

Abstract
Atrial fibrillation affects more than 33 million people worldwide and increases the risk of stroke, heart failure, and death.1,2 Fourteen genetic loci have been associated with atrial fibrillation in European and Asian ancestry groups.3–7 To further define the genetic basis of atrial fibrillation, we performed large-scale, multi-racial meta-analyses of common and rare variant association studies. The genome-wide association studies (GWAS) included 18,398 individuals with atrial fibrillation and 91,536 referents; the exome-wide association studies (ExWAS) and rare variant association studies (RVAS) involved 22,806 cases and 132,612 referents. We identified 12 novel genetic loci that exceeded genome-wide significance, implicating genes involved in cardiac electrical and structural remodeling. Our results nearly double the number of known genetic loci for atrial fibrillation, provide insights into the molecular basis of atrial fibrillation, and may facilitate new potential targets for drug discovery.8
Journal subject codes: Atrial fibrillation, population genetics, genome-wide association studies, gene-expression
Atrial fibrillation is a common cardiac arrhythmia that can cause serious complications such as stroke, heart failure, dementia, and death.1,2 The lifetime risk of atrial fibrillation is one in four9 and it has been estimated that more than 33 million individuals worldwide are affected.1 During the last decade, GWAS have identified 13 genetic loci associated with atrial fibrillation in Europeans and one Asian specific atrial fibrillation locus, of which a region near the gene encoding the transcription factor PITX2 has shown the strongest association.3–7 Recently, genome and exome sequencing studies have identified rare atrial fibrillation-associated mutations in MYL4,10 MYH6,11 CACNB2,12 and CACNA2D4.12 Given the incomplete understanding of the biology of atrial fibrillation and the modestly sized prior genetic association analyses, we sought to identify additional susceptibility loci by increasing the size and diversity of the atrial fibrillation studies.
We therefore investigated both common and rare variants in a large collection of individuals in the Atrial Fibrillation Genetics (AFGen) Consortium, by meta-analyses of GWAS, ExWAS, and RVAS in 33 studies, including 22,806 individuals with atrial fibrillation and 132,612 referents (Online methods). Fig. 1 illustrates our study design and Supplementary Tables 1 and 2 show baseline characteristics of the study participants.
Figure 1. Study flow-chart.
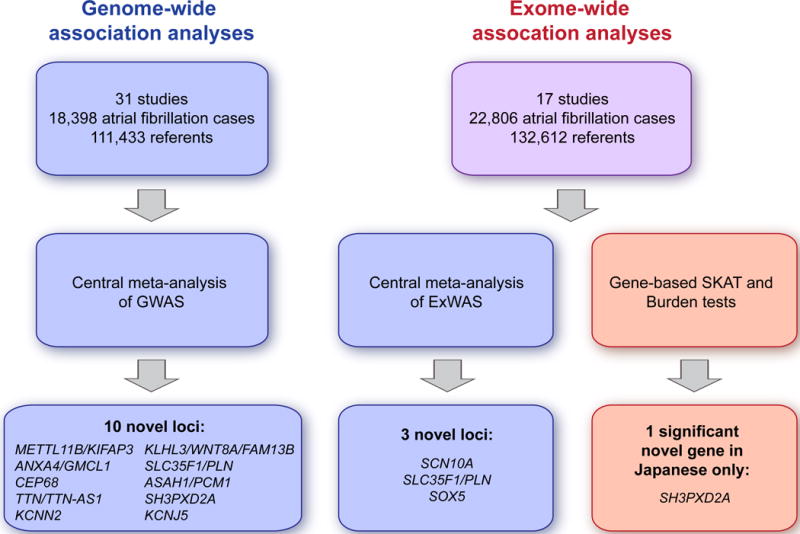
Overview of the approach employed for genome-wide and exome-wide association analyses.
In a meta-analysis of GWAS in 31 studies, we identified 10 new genetic loci associated with atrial fibrillation (P < 5×10−8) at METTL11B/KIFAP3, ANXA4/GMCL1, CEP68, TTN/TTN-AS1, KCNN2, KLHL3/WNT8A/FAM13B, SLC35F1/PLN, ASAH1/PCM1, SH3PXD2A, and KCNJ5 (Table 1, Figs. 2 and 3, Supplementary Fig. 1, Supplementary Table 3). The 13 genetic loci previously associated with atrial fibrillation in Europeans were again observed, while one locus previously reported in Asians only, did not reach genome-wide significance in our study (CUX2).
Table 1.
