

Abstract
Background
Clinical observations suggest that anaphylaxis is more common in adult women compared to adult men, although the mechanistic basis for this gender bias is not well understood
Objectives
To document gender dependent differences in a mouse model of anaphylaxis and explore the role of female sex hormones and the mechanisms responsible.
Methods
Passive systemic anaphylaxis was induced in female and male mice by histamine, as well as IgE or IgG receptor aggregation. Anaphylaxis was assessed by monitoring body temperature, release of mast cell mediators and/or hematocrit and lung weight as a measure of vascular permeability. A combination of ovariectomy, estrogen receptor antagonism, and estrogen administration techniques were used to establish estrogen involvement.
Results
Anaphylactic responses were more pronounced in female than in male mice. The enhanced severity of anaphylaxis in female mice was eliminated after pretreatment with an estrogen receptor antagonist or ovariectomy, but restored after administration of estradiol in ovariectomized mice, demonstrating that the sex-specific differences are due to the female steroid estradiol. Estrogen did not affect mast cell responsiveness or anaphylaxis onset. Instead, it increased tissue expression of endothelial nitric oxide synthase (eNOS). Blockage of NOS activity with the inhibitor L-NAME or genetic eNOS deficiency abolished the gender-related differences.
Conclusion
Our study defines a contribution of estrogen, through its regulation of eNOS expression and NO production, to vascular hyper-permeability and intensified anaphylactic responses in female mice, providing additional mechanistic insights into risk factors and possible implications for clinical management in the further exploration of human anaphylaxis.
Keywords: Anaphylaxis, estrogen, nitric oxide synthase, vascular permeability, mast cells
INTRODUCTION
A number of clinical studies have indicated gender differences in the incidence of systemic anaphylaxis,1, 2 showing that adult women suffer more frequently from anaphylaxis induced by food,3, 4 drugs,2, 3, 5 and radiocontrast agents6 as well as from idiopathic anaphylaxis7 compared to adult men. The fact that this gender-difference is observed during the reproductive but not the pre-puberty years,3, suggests that sex hormones may be involved in this sexual dimorphism. Additionally, clinical reports of catamenial (or cyclical) anaphylaxis, which is characterized by recurrent episodes of anaphylactic reactions occurring around the time of menstruation,8 suggest that female sex hormones, such as estrogens or progesterone, may be involved in susceptibility to anaphylaxis. However, other than in selected cases of suggested auto-immune progesterone sensitivity9, 10 there is little insight into how sex hormones influence anaphylaxis.
Anaphylaxis is usually triggered by an antigen (Ag) that recognizes and aggregates Ag-specific IgE bound to the high affinity receptor of IgE, FcεRI, in tissue-resident mast cells of sensitized individuals. Binding of Ag to FcεRI leads to the release and generation of various mediators including proteases, lipid-derived molecules, cytokines and histamine.11 These mediators act on the surrounding tissues leading to a number of biologic effects including vasodilation, plasma exudation and edema, causing anaphylaxis in the most severe cases. In experimental mouse models, anaphylaxis can also be elicited through IgE-independent mechanisms, including activation of IgG receptors (FcγR) present on mast cells, basophils, neutrophils and macrophages.12–14 Anaphylaxis in humans may also follow the administration of drugs or radiocontrast agents, whose mechanisms are not fully understood, but are believed to be independent from FcεR.
Here, we explore gender differences in severity of anaphylaxis employing a well-established mouse model of passive systemic anaphylaxis and demonstrate an enhanced severity and duration of anaphylaxis in female mice compared to male mice. The advantage of using this anaphylaxis model in male and female mice is that the differences in severity and duration at given concentrations of IgE and Ag can be attributed to gender and not to confounding factors, including previous exposure to Ag, type of Ag, antigen-specific IgE levels, underlying disease, and treatments that may affect severity and lethality in humans15. Differences in anaphylaxis severity found in this experimental model relate to incidence data only in that more severe reactions in individuals are more likely to be recognized and thus disproportionately contribute to the calculation of the incidence. We find that increased severity of anaphylaxis in female mice is dependent on estradiol-mediated up-regulation of endothelial nitric oxide synthase (eNOS), the enzyme responsible for production of nitric oxide (NO). The mechanisms that underlie human anaphylactic shock are not well understood in part because of the difficulties in designing appropriate clinical studies. However, we are hopeful that the insights from the mouse studies reported in this work will stimulate further research into gender differences and in the role of NO as a biologic mediator in severe systemic allergic reactions.
