

Abstract
Regulatory agencies require testing of chemicals and products to protect workers and consumers from potential eye injury hazards. Animal screening, such as the rabbit Draize test, for potential environmental toxicants is time-consuming and costly. Therefore, virtual screening using computational models to tag potential ocular toxicants is attractive to toxicologists and policy makers. We have developed quantitative structure–activity relationship (QSAR) models for a set of small molecules with animal ocular toxicity data compiled by the National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods. The data set was initially curated by removing duplicates, mixtures, and inorganics. The remaining 75 compounds were used to develop QSAR models. We applied both k nearest neighbor and random forest statistical approaches in combination with Dragon and Molecular Operating Environment descriptors. Developed models were validated on an external set of 34 compounds collected from additional sources. The external correct classification rates (CCR) of all individual models were between 72 and 87%. Furthermore, the consensus model, based on the prediction average of individual models, showed additional improvement (CCR = 0.93). The validated models could be used to screen external chemical libraries and prioritize chemicals for in vivo screening as potential ocular toxicants.
Graphical abstract
INTRODUCTION
The Draize test has been used as a standard testing protocol to evaluate ocular toxic potential of chemicals since it was developed in the 1940s.1 In this test, chemicals are applied to rabbit eyes, and the ocular responses are scored based on the damage to the cornea, iris, and conjunctiva. The Draize test has been applied by different regulatory agencies and pharmaceutical companies to evaluate the ocular toxicity of chemicals.2 Different agencies have their own scoring system to define the ocular toxicants. For example, the United Nation Globally Harmonized System (GHS),3 the U.S. Environmental Protection Agency (U.S. EPA) classification system,4 and the European Union (EU) classification system5 are three major regulatory criteria used for ocular hazard classification based on the Draize test results.
As one of the animal test protocols, the Draize test has the common disadvantages of other animal tests, such as being expensive and time-consuming. Furthermore, because the test rabbits need to be euthanized if the test uses irreversible damage to the eyes, the Draize test has been criticized for its cruelty.2 For this reason, alternative methods to evaluate the chemical ocular toxicity are in high demand. Since the 1990s, substantial efforts have been made to develop alternative in vitro methods to reproduce and predict eye irritation responses in the Draize test. For example, the National Toxicology Program (NTP) Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) and the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) gathered various in vitro methods to evaluate chemical ocular toxicity. The NICEATM and ICCVAM have also executed validating studies to assess the reliability of these testing protocols.6 Five of these methods were recently recommended by ICCVAM as potential alternatives to the Draize test.7 However, currently available in vitro toxicity assays have several limitations: they all require physical samples of compounds for testing, and despite significant technical advances of the ICCVAM ocular toxicity assays, they require animal eyes as the testing tissues and still remain time-consuming and resource-intensive.
As compared to experimental testing protocols, the computational tools that could be used to evaluate potential chemical toxicity are almost of no cost and applicable for virtual compounds before they are synthesized. Quantitative structure–activity relationship (QSAR) analysis is a widely used computational method to generate models and predict the toxicity of chemicals. In QSAR studies, the quality of the resulting models strongly depends on the chemical descriptors and the modeling approaches that are employed. Early efforts in the QSAR modeling of chemical ocular toxicity were based on the simple linear regression method and empirical descriptors (e.g., physicochemical properties).8,9 The models of this type are easy to implement and explain due to their simplicity, but their utility is limited to compounds that are highly similar to the modeling set. Later on, more sophisticated modeling approaches and descriptors have also been applied in this area. For example, Hopfinger and his co-workers used a membrane-simulated model to study ocular toxicity.10–13 They identified a set of empirical descriptors that strongly correlate to cornea permeability. Then, the same descriptor pool was used to develop an eye irritation model. For a summary of modeling studies in this area, please see the previously published reviews.14,15
Because of the limited availability of ocular toxicity data, very few of the previously published models were validated on external compounds. Most of the studies were based on the Draize test data instead of considering regulatory scoring systems. Furthermore, because only one type of descriptor and one modeling approach was used in most of the previous studies, the direct comparison of models is not possible. This paper addresses these challenges by applying the combinatorial QSAR (combi-QSAR) approach16 to an ocular toxicity data set recently compiled by ICCVAM. The compounds in this data set have been extensively studied by different regulatory agencies. We used several different combinations of various chemical descriptors and modeling approaches (so-called combi-QSAR approach). All of the resulting models were validated on a separately compiled external set. Previous combi-QSAR studies16–18 suggest that it is impossible to decide a priori which particular combination of a modeling method and a descriptor set will prove most successful. Thus, the consensus model (i.e., averaging of the results of all individual combi-QSAR models) is the best alternative, which usually outperforms individual models.16–18 In this study, the consensus model has clear improvement in predictivity and coverage. We expect to use the resulting models as a virtual screening tool to prioritize new compounds for future experimental testing.