Results from combined ancestry GWAS meta-analysis
| rsID | Chr | Gene(s) | Location relative to gene | Risk allele/reference allele | Risk allele frequency, % | OR | 95% CI | P-value | Mean imputation quality |
|---|---|---|---|---|---|---|---|---|---|
|
Novel associations
| |||||||||
| rs72700118 | 1q24 | METTL11B/KIFAP3 | Intergenic | A/C | 12 | 1.14 | 1.10–1.19 | 2.60×10−11 | 0.959 |
| rs3771537 | 2p13 | ANXA4/GMCL1 | Intronic | A/C | 53 | 1.09 | 1.06–1.12 | 7.92×10−12 | 0.987 |
| rs2540949 | 2p14 | CEP68 | Intronic | A/T | 61 | 1.08 | 1.06–1.11 | 2.93×10−10 | 0.991 |
| rs2288327 | 2q31 | TTN/TTN-AS1 | Intronic | G/A | 20 | 1.09 | 1.06–1.13 | 2.05×10−8 | 0.994 |
| rs337711 | 5q22 | KCNN2 | Intronic | T/C | 39 | 1.07 | 1.05–1.10 | 2.93×10−8 | 0.995 |
| rs2967791 | 5q31 | KLHL3/WNT8A/FAM13B | Intronic | T/C | 54 | 1.07 | 1.05–1.10 | 2.73×10−8 | 0.961 |
| rs4946333 | 6q22 | SLC35F1/PLN | Intronic | G/A | 50 | 1.08 | 1.05–1.10 | 1.89×10−9 | 0.995 |
| rs7508 | 8p22 | ASAH1/PCM1 | 3′UTR | A/G | 72 | 1.09 | 1.06–1.12 | 5.16×10−10 | 0.977 |
| rs35176054 | 10q24 | SH3PXD2A | Intronic | A/T | 13 | 1.14 | 1.10–1.18 | 8.63×10−12 | 0.939 |
| rs75190942 | 11q24 | KCNJ5 | Intronic | A/C | 8 | 1.17 | 1.11–1.24 | 1.59×10−8 | 0.744 |
|
Previously known associations | |||||||||
| rs11264280 | 1q21 | KCNN3 | Intergenic | T/C | 31 | 1.12 | 1.09–1.15 | 6.41×10−17 | 0.942 |
| rs520525 | 1q24 | PRRX1 | Intronic | A/G | 71 | 1.12 | 1.09–1.15 | 6.39×10−16 | 0.955 |
| rs11718898 | 3p25 | CAND2 | Exonic | C/T | 65 | 1.08 | 1.05–1.10 | 4.68×10−8 | 0.969 |
| rs6843082 | 4q25 | PITX2 | Intergenic | G/A | 25 | 1.45 | 1.41–1.49 | 3.41×10−155 | 0.989 |
| rs12664873 | 6q22 | GJA1 | Intergenic | T/G | 70 | 1.08 | 1.05–1.11 | 1.19×10−8 | 0.968 |
| rs1997572 | 7q31 | CAV1/2 | Intronic | G/A | 59 | 1.10 | 1.08–1.13 | 6.64×10−15 | 0.988 |
| rs7026071 | 9q22 | C9orf3 | Intronic | T/C | 40 | 1.09 | 1.07–1.12 | 1.31×10−12 | 0.970 |
| rs7915134 | 10q22 | SYNPO2L | Intergenic | C/T | 85 | 1.12 | 1.08–1.16 | 1.68×10−10 | 0.975 |
| rs11598047 | 10q24 | NEURL1 | Intronic | G/A | 16 | 1.18 | 1.14–1.21 | 1.67×10−22 | 0.971 |
| rs883079 | 12q24 | TBX5 | 3′UTR | T/C | 70 | 1.11 | 1.09–1.14 | 1.80×10−15 | 0.991 |
| rs1152591 | 14q23 | SYNE2 | Intronic | A/G | 46 | 1.09 | 1.06–1.11 | 1.04×10−10 | 0.960 |
| rs74022964 | 15q24 | HCN4 | Intergenic | T/C | 17 | 1.12 | 1.08–1.15 | 2.37×10−11 | 0.970 |
| rs2106261 | 16q22 | ZFHX3 | Intronic | T/C | 19 | 1.20 | 1.17–1.24 | 8.18×10−32 | 0.973 |
The most significant variant at each genetic locus associated with atrial fibrillation is listed. Gene names in bold font indicate that the variant is located within the gene, whereas additional gene names indicate eQTL gene or gene strongly suspected to be causal due to the function of the encoded protein. For intergenic variants, the closest gene(s) are listed. Chr, chromosome; CI, confidence interval; OR, odds ratio.
Figure 2. Manhattan plot of the combined ancestry GWAS meta-analyses.
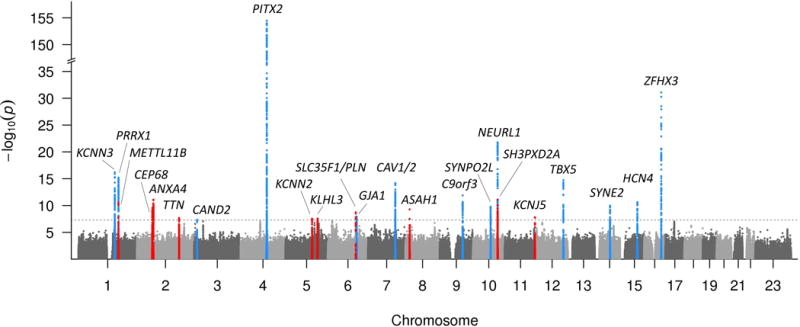
Manhattan plot showing novel (red) and replicated (blue) genetic loci associated with atrial fibrillation in the combined ancestry GWAS meta-analysis. The dotted line represents the threshold of statistical significance (5×10−8). The gene names represent the gene in closest proximity to the most significant variant at each locus. There is a break in the Y-axis to increase the resolution of the genetic loci near the genome-wide significance threshold.
Figure 3. Regional plots from combined ancestry GWAS meta-analysis.