METHODS
Further details can be found in the online repository.
Mice
Wild type (WT) C57Bl/6, Balb/c and NOS3−/− mice, as well as ovariectomized (OVX) and sham-operated mice were obtained from Jackson Laboratory (Bar Harbor, ME). Mice were used in accordance with NIH guidelines and animal study proposal approved by the NIH/NIAID Institutional Animal Care and Use Committee.
Systemic anaphylaxis
For IgE-induced passive systemic anaphylaxis (PSA), mice were sensitized with 3 μg of DNP-specific IgE and challenged 24 h later with 200 μg of antigen (Ag) i.v. (DNP-HSA, Sigma-Aldrich) in PBS. Anaphylactic responses were determined by measuring core body temperature. Alternatively, anaphylaxis was induced by a bolus of histamine dihydrochloride (5 μmol, Sigma Aldrich) or by 80 μg of anti-mouse CD16/32 2.4G2 clone in PBS i.v.. To investigate the involvement of NOS, mice were injected i.v. with 2.5 mg of L-NG-Nitroarginine Methyl Ester (L-NAME, Sigma-Aldrich) in 100 μl PBS, 1 h prior to DNP-challenge.
In some experiments, mice were euthanized 90 s after the induction of anaphylaxis and blood was collected. Plasma histamine and MCPT-1 levels were measured with specific ELISA kits (Beckman Coulter, Fullerton, CA and eBioscience, San Diego, CA, respectively).
To determine hematocrit levels, blood was collected before and 2 h after anaphylaxis with micro-hematocrit tubes (Jorvet, Loveland, CO) and the ratio of red blood cell volume compared to total volume of blood determined. For determination of lung fluid, left lung lobes were excised 2 h after Ag-injection. Excised lungs were weighed immediately (wet weight) as well as after drying at 55 °C overnight (dry weight) and the wet/dry weight ratio was calculated. The right lung lobes were excised, fixed, and embedded in paraffin. Histological sections of the paraffin embedded lungs were prepared and stained with H&E (American Histolabs).
Estrogen receptor blockage and estradiol administration in female mice
To block estrogen binding to its receptor (ER), mice received 5 injections with 20 mg/kg of the ER antagonist ICI182,780 (Tocris, Ellisville, MO).
To restore estrogen levels in OVX females, slow-releasing pellets (Innovative Research of America, Sarasota, FL) containing either 17β-estradiol (E2; 0.1 mg/pellet, 21 d release) or placebo (0.1 mg/pellet, 21 d release) were implanted and PSA was performed two weeks after implantation.
Statistics
All data are presented as means with SEM, or as boxplots with min/max range. Comparisons of temperature changes between groups were performed by a 2-way ANOVA. Differences between 2 variables were compared with two-tailed Student t test, and between multiple variables with 1-way ANOVA (Graph Pad Prism 4.01; San Diego, CA). A level of P < 0.05 was considered to be significant.
RESULTS
Gender differences in systemic anaphylaxis are estrogen-dependent
Resembling epidemiological studies on systemic anaphylaxis, IgE-mediated passive systemic anaphylaxis (PSA) was more severe in female C57Bl/6 mice than in male mice (Fig 1, A) as determined by a drop in core body temperature, which correlates with increases in plasma histamine, hematocrit values, hypotension and visible behavioral changes.16 The differential severity between age-matched males and females was also observed in Balb/c mice (E Fig1, A, left panel in the online repository) and was unrelated to differences in body weight because a similar difference in PSA was found in weight-matched instead of age-matched females and males (E Fig1, B in the online repository).
Fig 1. Enhanced anaphylaxis in female mice is estrogen dependent.
