

Abstract
Collectively, retinal diseases, including age-related macular degeneration, retinitis pigmentosa, and diabetic retinopathy, result in severe vision impairment worldwide. The absence and/or limited availability of successful drug therapies for these blinding disorders necessitates further understanding their pathobiology and identifying new targetable signaling pathways. Nuclear receptors are transcription regulators of many key aspects of human physiology, as well as pathophysiology, with reported roles in development, aging, and disease. Some of the pathways regulated by nuclear receptors include, but are not limited to, angiogenesis, inflammation, and lipid metabolic dysregulation, mechanisms also important in the initiation and development of several retinal diseases. Herein, we present an overview of the biology of three diseases affecting the posterior eye, summarize a growing body of evidence that suggests direct or indirect involvement of nuclear receptors in disease progression, and discuss the therapeutic potential of targeting nuclear receptors for treatment.
Keywords: nuclear receptors, retinal diseases, age-related macular degeneration, diabetic retinopathy, retinitis pigmentosa
Introduction
The human nuclear receptor (NR) superfamily is composed of 48 evolutionarily related transcription factors, which respond to endogenous ligands, including steroid hormones, fatty acids, bile acids, lipophilic vitamins, and cholesterol metabolites, and exogenous ligands, such as drugs and toxins.1–4 Functionally, NRs are critical regulators of a wide range of physiologic and developmental pathways. The myriad molecular pathways modulated by NRs include, but are not limited to, inflammation, lipid metabolism, apoptosis, extracellular matrix regulation, energy metabolism, and angiogenesis.5–7 These pathways are of particular interest to vision scientists, as these functions are also compromised in several retinal diseases, such as diabetic retinopathy (DR), age-related macular degeneration (AMD), and retinitis pigmentosa (RP)8–11 (Fig. 1). Treatment options currently available for the above-listed vision-impairing retinal diseases are either quite limited or unavailable. This unmet need classifies identification of new signaling pathways that contribute to initiation or progression of retinal diseases and potential therapeutic targets as a top priority. Of relevance is that many members of the NR superfamily have well-characterized ligands and serve as therapeutic targets for the development of drugs to treat diseases including cancer, diabetes, atherosclerosis, inflammation, and endocrine disorders.5,6,12,13 Accordingly, identification of NR-driven pathways altered in retinal diseases, for which there are drugs already available, would be highly advantageous to potentially decreasing the timeline to treatment in man for ocular indications. In light of the overlap between pathogenic pathways of several retinal disorders with other NR-regulated diseases, herein we provide an overview of a growing body of evidence that suggests NRs may play an overlooked role in the development of pathologies associated with retinal diseases. We begin with a brief overview of the NR superfamily. We follow with describing the known pathobiologies associated with several diseases affecting the posterior eye, including RP, DR, and AMD. Finally, we summarize recent progress in identifying the contribution of various NRs, either directly or indirectly, to the biology of these diseases. Collectively, these studies provide promising support for consideration of therapeutic targeting of this class of receptors for treatment of various retinal disorders.
Figure 1.
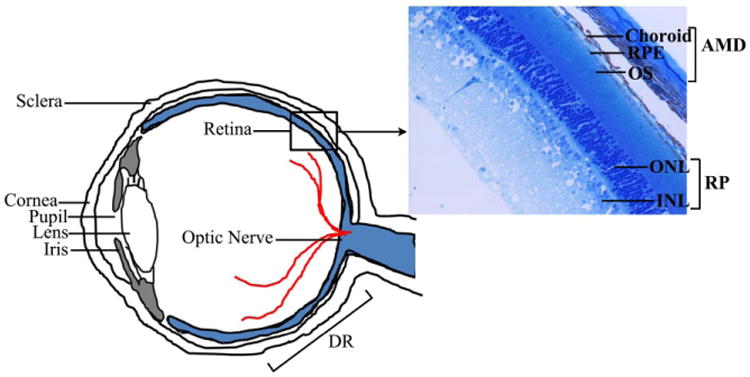
Schematic view of a cross section of the human eye highlighting the various cell types affected in the retinal diseases AMD, RP, and DR. INL, inner nuclear layer; ONL, outer nuclear layer; OS, outer segment; RPE, retinal pigment epithelium.
Nuclear Receptor Superfamily
NRs are primarily defined as ligand-regulated transcription factors that modulate target gene expression. The first NRs cloned were the glucocorticoid receptor (GR; NR3C1) and the estrogen receptor (ERα; NR3A1) in 1985 and 1986, respectively.14,15 This was closely followed by the discovery of receptors for mineralocorticoids, progesterone, androgens, and fat-soluble vitamins A and D.4 Comparison of sequences led to identification of an evolutionary conserved template and domain structure for the members of the NR superfamily. Overall, the conserved NR structure is modular and consists of six subregions (Fig. 2A, domains A–F). However, the sequence homologies of these subregions vary within the NR family.1,13 The amino terminus (also known as the A/B region) is the most divergent among receptors, and it contains a hormone-independent transactivation domain known as activation function 1 (AF-1), which is recognized by coactivators and/or other transcription factors. The region C is the most conserved domain, has two zinc-finger motifs, and contains a DNA-binding domain (DBD). Noteworthy are two exceptions: the dosage-specific sex reversal-adrenal hypoplasia congenital critical region on the X chromosome (DAX1; NR0B1) and short heterodimeric partner (SHP; NR0B2), both of which lack the DBD. The D region, which has a low degree of conservation, forms a “hinge domain,” as it is localized between the DBD and the ligand-binding domain (LBD). The E region contains the ligand-dependent transactivation domain (AF-2) and is well conserved between the various family members, but diverges sufficiently to ensure ligand selectivity. The F region is further toward the carboxyl terminus, present in only a few receptors to date, and its role remains unclear in NR function. Overall, the NR superfamily can functionally be categorized into three main groups on the basis of their ligand- and DNA-binding properties.16 The NRs of the first category, the classical steroid hormone receptors, activate transcription by binding to the response element located within the promoter region of their target genes (Fig. 2B). The second category of NRs is the orphan receptors. They are named as such because they have the conserved domain structure of NRs, but their endogenous ligands have not been identified to date. Orphan receptors for which the ligand has been identified make up the third family of NRs and are referred to as adopted orphan receptors (Table 1).
Figure 2.
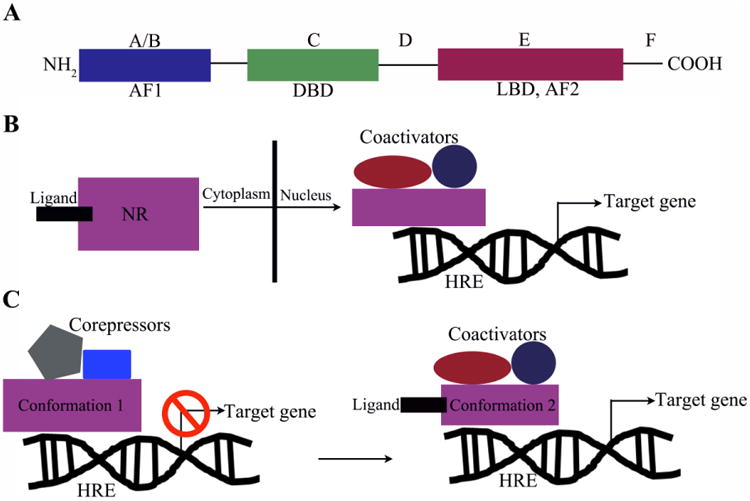
NR domain structure and mechanism of action. (A) NRs exhibit a conserved modular domain structure with an N-terminal conserved AF-1 region (A/B), followed by a zinc-finger DBD (C region), a hinge domain (D region), an LBD containing the AF-2 region (E region), and a C-terminal F region for some receptors. (B) Mechanistically, NRs are regulated by small-molecule ligands, which generally cause a conformational change, allowing binding of the receptor to the hormone response element and coregulator proteins. This, in turn, leads to transcription of target genes. (C) Alternatively, NRs can exist within the nucleus in a conformation (1) bound by corepressors, which blocks the transcription of target genes. Ligand binding induces a conformational change (2), which allows for binding of coactivators leading to transcription of target genes. AF, activation function; HRE, hormone response element.
Table 1.
