

Abstract
Type 1 diabetes mellitus (T1DM) is the most common autoimmune disease in pediatrics with a prevalence of roughly 1 in 500 children in the USA. Genome-wide association studies have identified more than 50 variants associated with increased risk for type 1 diabetes. Comparison of these variants with those identified in other autoimmune diseases reveals three important findings: (1) there is a high degree of overlap in implicated variants in diseases with similar patho-physiology, (2) in diseases with differing pathophysiology the same variants are often implicated in opposite roles, (3) in diseases with differing pathophysiology that have many non-overlapping or oppositely implicated variants there are still several variants which are overlapping or shared. Thus, the genetic overlap between T1DM and other autoimmune diseases forms the basis for our understanding of druggable targets in type 1 diabetes.
Keywords: Type 1 diabetes mellitus, Autoimmunity, Genomic, GWAS, ENCODE
Introduction
Type 1 diabetes mellitus (T1DM) is the third most common chronic disease of childhood and the most common autoimmune disease in pediatrics with a prevalence of approximately 1 in 500 children in the USA [1•]. In the past decade, the incidence of T1DM in the USA has increased by more than 20 % [1•]. This disease was once universally fatal, but is now managed with daily injections or continuous subcutaneous infusion of insulin. T1DM remains an active focus of research, but clinical progress aside from the development of new insulins (with longer or shorter half-lives) has been scant. Recent work has focused on three areas: (1) the construction of an artificial external pancreas [2, 3], (2) the use of stem cells to generate a new pancreas and/or pancreas transplant [4–8] and (3) genomic dissection of the genetic determinants of T1DM [9, 10, 11••, 12]. This first area is beginning to yield its first fruit, with recent publications on the use of an artificial pancreas showing improved HbA1c without increased hypoglycemia. This second area has had some recent notable successes; early outcomes from dual pancreas–kidney transplant patients with T1DM reveal that a significant number of patients have been able to stop daily injections of insulin. This third area has revealed unexpected contributors to genetic risk for T1DM; genomic approaches have allowed us to identify more than 50 variants that contribute to genetic risk for T1DM. Many of these variants have been found to be associated with other autoimmune diseases suggesting common pathological mechanisms between these illnesses [13•, 14, 15••].
Several lines of evidence implicate common pathways or mechanisms in disparate autoimmune diseases: (1) the common co-occurrence of some autoimmune diseases or phenotypes (e.g., T1DM and celiac disease or inflammatory bowel disease and arthritis), (2) epidemiological studies of populations and families describing high correlation of autoimmune disease rates [16] and (3) syndromes comprised of multiple autoimmune diseases (e.g., the autoimmune polyendocrine syndromes) [17]. This evidence is compelling, but does not, per se, aid in understanding disease etiology. Several recent studies attempt to use the overlap between autoimmune diseases to aid in pathogenic dissection and potentially point to intervention targets. This review will discuss this work.
Recent work determining the role of genes implicated in the similar phenotypes of the single-gene autoimmune polyendocrinopathies that include T1DM point to the structure of critical signaling pathways underlying self-tolerance [18]. These genes include the AIRE protein in autoimmune polyendocrinopathy type 1 and the FOX3 protein in X-linked polyendocrinopathy, immune dysfunction, and diarrhea, respectively. Similar approaches applied to the genes implicated in genome-wide association studies (GWAS) of non-syndromic T1DM promise to reveal a much broader and richer map of the genetic structure of immune signaling.
Comparing variants implicated in T1DM to variants identified in other autoimmune diseases reveals three surprising and important results. First, there is a very high degree (>40 % for several diseases and >50 % for at least one disease) of overlap that exceeds what would be expected based on the epidemiological data. Second, some of the overlap reveals that the same variants can be implicated in opposite roles in two different diseases (e.g., a high-risk variant for T1DM being a protective variant for inflammatory bowel disease (IBD)). Third, in diseases where most of the variants are non-overlapping or in opposing directions there are still several variants that are overlapping or shared.
