

Abstract
Here we report the draft genome sequence of Chitinophaga costai A37T2T (=CIP 110584T, =LMG 27458T), which was isolated from the endophytic community of Pinus pinaster tree. The total genome size of C. costaii A37T2T is 5.07 Mbp, containing 4204 coding sequences. Strain A37T2T encoded multiple genes likely involved in cellulolytic, chitinolytic and lipolytic activities. This genome showed 1145 unique genes assigned into 109 Cluster of Orthologous Groups in comparison with the complete genome of C. pinensis DSM 2588T. The genomic information suggests the potential of the strain A37T2T to interact with the plant metabolism. As there are only a few bacterial genomes related to Pine Wilt Disease, this work provides a contribution to the field.
Keywords: Chitinophaga costaii A37T2, Cellulase, Chitinase, Genome sequence
Introduction
The genus 10.1601/nm.8334 belongs to the family Chtiniphagaceae (phylum 10.1601/nm.7927) alongside with the genera 10.1601/nm.26356, 10.1601/nm.24507, 10.1601/nm.10262, 10.1601/nm.25037, 10.1601/nm.25347, 10.1601/nm.14773, 10.1601/nm.14303, 10.1601/nm.26907, 10.1601/nm.19904, 10.1601/nm.11311, 10.1601/nm.22146, 10.1601/nm.14308, 10.1601/nm.24623, 10.1601/nm.21260, 10.1601/nm.14305, 10.1601/nm.10412, 10.1601/nm.10256, 10.1601/nm.28007, 10.1601/nm.14937, 10.1601/nm.13147, 10.1601/nm.11309, 10.1601/nm.24621, 10.1601/nm.10067, 10.1601/nm.25337 and Vibriomonas. The genus 10.1601/nm.8334 is widely distributed in the environment and strains of this genus have been isolated from pine trees, soil, rhizosphere soil, roots, vermicompost and weathered rock [1]. Twenty-four species belonging to the genus 10.1601/nm.8334 have been described [2], and only the type species of the genus 10.1601/nm.8335 has the complete genome sequenced [3].
Pinus pinaster trees from Central Portugal present a diverse endophytic microbial community. Strain A37T2T was isolated as part of the endophytic microbiome of pine trees affected by Pine Wilt Disease (PWD) which is a world devastating disease, consequence of Bursaphelenchus xylophilus colonization in pine trees [4]. Here, we show the second genome of the genus 10.1601/nm.8334, a draft genome of 10.1601/nm.25336 A37T2T, previously isolated as endophyte of Pinus pinaster affected by PWD [1].
Organism information
Classification and features
The type strain A37T2T (=10.1601/strainfinder?urlappend=%3Fid%3DCIP+110584 T =10.1601/strainfinder?urlappend=%3Fid%3DLMG+27458 T), was isolated from tree trunk of a Pinus pinaster tree affected by PWD and it described as 10.1601/nm.25336 (family 10.1601/nm.14400, phylum 10.1601/nm.7927) [1]. It was Gram-stain-negative, facultative anaerobic, non-motile, formed rod-shaped cells, 0-5-1 μm in diameter and 1-8 μm in length after 48 h on R2A agar media (Fig. 1). Showed capacity to grow on R2A agar medium at 15-45 °C (optimum, 26-30 °C), at pH 5.5-8.0 (optimum, pH 7) and supplemented with up to 1% (w/v) NaCl (optimum without NaCl). The major fatty acids (>25%) showed by the strain A37T2T are saturated iso-C15: 0 and unsaturated C16: 1 ω5c. The major polar lipids were identified as phosphatidylethanolamine, two unidentified aminophospholipids and one unidentified lipid. No glycolipid was detected. The menaquinone 7 (MK-7) was shown as the major respiratory lipoquinone. The determined DNA G + C content of the 10.1601/nm.25336 A37T2T was 46.6 mol%. Key features of this microorganism are summarized in Table 1. A phylogenetic tree based on the 16S rRNA gene sequence of this strain and its closest relative members are given in Fig. 2. The sequences were aligned by SINA (v1.2.9) using the SILVA SEED as reference alignment [5]. Sequences were included in 16S rRNA-based Living Tree Project (LTP) release 115 database [6] by parsimony implemented in the ARB software package version 5.5 [7]. Evolutionary distances were calculated [8] and phylogenetic dendrograms were constructed using the neighbor-joining [9] and Randomized Axelerated Maximum Likelihood (RAxML) method with GTRGAMMA model [10] included in the ARB software [7]. Trees topologies were evaluated by performing bootstrap analysis [11] of 1000 data sets by using ARB software package.