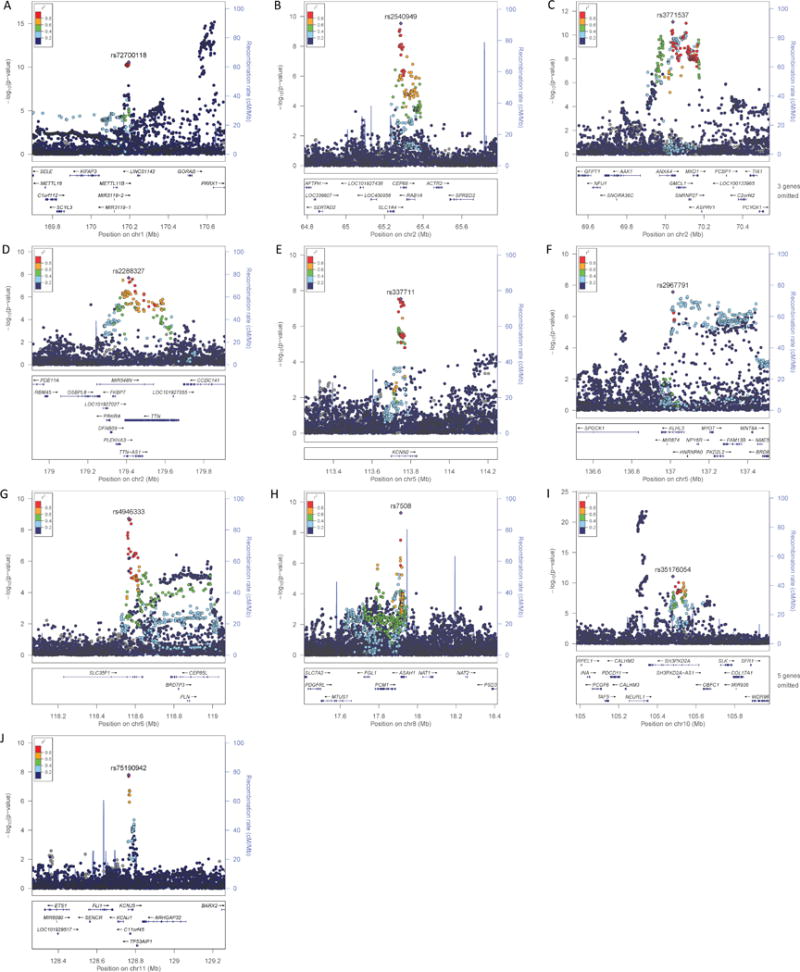
The most significant variant at each locus is plotted (purple, diamond-shaped) and identified with rsID. Each dot in the plots represent a single variant present in our results and the color of the dot indicates the degree of linkage disequilibrium with the most significant variant, as shown on the top left color chart on each panel. The lower part of each panel shows the locations of genes at the respective loci. r2, degree of linkage disequilibrium; chr, chromosome; Mb, megabases; cM, centiMorgan. Regional plots were created using LocusZoom.16
In a meta-analysis of ExWAS in 17 studies, we identified two additional novel genetic loci (SCN10A and SOX5, P < 1.04×10−6) as well as one new locus also identified in the GWAS meta-analysis (SLC35F1/PLN) (Table 2, Supplementary Fig. 2 and 3). Variants at each of these three loci have previously been associated with electrocardiographic traits (Supplementary Table 3).
Table 2.
Results from combined ancestry ExWAS meta-analysis
| rsID | Chr | Gene(s) | Location relative to gene | Risk allele/reference allele | Risk allele frequency, % | OR | 95% CI | P-value |
|---|---|---|---|---|---|---|---|---|
|
Novel associations
| ||||||||
| rs6800541 | 3p22 | SCN10A | Intronic | T/C | 61 | 1.08 | 1.05–1.12 | 8.79×10−7 |
| rs89107 | 6q22 | SLC35F1/PLN | Intronic | G/A | 58 | 1.07 | 1.04–1.10 | 9.51×10−7 |
| rs11047543 | 12p12 | SOX5 | Intergenic | G/A | 86 | 1.14 | 1.10–1.19 | 2.47×10−12 |
|
Previously known associations | ||||||||
| rs13376333 | 1q21 | KCNN3 | Intronic | T/C | 23 | 1.13 | 1.09–1.16 | 1.46×10−12 |
| rs17042171 | 4q25 | PITX2 | Intergenic | A/C | 21 | 1.64 | 1.59–1.69 | 8.31×10−227 |
| rs3807989 | 7q31 | CAV1 | Intronic | G/A | 58 | 1.09 | 1.06–1.12 | 6.52×10−8 |
| rs60632610 | 10q22 | SYNPO2L | Exonic; nonsyn | C/T | 85 | 1.12 | 1.08–1.15 | 1.54×10−10 |
| rs10151658 | 14q23 | SYNE2 | Exonic; nonsyn | C/A | 49 | 1.07 | 1.04–1.09 | 5.16×10−7 |
| rs2106261 | 16q22 | ZFHX3 | Intronic | A/G | 17 | 1.21 | 1.16–1.26 | 4.00×10−19 |
The most significant variant at each genetic locus associated with atrial fibrillation is listed. Gene names in bold font indicate that the variant is located within the gene, whereas additional gene names indicate eQTL gene or gene strongly suspected to be causal due to the function of the encoded protein. For intergenic variants, the closest gene(s) are listed. Chr, chromosome; CI, confidence interval; OR, odds ratio; nonsyn, nonsynonymous.
Finally, in an RVAS or burden test of rare variants, one gene, SH3PXD2A, reached genome-wide significance. This association was mainly driven by a rare coding variant that is unique to individuals of Asian ancestry (rs202011870, minor allele frequency (MAF) 0.18%, odds ratio (OR) 4.68, 95% confidence interval (CI) 2.97–7.39, P=3.3×10−11, Supplementary Tables 3–5) and the same locus was significantly associated with atrial fibrillation in the GWAS meta-analysis. Out of the 11 variants in the Asian ancestry burden test, rs149867987 also reached genome-wide significance and had an effect in the same direction as rs202011870. There was no genome-wide significant signal at SH3PXD2A in RVAS analyses in individuals of European or African American ancestry.
Ancestry-specific GWAS analysis revealed a significant association between African Americans (641 cases and 4956 referents) with atrial fibrillation and variants on chromosome 4q25 upstream of PITX2 (rs6843082, OR 1.40, 95% CI 1.24–1.58, P=4.31×10−8, Supplementary Table 6, Supplementary Fig. 4). Similarly, the 4q25/PITX2 region is the most significant locus for atrial fibrillation in individuals of Japanese ancestry (rs2723334, OR 1.94, 95% CI 1.68–2.25, P=8.46×10−19) and European ancestry (rs2129977, OR 1.45, 95% CI 1.41–1.49, P=7.25×10−136), and the lead SNPs in all three ancestry groups are in strong linkage disequilibrium, with an r2>0.94. Further ancestry-specific meta-analyses did not produce additional robust associations for atrial fibrillation (Supplementary Results, Supplementary Table 6–7, and Supplementary Figs. 4–6). Separate meta-analyses of incident and prevalent atrial fibrillation in Europeans did reveal one additional genome wide signal at chromosome 12p11/PKP2 that was only present in the prevalent atrial fibrillation analysis (Supplementary Results, Supplementary Tables 8–9, Supplementary Figs. 7–8); however, since this locus was not present in the combined analyses it was not pursued further.