Pathogenesis of T1DM
T1DM occurs as a result of autoimmune destruction of the insulin-producing beta cells of the pancreas. This destruction is thought to be mediated by T cells while being dependent on the production of antibodies interacting with the beta cell or its constituent proteins. Population risk for T1DM correlates with high-risk HLA allele prevalence. Once individuals with high-risk HLA alleles have more than one measurable beta cell antibody, the progression to T1DM occurs in more than 75 % within 20 years, 61 % within 10 years, and 35 % within 5 years. Thus, it is believed that antibody production is either one of the critical early initiators of T1DM or it is an indicator of progression past a committed checkpoint towards T1DM. Prior to antibody production, it appears that there is T cell infiltration of the pancreas and local inflammation called insulitis. This infiltration is thought to occur after development of anti-self repertoire T cells and B cells. The progression to T1DM then is conceptualized as starting with (1) the development of anti-self immune cells then progressing to (2) insulitis and then (3) destruction of the pancreas. It is believed that either this first step or this last step occurs after an infection with an agent that has antigens similar to those that are presented in the pancreas. Thus the infection either causes the generation of T cells and B cells specific to the pancreas or allows the expansion or activation of these cells to progress from low-grade subclinical insulitis to large-scale beta cell destruction. Because of the likely role of T cells in destruction of beta cells several recent studies have examined the ability of anti-CD-3 antibodies to prevent diabetes progression. While early small-scale studies of this approach seemed promising, larger studies have found no benefit to this approach [19].
Evidence Implicating Common Mechanisms of Autoimmune Disease
The pathogenesis of all of the autoimmune diseases would seem intuitively to reflect a failure in self-tolerance, but given the disparate phenotypes of autoimmune diseases, it is not necessary a priori to believe that the failure in self-tolerance is mediated by the same paths in different diseases. Several lines of evidence prior to GWAS results, however, implicate common mechanisms underlying autoimmune diseases; first, the common co-occurrence of some autoimmune diseases or phenotypes (e.g., T1DM and celiac disease or inflammatory bowel disease and arthritis) suggests that these diseases have a common genetic risk factor, second, epidemiological studies of populations and families [20] describing high correlation of autoimmune disease rates also suggests a common genetic risk factor between these diseases, and third, syndromes comprised of multiple autoimmune diseases (e.g., the autoimmune polyendocrine syndromes) where a genetic risk factor has been identified.
Epidemiological Evidence of Autoimmune Diseases That Include T1DM
As noted above, there are several lines of epidemiological evidence linking disparate autoimmune diseases. Only in the past two decades has the widespread use of the electronic medical record and improvements in disease registries allowed the emergence of some of this data. Autoimmune disease is observed to co-occur in individuals more often than we would expect to be the case if the co-occurrence were due to independent effects [16]. Also, different autoimmune diseases occur in families more often than would be expected if the co-occurrence were due to chance. For individuals diagnosed with T1DM, celiac disease and autoimmune thyroid disease are known to occur at an increased prevalence compared to the general population and are screened for annually in most centers.
Mendelian Autoimmune Diseases That Include T1DM
T1DM is also present in a number of individuals who have single-gene polyendocrine autoimmune syndromes. In humans, the gene mutation underlying X-linked polyendocrinopathy, immune dysfunction, and diarrhea (XPID—mutation in FOXP3) has been identified [21]. Individuals with XPID have mutations in FOXP3 and universally develop T1DM as well as widespread autoimmunity and chronic diarrhea starting as a neonate. FOXP3 is a transcription factor that controls the development of regulatory T cells. Regulatory T cells are critical to regulating self-tolerance and individuals with XPID completely lack these cells and have a florid multisystem autoimmunity. T1DM occurs in another polyendocrine syndrome, autoimmune polyendocrine syndrome type 1 (APS-1—mutation in AIRE), more than 20 % of the time [17]. Recent investigations of mouse models of APS-1, which are caused by a mutation in the AIRE protein, reveal that this protein plays a critical role in the generation of a developmentally specific kind of FOXP3 expressing T-regulatory cells [18]. Aire–/– mice do not lack all FOXP3 cells, but only a specific developmentally limited subset of all regulatory T cells. Expression of Aire for a short period of time early in development is sufficient to allow the generation of this population of cells and to prevent the development of APS-1. In humans, mutations that limit the function of Aire lead to APS-1 and high rates of T1DM. Thus, the work on these two kinds of single-gene autoimmune diabetes has revealed the structure of the pathway underlying the pathogenesis of T1DM in these syndromes. In these diseases, intriguingly, autoimmune diabetes does not appear to result from holes in the tolerance induction systems, but more from the breakdown of secondary systems (critical T-regulatory cells) that keep self-antigen-specific cells in check.