Human Nuclear Receptors.
| Subfamily | Name | Nomenclature | Ligand |
|---|---|---|---|
| Steroid hormone subfamily | Estrogen receptor | ERα (NR3A1) | Estradiol-17β, tamoxifen |
| ERβ (NR3A2) | Estradiol-17P | ||
| Estrogen receptor–related receptor | ERRα (NR3B1) | Orphan | |
| ERRβ (NR3B2) | 4-OH tamoxifen | ||
| ERRγ (NR3B3) | 4-OH tamoxifen | ||
| Glucocorticoid receptor | GR (NR3C1) | Cortisol dexamethasone | |
| Mineralocorticoid receptor | MR (NR3C2) | Aldosterone, spirolactone | |
| Progesterone receptor | PR (NR3C3) | Progesterone | |
| Androgen receptor | AR (NR3C4) | Testosterone | |
| Farnesoid X receptor | FXRα (NR1H4) | Bile acids, fexaramine | |
| FXRβ (NR1H5) | Lanosterol | ||
| Vitamin D receptor | VDR (NR1I1) | 1,25-Dihydroxy vitamin D3 | |
| Pregnane X receptor | PXR (NR1I2) | Xenobiotics | |
| Constitutive androstane receptor | CAR (NR1I3) | Xenobiotics, phenobarbital | |
| Orphan subfamily | Reverse erbA | Rev-erbα (NR1D1) | Orphan |
| Rev-erbβ (NR1D2) | Orphan | ||
| Human nuclear factor 4 | HNFα (NR2A1) | Orphan | |
| HNFγ (NR2A2) | Orphan | ||
| Testis receptor | TR2 (NR2C1) | Orphan | |
| TR4 (NR2C2) | Orphan | ||
| Tailless | TLL (NR2E2) | Orphan | |
| Photoreceptor-specific nuclear receptor | PNR (NR2E3) | Orphan | |
| Chicken ovalbumin upstream | COUP-TFI (NR2F1) | Orphan | |
| promoter transcription factor | COUP-TFII (NR2F2 | Orphan | |
| ErbA2-related gene 2 | EAR2 (NR2F6) | Orphan | |
| NGF-induced factor B | NGFIB (NR4A1) | Orphan | |
| Nur-related factor 1 | NURR1 (NR4A2) | Orphan | |
| Neuron-derived orphan receptor 1 | NOR1 (NR4A3) | Orphan | |
| Steroidogenic factor 1 | SF-1 (NR5A1) | Orphan | |
| Liver receptor homologous protein 1 | LRH1 (NR5A2) | Orphan | |
| Germ cell nuclear factor | GCNF (NR6A1) | Orphan | |
| DSS-AHC critical region on the chromosome, gene 1 | DAX1 (NR0B1) | Orphan | |
| Short heterodimeric partner | SHP (NR0B2) | Orphan | |
| Adopted orphan subfamily | Thyroid hormone receptor | TRα (NR1A1) | Thyroid hormone |
| TRβ (NR1A2) | Thyroid hormone | ||
| Retinoic acid receptor | RARα (NR1B1) | Retinoic acid | |
| RARβ (NR1B2) | Retinoic acid | ||
| RARγ NR1B2) | Retinoic acid | ||
| Peroxisome proliferator-activated receptor | PPARα (NR1C1) | Fatty acids, leukotriene | |
| PPARβ/δ (NR1C2) | B4, fibrates | ||
| PPARγ (NR1C2) | Fatty acids | ||
| Fatty acids, prostaglandin J2 | |||
| RAR-related orphan receptor | RORα (NR1F1) | Cholesterol, cholesteryl sulfate | |
| RORβ (NR1F2) | Retinoic acid | ||
| RORγ (NR1F3) | Retinoic acid | ||
| Liver X receptor | LXRα (NR1H3) | Oxysterols | |
| LXRβ (NR1H2) | Oxysterols |
NRs typically function as ligand-dependent transcription factors. Upon ligand binding, NRs recognize specific DNA-response elements called hormone receptor elements (HREs), located within the promoters of their cognate target genes, where the ligand-NR complex can modulate the ability of HREs to recruit a range of other transcriptional proteins that alter the rate of gene expression (Fig. 2B,C). Besides ligands and the sequence of HREs, an additional level of specificity is provided by the preferences of NRs for their binding partners. NRs can be organized into distinct oligomeric states, including homodimers (e.g., steroid receptors) and heterodimers with the “promiscuous” retinoid X receptor (RXR) (e.g., peroxisome proliferator-activated receptors [PPARα, β, γ; NR1C1, 2, 3],17 thyroid hormone receptors [TRα, β; NR1A1, 2], and several orphan receptors), or remain monomeric (e.g., steroidogenic factor 1 [SF-1; NR5A1]). In the absence of a ligand, NRs are either present in the cytoplasm, where they form a complex with heat shock proteins and immunophilin chaperones,18 or constitutively bound to the HREs in the nucleus, where they form complexes with corepressor proteins like SMRT/NCOR and HDAC, repressing transcription.19 In the first scenario, ligand binding leads to dissociation of NRs from the chaperones and translocation to the nucleus, where they bind to the HREs of their target genes (Fig. 2B). Conversely, in the second scenario, ligand binding leads to dissociation of the corepressor complex and recruitment of transcription factors/enhancers, leading to activation of gene expression (Fig. 2C). This highlights the highly selective and variable nature of NRs.
NRs and Ocular Diseases
A large number of studies have revealed that members of the NR superfamily serve a pivotal role in modulating gene regulatory programs that control metabolism, inflammation, extracellular matrix regulation, angiogenesis, and numerous other biological and physiological processes.5–7,20 The fundamental roles that NRs play are highlighted by the fact that many of the biological pathways regulated by NRs are altered in human diseases, including common metabolic and cardiovascular disease states, autoimmune diseases, certain dermatologic conditions, and various tumor formations, to name a few.5,21–23 Within the last 15 years, an almost certain significant role for NRs has also emerged in studies of retinal degenerations, including AMD, DR, and RP, in part through studies conducted in genetically modified mouse models.8,11,24–26 Further identification of potential candidate receptors in diseases affecting the outer retina stems from the development of an NR atlas of human retinal pigment epithelial cells, which resulted in a comprehensive evaluation of the expression of all human NRs in these cells.27 Similarly, candidate receptors in diseases affecting the inner retina arose from targeted evaluation of the expression of select receptors within the retina.11,28–31 Collectively, these studies have provided a great deal of information regarding the impact of dysregulated NR signaling on the molecular pathogenesis of various diseases of the eye. It is remarkable that several of the NRs identified in the retina belong to the “orphan receptor” category, without known ligands.11 This fact in and of itself opens up the field of ophthalmology to questions concerning unidentified receptor ligands and their role in basic eye physiology and disease. Importantly, it also fuels interest in synthetic ligands that may eventually find applications as therapeutic agents to treat retinal diseases. The rest of this review will focus on the role of NRs in regulating molecular pathways, which are important in the pathogenesis of retinal diseases, including RP, DR, and AMD, and the prospects of using NRs as therapeutic targets for drug development. Table 2 summarizes human NRs potentially involved in retinal diseases.
Table 2.
Nuclear Receptors Associated with Retinal Diseases.