Fig. 1.
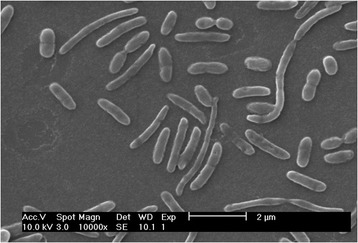
Scanning electron micrograph of C. costaii A37T2T after 48 h of growth on R2A agar plates at 30 °C
Table 1.
Classification and general features of Chitinophaga costaii A37T2T according to the MIGS recommendations [26]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [27] | |
| Phylum Bacteroidetes | TAS [28, 29] | ||
| Class Sphingobacteriia | TAS [28, 30] | ||
| Order Sphingobacteriales | TAS [28, 31] | ||
| Family Chitinophagaceae | TAS [32] | ||
| Genus Chitinophaga | TAS [33] | ||
| Species Chitinophaga costaii | TAS [1] | ||
| Type strain: A37T2T (=CIP 110584T, =LMG 27458T) | |||
| Gram stain | Negative | TAS [1] | |
| Cell shape | Rod | TAS [1] | |
| Motility | Non-motile | TAS [1] | |
| Sporulation | Not reported | NAS | |
| Temperature range | 15-45 °C | TAS [1] | |
| Optimum temperature | 26-30 °C | TAS [1] | |
| pH range; Optimum | 5.5-8.0; 7 | TAS [1] | |
| Carbon source | Glucose | TAS [1] | |
| MIGS-6 | Habitat | Endophyte of Pinus pinaster tree | TAS [1] |
| MIGS-6.3 | Salinity | 1.0% NaCl (w/v) | TAS [1] |
| MIGS-22 | Oxygen requirement | Facultative anaerobic | TAS [1] |
| MIGS-15 | Biotic relationship | Free-living | TAS [1] |
| MIGS-14 | Pathogenicity | Non-pathogen | NAS |
| MIGS-4 | Geographic location | Portugal | TAS [1] |
| MIGS-5 | Sample collection | July, 2009 | NAS |
| MIGS-4.1 | Latitude | 40.2962266 | NAS |
| MIGS-4.2 | Longitude | −7.9207357 | NAS |
| MIGS-4.4 | Altitude | 217 m | NAS |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [34]
Fig. 2.