MATERIALS AND METHODS
Data Sets
The ocular toxicity data set used in this study was obtained from the ICCVAM.6 The original data set contains the ocular toxicity results of 232 agents. All of the agents have been tested by the Draize test and have scores from at least one of the three regulatory scoring systems (GHS, U.S. EPA, and EU). However, about 50% of the agents are duplicates or substances without defined chemical structures. Furthermore, inorganic compounds and mixtures cannot be properly represented by chemical descriptors. After these compounds were removed as unsuitable for modeling, we had 75 unique organic compounds remaining out of the original ICCVAM data set.
In different regulatory scoring systems, different terminologies were used to represent the categories of ocular toxicants. Generally, the compounds could be classified as (1) severe ocular toxicants (category 1 of GHS, category I of U.S. EPA, and R41 of EU); (2) moderate ocular toxicants (category 2A of GHS, category II of U.S. EPA, and R36 of EU); (3) light ocular toxicants (category 2B of GHS and category III of U.S. EPA); and (4) nontoxicants (nonirritant of GHS, category IV of U.S. EPA, and nonirritant of EU). Because the “light ocular toxicants” defined by GHS (category 2B) and U.S. EPA (category III) are mostly “nonirritant” in EU results (except N-octanol in our data set), it is reasonable to define the severe and moderate ocular toxicants as “actives” and the light ocular toxicants and nontoxicants as “inactives”. On the basis of this rule, we defined a composite category for this study out of the three scoring systems (Table 1).
Table 1.
Transformation of Different Regulatory Scoring Systems of Chemical Ocular Toxicity into Binary Composite Classifications in This Study
| GHS | U.S. EPA | EU | composite | |
|---|---|---|---|---|
| severe ocular toxicants | category 1 | category I | R41 | “toxic” (active) |
| moderate ocular toxicants | category 2A | category II | R36 | “toxic” (active) |
| light ocular toxicants | category 2B | category III | “nontoxic” (inactive) | |
| nontoxicants | nonirritant | category IV | nonirritant | “nontoxic” (inactive) |
Using the composite classification as defined above, the ocular toxicity results from these three scoring systems are highly correlated with each other. Among the 75 ICCVAM compounds, 30 compounds are active, and 40 compounds are inactive in all of the sources (100 and 0% active data ratio, respectively, in Figure 1). The only five compounds that have conflicting results we defined as “actives” since all of them have at least one “moderate toxic” result (Figure 1). This 75 compound data set was then used to develop the QSAR models in this study.
Figure 1.

Experimental animal ocular toxicity results for 75 ICCVAM compounds.
From the previous ocular toxicity studies, we compiled an external set from two reports. Takahashi and his co-workers recently studied the correlation between the results of a short time bioassay and the GHS scores for a small set of compounds.19 Within this data set, 21 out of 44 compounds are not in the ICCVAM data set. The GHS scores of these 21 new compounds could be classified as 17 actives and four inactives based on the criteria mentioned above. In another study, Kulkarni and his co-workers developed a predictive ocular toxicity model of 37 compounds with their molar adjusted eye scores calculated from relevant Draize test results.10 There are 13 out of 37 compounds that are new to us, and all of them are inactives based on our definitions. As a result, we have a 34 compound external set (17 actives and 17 inactives) that could be used to validate our resulting models since all of these compounds do not exist in the ICCVAM data set and are “unknown” to the resulting models. The compounds in the modeling and external sets are available in Tables 1 and 2 in the Supporting Information.