| Retinal Disease | Nuclear Receptor | Molecular Pathway |
|---|---|---|
| Diabetic retinopathy | PPARα (NR1C1) | Neuroprotection115 |
| PPARβ/δ (NR1C2) | Angiogenesis80,82,116 | |
| PPARγ (NR1C2 | Neuroprotection,117 inflammation118,119 | |
| RORα (NR1F1) | Angiogenesis | |
| MR (NR3C2) | Angiogenesis,120 | |
| LXRα (NR1H3) | Vasoprotection,25 inflammation25 | |
| LXRβ (NR1H2) | ||
| Age-related macular degeneration | PPARα (NR1C1) | Angiogenesis121 |
| PPARβ/δ (NR1C2) | Angiogenesis,116 lipid absorption and processing122 | |
| PPARγ (NR1C2 | Fibrosis,123 oxidative stress,124 apoptosis,124 inflammation119 | |
| RORα (NR1F1) | Angiogenesis26 | |
| LXRα (NR1H3) | Inflammation,125 lipid processing8,24 | |
| LXRβ (NR1H2) | ||
| VDR (NR1I1) | Inflammation126 | |
| RXRα (NR2B1) | Apoptosis,127 oxidative stress127 | |
| RXRβ (NR2B2) | ||
| RXRγ (NR2B3) | ||
| PNR (NR2E3) | Neuroprotection128 | |
| ERα (NR3A1) | Linked to ARMS2129 | |
| ERβ (NR3A2) | ||
| GR (NR3C1) | Neuroprotection130 | |
| Retinitis pigmentosa | PNR (NR2E3) | Cone cell proliferation30,44,47 |
| Rev-erbα (NR1D1) | Cone cell differentiation31,48 | |
| Rev-erbβ (NR1D2) |
Retinitis Pigmentosa
RP is a group of heterogeneous inherited neurodegenerative retinal diseases. The prevalence of RP is approximately 1 in 4000 people, totaling close to 1.5 million affected individuals worldwide.32,33 It is characterized by progressive night blindness, gradual loss of the visual field, a decline in electroretinogram-recorded amplitudes, and compromised visual acuity.32 The pathogenesis of RP can be explained by a continuum of metabolic maladies that lead to morphological and functional compromise to the retina, including the death of rod photoreceptors, followed by progressive loss of cone photoreceptors; loss of the retinal pigment epithelial cells, which serve as vital support cells to the overlying photoreceptors; and eventually total blindness.34–38 The primary cause of photoreceptor degeneration, although influenced by various processes, is genetic and can be mapped to defects in multiple gene loci. To date, RP has been localized to a number of chromosomes, including 1, 3, 4, 6, 7, 8, 11, 14, 16, and 19 and X chromosome.36,39–41 A recent update of all the genes involved in retinal diseases can be found through the Retinal Information Network (RetNet, https://sph.uth.edu/retnet/). In general, four classes of mutations can be described in the pathogenesis of RP on the basis of the functional molecular mechanisms that are disrupted: (1) mutations that disturb visual transduction, (2) mutations affecting vitamin A metabolism, (3) mutations that impair photoreceptor differentiation and homeostasis, and (4) mutations affecting the turnover of photoreceptor outer segments.11,42
Several research groups have shown that select NRs are actively involved in photoreceptor physiology. For example, NR2E3, also known as photoreceptor cell–specific NR (PNR), plays a role in the regulation of photoreceptor-specific genes necessary for cell maintenance and photo-transduction and during development and differentiation in rod photoreceptor cells.29,43,44 With regards to RP, Coppieters et al. identified a heterozygous missense mutation (G56R) in the first zinc finger of the Nr2e3 gene in a large Belgian family with this disease.45 Furthermore, the autosomal recessive retinal degeneration 7 (rd7) mouse, identified at the Jackson Laboratory, has been presented as a murine model for NR2E3-associated retinal disease.30,46 These rd7 mice are characterized by retinal folding associated with retinal spots and late-onset retinal degeneration46 and exhibit phenotypes similar to those of patients with Nr2e3 mutations, namely, a significant increase in S-cones and progressive degeneration of rod and cone photoreceptor cells.30 Significant crosstalk is often found between various NRs.2 In fact, NR2E3 has been shown to interact with another orphan NR, NR1D1 (Rev-Erbα), in mice, and functions within the same transcriptional system.29,31,47 In support of this, shRNA-mediated knockdown of Nr1d1 in the mouse retina reportedly results in a retinal phenotype similar to that observed in the rd7 mice.31 Furthermore, the therapeutic strategy of gene delivery of Nr1d1 to the retina, performed in neonatal Nr2e3rd7/rd7 mice, was able to rescue retinal spotting and dysplasia associated with Nr2e3 loss. Finally, Nr1d1 gene delivery was also able to reregulate the key genes that are vital for photoreceptor homeostasis, which were misregulated by the absence of Nr2e3 in Nr2e3rd7/rd7 mice.48 This study established Nr1d1 as a potential therapeutic target that may be used as a tool for gene therapy. In addition to gene therapy, high-throughput screens for receptor ligands have identified small molecules that bind to the NR2E3.49 One of the ligands identified, compound 11a, was further tested in retinal explants and shown to induce expression of genes with DR1 and DR4 motifs in their promoters.50 Currently, there are no NR-based treatments for RP in clinical trials. This presents both a great challenge and an opportunity for the development of drugs or gene therapy–based therapeutic agents for RP.
Diabetic Retinopathy
DR is a common microvascular complication associated with diabetes, and is characterized by hyperglycemia, loss of pericytes, microaneurysms, and preretinal neovascularization, which can eventually lead to vision loss by hemorrhage and retinal detachment.51,52 Diabetes is now recognized as a global epidemic with a projected patient population of 380 million by 2025.53–55 As the global prevalence of diabetes increases, so will the incidence of DR. Approximately one-third of individuals with diabetes will develop some degree of retinopathy, which currently is the leading cause of blindness in patients aged 20–74 years, in the developed and developing countries.56 Broadly defined, DR can manifest in two forms: (1) nonproliferative DR and (2) proliferative DR. Nonproliferative DR can be further classified into mild, moderate, and severe stages that may or may not involve the development of diabetic macular edema, the most common cause of diabetes-related vision loss, while proliferative DR is characterized by retinal neovascularization.57,58 The possibility of developing DR can be decreased by early detection and control of blood glucose, blood pressure, and lipid intake.52,53 Although a strong link has been established between hyperglycemia and the development and progression of DR, the causal mechanism that leads to damage to the retinal microvasculature has yet to be clearly established.59,60
Similar to the brain, the retina is compartmentalized from the circulation by the inner blood–retina barrier, which is formed by the retinal microvasculature (retinal microvascular endothelial cells and pericytes), and by the outer blood–retina barrier, which is formed by the capillary endothelium and the tight junctions of retinal pigment epithelial cells.61 During DR, the inner blood–retina barrier is compromised, which may lead to diabetic macular edema. There is strong evidence that growth factors like vascular endothelial growth factor (VEGF), which regulates vascular permeability, play a key role in the development of edema.62 Existing treatment modalities for macular edema include laser photocoagulation, corticosteroids, and anti-VEGF agents, and their success is most likely due to targeting inflammation as well as VEGF-A.61,63,64 However, therapies for nonproliferative DR in the absence of macular edema are still lacking.
PPARγ (NR1C3) is a DR candidate receptor, as it has been shown to play an important role in a variety of biological processes dysregulated in diabetes and DR, including adipogenesis, glucose metabolism, angiogenesis, and inflammation.65 Thiazolidine derivatives like rosiglitazone and pioglitazone act as synthetic ligands for PPARγ, and are being used as oral antihyperglycemic agents for the therapy of non-insulin-dependent diabetes mellitus.66 Studies investigating the role of PPARγ activation on the pathogenesis of proliferative DR have shown that treatment with rosiglitazone can inhibit retinal leakage, as well as leukostasis, in the streptozotocin-induced model of DR, in part through inhibition of NF-κB and subsequent suppression of ICAM-1 expression, which aids in leukocyte adhesion to endothelial cells.65,67–69 Additionally, activation of PPARγ leads to suppression of pro-inflammatory gene expression and cytokine secretion.70–72 Furthermore, the PPARγ agonists rosiglitazone and troglitazone have been shown to inhibit retinal neovascularization in a murine model of oxygen-induced ischemia.73 In addition to regulating inflammation, PPARγ can negatively impact VEGF-mediated angiogenesis via modulation of cyclooxygenase 2, which is a key modulator of inflammatory angiogenesis.74–76 PPARγ agonists are already being used to treat diabetes and other metabolic disorders. These studies, along with PPARγ's established ability to inhibit inflammation and angiogenesis, portend of their potential utility as an alternative and/or complement to laser photocoagulation and anti-VEGF therapy in the treatment of DR.77
Another PPAR isoform, beta/delta (PPARβ/δ; NR1C2), may also be a promising target for DR. In addition to controlling oxidative metabolism at a ubiquitous level, PPARβ/δ has been shown to be a molecular modulator of inflammation,78 mechanistically through repression of NF-κB/AP-1-based transcription of the inflammatory response.79 A recent study reported that PPARβ/δ antagonism in a model of oxygen- induced retinopathy led to a decrease in preretinal neovascularization.80 GSK0660, a PPARβ/δ antagonist, was also shown to significantly decrease serum-induced human retinal microvascular endothelial cell proliferation and tube formation, two commonly used in vitro–based angiogenic assays, in a dose-dependent manner.80,81 Another study from the same group showed that GSK0060 stabilized tight junctions in human retinal microvascular endothelial cells treated with VEGF, concomitant with reducing VEGFR1/2 expression, suggesting a role in vascular permeability,82 and providing further support for pharmacologic inhibition of PPARβ/δ as the basis for therapeutic targeting of retinal neovascularization.