Genes Implicated in GWAS of T1DM
T1DM is unique among the common autoimmune diseases in that it is more common in children than adults and, within the spectra of autoimmune diseases, is relatively phenotypically homogenous. In general in GWAS, pediatric phenotypes require a smaller population to identify significant genetic contributors than adult populations. Thus, T1DM provides a potentially fertile starting point to understand autoimmune disease. That is because it occurs in children it may provide more power than other autoimmune diseases to identify variants associated with autoimmunity. Furthermore, T1DM has a disproportionately high genetically attributed risk, with a concordance rate in monozygotic twins of ∼50 % [22].
T1DM has been the focus of more than 10 full-scale GWAS and GWAS meta-analyses [23•, 24•, 25–32, 12, 13•] and nearly 60 variants have been identified as associated with T1DM risk. The first T1DM GWAS were published in 2007 [23•, 28]. Those studies identified several variants with strong associations with T1DM risk including variants implicating the following genes: HLA class II loci, the Insulin gene, PTPN22, CD25, CTLA4, and IFIH1. In addition to these candidate genes, several regions in the genome with multiple possible genes were identified, including 12q13 and 21q22. With increased numbers of subjects, increased collaboration, the widespread use of chips with more variants and improved imputation, the depth and breadth of the variants identified has increased dramatically. Subsequent research has identified likely candidate genes in the two regions 12q13 and 21q22, as well as increased the identified variants to nearly 60. By 2009, these identified variants were being used to predict disease [33, 34] with a receiver operator curve having an area under the curve of greater than 80 %.
Recent work has focused on pathway analysis as well as functional studies of the variants identified [35, 11••, 36, 10, 37, 38•]. Most of the variants identified to date are non-coding SNPs. Genes implicated by proximity to these SNPs in T1DM fall broadly into two categories: (1) pancreas-related and (2) immune-related. Variants in this first group include SNPs in the insulin gene. Variants in this second group include SNPs in many genes, most notably the HLA alleles in the MHC, PTPN22, STAT3, and the IL2RA. It is important to note, in the context of complex and not-entirely understood regulation of gene expression, that the genes implicated are not necessarily the most proximal gene to each SNP In addition to GWAS, several techniques have been used implicate specific genes; e.g., survey of expression quantitative trait loci or interrogation of chromatin state. Within immune-related genes, the function of the variants is mostly still not entirely understood, but the genes implicated can be broadly divided into (a) genes involved in innate immunity (e.g., IFIH1 and CLEC16A), (b) genes altering the strength of receptor signaling (e.g., PTPN22, PTPN2 and SH2B3), (c) genes altering the balance between immunity and tolerance, often via alteration of IL-2 signaling (e.g., IL-2, IL2RA, and PTPN2) and (d) other genes [10, 39, 29, 40–42].
From the perspective of pathway involvement in T1DM, the above provides an appealing rough outline of how T1DM might develop: (a) altered innate immunity gives rise to a change in the response to infection with a virus (i.e., an enterovirus), (b) this response is amplified by altered signaling leading to insulitis, (c) in the context of a bias away from tolerance this insulitis progresses to frank destruction of the islets and T1DM. It is not likely to be quite so simple, as each of the immune genes identified above is expressed in multiple cell types and alters signaling in a specific way in each cell type. Intervention or prevention will require us to have more granular knowledge about mechanism.