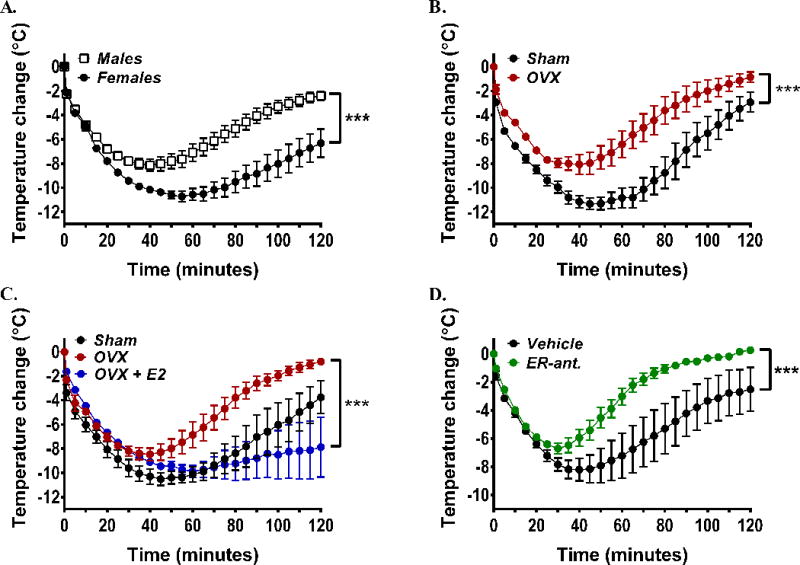
(A) Temperature changes in female and male C57Bl/6 mice during anaphylaxis. Mice were sensitized with DNP-specific IgE and challenged 24 h later with DNP-HSA. Data are from 3 independent experiments of n=6/group. (B–D) Temperature changes during anaphylaxis in ovariectomized (OVX) or sham-operated female mice (B), after implantation in OVX mice of 17β-estradiol-releasing (OVX+E2) or placebo pellets (OVX) (C), and in females after treatment with the estrogen receptor antagonist ICI182,780 (ER-ant) (D). In B–D, n=6 mice/group. *** P<0.001.
We next explored the basis for the gender disparity. Ablation of the major source of sex hormones in female mice by ovariectomy (OVX) resulted in a reduced PSA response compared to sham-operated females and appeared similar to that seen in male mice (Fig 1, B and E Fig 1, B, right panel in the online Repository). We then focused on 17β-estradiol (E2), which is the predominant circulating estrogen in females during the reproductive years.17 Subcutaneous implantation of estradiol-releasing pellets into OVX mice restored the presence of the hormone in circulation (E Fig1, C in the online repository) as well as the severity of the anaphylactic response (Fig 1, C), demonstrating a direct link between the presence of the female hormone estradiol and the gender disparities in anaphylaxis. Furthermore, there was a linear correlation between the circulating levels of estradiol and anaphylaxis severity and duration in female mice (E Fig 1, D in the online repository). Estradiol is known to mediate its actions by binding to the estrogen receptors (ER) α and β.18 Thus, to further confirm the involvement of estradiol in the severity of anaphylaxis, we blocked ER with the ER antagonist ICI182,780 (Fulvestrant).19 Treatment of female mice with ICI182,780 for 5 days was found to reduce the severity of the anaphylaxis, to essentially the response observed in male or OVX female mice (Fig 1 D, as compared to Fig 1, A–B). Taken together, our data point to a role for estradiol in determining the severity of anaphylactic shock in this model.