Chemical Descriptors
Chemical ocular toxicity models for the 75 ICCVAM compounds were developed with various types of 2D chemical descriptors, from Dragon (version 6)20 and MOE (version 2011).21 The types of MOE descriptors include topological indices, structural keys, E-state indices, physical properties (such as Log P, molecular weight, and molar refractivity), and topological polar surface area. The types of Dragon descriptors included in this study are E-state values and E-state counts, constitutional descriptors, topological descriptors, walk and path counts, connectivity and information indices, 2D autocorrelations, Burden eigenvalues, molecular properties (such as the octanol–water partition coefficient), Kappa, hydrogen bond acceptor/donor counts, molecular distance edge, molecular fragment counts, and chemical fingerprints. There are overlaps between Dragon descriptors and MOE descriptors, but both included unique types of descriptors as well. Initially, Dragon software yielded over 2000 chemical descriptors for the training set. Redundant descriptors were identified by analyzing correlations coefficients between all pairs of descriptors, and if the correlation coefficient between two descriptors was higher than 0.99, one of them was randomly removed. As a result, the total number of Dragon descriptors used in model building was reduced to 493. Because the number of MOE descriptors is much less than the Dragon descriptors, we used all 186 available MOE descriptors in our modeling process.
Universal Statistical Figures of Merit for All Models
Because we employed different modeling approaches and different descriptors in the modeling process (described below), universal statistical metrics are needed to evaluate the performance of models developed independently for the ICCVAM set. To harmonize the results of this study, we used sensitivity (the percentage of experimental toxicants that are predicted correctly), specificity (the percentage of experimental nontoxicants that are predicted correctly), and correct classification rate (CCR) to evaluate the predictions. These parameters are defined as follows:
| (1) |
| (2) |
| (3) |
k Nearest Neighbor (kNN)
The kNN QSAR method22 employs the kNN classification principle and the variable selection procedure. Briefly, a subset of nvar (number of selected variables) descriptors is selected randomly at the onset of the calculations. The nvar is set to different values, and the training set models are developed with leave-one-out cross-validation, where each compound is eliminated from the training set and its ocular toxicity is predicted as the average activity of k most similar molecules where the value of k is optimized as well (k = 1–5). The similarity is characterized by Euclidean distance between compounds in multidimensional descriptor space. A method of simulated annealing with the Metropolis-like acceptance criteria is used to optimize the selection of variables. The objective of this method is to obtain the best leave-one-out cross-validated (LOO-CV) CCR possible by optimizing the nvar and k. Additional details can be found elsewhere.22
Following our general QSAR modeling workflow methodology,23 all of the kNN models were extensively validated. The modeling compounds were divided multiple times into training/test sets using the Sphere Exclusion approach.24 The statistical significance of the models was characterized with the standard LOO-CV CCR for the training sets and the conventional CCR for the test sets. The model acceptability cutoff values of the LOO-CV accuracy of the training sets and the prediction accuracy for test sets were both set at 0.7. Models that did not meet both training and test set cutoff criteria were discarded. The discussion of the workflow used to develop validated QSAR models is given in a recent review.25
Random Forest (RF)
In machine learning, a RF is a predictor that consists of many decision trees and outputs the prediction that combines outputs from individual trees. The algorithm for inducing a random forest was developed by Breiman and Cutler.26 In this study, the implementation of the random forest algorithm available in R.2.15.127 was used. In the RF modeling procedure, n samples are randomly drawn from the original data. These samples were used to construct n training sets and to build n trees. For each node of the tree, m variables were randomly chosen from the all of the available chemical descriptors (e.g., the 493 Dragon descriptors). The best data split was calculated using the m variables for the modeling set. In this study, only the defined parameters (n = 500 and m = 13) were used for model development.
Combinatorial QSAR
The whole workflow of the modeling process is shown in Figure 2. The individual classification models were developed using the combination of one type of descriptors (Dragon or MOE) and one type of modeling tools (kNN or RF), resulting in four different models: Dragon-RF (D-R), Dragon-kNN (D-k), MOE-RF (M-R), and MOE-kNN (M-k). The consensus model was generated as the average of the prediction values of the individual models.
Figure 2.

Combinatorial QSAR modeling workflow.
RESULTS AND DISCUSSION
Overview of the ICCVAM and External Data Sets
We could analyze the chemical space of our data set, including both ICCVAM and external compounds, by performing principle component analysis (PCA) of the chemical descriptors used in this study. After PCA with the 186 MOE descriptors for all of the compounds in both modeling (ICCVAM) and external validation sets, we selected the first three most important components to generate a three-dimensional plot (Figure 3) for these 109 (75 modeling and 34 validation) compounds. This plot could be viewed as the chemical space covered by the compounds used in this study. We noticed two structural outliers in our data. The one in the modeling set is polyethylene glycol 400 (CAS Registry Number: 25322-68-3), widely used in a variety of pharmaceutical formulations due to its low toxicity. However, although it is a low molecular weight grade of polyethylene glycol, there is no other similar polymer in our data set. Similarly, there is a structural outlier, Acid red 92 (CAS Registry Number: 18472-87-2), existing in our external validation set. Excluding structural outliers from the modeling set may improve robustness of QSAR models,17 while outliers in the external set should be detected by model's applicability domain.18,28 This study, however, is limited by the relatively small size of the modeling set and could not demonstrate the advantage of using applicability domain (data not shown). However, when additional data becomes available in the future, this critical issue needs to be considered to develop enhanced models.
