandem mass spectra (MS/MS) were acquired in the Orbitrap mass analyzer. Data were acquired using Xcalibur software (version 2.0.6). MS/MS were converted into .mgf files using an in-house script. Database searches against the Swiss-Prot database were performed by Mascot (version 2.2; Matrix Science Inc.). MS/MS-based peptide and protein identifications were validated by Scaffold (version 4) and accepted at >95.0% probability (peptide) and >99.0% probability (protein).
Solute permeability with 10-kDa dextran and IgG
Effects of NMO-rAb on BBB integrity were assessed by measuring paracellular permeability (luminal to abluminal) to FITC-labeled 10-kDa dextran or human NMO rAb. Monolayer BMECs were cultured on 24-well collagen-coated Transwell tissue culture inserts (0.4-μm pore size; Corning Inc.) 3 days at 33°C and then 3 days at 37°C. Cell monolayers were exposed on the luminal side to NMO rAb or control rAb (50 μg/ml) for 24 hours at 37°C. Abluminal samples were then collected and replaced with equal volume of fresh medium. Solute permeability was assessed using FITC-dextran fluorescence. FITC-dextran fluorescence (10 kDa; Sigma-Aldrich) was added to the luminal insert (final concentration, 1 mg/ml), and 5 μl of medium was collected from the abluminal chamber over 40 min. Aliquots were diluted to 1 ml with PBS, 100 μl of each was transferred into 96-well black plates, and fluorescence signals were measured at 490/520 nm (absorption/emission) wavelengths using a SpectraMax M3e microplate reader (Molecular Devices). For IgG permeability, each rAb was incubated in the luminal chamber for 18 hours (the concentration of each rAb was 50 μg/ml; 100 μl of conditioned medium including rAb was incubated in the luminal chamber, and 200 μl of PBS was added to the abluminal chamber). Then, PBS in the abluminal chamber was collected, and the IgG concentration within the abluminal chamber was measured using Easy-Titer Human IgG (H + L) Assay Kit (Thermo Fisher Scientific).
Effect of commercial GRP78 Abs on NF-κB p65 nuclear translocation in BMECs
Seven commercial anti-GRP78 Abs (Santa Cruz Biotechnology, sc-376768, sc-1051, sc-1050, and sc-13968; Abcam, ab21685, ab12223; Sigma-Aldrich, G8918) were tested. Control IgGs were goat, rabbit, and mouse polyclonal Abs (Santa Cruz Biotechnology, sc-3887, sc-2027, and sc-2025). Cultured cells were incubated with these Abs or IgGs (10, 20, 40, and 80 μg/ml) for 1 hour and fixed with 4% PFA.
Removal of GRP78-IgG from pooled NMO-IgG
Plasmids containing the complementary DNAs for FLAG-tagged murine GRP78 or FLAG-tagged murine SNAP25 as a control (both from OriGene) were transfected into U87MG cells with Lipofectamine 2000 (Thermo Fisher Scientific) per manufacturer’s protocol. Transfected U87MG cells were collected 40 hours after transfection, washed in cold PBS, lysed in 1× radioimmunoprecipitation assay lysis buffer (Cell Signaling Technology), and pelleted by centrifugation (14,000g for 30 min at 4°C); supernatants were stored at −80°C. For immunoprecipitation, 200, 150, 100, and 25 μg/ml of pooled NMO-IgG or pooled control-IgG were incubated with 100 μg of either FLAG-tagged GRP or control FLAG-tagged SNAP25 protein, for 4 hours at 4°C, and then incubated with 40 μl of anti–FLAG-IgG coupling resin (EZview Red Anti-FLAG M2 Affinity Gel beads; Sigma-Aldrich, F2426), for 2 hours at 4°C. Immune complexes were pulled down, and supernatants were saved for analysis.
Statistics
Statistical analyses were performed using Prism 6 (GraphPad Software). In analyses that required only a single comparison, either a paired Student’s t test or unpaired Mann-Whitney U test was used to determine statistical significance (two-sided). For analyses requiring multiple comparisons, one-way ANOVA between individual groups were performed using Tukey multiple comparisons test, Dunnett’s multiple comparisons test, Sidak multiple comparisons test, or Holm-Sidak multiple comparisons test. All values are expressed as mean ± SEM. Degree of significance between groups is represented as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Material
Fig. S1. Effects of IgG from NMO or SLE patients or healthy controls on the activation of and binding to BMECs.