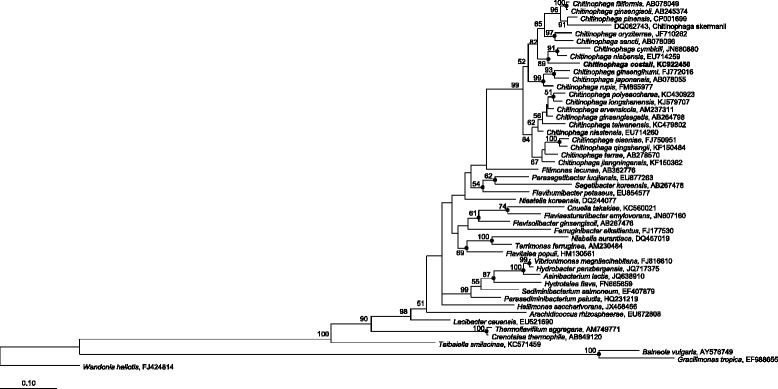
Phylogenetic tree based on a comparison of the 16S rRNA gene sequence of strain A37T2T and the other type strains within the family Chitinophagaceae. The tree was created using the maximum likelihood method (RAxML). The numbers on the tree indicate the percentages of bootstrap sampling, derived from 1000 replications; values below 50% are not shown. Symbol (•) indicates node branches conserved when the tree was reconstructed using the neighbor-joining method. The isolate characterized in this study is indicated in bold. Scale bar, 1 inferred nucleotide substitution per 100 nucleotides
Genome sequencing information
Genome project history
This Whole Genome Shotgun project has been deposited at ENA under the accession numbers FMAR01000001-FMAR01000056 and in the Integrated Microbial Genomes database (IMG) with Biosample ID SAMN05216457 [12]. The genome sequencing of this organism is part of the Genomic Encyclopedia of Bacteria and Archaea [13], 1000 Microbial Genomes project, phase III (KMG-III) [14], at the U.S. Department of Energy, Joint Genome Institute (JGI). The project information and its association with the MIGS is summarized in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Draft |
| MIGS-28 | Libraries used | Illumina Regular Fragment, 300 bp, Tubes |
| MIGS 29 | Sequencing platforms | Illumina HiSeq 2500-1 TB |
| MIGS 31.2 | Fold coverage | 297.2 |
| MIGS 30 | Assemblers | SPAdes |
| MIGS 32 | Gene calling method | NCBI Prokaryotic Genome Annotation Pipeline |
| Locus Tag | GA0116948 | |
| Genbank ID | FMAR00000000 | |
| GenBank Date of Release | August 3, 2016 | |
| GOLD ID | Gp0139259 | |
| BIOPROJECT | PRJNA322901 | |
| MIGS 13 | Source Material Identifier | A37T2T |
| Project relevance | GEBA-KMG |
Growth conditions and genomic DNA preparation
The strain A37T2T was grown on R2A agar media at 30 °C during 48 h and its genomic DNA was extracted using the E.Z.N.A. Bacterial DNA Kit (Omega Bio-Tek, Norcross, GA, USA) according to the manufacturer’s instructions.
Genome sequencing and assembly
The draft genome of 10.1601/nm.25336 A37T2T was generated at the DOE Joint Genome Institute (JGI) using the Illumina technology [15]. An Illumina 300 bp insert standard shotgun library was constructed and sequenced using the Illumina HiSeq–2500 1 TB platform, generating 9,965,394 reads totaling 1494.8 Mbp. All general aspects of library construction and sequencing performed at the JGI can be found at [16]. All raw Illumina sequence data was filtered using BBDuk [17], which removes known Illumina artifacts and PhiX. Reads with more than one “N” or with quality scores (before trimming) averaging less than 8 or reads shorter than 51 bp (after trimming) were discarded. Remaining reads were mapped to masked versions of human, cat and dog references using BBMAP [17] and discarded if identity exceeded 95%. Sequence masking was performed with BBMask [17]. Following steps were then performed for assembly: (1) artifact filtered Illumina reads were assembled using SPAdes (version 3.6.2) [18]; (2) assembled contigs were discarded if length was <1 kbp. Parameters for the SPAdes assembly were ––cov–cutoff auto ––phred–offset 33 –t 8 –m 40 ––careful –k 25,55,95 ––12.
Genome annotation
Protein-coding genes were identified using Prodigal [19], as part of the DOE-JGI genome annotation pipeline [20]. Additional gene prediction analysis and manual functional annotation were performed within the Integrated Microbial Genomes Expert Review system (IMG-ER), which provides tools for analyzing and reviewing the structural and functional annotations of genomes in a comparative context [12, 21]. Genome annotation procedures are detailed in Markowitz et al. [12] and references therein. Briefly, the predicted CDSs were translated and used to search the NCBI nonredundant database, UNIProt, TIGRFam, Pfam, KEGG, COG and InterPro databases. Transfer RNA genes were identified using the tRNAScan-SE tool and other non-coding RNAs were found using INFERNAL. Ribosomal RNA genes were predicted using hmmsearch against the custom models generated for each type of rRNA.