Figure 3.

Chemical space of the ICCVAM modeling set (purple, n = 75) and external validation set (red, n = 34) shown as first three principle components (57% explained variance) of 186 two-dimensional MOE descriptors using MOE 2011.
Model Characteristics
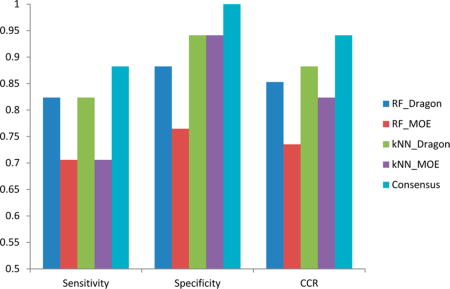
The prediction results of all four individual models and the consensus model for the 34 external compounds (17 toxicants and 17 nontoxicants) are shown in Figure 4. The sensitivity, specificity, and CCR were 0.82, 0.82–0.92, and 0.85–0.88, respectively, for all four individual models, respectively. Only the RF-MOE model has relatively low accuracy (CCR = 0.73). Because the number of nontoxicants is higher than the toxicants in our modeling set (40 nontoxicants vs 35 toxicants), the prediction accuracy of the external nontoxicants (specificity) by all of the resulting models is somewhat higher than the predictivity of the external toxicants (sensitivity) (Figure 4). In several of our previous studies, we indicated that a consensus model (based on the predictions averaged over all available individual models) has better performance when compared to most individual modes.17,18,28,29 Moreover, for the consensus model, there is no need to decide which model to use out of several equally performing ones. In this study, our consensus model was based on the average of all four individual models. The prediction result of the external set shows that the consensus model has better predictivity for both toxicants (sensitivity of 0.88) and nontoxicants (specificity of 1) (Figure 4).
Figure 4.

Performance on external set (n = 34) of four individual QSAR models and their consensus model.
To compare the resulting models with existing computational tools, we have used the eye irritation module of the current Organisation for Economic Co-operation and Development (OECD) QSAR Toolbox30 to predict the same external compounds. There are only two external compounds, 2-methyl-1-pentanol and 3-methoxy-1,2-propanediol, that were identified as having structural alerts of ocular toxicity. The 2-methyl-1-pentanol is toxic, but the 3-methoxy-1,2-propanediol is actually nontoxic. The remaining compounds were identified as having “undefined functional groups”. For this reason, the prediction results of external compounds by our models are better than the results obtained from the OECD QSAR Toolbox eye irritation module.
Interpretation of Predictive QSAR Models
To obtain relevant toxicity mechanisms from our modeling results, we chose to analyze Dragon descriptors for their diversity and kNN models for their ease of interpretation (by frequency of descriptor occurrence). Several descriptors were found to be most frequently used in the kNN models that satisfied our criterions (CCR > 0.7 for both training and relevant test sets), suggesting that they may play a critical role in predicting ocular toxicity of organic compounds. The most important Dragon descriptors used in the kNN modeling approach are shown in Table 2, along with their frequencies of occurrence in acceptable models and their interpretations (Table 2). The first important descriptor refers to alcohols, which may cause mild to severe eye irritation.31–34 There are three druglike index descriptors that are considered to be important in our models. These types of descriptors were created based on Ghose–Viswanadhan–Wendoloski's (GVW) consensus definitions of drug classes.35 They identified drug classes using calculated physiochemical properties. Many pharmaceutical drugs have eye-related side effects, including dry eye, pupil dilation, retinal damage, and reduced acuity.36 The descriptor 3 represents anti-infective drugs. Within this drug category, the ophthalmic solutions have been known to cause acute corneal epithelial cell membrane damage.37 Antidepressant drugs, which are represented by the descriptor 6, could induce ocular phototoxicity.38 The patients on antidepressants or undergoing neurological therapy are reported to be susceptible to retinal irritation and toxicity.38–40 There is no evidence about the relationship between ocular toxicity and esters (descriptor 4, Table 2). On the contrary, the esters are reported to be used to treat ocular infections.41 In our model, it could be considered as a negative modulator. As compared to esters, the carboxylic acids, which are represented by descriptor 8, can cause burns on the eyes.42 The remaining two descriptors (descriptors 5 and 7, Table 2) have obscure relationships with chemical ocular toxicity. Although ocular toxicity was observed with the amines (e.g., triethylamine43), this effect is most likely due not to a specific property of the chemical but rather to the alkaline nature of aliphatic amines.