Fig. S2. Binding of IgG from NMO or SLE patients or healthy controls to human astrocytes expressing AQP4 or human astrocytes.
Fig. S3. Effect of NMO-IgG or NMO-rAb on BMEC activation.
Fig. S4. Binding of NMO-IgG or NMO-rAb to human astrocytes expressing AQP4 or human astrocytes.
Fig. S5. Effects of non-AQP4 NMO-rAb on other MECs.
Fig. S6. Target of rAb ON-12-2-46 is widely expressed at both the cellular and tissue level.
Fig. S7. GRP78 expression on MECs in vitro and in vivo.
Fig. S8. Effect of commercial GRP78 Abs on BMEC activation.
Table S1. Effect of serum IgG from each patient with NMO and SLE and healthy controls on BMEC activation.
Table S2. Effect of pooled IgG and 14 rAbs from NMO patients and healthy controls on BMEC activation.
Table S3. Primary data (Microsoft Excel format).
Acknowledgments
We thank A. Navarro for assistance in animal injections and perfusions, K. Blauth and Y. Liu for assistance in confocal microscopy, and H. Schumann and K. Hickey for assistance in rAb production (all from the University of Colorado Denver, School of Medicine). We thank G. Marsh (Biogen) for providing us with primary MECs from human kidney or lung.
Funding: This work was funded with support from the Guthy-Jackson Charitable Foundation (to J.L.B. and R.M.R.), the NIH (EY022936 to J.L.B., NS072141 to G.P.O., K2471540 to R.M.R., and UM1AI110498 to J.L.B. and G.P.O.), and the National Multiple Sclerosis Society (to J.L.B. and G.P.O.).
Footnotes
Author contributions: F.S. and D.K. performed in vitro experiments with human BMECs in culture. K.L.S. and G.P.O. performed experiments with identification of GRP78. K.L.S. performed mouse injections and in vivo vasculature leakage studies. F.S., K.L.S., A.C.C., D.K., B.O., G.P.O., R.M.R., and J.L.B. analyzed and interpreted data. Y.T. contributed to the initial experimental design. T.J.K. and V.A.L. provided pooled NMO-IgG, control-IgG, and IgG from individual cases as well as input for experimental design. Y.S. and T.K. provided human BMEC lines as well as input for experimental design. F.S., K.L.S., R.M.R., and J.L.B. wrote the manuscript. V.A.L., B.O., R.M.R., and J.L.B. edited the manuscript. R.M.R. and J.L.B. conceived and supervised the study.
Competing interests: F.S., D.K., A.C.C., B.O., and R.M.R. are employees of Biogen, Cambridge, MA. Biogen and the University of Colorado have filed a provisional patent application. V.A.L. and T.J.K. have received royalties exceeding the federal threshold for significant financial interest from the licensing of a patent relating to AQP4 Ab as marker for NMO. They receive no royalties from the sale of tests performed by Mayo Medical Laboratories; however, Mayo Collaborative Services Inc. receives revenue for conducting these tests. J.L.B. and R.M.R. are inventors on a patent application (62/52,188) submitted by the University of Colorado and Biogen MA Inc. that covers, for example, the use of anti-HSPA5 Abs for delivery of agents to the CNS and disease monitoring in NMO and SLE.
Data and materials availability: NMO and control rAbs are available from J.L.B. under a material transfer agreement with the University of Colorado. BMEC TY10 cells are available from T.K. under a material transfer agreement with the Yamaguchi University.