The retinoic acid receptor-related orphan receptor alpha (RORα; NR1F1) is a lipid-sensing NR that has been shown to regulate the inflammatory response in allergy and autoimmune diseases.83–85 A recent report by Sun et al. demonstrated that the absence of RORα inhibited pathological angiogenesis in a model of oxygen-induced retinopathy, by directly impacting the expression of suppressor of cytokine signaling 3 (SOCS3), which is a critical regulator of tissue inflammation.26 Furthermore, treatment with an inverse agonist of RORα, SR1001, significantly repressed pathological angiogenesis in oxygen-induced retinopathy. Thus, RORα may also serve as a novel target for drug development to treat pathological retinal angiogenesis without directly altering levels of angiogenic growth factors, such as VEGF, which are essential for physiological angiogenesis.
Another NR of interest in DR is the liver x receptor (LXRα, β; NR1H2, 3). LXRs serve as cholesterol sensors and regulate cholesterol homeostasis.86 Importantly, they also appear to be at the intersection of lipid metabolism and inflammation, as activation of LXRs has also been shown to modulate macrophage function and result in potent anti-inflammatory effects.86,87 In a recent study of DR, a link between LXR expression in endothelial progenitor cells, critical for mediating vascular repair, and micro- and macrovascular complications was observed.25 Mice in which LXR was absent developed remarkable acellular capillaries and endothelial progenitor cell dysfunction similar to that observed in streptozotocin diabetic mice fed a high-fat diet. The most promising findings of this study were from the treatment of diabetic mice with the LXR agonist GW3965, which resulted in decreased micro- and macrovascular changes, including fewer acellular capillaries and reduced activated retinal glial cells, as demonstrated by glial fibrillary acidic protein (GFAP) immunoreactivity. Collectively, these studies point to the need to not only further consider targeting NRs such as the PPARs, ROR, and LXRs, as pharmacological targets for the treatment of DR, but also examine additional lipid- and immune-regulating NRs in the pathobiology of DR.
Age-Related Macular Degeneration
AMD is the leading cause of progressive vision loss in the elderly population, affecting 60 million people world-wide.88,89 The pathogenesis of early AMD involves accumulation of lipid and protein-filled extracellular deposits called drusen under the retinal pigment epithelial cells.90–94 Clinically, AMD progresses from an early to intermediate stage, and eventually to advanced stages, which can be classified into geographic atrophy or “late dry” and neovascular or “wet” AMD. The late dry form is characterized by the death of retinal pigment epithelial cells and subsequent loss of overlying photoreceptors in the central macula and vision loss.92,95–97 Wet AMD affects approximately 10% of the patients suffering from the disease, and is characterized by a breach through the retinal pigment epithelial cell layer and choroidal neovascularization within the outer retinal space, ultimately leading to scarring in the macular region.96,98 At present, there are no effective treatments available for the late dry clinical subtype, and success with anti-VEGF therapy of wet AMD has been seen in only a subset of patients.99 Therefore, there is a critical unmet need to identify new targets and develop alternate therapeutic approaches to help people suffering from vision loss due to AMD. The challenge with finding promising treatments for AMD is in part a reflection of the complexity of the disease, in which genetic, environmental, and systemic factors all play a role.8,96 The discovery of a large number of risk factors, in turn, has resulted in the identification of multiple molecular pathways that are believed to be dysregulated in AMD pathogenesis, including lipid metabolism, inflammation, extracellular matrix turnover, angiogenesis, complement dysregulation, and mitochondrial dysfunction, to name a few.96 Noteworthy is that interest in studying NRs in AMD stems from the fact that the NR superfamily members are known to regulate many of these AMD-associated pathways in other diseases, such as cancer, atherosclerosis, and autoimmune disease.5,6,8,22,24,96 The complexity of the disease can also be attributed to the tight interplay of cells, including the retinal pigment epithelial cells, photoreceptors, choroidal endothelial cells, and immune cells such as macrophages and microglia, the dysfunction of each of which contributes to AMD progression. Retinal pigment epithelial cell function and its role in drusen formation, and the secretion of angiogenic factors, are extensively studied, as the overall health of these cells is affected in all clinical subtypes of AMD.96 Because of this, recently, an NR atlas investigating the expression of all 48 human NRs in retinal pigment epithelial cells was developed, which led to the identification of several AMD candidate genes currently being studied.27
As mentioned earlier, LXRs are vital regulators of cholesterol homeostasis, glucose homeostasis, detoxification of bile acids, immunity, and neurological functions.100 A recent study elegantly demonstrated promising results in targeting the LXR receptor for the treatment of neovascular AMD. The investigators found that the expression of the cholesterol transporter, ABCA1, also an LXR target gene, decreases as a function of age in both human and mouse macro-phages,101 and in the absence of the ABCA1 receptor, mice develop larger and more severe experimentally induced neovascular lesions.101 Of significance, it was purported that the cholesterol content of macrophages specifically impacts macrophage phenotype and polarity, which consequently effects development of choroidal neovascularization. Though the impact of targeting LXR in dry AMD was not studied, the results support the hypothesis that targeting LXR may be effective in treating or removal of lipid-rich drusen, characteristic of early AMD. Overall, this study, in combination with other reports demonstrating activation of LXR, can attenuate inflammatory responses and support further pursuit of targeting LXRs for drug development in the treatment of choroidal neovascularization, as well as inflammatory mechanisms associated with AMD.102–105
Different isoforms of PPARs also have the potential to be viable targets for AMD, as they are involved in the regulation of established AMD pathogenic pathways, such as lipid metabolism, extracellular matrix remodeling, angiogenesis, and inflammation.8,24 Direct evidence comes from one study that examined the distribution of PPARγ in ocular samples from human dry and wet AMD patients and found increased immunoreactivity within the retina.106 Changes in the expression levels of PPARγ in retinal pigment epithelial cells, as a function of age and disease, were not reported. Interestingly, in cell cultures of ARPE19 cells, a spontaneously arising retinal pigment epithelial cell line from a 19-year-old donor, following treatment with hydrogen peroxide, there was an increase in PPARγ along with VEGF, heme-oxidase 1, and MMP-9 expression. Evidence is also available in support of potentially targeting PPARγ in wet AMD, based on in vivo treatment of rats following experimentally induced choroidal neovascularization.107 Conceding that there is an absence of reports that directly link the PPARα or PPARβ/δ isoforms with human AMD pathology, functional studies in ARPE19 and human primary retinal pigment epithelial cells have conjectured that these PPARs play a role in AMD pathology by regulating inflammation, oxidative stress, and lipid accumulation.8 While there are no PPAR Food and Drug Administration (FDA)–approved drugs for AMD, there are FDA-approved drugs for PPARγ and α. Additionally, recent development of highly specific and selective compounds for PPARβ/δ, such as GW501516, GW0742X, and GSK3787, has improved the study of the PPARβ/δ pathway in disease models such as obesity and atherosclerosis, which may provide improved insight into the regulation of inflammation and lipid metabolism through this pathway. Future studies utilizing these PPAR-targeted drugs in in vitro and in vivo models of AMD have the potential to lead to the development of new therapeutic approaches treating pathways associated with AMD progression.108
The aryl hydrocarbon receptor (AhR) is a member of the bHLH/PAS (basic helix–loop–helix/Per–Arnt–Sim) family of heterodimeric transcriptional regulators, with similar mechanisms of action to NRs. Although traditionally thought of as a receptor involved in toxin clearance, it has also been shown to play important roles in regulation of vascular development, angiogenesis, and inflammation.109–112 Several recent studies have reported a potential role for AhR in AMD.112–114 These studies found that genetic ablation of AhR in mice leads to development of ocular features of dry AMD. One study further reported an age-related decline of AhR activity in human retinal pigment epithelial cells, suggesting a potential age-related compromise in normal retinal epithelial cellular clearance mechanisms.113 Interestingly, the AhR knockout mice also displayed progressive choroidal thinning. A follow-up study identified a role of AhR in neovascularization. In aged mice, the absence of AhR resulted in an exacerbation of experimentally induced choroidal neovascularization concomitant with increased extracellular matrix dysregulation and inflammation.112 These studies solicit exploring the therapeutic benefits of targeting the AhR signaling pathway as a means for treating deposition of extracellular matrix material, angiogenesis, and fibrosis associated with AMD.