Genome properties
The draft genome sequence of 10.1601/nm.25336 strain A37T2T comprised 5,074,440 bp, based on 1494.8 Mbp of Illumina data with a mapped coverage of 297.2-fold of the genome. The final draft assembly contained 56 contigs in 56 scaffolds with more than 1052 bp. The G + C content was 47.6%. The genome encoded 4204 putative coding sequences (CDSs) (Table 3). Fifty four % of the CDSs, corresponding to 2284 proteins, could be assigned to Cluster of Orthologous Groups (COG) families [22] (Table 4). The draft genome sequence contained four ribosomal RNAs and 50 tRNAs loci (Table 3).
Table 3.
General genome features of Chitinophaga costaii A37T2T
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 5,074,440 | 100.00 |
| DNA coding (bp) | 4,431,743 | 87.33 |
| DNA G + C (bp) | 2,413,598 | 47.56 |
| DNA scaffolds | 56 | 100.00 |
| Total genes | 4274 | 100.00 |
| Protein coding genes | 4204 | 98.36 |
| RNA genes | 70 | 1.64 |
| Genes in internal clusters | 824 | 19.28 |
| Genes with function prediction | 3041 | 71.15 |
| Genes assigned to COGs | 2284 | 53.44 |
| Genes with Pfam domains | 1976 | 61.75 |
| Genes with signal peptides | 651 | 15.23 |
| Genes with transmembrane helices | 972 | 22.74 |
| CRISPR repeats | 3 | 0.00 |
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | %age | Description |
|---|---|---|---|
| J | 186 | 7.42 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.00 | RNA processing and modification |
| K | 215 | 8.58 | Transcription |
| L | 90 | 3.50 | Replication, recombination and repair |
| D | 19 | 0.76 | Cell cycle control, Cell division, chromosome partitioning |
| V | 98 | 3.91 | Defense mechanisms |
| T | 114 | 4.55 | Signal transduction mechanisms |
| M | 211 | 8.42 | Cell wall/membrane biogenesis |
| N | 12 | 0.48 | Cell motility |
| U | 20 | 0.80 | Intracellular trafficking and secretion |
| O | 136 | 5.42 | Posttranslational modification, protein turnover, chaperones |
| C | 126 | 5.03 | Energy production and conversion |
| G | 165 | 6.58 | Carbohydrate transport and metabolism |
| E | 196 | 7.82 | Amino acid transport and metabolism |
| F | 69 | 2.75 | Nucleotide transport and metabolism |
| H | 141 | 5.62 | Coenzyme transport and metabolism |
| I | 125 | 4.99 | Lipid transport and metabolism |
| P | 150 | 5.98 | Inorganic ion transport and metabolism |
| Q | 70 | 2.79 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 248 | 9.89 | General function prediction only |
| S | 104 | 4.15 | Function unknown |
| - | 1990 | 46.56 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
The Average Nucleotide Identity between 10.1601/nm.25336 A37T2T and 10.1601/nm.8335 10.1601/strainfinder?urlappend=%3Fid%3DDSM+2588 T was 70.9 based on 1593 of total Bidirectional Best Hits, using MiSI [23]. Figure 3 shows the circular graph of the genome of 10.1601/nm.25336 A37T2T query to the only available complete genome of the genus 10.1601/nm.8334, 10.1601/nm.8335 10.1601/strainfinder?urlappend=%3Fid%3DDSM+2588 T [2].
Fig. 3.