Estrogen does not affect mast cell responsiveness but promotes vascular leakage during anaphylaxis
The marked differences in PSA between OVX and sham-operated female mice were not accompanied by a difference in serum concentrations of the early released mast cell mediators histamine and mast cell protease-1 (MCPT-1) (Fig 2, A) or cytokines such as TNFα (E Fig 2, A and B in the online repository), suggesting that the effect of estradiol on anaphylaxis is not due to an enhancement of IgE/Ag-mediated mast cell responses. In agreement, degranulation (Fig 2, B) and release of TNFα (E Fig 2, C in the online repository) or IL-6 (data not shown) of mouse bone marrow derived mast cells (BMMCs) in response to maximal or submaximal concentrations of Ag was not affected by a wide range of estradiol concentrations (from physiological to supra-physiological) or by pretreatment with a physiological concentration of estradiol (35 pg/ml) hours to days prior to antigen stimulation (data not shown). Estradiol was also found ineffective in altering degranulation of human basophils,20 but it was reported to enhance IgE/Ag-induced degranulation by about 4% in transformed mast cells (RBL-2H3 and HMC-1).21 We confirmed that ERα, the ER found in mouse mast cells,21, 22 was expressed in BMMCs from male and female mice (Fig 2, B inset) suggesting that the lack of response to estradiol in mast cells is not due to loss of ER expression. Further evidence that the effect of estradiol on anaphylaxis is dissociated from mast cell effector responses comes from the finding that anaphylaxis induced by either a bolus of histamine, a key mediator of IgE/Ag-induced systemic anaphylaxis,16 or by crosslinking FcγRIIIA with 2.4G2, an anaphylaxis model that does not require neither FcεRI nor mast cells,23, 24 was also more severe in sham-operated than in OVX females (Fig 2, C–D, respectively). Collectively, the data suggest that the mechanism for estrogen potentiation on anaphylaxis is subsequent to mast cell mediator release.
Fig 2. Estrogen does not affect mast cell responses.
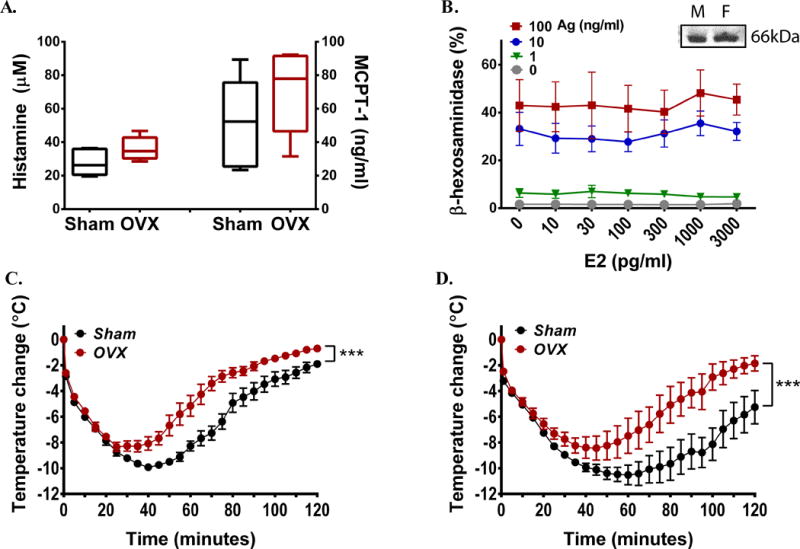
(A) Histamine and MCPT-1 released into circulation 90 s after challenging OVX or sham operated females with Ag (n=5/group). (B) β-hexosaminidase release from BMMC in the presence of different concentrations of estradiol (E2). Results are the average of 4 independent cultures (3 female and 1 male donor). Inset shows ERα expression in BMMCs from male (M) and female (F) mice. (C, D) Temperature changes during anaphylaxis induced by histamine (C) or anti-CD16/32 2.4G2 (D) in OVX and sham-operated mice. In C–D, n=6/group. *** P<0.001.
We hence investigated whether the presence of estrogens may enhance plasma exudation induced by vascular mediators. Hematocrit levels (Fig 3, A) and lung fluid accumulation, as measured by lung wet/dry ratios (Fig 3, B), which are both used to quantify vascular leakage during PSA,25 were significantly higher in females compared to males. Lung histology confirmed a more pronounced peri-bronchial edema as well as abundance of red blood cells in the airspace in female lungs (Fig 3, C). The data suggest that estrogens affect a pathway that regulates vascular permeability.
Fig 3. Presence of female sex hormones leads to increased vascular permeability during anaphylaxis.