Discussion
Though not exhaustive, we have provided an overview of the potential role of NRs in the pathobiology of several vision-impairing retinal diseases. This is a relatively young field of research with few studies to date. However, the abundance of NRs in the eye strongly support the concept of the eye as a specialized secondary endocrine organ, in which NRs most likely orchestrate complex events during development, cell differentiation, cell homeostasis, and by extension, disease. Furthermore, the overwhelming number of studies characterizing functions of various NRs in systemic and degenerative diseases that share common pathogenic pathways with retinal disorders underscore the need to examine the role of NRs in the initiation and progression of complex ocular diseases. Finally, it will be critical to fully tease out cell-specific activity of NRs, including their responses following treatment with classically defined modulators, including activators and inhibitors. A thorough evaluation of receptor activity may lead to identification of cell-specific responses reflecting the complex pharmacology of NRs. This will be critical in determining the utility of targeting NRs for treatment in the eye with selective modulators that have the potential to decrease detrimental off-target effects.
Acknowledgments
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Goldis Malek is the recipient of funding from the National Eye Institute (NEI EY02868), the Brightfocus Foundation for Macular Degeneration Research, and the Edward N. & Della L. Thome Memorial Foundation AMD Research Award. Additional funds come from an unrestricted Research to Prevent Blindness award and NEI core grant (NEI P30 EY005722) to the Department of Ophthalmology at Duke University.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Bookout AL, Jeong Y, Downes M, et al. Anatomical Profiling of Nuclear Receptor Expression Reveals a Hierarchical Transcriptional Network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, Thummel C, Beato M, et al. The Nuclear Receptor Superfamily: The Second Decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mangelsdorf DJ, Evans RM. The RXR Heterodimers and Orphan Receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 4.Evans RM. The Steroid and Thyroid Hormone Receptor Superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonoda J, Pei L, Evans RM. Nuclear Receptors: Decoding Metabolic Disease. FEBS Lett. 2008;582:2–9. doi: 10.1016/j.febslet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conzen SD. Minireview: Nuclear Receptors and Breast Cancer. Mol Endocrinol. 2008;22:2215–2228. doi: 10.1210/me.2007-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chawla A, Repa JJ, Evans RM, et al. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 8.Malek G, Lad EM. Emerging Roles for Nuclear Receptors in the Pathogenesis of Age-Related Macular Degeneration. Cell Mol Life Sci. 2014;71:4617–4636. doi: 10.1007/s00018-014-1709-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costa V, Ciccodicola A. Is PPARG the Key Gene in Diabetic Retinopathy? Br J Pharmacol. 2012;165:1–3. doi: 10.1111/j.1476-5381.2011.01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song MK, Roufogalis BD, Huang TH. Modulation of Diabetic Retinopathy Pathophysiology by Natural Medicines through PPAR-gamma-Related Pharmacology. Br J Pharmacol. 2012;165:4–19. doi: 10.1111/j.1476-5381.2011.01411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forrest D, Swaroop A. Minireview: The Role of Nuclear Receptors in Photoreceptor Differentiation and Disease. Mol Endocrinol. 2012;26:905–915. doi: 10.1210/me.2012-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tata JR. Signalling through Nuclear Receptors. Nat Rev Mol Cell Biol. 2002;3:702–710. doi: 10.1038/nrm914. [DOI] [PubMed] [Google Scholar]
- 13.Gronemeyer H, Gustafsson JA, Laudet V. Principles for Modulation of the Nuclear Receptor Superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 14.Hollenberg SM, Weinberger C, Ong ES, et al. Primary Structure and Expression of a Functional Human Glucocorticoid Receptor cDNA. Nature. 1985;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green S, Walter P, Kumar V, et al. Human Oestrogen Receptor cDNA: Sequence, Expression and Homology to v-erb-A. Nature. 1986;320:134–139. doi: 10.1038/320134a0. [DOI] [PubMed] [Google Scholar]
- 16.Sever R, Glass CK. Signaling by Nuclear Receptors. Cold Spring Harb Perspect Biol. 2013;5:a016709. doi: 10.1101/cshperspect.a016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pratt WB, Toft DO. Steroid Receptor Interactions with Heat Shock Protein and Immunophilin Chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 19.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and Signals: A Coactivator/Corepressor/Epigenetic Code for Integrating Signal-Dependent Programs of Transcriptional Response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 20.Giguere V. Orphan Nuclear Receptors: From Gene to Function. Endocr Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 21.Baek SH, Kim KI. Emerging Roles of Orphan Nuclear Receptors in Cancer. Annu Rev Physiol. 2014;76:177–195. doi: 10.1146/annurev-physiol-030212-183758. [DOI] [PubMed] [Google Scholar]
- 22.Racke MK, Gocke AR, Muir M, et al. Nuclear Receptors and Autoimmune Disease: The Potential of PPAR Agonists to Treat Multiple Sclerosis. J Nutr. 2006;136:700–703. doi: 10.1093/jn/136.3.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winterfield L, Cather J, Menter A. Changing Paradigms in Dermatology: Nuclear Hormone Receptors. Clin Dermatol. 2003;21:447–454. doi: 10.1016/j.clindermatol.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 24.Malek G. Nuclear Receptors as Potential Therapeutic Targets for Age-Related Macular Degeneration. Adv Exp Med Biol. 2014;801:317–321. doi: 10.1007/978-1-4614-3209-8_40. [DOI] [PubMed] [Google Scholar]
- 25.Hazra S, Rasheed A, Bhatwadekar A, et al. Liver X Receptor Modulates Diabetic Retinopathy Outcome in a Mouse Model of Streptozotocin-Induced Diabetes. Diabetes. 2012;61:3270–3279. doi: 10.2337/db11-1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y, Liu CH, SanGiovanni JP, et al. Nuclear Receptor RORalpha Regulates Pathologic Retinal Angiogenesis by Modulating SOCS3-Dependent Inflammation. Proc Natl Acad Sci U S A. 2015;112:10401–10406. doi: 10.1073/pnas.1504387112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dwyer MA, Kazmin D, Hu P, et al. Research Resource: Nuclear Receptor Atlas of Human Retinal Pigment Epithelial Cells: Potential Relevance to Age-Related Macular Degeneration. Mol Endocrinol. 2011;25:360–372. doi: 10.1210/me.2010-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wright AF, Reddick AC, Schwartz SB, et al. Mutation Analysis of NR2E3 and NRL Genes in Enhanced S Cone Syndrome. Hum Mutat. 2004;24:439. doi: 10.1002/humu.9285. [DOI] [PubMed] [Google Scholar]
- 29.Cheng H, Khanna H, Oh EC, et al. Photoreceptor-Specific Nuclear Receptor NR2E3 Functions as a Transcriptional Activator in Rod Photoreceptors. Hum Mol Genet. 2004;13:1563–1575. doi: 10.1093/hmg/ddh173. [DOI] [PubMed] [Google Scholar]
- 30.Haider NB, Naggert JK, Nishina PM. Excess Cone Cell Proliferation Due to Lack of a Functional NR2E3 Causes Retinal Dysplasia and Degeneration in rd7/rd7 Mice. Hum Mol Genet. 2001;10:1619–1626. doi: 10.1093/hmg/10.16.1619. [DOI] [PubMed] [Google Scholar]