For the insulin gene and the HLA class II genes implicated to date, the function of the alterations appears to be either decreased presentation of insulin during negative selection of immune cells (and thus decreased tolerance) or increased activation of T cells by a particularly activating presentation of fragments of insulin. It is not clear how altered presentation insulin is likely to predispose to other diseases, however, it is possible that some of the associations at the MHC alter epitope presentation for other self-antigens and similarly predispose to other autoimmune diseases.
Development of the Immunochip
In addition to the epidemiological evidence detailed briefly above and the single-gene disorders that include T1DM and other autoimmune diseases, comparison of GWAS results (as detailed in the publically available Immunobase) reveals many shared loci between disparate autoimmune diseases. Recognition of the loci shared between autoimmune diseases prompted the Wellcome Trust Case-Control Consortium to motivate the creation of the “Immunochip consortium”. This consortium is composed of leading investigators covering all of the major autoimmune and seronegative diseases (including rheumatoid arthritis, ankylosing spondylitis, systemic lupus erythematosus, T1DM, autoimmune thyroid disease, celiac disease, multiple sclerosis, ulcerative colitis, Crohn's disease, and psoriasis). This consortium selected roughly 3000 SNPs from GWAS results for each disease for deep replication and coverage of putative candidate genes to generate the Immunochip. The Immunochip was generated in a large volume (∼150,000) allowing it to benefit from extremely low cost (∼$39). This chip has allowed and will continue to allow fine-mapping of many alleles across autoimmune diseases that would otherwise not be possible at this cost.
The low-hanging fruit in genomic studies are (1) to increase the number of individuals analyzed to improve the power of these studies and identify novel alleles and (2) to improve the resolution of current studies, or finely map the positive loci already identified to determine the causative variants mechanistically responsible disease. The Immunochip advances both of these aims.
The first publication using the Immunochip in 2011, focusing on celiac disease, revealed (1) several new loci and (2) that several of the individual loci implicated in celiac disease could be divided into two nearby positive signals [14]. The Immunochip has now been used as one element of an investigation into shared genes across many autoimmune diseases, which we will describe in the next section [43, 44••, 15••, 38•, 45, 11••].
Genetic Overlap of T1DM and Other Autoimmune Diseases
Previous GWAS identified SNPs associated with the risk for autoimmune diseases including T1DM. Identified SNPs are highly overlapping between autoimmune diseases (Fig. 1). These SNPs are mostly not the causal variants responsible for functional changes underlying disease but are haplotype tags. That is, these SNPs do not have a functional role, but are in linkage disequilibrium with the variants that are themselves causal. The Immunochip is the result of a large effort to more finely map the causal variants using improved genetic data. Several groups have used the Immunochip, in concert with other kinds of systems-scale or bioinformatic data to identify causal variants within and across several diseases [11••, 44••].
Fig. 1.
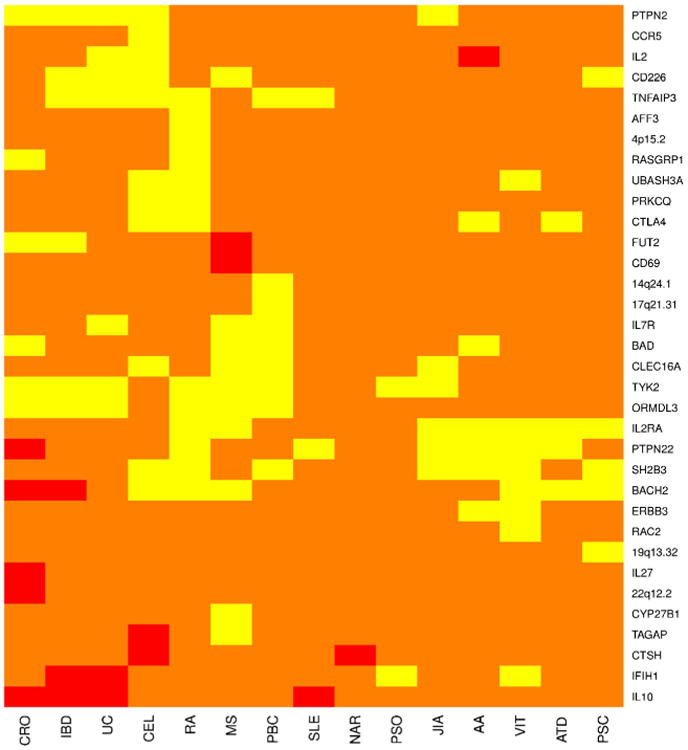