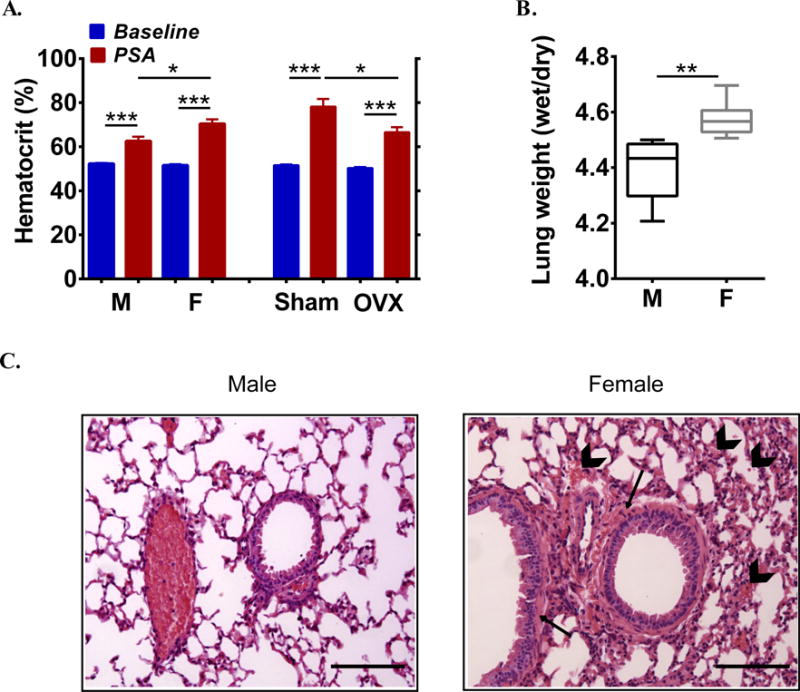
(A) Hematocrit values at baseline and after PSA in males (M), females (F), sham-operated and OVX mice. (B) Wet/dry lung weight ratios of male and female mice 2 h after anaphylaxis. In A–B, n=6/group. (C) Representative H&E stained lung slides from a male and female mouse 2 h after IgE/Ag-induced PSA showing peri-bronchial edema (arrows) and red blood cells accumulated in airspace (arrowheads). Scale bar: 100μm. *P<0.05, **P<0.01, *** P<0.001.
Increased severity to anaphylaxis by estrogen is mediated by eNOS
Endothelial nitric oxide synthase (eNOS) and the product of its activity, nitric oxide (NO), have been reported to regulate vascular permeability in mice induced by vascular endothelial growth factor (VEGF)26 and vascular reactivity in female mice undergoing anaphylaxis.27 As eNOS expression and/or activity are susceptible to estrogen regulation,28–30 we investigated whether levels of this enzyme might relate to the gender-related differences observed in IgE- mediated anaphylaxis in our mouse model. Both lungs (Fig 4, A and B upper panels) and aortas (E Fig3, A in the online repository) of female mice showed higher expression of eNOS than tissues from male or OVX mice, at baseline as well as 10 min after induction of PSA. Although eNOS expression in OVX females was not statistically different from that in males, there was a slightly higher trend in OVX females, which may suggest compensatory mechanisms in OVX mice or a minor participation of non-estrogen factors to the elevated eNOS expression in females compared to males. The finding that treatment of cultured mouse lung endothelial cells (MLEC) with estradiol increased eNOS expression (E Fig2, B in the online repository) further supports the conclusion that the increased eNOS expression in females is indeed mediated through estradiol. Mirroring eNOS levels, phosphorylation of eNOS at Ser1177, which has been shown to enhance eNOS activity,31 was increased in the lungs of female mice as compared to those in male or OVX mice (Fig 4, A and B lower panels). We did not find an increase in the ratio of phospho Ser1177-eNOS to eNOS 10 min after antigen challenge as described after administration of platelet activating factor (PAF),27, 31 a potent and usually lethal anaphylaxis mediator with a minor role in IgE/Ag-induced systemic anaphylaxis in mice,12, 32 which may suggest differences in the mechanism of action or differences in the time course or robustness of eNOS phosphorylation.
Fig 4. Estrogen regulates eNOS expression in vivo.