- 31.Mollema NJ, Yuan Y, Jelcick AS, et al. Nuclear Receptor Rev-erb alpha (Nr1d1) Functions in Concert with Nr2e3 to Regulate Transcriptional Networks in the Retina. PLoS One. 2011;6:e17494. doi: 10.1371/journal.pone.0017494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamel C. Retinitis Pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shintani K, Shechtman DL, Gurwood AS. Review and Update: Current Treatment Trends for Patients with Retinitis Pigmentosa. Optometry. 2009;80:384–401. doi: 10.1016/j.optm.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 34.Hartong DT, Berson EL, Dryja TP. Retinitis Pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 35.Berson EL. Retinitis Pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34:1659–1676. [PubMed] [Google Scholar]
- 36.Haim M. Epidemiology of Retinitis Pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002:1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 37.Baumgartner WA. Etiology, Pathogenesis, and Experimental Treatment of Retinitis Pigmentosa. Med Hypotheses. 2000;54:814–824. doi: 10.1054/mehy.1999.0957. [DOI] [PubMed] [Google Scholar]
- 38.John SK, Smith JE, Aguirre GD, et al. Loss of Cone Molecular Markers in Rhodopsin-Mutant Human Retinas with Retinitis Pigmentosa. Mol Vis. 2000;6:204–215. [PubMed] [Google Scholar]
- 39.Ammann F, Klein D, Franceschetti A. Genetic and Epidemiological Investigations on Pigmentary Degeneration of the Retina and Allied Disorders in Switzerland. J Neurol Sci. 1965;2:183–196. doi: 10.1016/0022-510x(65)90079-1. [DOI] [PubMed] [Google Scholar]
- 40.Boughman JA, Conneally PM, Nance WE. Population Genetic Studies of Retinitis Pigmentosa. Am J Hum Genet. 1980;32:223–235. [PMC free article] [PubMed] [Google Scholar]
- 41.van Soest S, Westerveld A, de Jong PT, et al. Retinitis Pigmentosa: Defined from a Molecular Point of View. Surv Ophthalmol. 1999;43:321–334. doi: 10.1016/s0039-6257(98)00046-0. [DOI] [PubMed] [Google Scholar]
- 42.Wert KJ, Lin JH, Tsang SH. General Pathophysiology in Retinal Degeneration. Dev Ophthalmol. 2014;53:33–43. doi: 10.1159/000357294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi M, Takezawa S, Hara K, et al. Identification of a Photoreceptor Cell-Specific Nuclear Receptor. Proc Natl Acad Sci U S A. 1999;96:4814–4819. doi: 10.1073/pnas.96.9.4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haider NB, Jacobson SG, Cideciyan AV, et al. Mutation of a Nuclear Receptor Gene, NR2E3, Causes Enhanced S Cone Syndrome, a Disorder of Retinal Cell Fate. Nat Genet. 2000;24:127–131. doi: 10.1038/72777. [DOI] [PubMed] [Google Scholar]
- 45.Coppieters F, Leroy BP, Beysen D, et al. Recurrent Mutation in the First Zinc Finger of the Orphan Nuclear Receptor NR2E3 Causes Autosomal Dominant Retinitis Pigmentosa. Am J Hum Genet. 2007;81:147–157. doi: 10.1086/518426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akhmedov NB, Piriev NI, Chang B, et al. A Deletion in a Photoreceptor-Specific Nuclear Receptor mRNA Causes Retinal Degeneration in the rd7 Mouse. Proc Natl Acad Sci U S A. 2000;97:5551–5556. doi: 10.1073/pnas.97.10.5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haider NB, Mollema N, Gaule M, et al. Nr2e3-Directed Transcriptional Regulation of Genes Involved in Photoreceptor Development and Cell-Type Specific Phototransduction. Exp Eye Res. 2009;89:365–372. doi: 10.1016/j.exer.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cruz NM, Yuan Y, Leehy BD, et al. Modifier Genes as Therapeutics: The Nuclear Hormone Receptor Rev Erb alpha (Nr1d1) Rescues Nr2e3 Associated Retinal Disease. PLoS One. 2014;9:e87942. doi: 10.1371/journal.pone.0087942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolkenberg SE, Zhao Z, Kapitskaya M, et al. Identification of Potent Agonists of Photoreceptor-Specific Nuclear Receptor (NR2E3) and Preparation of a Radioligand. Bioorg Med Chem Lett. 2006;16:5001–5004. doi: 10.1016/j.bmcl.2006.07.056. [DOI] [PubMed] [Google Scholar]
- 50.Webber AL, Hodor P, Thut CJ, et al. Dual Role of Nr2e3 in Photoreceptor Development and Maintenance. Exp Eye Res. 2008;87:35–48. doi: 10.1016/j.exer.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 51.Lee R, Wong TY, Sabanayagam C. Epidemiology of Diabetic Retinopathy, Diabetic Macular Edema and Related Vision Loss. Eye Vis (Lond) 2015;2:17. doi: 10.1186/s40662-015-0026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wan TT, Li XF, Sun YM, et al. Recent Advances in Understanding the Biochemical and Molecular Mechanism of Diabetic Retinopathy. Biomed Pharmacother. 2015;74:145–147. doi: 10.1016/j.biopha.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Yau JW, Rogers SL, Kawasaki R, et al. Global Prevalence and Major Risk Factors of Diabetic Retinopathy. Diabetes Care. 2012;35:556–564. doi: 10.2337/dc11-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wild S, Roglic G, Green A, et al. Global Prevalence of Diabetes: Estimates for the Year 2000 and Projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 55.Whiting DR, Guariguata L, Weil C, et al. IDF Diabetes Atlas: Global Estimates of the Prevalence of Diabetes for 2011 and 2030. Diabetes Res Clin Pract. 2011;94:311–321. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 56.Ruta LM, Magliano DJ, Lemesurier R, et al. Prevalence of Diabetic Retinopathy in Type 2 Diabetes in Developing and Developed Countries. Diabet Med. 2013;30:387–398. doi: 10.1111/dme.12119. [DOI] [PubMed] [Google Scholar]
- 57.Tarr JM, Kaul K, Chopra M, et al. Pathophysiology of Diabetic Retinopathy. ISRN Ophthalmol. 2013;2013:343560. doi: 10.1155/2013/343560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burditt AG, Caird FI, Draper GJ. The Natural History of Diabetic Retinopathy. Q J Med. 1968;37:303–317. [PubMed] [Google Scholar]
- 59.White NH, Cleary PA, Dahms W, et al. Beneficial Effects of Intensive Therapy of Diabetes during Adolescence: Outcomes after the Conclusion of the Diabetes Control and Complications Trial (DCCT) J Pediatr. 2001;139:804–812. doi: 10.1067/mpd.2001.118887. [DOI] [PubMed] [Google Scholar]
- 60.Matthews DR, Stratton IM, Aldington SJ, et al. Risks of Progression of Retinopathy and Vision Loss Related to Tight Blood Pressure Control in Type 2 Diabetes Mellitus: UKPDS 69. Arch Ophthalmol. 2004;122:1631–1640. doi: 10.1001/archopht.122.11.1631. [DOI] [PubMed] [Google Scholar]
- 61.Klaassen I, Van Noorden CJ, Schlingemann RO. Molecular Basis of the Inner Blood-Retinal Barrier and Its Breakdown in Diabetic Macular Edema and Other Pathological Conditions. Prog Retin Eye Res. 2013;34:19–48. doi: 10.1016/j.preteyeres.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 62.Kim LA, D'Amore PA. A Brief History of Anti-VEGF for the Treatment of Ocular Angiogenesis. Am J Pathol. 2012;181:376–379. doi: 10.1016/j.ajpath.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wenick AS, Bressler NM. Diabetic Macular Edema: Current and Emerging Therapies. Middle East Afr J Ophthalmol. 2012;19:4–12. doi: 10.4103/0974-9233.92110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Witkin AJ, Brown GC. Update on Nonsurgical Therapy for Diabetic Macular Edema. Curr Opin Ophthalmol. 2011;22:185–189. doi: 10.1097/ICU.0b013e3283459724. [DOI] [PubMed] [Google Scholar]