A heatmap showing the shared genetics of 15 autoimmune diseases with T1D at 34 non-MHC loci gathered from information contained at the Immunobase website (www.immunobase.org). All relevant publications can be found in a curated list at the Immunobase website. The regions are named for potential causative genes located at the locus and do not represent the actual causal gene. The regions are defined by the location of the index SNP at the locus and extending out +/–0.1 cM from that location. Yellow indicates that a SNP in this region has odds ratios that are in the same direction for both T1D and the disease in question or the direction of effect was unknown for one the diseases. Red indicates that a SNP in this region has odds ratios that are opposing each other in direction for both diseases. Orange represents a region with no known SNP sharing significance with T1D at this time. For a SNP to be considered shared it must be genome-wide significant (p<5.0×10–8) in one of the diseases and at least suggestive of significance (p<1.0×10– 5) in the other diseases. AA=alopecia areata, ATD=autoimmune thyroid disease, CEL=celiac disease, CRO=Crohn's disease, JIA=juvenile idio-pathic arthritis, MS=multiple sclerosis, PBC=primary biliary cirrhosis, PSO=psoriasis, RA=rheumatoid arthritis, SLE=systemic lupus erythematosis, T1D=type 1 diabetes, UC=ulcerative colitis, IBD=in-flammatory bowel disease, NAR=narcolepsy, PSC=primary sclerosing cholangitis, VIT=vitiligo
One group has developed a method they term probabilistic identification of causal SNPs (PICS) [44••] to identify likely causal variants across autoimmune diseases; this method uses a Baysean approach to integrate Immunochip data with transcriptional data and epigenomic data on active regulatory elements in primary immune cells in resting and stimulated conditions. By comparing these three kinds of data in these primary cells with the variants identified in the Immunochip, they have generated a set of SNPs (PICS SNPs) believed to be highly enriched in causal variants. Overlapping this set of SNPs with data from the Epigenomics Roadmap and Encyclopedia of DNA Elements (ENCODE) project for transcription binding sites and DNase hypersensitivity sites shows that candidate causal SNPs are strongly enriched nearby binding sites for immune-related transcription factors, including NF-kB, SPI1, IRF4, and BATF. However, there is a paucity of recognized transcription factor motifs at the variants themselves; thus, their results suggest that many causal SNPs confer disease risk by modulating transcription factor dependent enhancer activity in a way that is not explainable by current gene regulatory models.
A second group has used a similar approach [11••] to identify likely causal SNPs, by assessing enrichment of Immunochip-identified SNPs for likely causal SNPs also using a Bayesian approach [46•]. They used these SNPs to interrogate chromatin states across 127 tissues derived from the Epigenomics Roadmap and Encyclopedia of DNA Elements (ENCODE) project. This work described a strong enrichment of SNPs in enhancer chromatin states, rather than promoter chromatin states, in immunologically relevant tissues such as the thymus, CD4+ and CD8+ T cells, B cells and CD34+ stem cells. That is, the likely causal variants in autoimmune disease alter enhancer activity outside of recognized transcription factor binding sites.
This work additionally revealed fundamental similarity in the genetic variants underlying diseases that are characterized by auto-antibody positivity (e.g., alopecia areata, autoimmune thyroid disease, celiac disease, multiple sclerosis, primary biliary cirrhosis, primary sclerosing cholangitis, rheumatoid arthritis and systemic lupus erythematosus), and significant differences in the clustering of gene variants between these diseases and diseases that are not characterized by auto-antibody positivity (ankylosing spondylitis, Sjogren's syndrome, psoriasis, Crohn's disease and ulcerative colitis). Specifically, many of the variants that predispose towards auto-antibody positive diseases are protective against auto-antibody negative diseases. Such a dichotomy was predicted by previous results comparing diseases of these two classes [13•], but is nonetheless interesting. This result implies that attempting to prevent T1DM, or prevent progression from insulitis to clinical T1DM by manipulating pathways involving variants that alter risk for auto-antibody-positive diseases and auto-antibody-negative diseases in opposite directions could increase risk for auto-antibody-Negative disease autoimmunity.