We then performed an in silico replication of our results using two ethnically distinct studies. First, we replicated the atrial fibrillation associated variants in 8,180 cases and 28,612 referents from the Biobank Japan study (Online methods, Supplementary Table 10). The novel atrial fibrillation variant intronic to CEP68 reached genome-wide significance among Japanese, whereas the atrial fibrillation variants at KCNN2 and SOX5 achieved significance when correcting for multiple testing of 33 variants (P<1.5×10−3). The loci at ASAH1, TTN, and METTL11B reached nominal significance in Japanese (P<0.05). Of note, approximately 10% of the cases in the GWAS discovery analysis and Japanese replication analysis were overlapping (837 cases and 3293 referents). The lack of replication of the remaining loci likely reflects the heterogeneous nature of atrial fibrillation across different ancestries.
Second, we performed replication in 3,366 cases and 139,852 referents of mainly European ancestry in the UK Biobank (Online methods, Supplementary Table 11). The atrial fibrillation locus at SH3PXD2A reached genome-wide significance in the UK Biobank, whereas the loci METTL11B, CEP68, and KLHL3/WNT8A/FAM13B were significantly associated when correcting for multiple testing of 31 variants (P<1.6×10−3), and the loci at TTN, ASAH1, KCNJ5, and SCN10A reached nominal significance (P<0.05). The lack of replication of all of the atrial fibrillation loci is likely caused by reduced statistical power due to decreased sample size in the replication sample (18,398 versus 3,366 atrial fibrillation cases). However, there was a consistent direction of effects for all atrial fibrillation loci in the discovery and replication analyses.
Conditional analyses based on the summary level results of the GWAS meta-analysis were performed to identify multiple, independent signals on each chromosome containing atrial fibrillation loci (Online Methods). We confirmed that the two loci METTL11B/KIFAP3 and PRRX1, located ~350 kilobases (kb) apart on chromosome 1, were independent signals, as were the two loci SH3PXD2A and NEURL1, ~200 kb apart on chromosome 10 (Supplementary Table 12, Supplementary Fig. 9).
We found that seven of the known or new atrial fibrillation loci were associated with atrial fibrillation-related phenotypes, such as electrocardiographic traits, left ventricle internal diastolic diameter, and stroke (Supplementary Table 3 and 13, Supplementary Fig. 10). Given the close relation between atrial fibrillation and cardioembolic stroke, we then sought to determine whether the novel atrial fibrillation variants were associated with stroke risk. We performed an in silico lookup in GWAS data for stroke subtypes from the Neuro-CHARGE and METASTROKE consortia. None of the novel loci for atrial fibrillation were associated with ischemic stroke, cardioembolic stroke, small, or large vessel disease (Supplementary Tables 14–15).
Next, we performed an in silico evaluation of the known and newly identified atrial fibrillation associated loci (Online Methods, Supplementary Results). We compared the atrial fibrillation loci (n=24) to other trait-associated loci from the NHGRI-EBI GWAS catalog (n=3,381) and matching control loci selected for similar architectural properties (n=9,093). Interestingly, the atrial fibrillation loci were significantly conserved across species, and were also significantly enriched for active enhancers in cardiac tissues as denoted by H3K27ac marks, compared to other trait-associated loci from the NHGRI-EBI GWAS catalog and matching control loci (Supplementary Fig. 11). Moreover, the genes at atrial fibrillation loci displayed enrichment for Gene Ontology terms important for cardiac action potential propagation and cardiac contractility compared to the control loci, although this enrichment was not significant when corrected for multiple hypothesis testing (Supplementary Table 16).
We also performed expression quantitative trait locus (eQTL) analyses of the atrial fibrillation-associated genetic loci using two additional approaches (Online Methods). We identified significant eQTLs for seven of the twelve novel atrial fibrillation associated loci (closest gene;eQTL gene: METTL11B;KIFAP3, ANXA4;ANXA4/GMCL1/PCYOX1/SNRNP27, CEP68;CEP68, KCNN2;KCNN2, KLHL3;FAM13B/REEP2, ASAH1;ASAH1/PCM1/RP11-806O11.1, and KCNJ5;KCNJ5/C11orf45) and eight of the thirteen previously reported atrial fibrillation loci (Supplementary Tables 17–20, Supplementary Fig. 12).
In the current work, we have identified 12 novel genetic loci for atrial fibrillation in our large-scale analyses of common, coding, and rare genetic variation for atrial fibrillation (Supplementary Table 3). When considered together with the known atrial fibrillation loci, the genes at these loci broadly encode ion channels, sarcomeric proteins, and transcription factors that underlie this common arrhythmia. Genes at five of the genetic loci identified encode potassium or sodium channels, including two novel loci at the genes KCNN2 and KCNJ5 that are known to be involved in the maintenance of the atrial cardiac action potential. Since the cellular hallmark of atrial fibrillation is shortening of the atrial action potential duration and calcium overload, the KCNN2 and KCNN3 genes are particularly interesting. The lead variant at chromosome 5q22 is located intronic to and has a significant eQTL with KCNN2, which encodes the calcium dependent potassium channel SK2. The SK2 protein is known to form heteromeric channel complexes with SK3, which is a product of the KCNN3 gene that is strongly associated with atrial fibrillation in the present and previous atrial fibrillation GWAS meta-analyses.5,6
Similarly, KCNJ5 encodes the potassium channel Kir3.4 or GIRK4 that is known to form heteromeres with Kir3.1/GIRK1/KCNJ3 and assemble to form the inwardly rectifying, IKAch channel complex. The IKAch complex is regulated by G protein signaling, is well-known to regulate the membrane potential in the sinoatrial node and atria, and has been considered as a therapeutic target for atrial fibrillation.