Table 2.
Most Important Dragon Chemical Descriptors Used To Predict Chemical Ocular Toxicity in kNN QSAR Models
| No. | Descriptor Name |
Description | Illustration | Freq. | Z- Score |
|---|---|---|---|---|---|
| 1 | nROH | number of alcohol groups |
|
3.4 | 6.7 |
| 2 | GVWAI-80 | Consensus Definition of Drug-Like-Molecule | AlogP = −0.4 – 5.6; molar refractivity = 40 – 130; molecular weight = 160 – 480; atom number = 20 – 70 | 1.9 | 3.3 |
| 3 | Infective-80 | Anti-infective drug like index at 80% | AlogP = −0.3 – 5.1; molar refractivity = 44 – 144; molecular weight = 145 – 455; atom number = 12 – 64 | 1.5 | 3.1 |
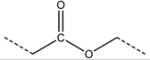
| 4 | nRCOOR | number of esters (aliphatic) |
|
1.4 | 2.2 |
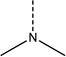
| 5 | N-075 | R--N—X or R--N--X (number of nitrogen atoms with two substitutients) |
|
1.2 | 1.8 |
| 6 | Depressant-80 | GVW anti-depressant drug like index at 80% | AlogP = 1.4 – 4.9; molar refractivity = 62 – 114; molecular weight = 210 – 380; atom number = 32 – 56 | 1.1 | 1.7 |
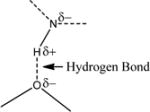
| 7 | nHDon | number of donor atoms for H-bonds (N and O) |
|
1.1 | 1.7 |
| 8 | nRCOOH | number of carboxylic acids (aliphatic) |
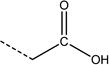
|
1.1 | 1.7 |
Advantages and Pitfalls of the Consensus Model
To stress the advantage of the consensus model using the combi-QSAR technique, it should be made clear that from the viewpoint of toxicologists and other biologists that QSAR modeling is always a tool to analyze how the change of functional groups affects biological activity for a generic set of compounds. On the basis of this hypothesis, most of the traditional QSAR studies used a single modeling approach to develop a single model based on one type of descriptors. The assumption that “similar structures yield similar properties” runs contrary to the use of the traditional QSAR workflow for data sets with diverse compounds. As compared to traditional data mining and data modeling procedures (e.g., modeling by using one statistical tool and one type of descriptors), this study focuses on prediction based on a combination of various types of models (by using different statistical tools and different types of chemical descriptors). Consensus modeling based on the combi-QSAR workflow will take advantage of the output from each individual model and utilize most optimally the chemical and/or biological information of the diverse chemical data set. On the other hand, the combi-QSAR modeling is more time-consuming than the development of a single model.
It needs to be noted that the prediction values of all individual models and that of the consensus model range from 0 to 1, and we set the classification threshold as 0.5 to determine toxic and nontoxic predictions. Table 3 shows the experimental and predicted ocular toxicity results of several sample compounds from the external set. In most cases, the consensus model could compensate for the errors from individual models (e.g., the predictions of compounds #2 and #3). However, the last compound was predicted incorrectly by all individual models, and the consensus model failed as well. In this case, our modeling results indicate that we are lacking necessary information in the modeling set for compounds with such a chemical scaffold. Therefore, compounds similar to compound #4 in Table 3 should be tested experimentally with higher priority in the future as this would allow knowledge gaps to be addressed and for the improvement of computational models.
Table 3.
Experimental and Predicted Ocular Toxicity Results of Four External Compoundsa
| No. | Compounds | Exp. Results |
Pred- 1 |
Pred- 2 |
Pred- 3 |
Pred- 4 |
Pred- consensus |
|---|---|---|---|---|---|---|---|
| 1 |
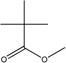
|
Non-toxic | Non-toxic (0.48) | Non-toxic (0.28) | Toxic (0.58) | Non-toxic (0.27) | Non-toxic (0.40) |
| 2 |
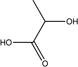
|
Toxic | Toxic (0.76) | Toxic (0.70) | Toxic (0.74) | Non-toxic (0.16) | Toxic (0.60) |
| 3 |

|
Toxic | Toxic (0.68) | Non-toxic (0.16) | Toxic (0.88) | Non-toxic (0.48) | Toxic (0.55) |
| 4 |
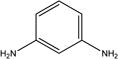
|
Toxic | Non-toxic (0.19) | Non-toxic (0.41) | Non-toxic (0.21) | Non-toxic (0.03) | Non-toxic (0.21) |
The prediction results are determined by the actual prediction values in the parentheses (lower than 0.5 is nontoxic, and above 0.5 is toxic).