- 65.Rosen ED, Spiegelman BM. PPARgamma: A Nuclear Regulator of Metabolism, Differentiation, and Cell Growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 66.Della-Morte D, Palmirotta R, Rehni AK, et al. Pharmacogenomics and Pharmacogenetics of Thiazolidinediones: Role in Diabetes and Cardiovascular Risk Factors. Pharmacogenomics. 2014;15:2063–2082. doi: 10.2217/pgs.14.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim EK, Kwon KB, Koo BS, et al. Activation of Peroxisome Proliferator-Activated Receptor-gamma Protects Pancreatic beta-Cells from Cytokine-Induced Cytotoxicity via NF kappaB Pathway. Int J Biochem Cell Biol. 2007;39:1260–1275. doi: 10.1016/j.biocel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 68.Lee KS, Kim SR, Park SJ, et al. Peroxisome Proliferator Activated Receptor-gamma Modulates Reactive Oxygen Species Generation and Activation of Nuclear Factor-kappaB and Hypoxia-Inducible Factor 1alpha in Allergic Airway Disease of Mice. J Allergy Clin Immunol. 2006;118:120–127. doi: 10.1016/j.jaci.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 69.Sung B, Park S, Yu BP, et al. Amelioration of Age-Related Inflammation and Oxidative Stress by PPARgamma Activator: Suppression of NF-kappaB by 2,4-Thiazolidinedione. Exp Gerontol. 2006;41:590–599. doi: 10.1016/j.exger.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 70.Uchimura K, Nakamuta M, Enjoji M, et al. Activation of Retinoic X Receptor and Peroxisome Proliferator-Activated Receptor-gamma Inhibits Nitric Oxide and Tumor Necrosis Factor-alpha Production in Rat Kupffer Cells. Hepatology. 2001;33:91–99. doi: 10.1053/jhep.2001.21145. [DOI] [PubMed] [Google Scholar]
- 71.Wong A, Dukic-Stefanovic S, Gasic-Milenkovic J, et al. Anti-Inflammatory Antioxidants Attenuate the Expression of Inducible Nitric Oxide Synthase Mediated by Advanced Glycation Endproducts in Murine Microglia. Eur J Neurosci. 2001;14:1961–1967. doi: 10.1046/j.0953-816x.2001.01820.x. [DOI] [PubMed] [Google Scholar]
- 72.Chawla A, Barak Y, Nagy L, et al. PPAR-gamma Dependent and Independent Effects on Macrophage-Gene Expression in Lipid Metabolism and Inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 73.Murata T, Hata Y, Ishibashi T, et al. Response of Experimental Retinal Neovascularization to Thiazolidinediones. Arch Ophthalmol. 2001;119:709–717. doi: 10.1001/archopht.119.5.709. [DOI] [PubMed] [Google Scholar]
- 74.Scoditti E, Massaro M, Carluccio MA, et al. PPARgamma Agonists Inhibit Angiogenesis by Suppressing PKCalpha- and CREB-Mediated COX-2 Expression in the Human Endothelium. Cardiovasc Res. 2010;86:302–310. doi: 10.1093/cvr/cvp400. [DOI] [PubMed] [Google Scholar]
- 75.Wu G, Mannam AP, Wu J, et al. Hypoxia Induces Myocyte-Dependent COX-2 Regulation in Endothelial Cells: Role of VEGF. Am J Physiol Heart Circ Physiol. 2003;285:H2420–H2429. doi: 10.1152/ajpheart.00187.2003. [DOI] [PubMed] [Google Scholar]
- 76.Gately S, Li WW. Multiple Roles of COX-2 in Tumor Angiogenesis: A Target for Antiangiogenic Therapy. Semin Oncol. 2004;31:2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 77.Stahl A, Sapieha P, Connor KM, et al. Short Communication: PPAR gamma Mediates a Direct Antiangiogenic Effect of omega 3-PUFAs in Proliferative Retinopathy. Circ Res. 2010;107:495–500. doi: 10.1161/CIRCRESAHA.110.221317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Escher P, Braissant O, Basu-Modak S, et al. Rat PPARs: Quantitative Analysis in Adult Rat Tissues and Regulation in Fasting and Refeeding. Endocrinology. 2001;142:4195–4202. doi: 10.1210/endo.142.10.8458. [DOI] [PubMed] [Google Scholar]
- 79.Schnegg CI, Robbins ME. Neuroprotective Mechanisms of PPARdelta: Modulation of Oxidative Stress and Inflammatory Processes. PPAR Res. 2011;2011:373560. doi: 10.1155/2011/373560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Capozzi ME, McCollum GW, Savage SR, et al. Peroxisome Proliferator-Activated Receptor-beta/delta Regulates Angiogenic Cell Behaviors and Oxygen-Induced Retinopathy. Invest Ophthalmol Vis Sci. 2013;54:4197–4207. doi: 10.1167/iovs.13-11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shearer BG, Steger DJ, Way JM, et al. Identification and Characterization of a Selective Peroxisome Proliferator-Activated Receptor beta/delta (NR1C2) Antagonist. Mol Endocrinol. 2008;22:523–529. doi: 10.1210/me.2007-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suarez S, McCollum GW, Bretz CA, et al. Modulation of VEGF-Induced Retinal Vascular Permeability by Peroxisome Proliferator-Activated Receptor-beta/delta. Invest Ophthalmol Vis Sci. 2014;55:8232–8240. doi: 10.1167/iovs.14-14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bitsch F, Aichholz R, Kallen J, et al. Identification of Natural Ligands of Retinoic Acid Receptor-Related Orphan Receptor alpha Ligand-Binding Domain Expressed in Sf9 cells—A Mass Spectrometry Approach. Anal Biochem. 2003;323:139–149. doi: 10.1016/j.ab.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 84.Halim TY, MacLaren A, Romanish MT, et al. Retinoic-Acid-Receptor-Related Orphan Nuclear Receptor alpha Is Required for Natural Helper Cell Development and Allergic Inflammation. Immunity. 2012;37:463–474. doi: 10.1016/j.immuni.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 85.Solt LA, Kumar N, Nuhant P, et al. Suppression of TH17 Differentiation and Autoimmunity by a Synthetic ROR Ligand. Nature. 2011;472:491–494. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee SD, Tontonoz P. Liver X Receptors at the Intersection of Lipid Metabolism and Atherogenesis. Atherosclerosis. 2015;242:29–36. doi: 10.1016/j.atherosclerosis.2015.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ito A, Hong C, Rong X, et al. LXRs Link Metabolism to Inflammation through Abca1-Dependent Regulation of Membrane Composition and TLR Signaling. Elife. 2015;4:e08009. doi: 10.7554/eLife.08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rein DB, Wittenborn JS, Zhang X, et al. Forecasting Age-Related Macular Degeneration through the Year 2050: The Potential Impact of New Treatments. Arch Ophthalmol. 2009;127:533–540. doi: 10.1001/archophthalmol.2009.58. [DOI] [PubMed] [Google Scholar]
- 89.Klein R, Cruickshanks KJ, Myers CE, et al. The Relationship of Atherosclerosis to the 10-Year Cumulative Incidence of Age-Related Macular Degeneration: The Beaver Dam Studies. Ophthalmology. 2013;120:1012–1019. doi: 10.1016/j.ophtha.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sohrab MA, Smith RT, Fawzi AA. Imaging Characteristics of Dry Age-Related Macular Degeneration. Semin Ophthalmol. 2011;26:156–166. doi: 10.3109/08820538.2011.570848. [DOI] [PubMed] [Google Scholar]
- 91.Green WR, Enger C. Age-Related Macular Degeneration Histopathologic Studies The 1992 Lorenz E Zimmerman Lecture. Ophthalmology. 1993;100:1519–1535. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- 92.Curcio CA, Millican CL. Basal Linear Deposit and Large Drusen Are Specific for Early Age-Related Maculopathy. Arch Ophthalmol. 1999;117:329–339. doi: 10.1001/archopht.117.3.329. [DOI] [PubMed] [Google Scholar]
- 93.Loffler KU, Lee WR. Basal Linear Deposit in the Human Macula. Graefes Arch Clin Exp Ophthalmol. 1986;224:493–501. doi: 10.1007/BF02154735. [DOI] [PubMed] [Google Scholar]
- 94.van der Schaft TL, Mooy CM, de Bruijn WC, et al. Early Stages of Age-Related Macular Degeneration: An Immunofluorescence and Electron Microscopy Study. Br J Ophthalmol. 1993;77:657–661. doi: 10.1136/bjo.77.10.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bhutto I, Lutty G. Understanding Age-Related Macular Degeneration (AMD): Relationships between the Photoreceptor/Retinal Pigment Epithelium/Bruch's Membrane/Choriocapillaris Complex. Mol Aspects Med. 2012;33:295–317. doi: 10.1016/j.mam.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ambati J, Fowler BJ. Mechanisms of Age-Related Macular Degeneration. Neuron. 2012;75:26–39. doi: 10.1016/j.neuron.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Curcio CA, Medeiros NE, Millican CL. Photoreceptor Loss in Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 1996;37:1236–1249. [PubMed] [Google Scholar]
- 98.Lim LS, Mitchell P, Seddon JM, et al. Age-Related Macular Degeneration. Lancet. 2012;379:1728–1738. doi: 10.1016/S0140-6736(12)60282-7. [DOI] [PubMed] [Google Scholar]
- 99.Ng EW, Adamis AP. Targeting Angiogenesis, the Underlying Disorder in Neovascular Age-Related Macular Degeneration. Can J Ophthalmol. 2005;40:352–368. doi: 10.1016/S0008-4182(05)80078-X. [DOI] [PubMed] [Google Scholar]