Interestingly, the gene identified in our rare and common variant analyses, SH3PXD2A, is expressed in human atria and ventricles and encodes TKS5, a tyrosine kinase substrate. The rare variant association was largely driven by the variant rs202011870, which results in a leucine to arginine substitution at position 396. TKS5 has been shown to be important in determining the invasiveness of cancer cells13 and has been suggested to mediate the neurotoxic effect of beta-amyloid in Alzheimer disease in association with the matrix metalloproteinase gene ADAM12.14 Developmentally, SH3PXD2A is important for neural crest migration; homozygous knockout in mice result in complete cleft in the secondary palate and neonatal death;15 however, the relation between SH3PXD2A and atrial fibrillation is unclear and as with any rare variant association, replication in a large, independent dataset will ultimately be required.
Finally, we found that the atrial fibrillation loci have significant conservation across species, and are enriched for active enhancers in cardiac tissues, compared to other GWAS or control loci. Since many of the identified atrial fibrillation loci include genes that encode transcription factors (PITX2, ZFHX3, PRRX1, SOX5, and TBX5), we hypothesize that these loci may be more conserved, because they may underlie a canonical program for left atrial and/or pulmonary venous development.
While the strengths of our study include the large sample sizes, analyses of common and rare genetic variation, and the inclusion of different races and ethnicities, our study was subject to some limitations. Specifically, it is important to note that the estimates of variance explained by genetic variation can be challenging for qualitative traits such as atrial fibrillation, particularly given the marked variability in prevalence of the disease according to age. Thus, as with GWAS for other common conditions, we anticipate that the newly described loci for atrial fibrillation would only explain a small portion of the variance of atrial fibrillation.
In conclusion, we have nearly doubled the number of known genetic loci associated with atrial fibrillation through meta-analysis of more than 22,000 individuals with atrial fibrillation. We have identified a series of novel atrial fibrillation-associated variants, which lie proximal to genes involved in atrial electrical and mechanical function. Our results will facilitate downstream research establishing the mechanistic links between identified genetic loci and atrial fibrillation pathogenesis, potentially aiding in the discovery of new therapeutic targets for the treatment of atrial fibrillation.8
Code availability
The computer code that support the results of the present study are available from the corresponding author upon request.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Online METHODS
Study population
The Atrial Fibrillation Genetics Consortium (AFGen) is a collaboration between multiple studies with the aim of investigating the genetic causes of atrial fibrillation. In this study, we included 33 studies from AFGen, of which 31 participated in the GWAS meta-analysis, whereas 17 studies were part of the exome chip analyses. Supplementary Table 21 shows per study overlap of samples between the GWAS and exome chip analyses. The majority of the participants were of European ancestry (15,993 cases, 113,719 referents). We also included studies with African-American (3 studies; 641 cases, 4956 referents), Japanese (1 study; 837 cases, 2456 referents), Hispanic (1 study; 277 cases, 3081 referents), and Brazilian (1 study; 187 cases, 550 referents) ancestry (Supplementary Table 1). The ExWAS and RVAS involved 22,806 cases and 132,612 referents of European (13,496 cases, 96,273 referents), African American (681 cases, 4,871 referents), and Asian (8,180 cases, 28,612 referents) ethnicities (Supplementary Table 2). Overall, adjudication of atrial fibrillation included either documented atrial fibrillation on an electrocardiogram and/or one in-patient or two out-patient diagnoses of atrial fibrillation. Referents were free of atrial fibrillation. All participating studies had obtained informed consent from all cases and referents and had obtained approval from their respective ethics committees or institutional review boards.
GWAS meta-analyses
Each study performed genotyping and imputation to the 1000 Genomes Project Phase 1 reference panel (March 2012 release). Detailed methods for each study are described in the Supplementary Note and in Supplementary Table 22. Cox proportional hazards models were used for incident data with time-to-event from study enrollment. Logistic regression models were used for prevalent and case-control data. Models were adjusted for age and sex if available, and if appropriate, for principal components of the genotype matrix to control for population stratification. For studies with prevalent cases at time of enrollment (or blood draw) and incident cases identified during follow up, two analyses were performed: 1) Prevalent analysis at baseline/blood draw: all individuals who were diagnosed with atrial fibrillation prior to baseline were defined as cases, and all individuals who were not diagnosed with AF prior to baseline were defined as referents in a logistic regression analysis (future cases were controls in this analysis); 2) Incident analysis looking forward from baseline: prevalent cases were excluded and time-to-atrial fibrillation diagnosis was analyzed, using Cox proportional hazards models, with censoring at last follow-up. The two analyses are approximately independent, because they consider different periods of risk, as described by Benjamin et al.1 Pre- and post-GWAS filtering was performed according to predefined quality control filters (Supplementary Table 23). Briefly, variants with MAF <1%, imputation quality <0.3 (IMPUTE), or that were present in <2 studies were excluded.
We meta-analyzed summary level GWAS results using an inverse variance-weighted fixed-effects model with METAL software.2 For the combined ancestry GWAS meta-analysis, we tested 11,795,432 variants. The traditional Bonferroni correction for number of variants tested is often regarded as too conservative, because the tests are not independent due to LD. Thus, we chose the most widely used and accepted significance threshold for GWAS in our GWAS meta-analyses.3–6 Variants that reached a genome-wide P-value <5×10−8 were considered statistically significant. Meta-analyses were also performed separately for each ethnicity group and for incident and prevalent atrial fibrillation to identify potentially differential associations and effects.