Future Directions
To understand the mechanisms underlying the risk-associated variants identified in GWAS we must use other biological data and experimental manipulation to determine function. In addition to the work described above comparing the variants implicated in multiple autoimmune diseases there have been great strides in dissecting the function of these variants or the mechanisms underlying the associations already identified [37, 44••]. Some of this work starts from the pathways implicated in GWAS and attempts to examine the effects of alteration in pathway activity [37], while other work attempts to identify the actual variants responsible for the functional effects (as opposed to the variants identified in GWAS) [44••] to use in further experimentation.
The opposite effect of some gene variants on different autoimmune disease types suggests that in a high-risk context for one kind of disease (e.g., auto-antibody positive T1DM) and a low risk context for the other type (e.g., auto-antibody negative) it might be possible to antagonize the effects of one variant without markedly increasing the risk for the second disease. That is, in a person with high-risk T1DM HLA alleles, mimicking the effect of the IL2RA SNP that only slightly increases risk for ulcerative colitis (and decreases risk for diabetes) might have an overall positive effect.
In a moderate-risk context, the work above using the Immunochip points towards two productive avenues for future T1DM prevention; first, to aim towards manipulating variants in pathways that are limited to T1DM and do not have any effect on risk for auto-antibody-negative disease, and second, to attempt to manipulate pathways where risk in auto-antibody-positive disease and auto-antibody-negative disease are affected in the same direction (e.g., opposing the effect of the identified variants in TYK2 or PTPN2).
Conclusion
Recent work comparing the genetics of T1DM to other autoimmune diseases reveals fundamental similarities and identifies specific pathways for manipulation to possibly prevent disease in high-risk individuals. Using Immunochip data to interrogate other modalities of systems-scale data appears to dramatically improve the identification of causative variants for T1DM and other autoimmune diseases. Analysis of T1DM data in the context of other autoimmune diseases increases our understanding of pathway level data as well as the possible interactions between pathways. The possibility that shared pathways underlie multiple autoimmune diseases may mean that progress in one disease may be easily and rapidly applicable to other autoimmune diseases. Thus this work indicates that the rapid pace of our knowledge accumulation in multiple diseases may soon lead to progress in prevention of several diseases. Some of these same variants may confer risk for non-autoimmune diseases [47•] like lipid disorders and thus, progress in autoimmune disease may also fundamentally improve our ability to understand and treat cancer and heart disease. The recent groundbreaking work outlined above is the prelude to physiologic work [11••, 44••] and will provide the basis to focus this work on causal variants rather than haplotype tags.