REFERENCES AND NOTES
- 1.Jarius S, Wildemann B. The history of neuromyelitis optica. J Neuroinflammation. 2013;10:8. doi: 10.1186/1742-2094-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brimberg L, Mader S, Fujieda Y, Arinuma Y, Kowal C, Volpe BT, Diamond B. Antibodies as mediators of brain pathology. Trends Immunol. 2015;36:709–724. doi: 10.1016/j.it.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 4.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 5.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, Nytrova P, Sobek O, Nielsen HH, Barington T, Lillevang ST, Illes Z, Rentzsch K, Berthele A, Berki T, Granieri L, Bertolotto A, Giometto B, Zuliani L, Hamann D, van Pelt ED, Hintzen R, Höftberger R, Costa C, Comabella M, Montalban X, Tintoré M, Siva A, Altintas A, Deniz G, Woodhall M, Palace J, Paul F, Hartung HP, Aktas O, Jarius S, Wildemann B, Vedeler C, Ruiz A, Leite MI, Trillenberg P, Probst M, Saschenbrecker S, Vincent A, Marignier R. Multicentre comparison of a diagnostic assay: Aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2016;87:1005–1015. doi: 10.1136/jnnp-2015-312601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, Palace J, Mandrekar JN, Vincent A, Bar-Or A, Pittock SJ. Serologic diagnosis of NMO: A multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665–671. doi: 10.1212/WNL.0b013e318248dec1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, Lucchinetti CF, Zéphir H, Moder K, Weinshenker BG. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol. 2008;65:78–83. doi: 10.1001/archneurol.2007.17. [DOI] [PubMed] [Google Scholar]
- 9.Jarius S, Paul F, Franciotta D, de Seze J, Münch C, Salvetti M, Ruprecht K, Liebetrau M, Wandinger KP, Akman-Demir G, Melms A, Kristoferitsch W, Wildemann B. Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: Ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler J. 2011;18:1135–1143. doi: 10.1177/1352458511431728. [DOI] [PubMed] [Google Scholar]
- 10.Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, Gilden D, Nessler S, Stadelmann C, Hemmer B. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66:617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Bennett JL, Verkman AS. Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol. 2011;70:943–954. doi: 10.1002/ana.22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, Fallier-Becker P, Noell S, Lennon VA. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci USA. 2012;109:1245–1250. doi: 10.1073/pnas.1109980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossi A, Ratelade J, Papadopoulos MC, Bennett JL, Verkman AS. Neuromyelitis optica IgG does not alter aquaporin-4 water permeability, plasma membrane M1/M23 isoform content, or supramolecular assembly. Glia. 2012;60:2027–2039. doi: 10.1002/glia.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, Mandler RN, Weinshenker BG, Pittock SJ, Wingerchuk DM, Lucchinetti CF. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130:1194–1205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- 16.Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: State-of-the-art and emerging therapies. Nat Rev Neurol. 2014;10:493–506. doi: 10.1038/nrneurol.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, Reuss R, Probst C, Kristoferitsch W, Wandinger KP, Wildemann B. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: Frequency, origin, and diagnostic relevance. J Neuroinflammation. 2010;7:52. doi: 10.1186/1742-2094-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowarik MC, Dzieciatkowska M, Wemlinger S, Ritchie AM, Hemmer B, Owens GP, Bennett JL. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J Neuroinflammation. 2015;12:19. doi: 10.1186/s12974-015-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majed M, Fryer JP, McKeon A, Lennon VA, Pittock SJ. Clinical utility of testing AQP4-IgG in CSF: Guidance for physicians. Neurol Neuroimmunol Neuroinflamm. 2016;3:e231. doi: 10.1212/NXI.0000000000000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Apiwattanakul M, Popescu BF, Matiello M, Weinshenker BG, Lucchinetti CF, Lennon VA, McKeon A, Carpenter AF, Miller GM, Pittock SJ. Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol. 2010;68:757–761. doi: 10.1002/ana.22121. [DOI] [PubMed] [Google Scholar]