ExWAS and rare variant meta-analyses
Each study performed exome variant genotyping and association analyses locally, using a logistic model that combined incident and prevalent cases and referents (Supplementary Table 24). Individual variants that passed quality control filters and were present in at least 2 studies with average MAF≥0.5% (Supplementary Table 23), were meta-analyzed using the score test implemented in the seqMeta package of R statistical software.7 For the combined ancestry ExWAS meta-analysis, we tested 48,133 variants and used a significance level of 1.04×10−6, which is approximately a Bonferroni adjustment of 0.05/48,133. For MAF > 0.5%, we had approximately 80% power to detect variants with a multiplicative genotype relative risk of 1.4. RVAS was performed on rare variants from the exome chip array using SKAT8 and burden tests with three approaches: 1) all non-synonymous and splice site variants, 2) non-synonymous variants annotated as possibly damaging, and 3) loss-of-function variants only. For each gene-based test we excluded variants with MAF >5% and excluded genes with cumulative MAF <0.05%.
Approximate joint and conditional analysis
To identify independent variants within the 12 significant genetic loci, we performed an approximate joint and conditional association analysis implemented in the software GCTA9 using summary level statistics from the meta-analysis. We used a stepwise procedure for detecting additional independent variants with a European ancestry reference panel from the Framingham Heart Study (n=2764 unrelated individuals).
Functional annotation
Functional element enrichment
Loci were defined as regions encompassing variants that were in linkage disequilibrium with the query variant (r2>0.8 in CEU population) and that were no greater than 500 kb from the query variant. Loci had to encompass at least 5 kb both upstream and downstream of the query variant. Overlapping loci were merged. The GWAS control loci were calculated from unique variants from the NHGRI-EBI GWAS catalog (as of May 31, 2016) that had a P-value <5×10−8. The 1000 Genomes control loci were calculated using 24,000 matched variants based on MAF, gene density, distance to nearest gene, and number of nearby variants in linkage disequilibrium determined by the SNPsnap tool.10 The SNPsnap matched variants were calculated using the European population and an r2 cutoff of 0.8, but otherwise default parameters. Each locus in each experimental set was intersected with various markers for functional elements to determine the median percent overlap of each experimental set. The markers included phastCons 46-way primate and mammalian conserved elements, Roadmap Epigenome H3K27ac gapped peaks, and ENCODE DNaseHS sites. Statistical significance was calculated by one-tailed bootstrapping for enrichment with 1,000 random sub-samplings of each control set.
Gene ontology analysis of atrial fibrillation loci
RefSeq genes that overlapped atrial fibrillation-associated loci as well as genes that overlapped the GWAS catalog control loci and the 1000 Genomes matched control loci were used for gene ontology enrichment analysis. The genes that overlapped the control loci were used as two separate background sets. Enrichment calculations were provided by the GOrilla tool.11
In silico database interrogation
All statistically significant variants and genes from GWAS and RVAS analyses were selected for an in silico assessment through lookups in the following databases: The Gene Tissue Expression database (GTEx),12 RegulomeDB,13 HaploREG,14 GeneCards (www.genecards.org/), dbSNP.15 From the GTEx search, we report statistically significant eQTLs in cardiac and skeletal muscle tissues. The NHGRI-EBI GWAS catalog16 was interrogated with the aim of identifying possible pleiotropy with other cardiovascular phenotypes. At each locus, we defined a region based on LD span (r2 > 0.2) with the lead SNP. We searched the GWAS catalog for all SNPs within these regions and report LD of proxies with the lead SNP when available. LD information was identified using the SNiPA tool17 (Available at http://www.snipa.org. Accessed 6-24-2016.)
Expression Quantitative Trait Locus analyses
1. eQTL analyses in the Cleveland Clinic Atrial Tissue Bank and Arrhythmia Biorepository
We performed analyses of gene expression in human left atrial tissue samples obtained from the Cleveland Clinic Atrial Tissue Bank and Arrhythmia Biorepository. Genotypes were determined using the Illumina Human Hap550 v3 or Hap610 v1 chips; whereas RNA expression levels were determined using the Illumina HumanHT-12 v3 or v4 chips. The atrial samples were obtained from 289 individuals of European American (EA) ethnicity and 40 individuals of African American (AA) ethnicity. Of the EA individuals, 80 were female, 70 had no history of atrial fibrillation, and 136 were in atrial fibrillation at the time of tissue acquisition; 266 samples were from left atrial appendage (LAA) tissue and 23 the left atrial pulmonary vein junction tissue (LA-PV). Of the AA individuals, 25 were female, 16 had no history of atrial fibrillation, and 12 were in atrial fibrillation at the time of tissue acquisition; 34 samples were from LAA and 6 from LA-PV tissue. Methods have previously been described in depth by Deshmukh et al.18 We performed cis-eQTL analyses for all statistically significant genetic variants identified in GWAS analyses. The Benjamini and Hochberg adjustment was applied to the results to control the false discovery rate (FDR).19 P-values were adjusted based on the FDR of both genome-wide testing and specific variant sets, respectively. Probe-variant pairs with a genome-wide adjusted P-value less than 0.05 were deemed significant.
2. Examination of eQTLs in cardiac and skeletal muscle tissues from the GTEx database
The GTEx database was interrogated for all genetic loci associated with atrial fibrillation in the present meta-analyses. We selected the index variants and all proxies at the atrial fibrillation loci and looked for eQTLs in a subset of the GTEx database for right atrial, left ventricular, and skeletal muscle tissues that are most relevant to atrial fibrillation.