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
Footnotes
This article is part of the Topical Collection on Genetics
Compliance with Ethics Guidelines: Conflict of Interest Jeffrey D. Roizen, Jonathan P. Bradfield, and Hakon Hakonarson declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1•.Dabelea D, Mayer-Davis EJ, Saydah S, et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA. 2014;311(17):1778–86. doi: 10.1001/jama.2014.3201. Recent work describing the increasing prevalence of diabetes (both type 1 and type 2) in the pediatric population from 2001–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dassau E, Brown SA, Basu A, et al. Adjustment of open-loop settings to improve closed-loop results in type 1 diabetes: a multi-center randomized trial. J Clin Endocrinol Metab. 2015 doi: 10.1210/jc.2015-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gingras V, Rabasa-Lhoret R, Messier V, et al. Efficacy of dual-hormone artificial pancreas to alleviate the carbohydrate-counting burden of type 1 diabetes: A randomized crossover trial. Diabete Metab. 2015 doi: 10.1016/j.diabet.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Pagliuca FW, Mllman JR, Gurtler M, et al. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159(2):428–39. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khosravi-Maharlooei M, Hajizadeh-Saffar E, Tahamtani Y, et al. Islet transplantation for type 1 diabetes: so close and yet so far away. Eur J Endocrinol. 2015 doi: 10.1530/eje-15-0094. [DOI] [PubMed] [Google Scholar]
- 6.Tomei AA, Villa C, Ricordi C. Development of an encapsulated stem cell-based therapy for diabetes. Expert Opin Biol Ther. 2015:1–16. doi: 10.1517/14712598.2015.1055242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehmann R, Graziano J, Brockmann J, et al. Glycemic control in simultaneous islet-kidney versus pancreas-kidney transplantation in type 1 diabetes: a prospective 13-year follow-up. Diabetes Care. 2015;38(5):752–9. doi: 10.2337/dc14-1686. [DOI] [PubMed] [Google Scholar]
- 8.Park KT, Jun H, Kim MG, et al. Simultaneous pancreas-kidney transplantation from living donor using hand-assisted laparoscopic donor surgery: single-center experience. Transplant Proc. 2015;47(4):1096–8. doi: 10.1016/j.transproceed.2014.10.063. [DOI] [PubMed] [Google Scholar]
- 9.Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nat Rev Genet. 2011;12(11):781–92. doi: 10.1038/nrg3069. [DOI] [PubMed] [Google Scholar]
- 10.Tomlinson MJ, Pitsillides A, Pickin R, et al. Fine mapping and functional studies of risk variants for type 1 diabetes at chromosome 16p13.13. Diabetes. 2014;63(12):4360–8. doi: 10.2337/db13-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11••.Onengut-Gumuscu S, Chen WM, Burren O, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet. 2015;47(4):381–6. doi: 10.1038/ng.3245. Recent study that used the overlap of finely mapped SNPs and ENCODE data to identify likely causal SNPs for T1DM and other autoimmune diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradfield JP, Qu HQ, Wang K, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011;7(9):e1002293. doi: 10.1371/journal.pgen.1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13•.Wang K, Baldassano R, Zhang H, et al. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum Mol Genet. 2010;19(10):2059–67. doi: 10.1093/hmg/ddq078. Early comparative study that identified oppositely oriented associations in T1DM and inflammatory bowel disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trynka G, Hunt KA, Bockett NA, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43(12):1193–201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15••.Fortune MD, Guo H, Burren O, Schofield E, Walker NM, Ban M, et al. Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nat Genet. 2015 doi: 10.1038/ng.3330. Uses baysean inference to identify likely causal SNPs across several autoimmune diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eaton WW, Rose NR, Kalaydjian A, et al. Epidemiology of autoimmune diseases in Denmark. J Autoimmun. 2007;29(1):1–9. doi: 10.1016/j.jaut.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. N Engl J Med. 2004;350(20):2068–79. doi: 10.1056/NEJMra030158.. [DOI] [PubMed] [Google Scholar]
- 18.Yang S, Fujikado N, Kolodin D, et al. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science. 2015;348(6234):589–94. doi: 10.1126/science.aaa7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aronson R, Gottlieb PA, Christiansen JS, et al. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care. 2014;37(10):2746–54. doi: 10.2337/dc13-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Criswell LA, Pfeiffer KA, Lum RF, et al. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet. 2005;76(4):561–71. doi: 10.1086/429096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 22.Huber A, Menconi F, Corathers S, et al. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: from epidemiology to mechanisms. Endocr Rev. 2008;29(6):697–725. doi: 10.1210/er.2008-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23•.Hakonarson H, Grant SF, Bradfield JP, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448(7153):591–4. doi: 10.1038/nature06010. One of the two first GWAS of T1DM. [DOI] [PubMed] [Google Scholar]