CONCLUSIONS
In this study, several QSAR approaches have been used to develop predictive models of the largest publicly available regulatory ocular toxicity set of diverse organic compounds. Every compound in this data set has been extensively evaluated for its eye irritation effect by the Draize test. Three regulatory agencies categorized these compounds using their own scoring system based on Draize test results. We used a composite score to classify all of the compounds into ocular toxicants and nontoxicants. This composite score has high correlation with all three regulatory scoring systems. The resulting models were validated by predicting the toxicity of an external validation set compiled from different sources. It was observed that all models showed comparable performance for the external validation set. The most significant result of our studies is the demonstrated superior performance of the consensus modeling approach when all models are used concurrently and predictions from individual models are averaged. The predictive accuracy of the consensus QSAR models was shown to be superior to any individual models when predicting the same set of external compounds. The models reported in this paper could be used to evaluate the ocular toxicity of new organic compounds. Meanwhile, we invite all interested researchers to send us compounds for ocular toxicity prediction.
Supplementary Material
Acknowledgments
Funding
The work was supported by the Society of Toxicology (Grant no. 11-0897).
We thank Kimberlee Moran of Rutgers-Camden for her help with the manuscript preparation and Dr. Judy Strickland of ILS, Inc., for her help and comments on the entire project.
ABBREVIATIONS
- GHS
Globally Harmonized System
- U.S. EPA
U.S. Environmental Protection Agency
- EU
European Union
- NTP
National Toxicology Program
- NICEATM
NTP Interagency Center for the Evaluation of Alternative Toxicological Methods
- ICCVAM
Interagency Coordinating Committee on the Validation of Alternative Methods
- QSAR
quantitative structure–activity relationship
- combi-QSAR
combinatorial QSAR
- CCR
correct classification rate
- LOO-CV
leave-one-out cross-validated
- kNN
k nearest neighbor
- RF
random forest
- PCA
principle component analysis
- OECD
Organisation for Economic Co-operation and Development
Footnotes
ASSOCIATED CONTENT
Tables of compounds in the modeling and external sets. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Draize JH, Woodard G, Calvery HO. Methods for the study of irritation and toxicity of substances applied topically to the skin and mucous membranes. J. Pharmacol. Exp. Ther. 1944;82:377–390. [Google Scholar]
- 2.Wilhelmus KR. The Draize eye test. Surv. Ophthalmol. 2001;45:493–515. doi: 10.1016/s0039-6257(01)00211-9. [DOI] [PubMed] [Google Scholar]
- 3.United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS) United Nations Publications; New York & Geneva: 2007. [Google Scholar]
- 4.Office of Prevention, P. &. S. O., editor. EPA. Label Review Manual: EPA735-B-03-001. U.S. Environmental Protection Agency; Washington, DC: 2012. [Google Scholar]
- 5.European Union. Commission Directive 2001/59/EC of 6 August 2001 adapting to technical progress for the 28th time Council Directive 67/548/EEC on the approximation of the laws, regulations and administrative provisions relating to the classification, packaging and labelling of dangerous substances. Official Journal of the European Communities; 2001. pp. 1–333. [Google Scholar]
- 6.ICCVAM and NICEATM. ICCVAM Test Method Evaluation Report: Current Validation Status of In Vitro Test Methods Proposed for Identifying Eye Injury Hazard Potential of Chemicals and Products, NIH Document No. 10-7553. National Toxicology Program; Research Triangle Park, NC: 2010. [Google Scholar]
- 7.ICCVAM and NICEATM. Independent Scientific Peer Review Panel Report: Evaluation of the Validation Status of Alternative Ocular Safety Testing Methods and Approaches. National Toxicology Program; Research Triangle Park, NC: 2009. [Google Scholar]
- 8.Abraham MH, Kumarsingh R, Cometto-Muniz JE, Cain WS. Draize eye scores and eye irritation thresholds in man can be combined into one QSAR. Ann. N.Y. Acad. Sci. 1998;855:652–656. doi: 10.1111/j.1749-6632.1998.tb10641.x. [DOI] [PubMed] [Google Scholar]
- 9.Abraham MH, Kumarsingh R, Cometto-Muniz JE, Cain WS. A quantitative structure-activity relationship (QSAR) for a draize eye irritation database. Toxicol. In Vitro. 1998;12:201–207. doi: 10.1016/s0887-2333(97)00117-3. [DOI] [PubMed] [Google Scholar]