3. GTEx region based analyses
GTEx region based analyses were performed by comparing the percent of atrial fibrillation loci with at least one eQTL to the percent of control loci with at least one eQTL. All tissues in the GTEx database were used for this analysis. Atrial fibrillation loci and control loci were defined as described in the “Functional element enrichment” section above. Statistical significance was calculated by a one-tailed test based on 1,000 bootstrap samples from each set of control loci.
Replication of genetic variants specific to African American ancestry GWAS meta-analysis
We sought to replicate variants specific to the African American ancestry GWAS meta-analysis in 447 atrial fibrillation cases and 442 referents of African American ancestry. Custom TaqMan® genotyping probes for rs115339321 and rs79433233 were obtained from Life Technologies. Genotyping was performed on 5 ng of DNA input using the TaqMan® genotyping master mix on a Bio-Rad CFX384 real time PCR instrument. Genotyping was performed in 447 atrial fibrillation cases and 442 referents obtained from four studies (BioVU, Duke Biobank, MGH, and Penn Biobank), with genotype calls being performed by end state fluorescence after 40 cycles. See Supplementary Results and Supplementary Tables 25–26 for further details.
In silico replication in the BioBank Japan (BBJ) study
The variant with the lowest P-value at each independent novel atrial fibrillation locus was selected for in silico replication in the results from GWAS analysis in 8180 individuals with atrial fibrillation and 28,612 referents from the BioBank Japan study. The cases were selected from the Biobank Japan which contains DNA and serum samples collected throughout Japan and atrial fibrillation was defined as persistent or paroxysmal atrial fibrillation diagnosed by a physician. The referents were selected from the Tohoku Medical Megabank organization,20 the Japan Public Health Centre-based Prospective study, and the Japan Multi-institutional Collaborative Cohort (J-MICC) Study. Samples were genotyped using the Illumina Human OmniExpress BeadChip Kit and Infinium OmniExpressExome BeadChip Kit. Only autosomal variants were included in the GWAS. Variants with call rate <99%, variants that deviated from Hardy-Weinberg equilibrium among control samples (<1×10−6), and non-polymorphic variants were excluded.
In silico replication in the UK Biobank study
Replication was performed using 143,218 unrelated adults of primarily European ancestry (>80%), aged 40–69 years old between 2006 and 2010, from the UK Biobank interim dataset released in May 2015. We defined atrial fibrillation as reported during a baseline interview; presence of a procedure code for cardioversion, atrial flutter or fibrillation ablation, or atrioventricular node ablation; billing code for atrial fibrillation; or atrial fibrillation reported on a death record (specific codes used in the definition are available upon request). Of the 143,218 individuals in the replication dataset, we identified 3366 individuals with atrial fibrillation, according to the criteria above. Details of genotyping, imputation, and calculation of principal components of ancestry in the UK biobank interim dataset can be found on the UK biobank website (http://www.ukbiobank.ac.uk/). Briefly, samples were genotyped either by UK BiLEVE Axiom array (UKBL) or UK Biobank Axiom array (UKBB). Both arrays include ~800,000 SNPs and more than 95% of common marker contents are similar. Imputation was phased by modified version of SHAPEIT2 and imputed by IMPUTE2, using a combined panel of UK10K haplotype and 1000G phase 3 as the reference panel. All significant variants detected in the discovery study passed quality control filters in the UK biobank data (imputation quality info ≥ 0.4, variant missing rate < 5%, individual missing rate < 10%, and variant genotype probability > 0.9 in > 90% of the individuals). Variants were then transformed to hard-called genotypes (probability threshold ≥ 0.9, minor allele frequency (MAF) ≥ 0.01, and missing rate per variant <5%). We used logistic regression to test the association between each hard-called variant and risk of atrial fibrillation using an additive genetic model, adjusting for baseline age, sex, array, and the first 15 principal components of ancestry. Quality control, transformation and analyses were performed by QCTOOL and Plink v1.90b. Since we performed an in silico replication of 31 variants, we set a conservative significance threshold of 1.6 × 10−3 (0.05/31).
Pathway analyses
Pathway analyses provide a potential route to investigate the collective effects of multiple genetic variants on biological systems (see Supplementary Results and Supplementary Tables 27–29). We utilized two different methods for pathway analysis:
1. DEPICT
We ran the analysis DEPICT,21 which integrates multiple layers of evidence to identify causal genes at GWAS loci. From meta-analysis results, we first performed clumping to identify independent loci using plink.22 We then performed analysis using DEPICT with the default settings.
2. Ingenuity Pathway Analysis (IPA)
Data were analyzed through the use of QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity). For each of the tested genetic variants, we mapped it back to the reference human genome (NCBI Build 37, 2009) and examined its location relative to RefSeq genes (May 15, 2016). The gene score was defined as the most significant variants that were located within 110kb upstream and 40kb downstream of the gene’s most extreme transcript boundaries. Of the 27,011 genes evaluated, 338 reached a score less than 5×10−6. These genes were then imported into IPA analysis. Fisher’s exact test was used to justify the enrichment of each of the canonical pathways.
Assessment of pleiotropy with the ischemic stroke phenotype
In order to evaluate pleiotropy with the ischemic stroke phenotype, we selected the variant with the lowest P-value at each independent novel atrial fibrillation locus and performed a lookup in the results from 1000 Genomes imputed GWAS meta-analyses from the Neurology Working Group of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium (4348 stroke patients and 80,613 referents)23 and the METASTROKE consortium (10,307 ischemic stroke cases and 19,326 referents) of the International Stroke Genetics Consortium (ISGC).24
Supplementary Material
Acknowledgments
A full list of acknowledgments appears in the Supplementary Note.
Dr. Ellinor is the PI on a grant from Bayer HealthCare to the Broad Institute focused on the genetics and therapeutics of atrial fibrillation.