- 21.Carvalho D, Savage COS, Isenberg D, Pearson JD. IgG anti-endothelial cell autoantibodies from patients with systemic lupus erythematosus or systemic vasculitis stimulate the release of two endothelial cell-derived mediators, which enhance adhesion molecule expression and leukocyte adhesion in an autocrine manner. Arthritis Rheum. 1999;42:631–640. doi: 10.1002/1529-0131(199904)42:4<631::AID-ANR5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 22.Renaudineau Y, Dugué C, Dueymes M, Youinou P. Antiendothelial cell antibodies in systemic lupus erythematosus. Autoimmun Rev. 2002;1:365–372. doi: 10.1016/s1568-9972(02)00063-0. [DOI] [PubMed] [Google Scholar]
- 23.Yu YJ, Watts RJ. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics. 2013;10:459–472. doi: 10.1007/s13311-013-0187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones AR, Shusta EV. Blood–brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24:1759–1771. doi: 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watts RJ, Dennis MS. Bispecific antibodies for delivery into the brain. Curr Opin Chem Biol. 2013;17:393–399. doi: 10.1016/j.cbpa.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 26.Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ, Solanoy H, Tong RK, Hoyte K, Luk W, Lu Y, Gadkar K, Prabhu S, Ordonia BA, Nguyen Q, Lin Y, Lin Z, Balazs M, Scearce-Levie K, Ernst JA, Dennis MS, Watts RJ. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med. 2013;5:183ra57. doi: 10.1126/scitranslmed.3005338. [DOI] [PubMed] [Google Scholar]
- 27.Stanimirovic D, Kemmerich K, Haqqani AS, Farrington GK. Engineering and pharmacology of blood–brain barrier-permeable bispecific antibodies. Adv Pharmacol. 2014;71:301–335. doi: 10.1016/bs.apha.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett JL, O’Connor KC, Bar-Or A, Zamvil SS, Hemmer B, Tedder TF, von Büdingen HC, Stuve O, Yeaman MR, Smith TJ, Stadelmann C. B lymphocytes in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. 2015;2:e104. doi: 10.1212/NXI.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimizu F, Sano Y, Takahashi T, Haruki H, Saito K, Koga M, Kanda T. Sera from neuromyelitis optica patients disrupt the blood–brain barrier. J Neurol Neurosurg Psychiatry. 2012;83:288–297. doi: 10.1136/jnnp-2011-300434. [DOI] [PubMed] [Google Scholar]
- 31.Lee AS. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–818. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-κB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem. 2006;281:13694–13707. doi: 10.1074/jbc.M511694200. [DOI] [PubMed] [Google Scholar]
- 35.Quinones QJ, de Ridder GG, Pizzo SV. GRP78: A chaperone with diverse roles beyond the endoplasmic reticulum. Histol Histopathol. 2008;23:1409–1416. doi: 10.14670/HH-23.1409. [DOI] [PubMed] [Google Scholar]
- 36.Jakobsen CG, Rasmussen N, Laenkholm AV, Ditzel HJ. Phage display derived human monoclonal antibodies isolated by binding to the surface of live primary breast cancer cells recognize GRP78. Cancer Res. 2007;67:9507–9517. doi: 10.1158/0008-5472.CAN-06-4686. [DOI] [PubMed] [Google Scholar]
- 37.Birukova AA, Singleton PA, Gawlak G, Tian X, Mirzapoiazova T, Mambetsariev B, Dubrovskyi O, Oskolkova OV, Bochkov VN, Birukov KG. GRP78 is a novel receptor initiating a vascular barrier protective response to oxidized phospholipids. Mol Biol Cell. 2014;25:2006–2016. doi: 10.1091/mbc.E13-12-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venugopal S, Chen M, Liao W, Er SY, Wong WSF, Ge R. Isthmin is a novel vascular permeability inducer that functions through cell-surface GRP78-mediated Src activation. Cardiovasc Res. 2015;107:131–142. doi: 10.1093/cvr/cvv142. [DOI] [PubMed] [Google Scholar]
- 39.Takeshita Y, Ransohoff RM. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol Rev. 2012;248:228–239. doi: 10.1111/j.1600-065X.2012.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hadad N, Tuval L, Elgazar-Carmom V, Levy R, Levy R. Endothelial ICAM-1 protein induction is regulated by cytosolic phospholipase A2α via both NF-κB and CREB transcription factors. J Immunol. 2011;186:1816–1827. doi: 10.4049/jimmunol.1000193. [DOI] [PubMed] [Google Scholar]