- 10.Kulkarni A, Hopfinger AJ, Osborne R, Bruner LH, Thompson ED. Prediction of eye irritation from organic chemicals using membrane-interaction QSAR analysis. Toxicol. Sci. 2001;59:335–345. doi: 10.1093/toxsci/59.2.335. [DOI] [PubMed] [Google Scholar]
- 11.Kulkarni AS, Hopfinger AJ. Membrane-interaction QSAR analysis: application to the estimation of eye irritation by organic compounds. Pharm. Res. 1999;16:1245–1253. doi: 10.1023/a:1014853731428. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Liu J, Pan D, Hopfinger AJ. A study of the relationship between cornea permeability and eye irritation using membrane-interaction QSAR analysis. Toxicol. Sci. 2005;88:434–446. doi: 10.1093/toxsci/kfi319. [DOI] [PubMed] [Google Scholar]
- 13.Patel HC, Duca JS, Hopfinger AJ, Glendening CD, Thompson ED. Quantitative component analysis of mixtures for risk assessment: application to eye irritation. Chem. Res. Toxicol. 1999;12:1050–1056. doi: 10.1021/tx990098z. [DOI] [PubMed] [Google Scholar]
- 14.Somps CJ, Greene N, Render JA, Aleo MD, Fortner JH, Dykens JA, Phillips G. A current practice for predicting ocular toxicity of systemically delivered drugs. Cutaneous Ocul. Toxicol. 2009;28:1–18. doi: 10.1080/15569520802618585. [DOI] [PubMed] [Google Scholar]
- 15.Patlewicz G, Rodford R, Walker JD. Quantitative structure-activity relationships for predicting skin and eye irritation. Environ. Toxicol. Chem. 2003;22:1862–1869. doi: 10.1897/01-439. [DOI] [PubMed] [Google Scholar]
- 16.Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, Gramatica P, Oberg T, Dao P, Cherkasov A, Tetko IV. Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. J. Chem. Inf. Model. 2008;48:766–784. doi: 10.1021/ci700443v. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H, Martin TM, Ye L, Sedykh A, Young DM, Tropsha A. Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure. Chem. Res. Toxicol. 2009;22:1913–1921. doi: 10.1021/tx900189p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Zhu H, Oprea TI, Golbraikh A, Tropsha A. QSAR modeling of the blood-brain barrier permeability for diverse organic compounds. Pharm. Res. 2008;25:1902–1914. doi: 10.1007/s11095-008-9609-0. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi Y, Hayashi T, Watanabe S, Hayashi K, Koike M, Aisawa N, Ebata S, Sakaguchi H, Nakamura T, Kuwahara H, Nishiyama N. Inter-laboratory study of short time exposure (STE) test for predicting eye irritation potential of chemicals and correspondence to globally harmonized system (GHS) classification. J. Toxicol. Sci. 2009;34:611–626. doi: 10.2131/jts.34.611. [DOI] [PubMed] [Google Scholar]
- 20.DRAGON. DRAGON for Windows (Software for Molecular Descriptor Calculations) Talete s.r.l.; Milan (Italy): 2011. [Google Scholar]
- 21.Chemical Computing Group. MOE. Chemical Computing Group; Montreal, Quebec, Canada: 2011. [Google Scholar]
- 22.Zheng W, Tropsha A. Novel variable selection quantitative structure–property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci. 2000;40:185–194. doi: 10.1021/ci980033m. [DOI] [PubMed] [Google Scholar]
- 23.Tropsha A, Golbraikh A. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 2007;13:3494–3504. doi: 10.2174/138161207782794257. [DOI] [PubMed] [Google Scholar]
- 24.Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput.-Aided Mol. Des. 2003;17:241–253. doi: 10.1023/a:1025386326946. [DOI] [PubMed] [Google Scholar]
- 25.Tropsha A, Golbraikh A. Predictive QSAR Modeling Workflow, Model Applicability Domains, and Virtual Screening. Curr. Pharm. Des. 2007;13:3494–3504. doi: 10.2174/138161207782794257. [DOI] [PubMed] [Google Scholar]
- 26.Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 27.Dalgaard P. Introductory Statistics with R. Springer; New York: 2008. [Google Scholar]
- 28.Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, Gramatica P, Oberg T, Dao P, Cherkasov A, Tetko IV. Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. J. Chem. Inf. Model. 2008;48:766–784. doi: 10.1021/ci700443v. [DOI] [PubMed] [Google Scholar]
- 29.Tetko IV, Sushko I, Pandey AK, Zhu H, Tropsha A, Papa E, Oberg T, Todeschini R, Fourches D, Varnek A. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: Focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 2008;48:1733–1746. doi: 10.1021/ci800151m. [DOI] [PubMed] [Google Scholar]
- 30.OECD (Q)SAR Application Toolbox. http://www.oecd.org/chemicalsafety/assessmentofchemicals/theoecdqsartoolbox.htm.