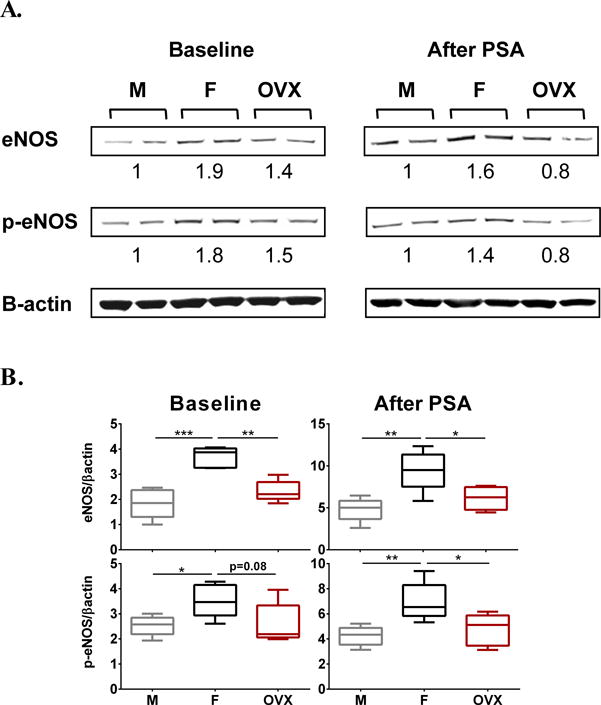
(A, B) Expression levels of eNOS and Ser1177-phosphorylated eNOS (p-eNOS) in female (F), male (M) and OVX female lung lysates at baseline and after IgE/Ag-induced PSA. In A, representative Western blots of lung lysates are shown. Numbers below indicate the fold increases in eNOS or p-eNOS band intensities (corrected to their respective β-actin loading control) as compared to males. In B the band intensitites in lung eNOS and p-eNOS (n=5/group) are shown. *P<0.05, **P<0.01,*** P<0.001. No statistical differences were found in the levels of eNOS or p-eNOS between males and OVX females.
We then examined whether the higher constitutive expression and phosphorylation of eNOS in female tissues relates functionally to the gender bias in anaphylaxis. We first injected the NOS inhibitor, L-NG-Nitroarginine Methyl Ester (L-NAME) 1 h prior to Ag-challenge and found that inhibition of NO production obliterated the differences between females and males in the PSA response (Fig 5, A). Further, the effect of L-NAME was specific to eNOS inhibition since female and male anaphylactic responses were indistinguishable in mice genetically deficient in eNOS (NOS3−/−) (Fig 5, B). Similarly, the protective effect of L-NAME in female mice (Fig 5, C, lower panel) was abolished when estradiol function was blocked by pretreatment with the ER-antagonist ICI182,780 (Fig 5, C, upper panel). The effect of L-NAME on body temperature changes in the absence or presence of the ER-antagonist (Fig 5, C) also correlated with corresponding effects on hematocrit values (i.e protective in the absence of ER-antagonist and ineffective in the presence of the ER-antagonist) (Fig 5, D lower and upper panels, respectively) and behavioral changes, suggesting that the effect of eNOS inhibition does not reflect an action on thermal regulation33 but a general effect on the anaphylactic response. Collectively, the data provides evidence that estradiol, signaling through its receptors, up-regulates eNOS expression and subsequent NO production which are critically important for the increased vascular reactivity and worsening of the anaphylactic response in female mice.
Fig 5. eNOS activity is crucial for determining female susceptibility to anaphylaxis.