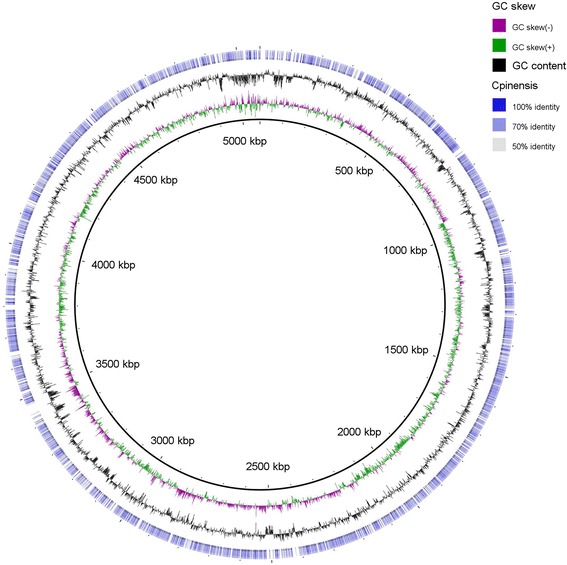
The genome of Chitinophaga costaii A37T2T. From outside to the center: genes of genome of C. pinensis DSM 2588T and its similarity with the genome of C. costaii (50-100%), GC content of C. costaii A37T2T, GC skew of C. costaii A37T2T, genome of C. costaii A37T2T
The comparison between the draft genome of 10.1601/nm.25336 A37T2T and the complete genome of 10.1601/nm.8335 10.1601/strainfinder?urlappend=%3Fid%3DDSM+2588 T showed 1145 unique genes only present in the genome of 10.1601/nm.25336 A37T2T and 3493 unique genes only present in the genome of 10.1601/nm.8335 10.1601/strainfinder?urlappend=%3Fid%3DDSM+2588 T. Focused on the unique genes present on the genome of strain A37T2T it was possible to assigned 109 COG, summarized in Table 5.
Table 5.
Unique Cluster Orthologous Groups present in the genome of C. costaii A37T2T
| Catergory Code | Catergory | COG ID |
|---|---|---|
| C | Energy production and conversion | COG0280, COG0374, COG0680, COG1740 |
| E | Amino acid transport and metabolism | COG1027, COG1586, COG2355, COG3104 |
| F | Nucleotide transport and metabolism | COG0027 |
| G | Carbohydrate transport and metabolism | COG0021, COG0058, COG0588, COG0662, COG0837, COG1080, COG1803, COG1925, COG2079, COG2893, COG3444, COG3716, COG3934 |
| H | Coenzyme transport and metabolism | COG0561, COG1056, COG2091, COG2227, COG2329 |
| I | Lipid transport and metabolism | COG0671, COG0821, COG2246 |
| J | Translation, ribosomal structure and biogenesis | COG0060, COG0255, COG0257, COG0267, COG0268, COG0333, COG4680 |
| K | Transcription | COG1476, COG4933 |
| L | Replication, recombination and repair | COG0863, COG1722 |
| M | Cell wall/membrane/envelope biogenesis | COG1083, COG1922, COG2089, COG2829, COG2982, COG3511, COG3637 |
| O | Posttranslational modification, protein turnover, chaperones | COG0068, COG0298, COG0309, COG0409 |
| P | Inorganic ion transport and metabolism | COG0428, COG1218, COG1230, COG1416, COG4772 |
| Q | Secondary metabolites biosynthesis, transport and catabolism | COG2130, COG2162, COG3733, COG4242 |
| R | General function prediction only | COG0312, COG0375, COG0429, COG0457, COG1062, COG1373, COG2320, COG3153, COG3488, COG4674, COG0561, COG2130, COG4242 |
| S | Function unknown | COG0393, COG1286, COG2442, COG2962, COG3219, COG3247, COG3310, COG3361, COG3461, COG3477, COG3487, COG3489, COG3528, COG3548, COG3918, COG3943, COG4487, COG4700, COG4859, COG4924 |
| T | Signal transduction mechanisms | COG0517, COG2184, COG2203, COG3292, COG1925 |
| U | Intracellular trafficking, secretion, and vesicular transport | COG1272, COG1826, COG3451 |
| V | Defense mechanisms | COG0286, COG0610, COG0732, COG3512, COG3513, COG4823, COG5499 |
| X | Mobilome: prophages, transposons | COG3385, COG3436, COG3600, COG3654 |
Insights from the genome sequence
The draft genome sequence of 10.1601/nm.25336 A37T2T carries multiple genes involved in cellulolytic activity, including one gene encoding the enzyme cellulase (SCC15587) and six genes encoding for β-glucosidase (SCB82491, SCB92249, SCB95191, SCC15475, SCC57293, SCC61957), which might be involved in cellulose degradation in the environment and in biotechnological processes [24]. As expected for this genus, four genes encoding chitinases (SCC19468, SCC19522, SCC23114, SCC34676) were found. Six genes encoded lysophospholipase L1, including representatives of both of size groups, i.e. less than 300aa (SCB77875, SCC28514, SCC37316, SCC54197) and less than 500aa (SCB98645, SCC50813). Moreover, the genome of strain A37T2T encoded 1-aminocyclopropane-1-carboxylate deaminase (SCB80758), a hydrolase that might be involved in lowering ethylene levels in the plant [25]. In summary, the genome sequence suggested multiple potentials for the strain to interact with the plant metabolism.