- 24•.Hakonarson H, Qu HQ, Bradfield JP, et al. A novel susceptibility locus for type 1 diabetes on Chr12q13 identified by a genome-wide association study. Diabetes. 2008;57(4):1143–6. doi: 10.2337/db07-1305. Used larger sample size to identify rare risk-associated variant in T1D. [DOI] [PubMed] [Google Scholar]
- 25.Concannon P, Onengut-Gumuscu S, Todd JA, et al. A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes. 2008;57(10):2858–61. doi: 10.2337/db08-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant SF, Qu HQ, Bradfield JP, et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes. 2009;58(1):290–5. doi: 10.2337/db08-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39(7):857–64. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrett JC, Clayton DG, Concannon P, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–7. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qu HQ, Bradfield JP, Li Q, et al. In silico replication of the genome-wide association results of the Type 1 Diabetes Genetics Consortium. Hum Mol Genet. 2010;19(12):2534–8. doi: 10.1093/hmg/ddq133. [DOI] [PubMed] [Google Scholar]
- 31.Cooper JD, Smyth DJ, Smiles AM, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet. 2008;40(12):1399–401. doi: 10.1038/ng.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallace C, Smyth DJ, Maisuria-Armer M, et al. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet. 2010;42(1):68–71. doi: 10.1038/ng.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei Z, Wang K, Qu HQ, et al. From disease association to risk assessment: an optimistic view from genome-wide association studies on type 1 diabetes. PLoS Genet. 2009;5(10):e1000678. doi: 10.1371/journal.pgen.1000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eleftherohorinou H, Wright V, Hoggart C, et al. Pathway analysis of GWAS provides new insights into genetic susceptibility to 3 inflammatory diseases. PLoS One. 2009;4(11):e8068. doi: 10.1371/journal.pone.0008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tuller T, Atar S, Ruppin E, et al. Common and specific signatures of gene expression and protein-protein interactions in autoimmune diseases. Genes Immun. 2013;14(2):67–82. doi: 10.1038/gene.2012.55. [DOI] [PubMed] [Google Scholar]
- 36.Ferreira RC, Simons HZ, Thompson WS, et al. IL-21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia. 2015;58(4):781–90. doi: 10.1007/s00125-015-3509-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dendrou CA, Plagnol V, Fung E, et al. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat Genet. 2009;41(9):1011–5. doi: 10.1038/ng.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38•.Guo H, Fortune MD, Burren OS, et al. Integration of disease association and eQTL data using a Bayesian colocalisation approach highlights six candidate causal genes in immune-mediated diseases. Hum Mol Genet. 2015;24(12):3305–13. doi: 10.1093/hmg/ddv077. Integrates eQTL data to identify likely causal variants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smyth DJ, Cooper JD, Bailey R, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38(6):617–9. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 40.Bell GI, Horita S, Karam JH. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes. 1984;33(2):176–83. doi: 10.2337/diab.33.2.176. [DOI] [PubMed] [Google Scholar]
- 41.Bottini N, Musumeci L, Alonso A, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36(4):337–8. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 42.Lowe CE, Cooper JD, Brusko T, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39(9):1074–82. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 43.Eyre S, Bowes J, Diogo D, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. 2012;44(12):1336–40. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44••.Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–43. doi: 10.1038/nature13835. Generated and used an algorhythm (PICS) to identify likely causal variants in automimmune disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hrdlickova B, Westra HJ, Franke L, Wijmenga C. Celiac disease: moving from genetic associations to causal variants. Clin Genet. 2011;80(3):203–313. doi: 10.1111/j.1399-0004.2011.01707.x. [DOI] [PubMed] [Google Scholar]
- 46•.Wellcome Trust Case Control C. Maller JB, McVean G, et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet. 2012;44(12):1294–301. doi: 10.1038/ng.2435. This is an early paper looking at specific shared loci underlying auto-immunoe disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Andreassen OA, Desikan RS, Wang Y, et al. Abundant genetic overlap between blood lipids and immune-mediated diseases indicates shared molecular genetic mechanisms. PLoS One. 2015;10(4):e0123057. doi: 10.1371/journal.pone.0123057. Identifies common loci shared between autoimmune disease and lipid dysregulation. [DOI] [PMC free article] [PubMed] [Google Scholar]