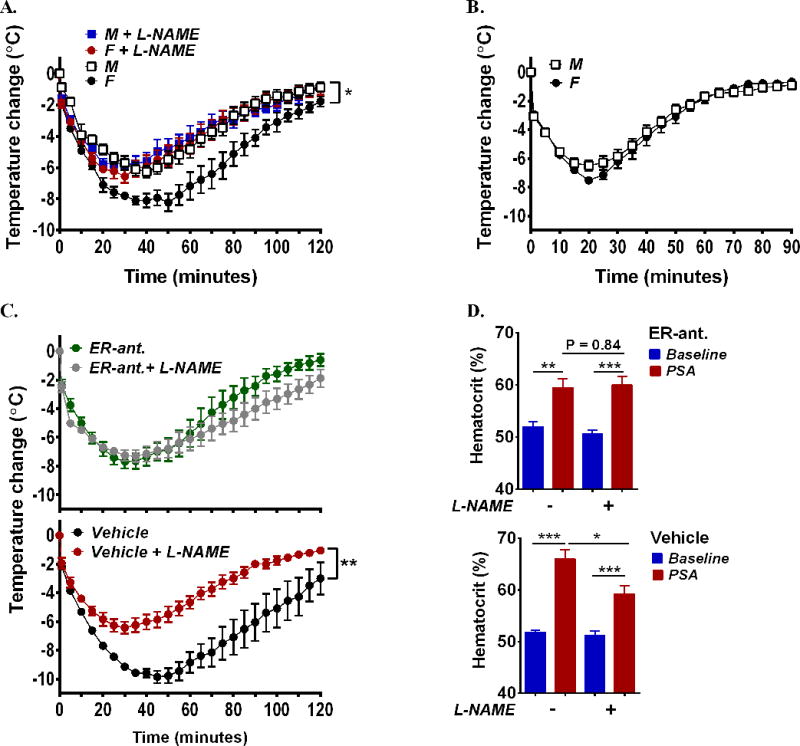
(A, B) Temperature changes during anaphylaxis induced by IgE/Ag in (A) female and male mice pretreated with the NOS inhibitor L-NAME or vehicle or (B) in NOS3−/− female and male mice. (C–D) Effect of L-NAME on temperature (C) and hematocrit changes (D) during IgE/Ag-induced anaphylaxis in female mice pretreated with ER-antagonist ICI182,780 (upper panels) or vehicle (lower panels). In A–D, n=5–8/group. *P<0.05, **P<0.01,*** P<0.001.
DISCUSSION
The increased incidence of anaphylaxis in women has been recognized in the clinic for years.34–36 Our study shows that sex-specific differences in systemic anaphylaxis are replicated in mice and, as in humans, are not restricted to IgE-mediated responses. The incidence, onset and/or severity of many human diseases, including allergic diseases such as asthma, show a gender bias that is likewise conserved across species.37, 38 Although in instances, these gender differences have been linked to particular sex hormones that act exacerbating or protecting against the disease, a direct demonstration of such a link and the specific mechanisms involved have mostly remained elusive.38 Here we provide evidence that a critical mediator exacerbating the anaphylactic response in female mice is the sex steroid estradiol. As shown by the data presented in this paper, the presence of estradiol leads to up-regulation of eNOS levels and activity that are critical in determining the gender bias in anaphylaxis severity in this experimental model. This study should thus help us to address gender disparities in anaphylaxis in humans as well as provoke inspection of anaphylaxis linked to hormonal replacement therapy and to the menstrual cycle; and support the concept that the effectiveness of treatment strategies should also be evaluated taking gender into consideration.
The conclusion that the sex hormone estradiol potentiates anaphylaxis and is responsible for the gender differences in a mouse model of IgE-mediated passive systemic anaphylaxis, is supported by the findings that blocking its receptors or chronic sex hormone depletion by ovariectomy obliterated the exacerbated responses in females, an effect that was reverted in the latter after administration of exogenous estradiol. The differences in anaphylaxis severity between ovariectomized and sham-operated females were also apparent when anaphylaxis was induced by either FcγRIII-crosslinking or histamine, which do not require FcεRI or mast cells.23 In addition, we found no evidence for an effect of estradiol on FcεRI-mediated mast cell responses, indicating that the estrogen-mediated potentiation of anaphylaxis is subsequent to mast cell activation and occurs regardless of the trigger mechanism. This conclusion is in agreement with clinical reports describing a female preponderance not only in IgE-mediated but also in anaphylactic reactions evoked by certain drugs and radiocontrast media,3, 5, 6 which do not seem to involve FcεRI.