Conclusions
This work contributed to the knowledge of the genome sequence of the type species of 10.1601/nm.25336 A37T2T (=10.1601/strainfinder?urlappend=%3Fid%3DCIP+110584 T, =10.1601/strainfinder?urlappend=%3Fid%3DLMG+27458 T), an endophyte of P. pinaster affected by PWD. The genome encoded multiple genes involved in cellulolytic activity and the sequence provided insights into the role of bacteria in PWD. As there are only a few bacterial genomes related to PWD, this work provides a contribution to this field.
Acknowledgments
Funding
We thank to Ana Paula Piedade for SEM analysis. This work was supported by CEMMPRE and by Fundação para a Ciência e a Tecnologia (FCT) under the project UID/EMS/00285/2013. D.N.P. was supported by FCT, postdoctoral fellowship SFRH/BPD/100721/2014. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Abbreviations
- PWD
Pine wilt disease
- PWN
Pinewood nematode
Authors’ contributions
DNP isolated the strain, extracted the DNA, performed laboratory experiments, analyzed all the data, and with PVM wrote the manuscript. WBW, NS, TW and NCK did the genome sequencing, assembly and annotation. WBW, NS, TW and NCK revise the manuscript. All the authors read and approved the final manuscript.
Competing interests
The authors have no competing of interests to declare.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Proença DN, Nobre MF, Morais PV. Chitinophaga costaii sp. nov., an endophyte of Pinus pinaster, and emended description of Chitinophaga niabensis. Int J Syst Evol Microbiol. 2014;64:1237–1243. doi: 10.1099/ijs.0.053454-0. [DOI] [PubMed] [Google Scholar]
- 2.List of prokaryotic names with standing in nomenclature. http://www.bacterio.net. Accessed 2 Dec 2016.
- 3.Glavina Del Rio T, Abt B, Spring S, Lapidus A, Nolan M, Tice H, et al. Complete genome sequence of Chitinophaga pinensis type strain (UQM 2034T) Stand Genomic Sci. 2010;2:87–95. doi: 10.4056/sigs.661199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Proença DN, Grass G, Morais PV. Understanding pine wilt disease: roles of the pine endophytic bacteria and of the bacteria carried by the disease-causing pinewood nematode. Microbiology 2016;0:1–20. [DOI] [PMC free article] [PubMed]
- 5.Pruesse E, Peplies J, Glöckner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.“The All-Species Living Tree” Project. http://www.arb-silva.de/projects/living-tree. Accessed 15 Jan 2016.