- 41.Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood–brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 42.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M, Arap MA, Hong WK, Troncoso P, Logothetis CJ, Pasqualini R, Arap W. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21:57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 44.Raiter A, Vilkin A, Gingold R, Levi Z, Halpern M, Niv Y, Hardy B. The presence of anti-GRP78 antibodies in the serum of patients with colorectal carcinoma: A potential biomarker for early cancer detection. Int J Biol Markers. 2014;29:431–435. doi: 10.5301/jbm.5000086. [DOI] [PubMed] [Google Scholar]
- 45.Tsunemi S, Nakanishi T, Fujita Y, Bouras G, Miyamoto Y, Miyamoto A, Nomura E, Takubo T, Tanigawa N. Proteomics-based identification of a tumor-associated antigen and its corresponding autoantibody in gastric cancer. Oncol Rep. 2010;23:949–956. doi: 10.3892/or_00000719. [DOI] [PubMed] [Google Scholar]
- 46.Bläß S, Union A, Raymackers J, Schumann F, Ungethüm U, Müller-Steinbach S, De Keyser F, Engel JM, Burmester GR. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. 2001;44:761–771. doi: 10.1002/1529-0131(200104)44:4<761::AID-ANR132>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 47.Nishioku T, Yamauchi A, Takata F, Watanabe T, Furusho K, Shuto H, Dohgu S, Kataoka Y. Disruption of the blood–brain barrier in collagen-induced arthritic mice. Neurosci Lett. 2010;482:208–211. doi: 10.1016/j.neulet.2010.07.035. [DOI] [PubMed] [Google Scholar]
- 48.Jeltsch-David H, Muller S. Neuropsychiatric systemic lupus erythematosus: Pathogenesis and biomarkers. Nat Rev Neurol. 2014;10:579–596. doi: 10.1038/nrneurol.2014.148. [DOI] [PubMed] [Google Scholar]
- 49.Owens GP, Bennett JL, Lassmann H, O’Connor KC, Ritchie AM, Shearer A, Lam C, Yu X, Birlea M, DuPree C, Williamson RA, Hafler DA, Burgoon MP, Gilden D. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann Neurol. 2009;65:639–649. doi: 10.1002/ana.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burgoon MP, Williamson RA, Owens GP, Ghausi O, Bastidas RB, Burton DR, Gilden DH. Cloning the antibody response in humans with inflammatory CNS disease: Isolation of measles virus-specific antibodies from phage display libraries of a subacute sclerosing panencephalitis brain. J Neuroimmunol. 1999;94:204–211. doi: 10.1016/s0165-5728(98)00243-4. [DOI] [PubMed] [Google Scholar]
- 51.Sano Y, Shimizu F, Abe M, Maeda T, Kashiwamura Y, Ohtsuki S, Terasaki T, Obinata M, Kajiwara K, Fujii M, Suzuki M, Kanda T. Establishment of a new conditionally immortalized human brain microvascular endothelial cell line retaining an in vivo blood-brain barrier function. J Cell Physiol. 2010;225:519–528. doi: 10.1002/jcp.22232. [DOI] [PubMed] [Google Scholar]
- 52.Ligresti G, Nagao RJ, Xue J, Choi YJ, Xu J, Ren S, Aburatani T, Anderson SK, MacDonald JW, Bammler TK, Schwartz SM, Muczynski KA, Duffield JS, Himmelfarb J, Zheng Y. A novel three-dimensional human peritubular microvascular system. J Am Soc Nephrol. 2016;27:2370–2381. doi: 10.1681/ASN.2015070747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haruki H, Sano Y, Shimizu F, Omoto M, Tasaki A, Oishi M, Koga M, Saito K, Takahashi T, Nakada T, Kanda T. NMO sera down-regulate AQP4 in human astrocyte and induce cytotoxicity independent of complement. Neurol Sci. 2013;331:136–144. doi: 10.1016/j.jns.2013.05.035. [DOI] [PubMed] [Google Scholar]
- 54.Jordan I, Briese T, Averett DR, Lipkin WI. Inhibition of Borna disease virus replication by ribavirin. J Virol. 1999;73:7903–7906. doi: 10.1128/jvi.73.9.7903-7906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effects of IgG from NMO or SLE patients or healthy controls on the activation of and binding to BMECs.
Fig. S2. Binding of IgG from NMO or SLE patients or healthy controls to human astrocytes expressing AQP4 or human astrocytes.
Fig. S3. Effect of NMO-IgG or NMO-rAb on BMEC activation.
Fig. S4. Binding of NMO-IgG or NMO-rAb to human astrocytes expressing AQP4 or human astrocytes.
Fig. S5. Effects of non-AQP4 NMO-rAb on other MECs.
Fig. S6. Target of rAb ON-12-2-46 is widely expressed at both the cellular and tissue level.
Fig. S7. GRP78 expression on MECs in vitro and in vivo.
Fig. S8. Effect of commercial GRP78 Abs on BMEC activation.
Table S1. Effect of serum IgG from each patient with NMO and SLE and healthy controls on BMEC activation.
Table S2. Effect of pooled IgG and 14 rAbs from NMO patients and healthy controls on BMEC activation.
Table S3. Primary data (Microsoft Excel format).