- 100.Baranowski M. Biological Role of Liver X Receptors. J Physiol Pharmacol. 2008;59(Suppl 7):31–55. [PubMed] [Google Scholar]
- 101.Sene A, Khan AA, Cox D, et al. Impaired Cholesterol Efflux in Senescent Macrophages Promotes Age-Related Macular Degeneration. Cell Metab. 2013;17:549–561. doi: 10.1016/j.cmet.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang YY, Dahle MK, Steffensen KR, et al. Liver X Receptor Agonist GW3965 Dose-Dependently Regulates LPS-Mediated Liver Injury and Modulates Posttranscriptional TNF-alpha Production and p38 Mitogen-Activated Protein Kinase Activation in Liver Macrophages. Shock. 2009;32:548–553. doi: 10.1097/SHK.0b013e3181a47f85. [DOI] [PubMed] [Google Scholar]
- 103.Liu Y, Qiu de K, Ma X. Liver X Receptors Bridge Hepatic Lipid Metabolism and Inflammation. J Dig Dis. 2012;13:69–74. doi: 10.1111/j.1751-2980.2011.00554.x. [DOI] [PubMed] [Google Scholar]
- 104.Cui G, Qin X, Wu L, et al. Liver X Receptor (LXR) Mediates Negative Regulation of Mouse and Human Th17 Differentiation. J Clin Invest. 2011;121:658–670. doi: 10.1172/JCI42974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hindinger C, Hinton DR, Kirwin SJ, et al. Liver X Receptor Activation Decreases the Severity of Experimental Autoimmune Encephalomyelitis. J Neurosci Res. 2006;84:1225–1234. doi: 10.1002/jnr.21038. [DOI] [PubMed] [Google Scholar]
- 106.Herzlich AA, Tuo J, Chan CC. Peroxisome Proliferator-Activated Receptor and Age-Related Macular Degeneration. PPAR Res. 2008;2008:389507. doi: 10.1155/2008/389507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Murata T, He S, Hangai M, et al. Peroxisome Proliferator-Activated Receptor-gamma Ligands Inhibit Choroidal Neovascularization. Invest Ophthalmol Vis Sci. 2000;41:2309–2317. [PubMed] [Google Scholar]
- 108.Graham TL, Mookherjee C, Suckling KE, et al. The PPARdelta Agonist GW0742X Reduces Atherosclerosis in LDLR(–/–) Mice. Atherosclerosis. 2005;181:29–37. doi: 10.1016/j.atherosclerosis.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 109.Wu D, Nishimura N, Kuo V, et al. Activation of Aryl Hydrocarbon Receptor Induces Vascular Inflammation and Promotes Atherosclerosis in Apolipoprotein E–/– Mice. Arterioscler Thromb Vasc Biol. 2011;31:1260–1267. doi: 10.1161/ATVBAHA.110.220202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gomez-Duran A, Carvajal-Gonzalez JM, Mulero-Navarro S, et al. Fitting a Xenobiotic Receptor into Cell Homeostasis: How the Dioxin Receptor Interacts with TGFbeta Signaling. Biochem Pharmacol. 2009;77:700–712. doi: 10.1016/j.bcp.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 111.Ishimura R, Kawakami T, Ohsako S, et al. Dioxin-Induced Toxicity on Vascular Remodeling of the Placenta. Biochem Pharmacol. 2009;77:660–669. doi: 10.1016/j.bcp.2008.10.030. [DOI] [PubMed] [Google Scholar]
- 112.Choudhary M, Kazmin D, Hu P, et al. Aryl Hydrocarbon Receptor Knock-Out Exacerbates Choroidal Neovascularization via Multiple Pathogenic Pathways. J Pathol. 2015;235:101–112. doi: 10.1002/path.4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hu P, Herrmann R, Bednar A, et al. Aryl Hydrocarbon Receptor Deficiency Causes Dysregulated Cellular Matrix Metabolism and Age-Related Macular Degeneration-Like Pathology. Proc Natl Acad Sci U S A. 2013;110:E4069–4078. doi: 10.1073/pnas.1307574110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim SY, Yang HJ, Chang YS, et al. Deletion of Aryl Hydrocarbon Receptor AHR in Mice Leads to Subretinal Accumulation of Microglia and RPE Atrophy. Invest Ophthalmol Vis Sci. 2014;55:6031–6040. doi: 10.1167/iovs.14-15091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bordet R, Ouk T, Petrault O, et al. PPAR: A New Pharmacological Target for Neuroprotection in Stroke and Neurodegenerative Diseases. Biochem Soc Trans. 2006;34:1341–1346. doi: 10.1042/BST0341341. [DOI] [PubMed] [Google Scholar]
- 116.Bishop-Bailey D. A Role for PPARbeta/delta in Ocular Angiogenesis. PPAR Res. 2008;2008:825970. doi: 10.1155/2008/825970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Aoun P, Simpkins JW, Agarwal N. Role of PPAR-gamma Ligands in Neuroprotection against Glutamate-Induced Cytotoxicity in Retinal Ganglion Cells. Invest Ophthalmol Vis Sci. 2003;44:2999–3004. doi: 10.1167/iovs.02-1060. [DOI] [PubMed] [Google Scholar]
- 118.Bernardo A, Ajmone-Cat MA, Gasparini L, et al. Nuclear Receptor Peroxisome Proliferator-Activated Receptor-gamma Is Activated in Rat Microglial Cells by the Anti-Inflammatory Drug HCT1026, a Derivative of Flurbiprofen. J Neurochem. 2005;92:895–903. doi: 10.1111/j.1471-4159.2004.02932.x. [DOI] [PubMed] [Google Scholar]
- 119.Malchiodi-Albedi F, Matteucci A, Bernardo A, et al. PPAR-gamma, Microglial Cells, and Ocular Inflammation: New Venues for Potential Therapeutic Approaches. PPAR Res. 2008;2008:295784. doi: 10.1155/2008/295784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wilkinson-Berka JL, Tan G, Jaworski K, et al. Identification of a Retinal Aldosterone System and the Protective Effects of Mineralocorticoid Receptor Antagonism on Retinal Vascular Pathology. Circ Res. 2009;104:124–133. doi: 10.1161/CIRCRESAHA.108.176008. [DOI] [PubMed] [Google Scholar]
- 121.Del VCM, Gehlbach PL. PPAR-alpha Ligands as Potential Therapeutic Agents for Wet Age-Related Macular Degeneration. PPAR Res. 2008;2008:821592. doi: 10.1155/2008/821592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Luquet S, Gaudel C, Holst D, et al. Roles of PPAR delta in Lipid Absorption and Metabolism: A New Target for the Treatment of Type 2 Diabetes. Biochim Biophys Acta. 2005;1740:313–317. doi: 10.1016/j.bbadis.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 123.Hatanaka H, Koizumi N, Okumura N, et al. Epithelial-Mesenchymal Transition-Like Phenotypic Changes of Retinal Pigment Epithelium Induced by TGF-beta Are Prevented by PPAR-gamma Agonists. Invest Ophthalmol Vis Sci. 2012;53:6955–6963. doi: 10.1167/iovs.12-10488. [DOI] [PubMed] [Google Scholar]
- 124.Rodrigues GA, Maurier-Mahe F, Shurland DL, et al. Differential Effects of PPARgamma Ligands on Oxidative Stress-Induced Death of Retinal Pigmented Epithelial Cells. Invest Ophthalmol Vis Sci. 2011;52:890–903. doi: 10.1167/iovs.10-5715. [DOI] [PubMed] [Google Scholar]
- 125.Choudhary M, Malek G. A Brief Discussion on Lipid Activated Nuclear Receptors and Their Potential Role in Regulating Microglia in Age-Related Macular Degeneration (AMD) Adv Exp Med Biol. 2016;854:45–51. doi: 10.1007/978-3-319-17121-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Morrison MA, Silveira AC, Huynh N, et al. Systems Biology-Based Analysis Implicates a Novel Role for Vitamin D Metabolism in the Pathogenesis of Age-Related Macular Degeneration. Hum Genomics. 2011;5:538–568. doi: 10.1186/1479-7364-5-6-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ayala-Pena VB, Pilotti F, Volonte Y, et al. Protective Effects of Retinoid X Receptors on Retina Pigment Epithelium Cells. Biochim Biophys Acta. 2016;1863:1134–1145. doi: 10.1016/j.bbamcr.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 128.Qin Q, Knapinska A, Dobri N, et al. In Pursuit of Synthetic Modulators for the Orphan Retina-Specific Nuclear Receptor NR2E3. J Ocul Pharmacol Ther. 2013;29:298–309. doi: 10.1089/jop.2012.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Edwards DR, Gallins P, Polk M, et al. Inverse Association of Female Hormone Replacement Therapy with Age-Related Macular Degeneration and Interactions with ARMS2 Polymorphisms. Invest Ophthalmol Vis Sci. 2010;51:1873–1879. doi: 10.1167/iovs.09-4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vojnikovic B, Spanjol J. Prednisolone Neuroprotective Therapy in Age-Related Macular Degeneration. Coll Antropol. 2007;31(Suppl 1):69–70. [PubMed] [Google Scholar]