- 31.Ahn JH, Eum KH, Kim YK, Oh SW, Kim YJ, Lee M. Assessment of the dermal and ocular irritation potential of alcohol hand sanitizers containing aloe vera with in vitro and in vivo methods. Mol. Cell. Toxicol. 2010;6:401–408. [Google Scholar]
- 32.aaJester JV, Li L, Molai A, Maurer JK. Extent of initial corneal injury as a basis for alternative eye irritation tests. Toxicol. In Vitro. 2001;15:115–130. doi: 10.1016/s0887-2333(00)00065-5. [DOI] [PubMed] [Google Scholar]
- 33.Zhang P, Liu M, Liao RF. Toxic effect of using twenty percent alcohol on corneal epithelial tight junctions during LASEK. Mol. Med. Rep. 2012;6:33–38. doi: 10.3892/mmr.2012.880. [DOI] [PubMed] [Google Scholar]
- 34.Okamoto Y, Ohkoshi K, Itagaki H, Tsuda T, Kakishima H, Ogawa T, Kasai Y, Ohuchi J, Kojima H, Kurishita A, Kaneko T, Matsushima Y, Iwabuchi Y, Ohno Y. Interlaboratory validation of the in vitro eye irritation tests for cosmetic ingredients. (3) evaluation of the haemolysis test. Toxicol. In Vitro. 1999;13:115–124. doi: 10.1016/s0887-2333(98)00066-6. [DOI] [PubMed] [Google Scholar]
- 35.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 36.Jaanus SD. Ocular side effects of selected systemic drugs. Optom. Clin. 1992;2:73–96. [PubMed] [Google Scholar]
- 37.Matsumoto S, Stern ME. Effect of anti-infective ophthalmic solutions on corneal cells in vitro. Adv. Ther. 2000;17:148–151. doi: 10.1007/BF02853156. [DOI] [PubMed] [Google Scholar]
- 38.Wang RH, Dillon J, Reme C, Whitt R, Roberts JE. The potential ocular phototoxicity of antidepressant drugs. Lens Eye Toxic. Res. 1992;9:483–491. [PubMed] [Google Scholar]
- 39.Hadjikoutis S, Morgan JE, Wild JM, Smith PE. Ocular complications of neurological therapy. Eur. J. Neurol. 2005;12:499–507. doi: 10.1111/j.1468-1331.2005.01025.x. [DOI] [PubMed] [Google Scholar]
- 40.Siu TL, Morley JW, Coroneo MT. Toxicology of the retina: Advances in understanding the defence mechanisms and pathogenesis of drug- and light-induced retinopathy. Clin. Exp. Ophthalmol. 2008;36:176–185. doi: 10.1111/j.1442-9071.2008.01699.x. [DOI] [PubMed] [Google Scholar]
- 41.Katragadda S, Gunda S, Hariharan S, Mitra AK. Ocular pharmacokinetics of acyclovir amino acid ester prodrugs in the anterior chamber: evaluation of their utility in treating ocular HSV infections. Int. J. Pharm. 2008;359:15–24. doi: 10.1016/j.ijpharm.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patnaik P. Acids, Carboxylic. In: Patnaik P, editor. A Comprehensive Guide to the Hazardous Properties of Chemical Substances. Wiley; New York: 2007. pp. 105–114. [Google Scholar]
- 43.Jarvinen P, Engstrom K, Riihimaki V, Ruusuvaara P, Setala K. Effects of experimental exposure to triethylamine on vision and the eye. Occup. Environ. Med. 1999;56:1–5. doi: 10.1136/oem.56.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.