- 7.Ludwig W, Strunk O, Westram R, Richter L, Meier H. Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mamm. Protein Metab. New York: Academic Press; 1969. pp. 21–132. [Google Scholar]
- 9.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 10.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 11.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution (N Y) 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 12.Markowitz VM, Chen IMA, Chu K, Szeto E, Palaniappan K, Pillay M, et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 2014;42:568–573. doi: 10.1093/nar/gkt919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kyrpides NC, Hugenholtz P, Eisen JA, Woyke T, Göker M, Parker CT, et al. Genomic encyclopedia of bacteria and Archaea: sequencing a myriad of type strains. PLoS Biol. 2014;12:1–7. doi: 10.1371/journal.pbio.1001920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitman WB, Woyke T, Klenk H-P, Zhou Y, Lilburn TG, Beck BJ, et al. Genomic encyclopedia of bacterial and Archaeal type strains, phase III: the genomes of soil and plant-associated and newly described type strains. Stand Genomic Sci. 2015;10:26. doi: 10.1186/s40793-015-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett S. Solexa Ltd Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 16.Joint Genome Institute. http://www.jgi.doe.gov. Accessed 2 Jan 2017.
- 17.BBMap short read aligner, and other bioinformatic tools. http://sourceforge.net/projects/bbmap. Accessed 15 Apr 2016.
- 18.Bankevich A, Nurk S, Antipov D, Gurevich A a, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huntemann M, Ivanova NN, Mavromatis K, Tripp HJ, Paez-Espino D, Palaniappan K, et al. The standard operating procedure of the DOE-JGI microbial genome annotation pipeline (MGAP v.4) Stand Genomic Sci. 2015;10:86. doi: 10.1186/s40793-015-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen I-MA, Markowitz VM, Palaniappan K, Szeto E, Chu K, Huang J, et al. Supporting community annotation and user collaboration in the integrated microbial genomes (IMG) system. BMC Genomics BMC Genomics. 2016;17:307. doi: 10.1186/s12864-016-2629-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC, et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015;43:6761–6771. doi: 10.1093/nar/gkv657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adrio JL, Demain AL. Microbial enzymes: tools for biotechnological processes. Biomol Ther. 2014;4:117–139. doi: 10.3390/biom4010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glick BR. Plant growth-promoting bacteria: mechanisms and applications. Scientifica (Cairo) 2012;2012:1–15. doi: 10.6064/2012/963401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Editor L. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2012;62:1–4. doi: 10.1099/ijs.0.039487-0. [DOI] [PubMed] [Google Scholar]
- 29.Krieg N, Ludwig W, Euzéby J, Whitman W, et al. Phylum XIV. Bacteroidetes phyl. Nov. In: Krieg N, Staley J, Brown D, Hedlund B, Paster B, Ward N, et al., editors. Bergey’s man. Syst. Bacteriol. Second Edi. New York: Springer; 2011. p. 25. [Google Scholar]
- 30.Kämpfer P, Class III, et al. Sphingobacteriia class. Nov. In: Krieg NR, Staley J, Brown D, Hedlund B, Paster B, Ward N, et al., editors. Bergey’s man. Syst. Bacteriol. Second Edi. New York: Springer; 2011. p. 330. [Google Scholar]
- 31.Kämpfer P. Order I. Sphingobacteriales ord. Nov. In: Krieg N, Staley J, Brown D, Hedlund B, Paster B, Ward N, et al., editors. Bergey’s Man. Syst. Bacteriol. Second ed. New York: Springer; 2011. p. 330.
- 32.Kämpfer P, Lodders N, Falsen E. Hydrotalea flava gen. Nov., sp. nov., a new member of the phylum Bacteroidetes and allocation of the genera Chitinophaga, Sediminibacterium, Lacibacter, Flavihumibacter, Flavisolibacter, Niabella, Niastella, Segetibacter, Parasegetibacter, Terrimonas, Fer. Int J Syst Evol Microbiol. 2011;61:518–523. doi: 10.1099/ijs.0.023002-0. [DOI] [PubMed] [Google Scholar]
- 33.Sangkhobol V, Skerman VBD. Chitinophaga, a new genus of chitinolytic myxobacteria. Int J Syst Bacteriol. 1981;31:285–293. doi: 10.1099/00207713-31-3-285. [DOI] [Google Scholar]
- 34.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]