Footnotes
Author Contributions
I.E.C., C.R., X.Y., T.T., K.L.L., E.B.J, S.A.L., M.R., B.G., P.T.E. wrote and edited the manuscript. All authors contributed to and discussed the results, and commented on the manuscript. GWAS and ExWAS analyses: A.V.S, N.A.B., M.M-N., I.S., C.S., P.E.W., S.A., S.T., J.A.B., J.C.B., H.L., J.H., J.Y., X.G., F.R., M.N.N., D.E.A., G.P., S-K.L., Y.K., M.K., A.C.P., A.R.H., J.S., L-P.L., M.A., M.E.K., J.G.S., R.M., S.G., S.T., M.D., S.W., J.W., D.I.C., M.V.P., Q.Y., T.B.H., M.F.S., J.S., D.v.W., M.K. Individual dataset quality control and GWAS and ExWAS meta-analyses: I.E.C., K.L.L., C.R., X.Y., M.R., B.G., Y.P.H., N.V., J.E.S. Replication in METASTROKE and Neuro-CHARGE: Q.Y., J.H., S.D., G.C., B.B.W. Replication in UK Biobank: S.K., D.K., C.N-C. Replication in Biobank Japan: S-K.L., Y.K., M.K., T.T. Replication in African American population: R.D., D.J.R., S.S., A.S. CCAF eQTL analyses: J.B., M.K.C., D.v.W., J.D.S. Functional annotation: I.E.C., S.H.C., L-C.W., M.L., C.R., M.C., N.R.T., S.C. Pathway analyses: H.L.
Competing Financial Interests Statement
Dr. Ellinor is the PI on a grant from Bayer HealthCare to the Broad Institute focused on the genetics and therapeutics of atrial fibrillation. The remaining authors have no disclosures.
Disclosures: The remaining authors have no disclosures.
References
- 1.Chugh SS, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014;129:837–47. doi: 10.1161/CIRCULATIONAHA.113.005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.January CT, et al. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:2246–2280. doi: 10.1016/j.jacc.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 3.Gudbjartsson DF, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41:879–81. doi: 10.1038/ng.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellinor PT, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010;42:240–4. doi: 10.1038/ng.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellinor PT, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–5. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinner MF, et al. Integrating genetic, transcriptional, and functional analyses to identify 5 novel genes for atrial fibrillation. Circulation. 2014;130:1225–35. doi: 10.1161/CIRCULATIONAHA.114.009892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson MR, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–860. doi: 10.1038/ng.3314. [DOI] [PubMed] [Google Scholar]
- 9.Lloyd-Jones DM, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. 2004;110:1042–1046. doi: 10.1161/01.CIR.0000140263.20897.42. [DOI] [PubMed] [Google Scholar]
- 10.Gudbjartsson DF, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. 2015;47:435–444. doi: 10.1038/ng.3247. [DOI] [PubMed] [Google Scholar]
- 11.Holm H, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 2011;43:316–20. doi: 10.1038/ng.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weeke P, et al. Exome sequencing implicates an increased burden of rare potassium channel variants in the risk of drug-induced long QT interval syndrome. J Am Coll Cardiol. 2014;63:1430–7. doi: 10.1016/j.jacc.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seals DF, et al. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155–65. doi: 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Laumet G, et al. A study of the association between the ADAM12 and SH3PXD2A (SH3MD1) genes and Alzheimer’s disease. Neurosci Lett. 2010;468:1–2. doi: 10.1016/j.neulet.2009.10.040. [DOI] [PubMed] [Google Scholar]
- 15.Cejudo-Martin P, et al. Genetic disruption of the sh3pxd2a gene reveals an essential role in mouse development and the existence of a novel isoform of tks5. PLoS One. 2014;9:e107674. doi: 10.1371/journal.pone.0107674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pruim RJ, et al. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Online methods references
- 1.Benjamin EJ, et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41:879–81. doi: 10.1038/ng.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willer CJ, Li Y, Abecasis GR. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 4.Hoggart CJ, Clark TG, De Iorio M, Whittaker JC, Balding DJ. Genome-wide significance for dense SNP and resequencing data. Genet Epidemiol. 2008;32:179–185. doi: 10.1002/gepi.20292. [DOI] [PubMed] [Google Scholar]
- 5.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32:227–234. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. 2014;15:335–346. doi: 10.1038/nrg3706. [DOI] [PubMed] [Google Scholar]
- 7.Lumley T, Brody J, Dupuis J, Cupples A. Meta-analysis of a rare-variant association test. 2012 [Google Scholar]
- 8.Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–775. doi: 10.1093/biostatistics/kxs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pers TH, Timshel P, Hirschhorn JN. SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics. 2015;31:418–20. doi: 10.1093/bioinformatics/btu655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyle AP, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold M, Raffler J, Pfeufer A, Suhre K, Kastenmüller G. SNiPA: An interactive, genetic variant-centered annotation browser. Bioinformatics. 2015;31:1334–1336. doi: 10.1093/bioinformatics/btu779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deshmukh A, et al. Left atrial transcriptional changes associated with atrial fibrillation susceptibility and persistence. Circ Arrhythm Electrophysiol. 2015;8:32–41. doi: 10.1161/CIRCEP.114.001632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995:289–300. [Google Scholar]
- 20.Kuriyama S, et al. The Tohoku Medical Megabank Project: Design and Mission. J Epidemiol. 2016;26:493–511. doi: 10.2188/jea.JE20150268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pers TH, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2015;6:5890. doi: 10.1038/ncomms6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neurology Working Group of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium, Stroke Genetics Network (SiGN) & International Stroke Genetics Consortium (ISGC) Identification of additional risk loci for stroke and small vessel disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2016;15:695–707. doi: 10.1016/S1474-4422(16)00102-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malik R, et al. Low-frequency and common genetic variation in ischemic stroke: The METASTROKE collaboration. Neurology. 2016;86:1217–26. doi: 10.1212/WNL.0000000000002528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.