Instead, estrogen-mediated gender differences in anaphylaxis are linked to the function of eNOS since they were abolished by pharmacological inhibition or genetic deficiency of eNOS and the beneficial effect of NOS inhibition in females was prevented by ER antagonists. This implies that the effect of estradiol takes center stage at the endothelial lining of vessels, where eNOS is predominantly expressed.39 The product of eNOS activity, NO, is an endogenous vasodilator40 that preserves endothelial function and homeostasis and thus protects against endothelial dysfunction in diseases such as hypertension, heart failure, diabetes and atherosclerosis,41 all of which are generally less prevalent in women than in men.42 ERs are also associated with protective cardiovascular effects43 and estrogens regulate the expression of eNOS and/or NO-production44 in rat tissues45, 46 and human endothelial cell lines.29, 30 In agreement, we find that lungs and aortas from female mice express higher levels of eNOS, with concomitant higher baseline phosphorylation in Ser1177 than the male or ovariectomized counterparts, and that estradiol increases eNOS expression in mouse endothelial cells (E Fig 3, B in the online repository). Preliminary results suggest that eNOS expression levels in primary human pulmonary artery endothelial cells (PAEC) from female donors are also higher than those in male donors (E Fig 3, C in the online repository). Altogether, the data presented herein and elsewhere suggest that estradiol-mediated eNOS up-regulation in females is conserved across species.
NO is a known regulator of many processes involved in anaphylactic shock such as vasodilation and vascular leakage.47 A study exclusively performed in female mice reported that eNOS deficiency protected against PAF- or Ag-induced shock,27 while in another study using male wild-type mice the protective effect of NO in PAF-induced anaphylaxis was seemingly less pronounced.31 However, the relationship of NO and anaphylaxis to gender as we examine here was not directly investigated in these previous studies. Our results indicate that the protective effect of NOS inhibition during non-lethal IgE/Ag-induced anaphylactic shock appears restricted to female mice. In humans, the role of NO in anaphylaxis is not well understood and its relationship with gender has not been explored. Based on our mouse study and given that NOS inhibitors such as L-NAME have helped patients suffering from cardiogenic shock,48 we suggest that the use of these inhibitors might deserve some consideration for the treatment of anaphylaxis in selected instances such in highly susceptible females. Treatment with methylene blue, a dye that competitively inhibits guanylate cyclase, thus interfering with the vasodilatory actions of NO, has shown life-saving effects in female and male patients with severe anaphylaxis,49 supporting the possibility that NO inhibition could be explored as an option in certain cases of anaphylaxis regardless of gender. Further studies are clearly needed to determine the usefulness of such drugs in the management of patients with recurrent severe anaphylaxis.
In the context of the known impact of estrogen on NO and cardiovascular health, our findings imply that, although a higher availability of NO in females may be beneficial for cardiovascular function,50 under circumstances of acute release of vasoactive mediators during an anaphylactic reaction, NO may predispose females to greater and detrimental vascular responses. In addition, our results support consideration of whether estrogen-containing hormonal therapy and contraceptives should be examined for their consequences in women susceptible to anaphylaxis. Lastly, our study reinforces the concern that preferential use of males in some disease animal models may overlook critical interactions between a particular gene or mutation and female-predominant pathways (i.e. the NO pathway) that may be dramatically consequential for sex-specific disease risk.
Supplementary Material
KEY MESSAGES.
The increased susceptibility to anaphylaxis in women noted in clinical studies is reproduced in a mouse model of systemic anaphylaxis and the gender differences are dependent on estradiol.
The effect of estradiol on anaphylaxis does not relate to alterations in the responsiveness of mast cells but rather to enhanced vascular permeability, which is linked to estradiol-mediated up-regulation of eNOS and NO production.
Our findings provide insight into the mechanistic basis of gender bias in anaphylaxis and infer that, although increased eNOS action by estradiol may be beneficial for women in the context of cardiovascular disease, it may contribute to enhanced vascular responses during anaphylaxis
CAPSULE SUMMARY.
Anaphylaxis appears to occur more frequently in women. Using a mouse model of passive systemic anaphylaxis we demonstrate an increased severity in females attributable to estradiol, which increases eNOS expression and NO production resulting in vascular hyper-permeability.
Acknowledgments
Funding sources: This work was supported by the Division of Intramural Research Program within NIAID, NIH and by the Research Foundation Flanders (F.W.O.). V.H. was a research fellow and the recipient of a Travel Grant from F.W.O. as well as a Gustave Boël – Sofina – Belgian American Educational Foundation (B.A.E.F.) fellow.
ABBREVIATIONS
- Ag
antigen
- BMMC
bone marrow derived mast cells
- Ig
immunoglobulin
- E2
17β-estradiol
- eNOS
endothelial nitric oxide synthase
- ER
estrogen receptor
- FcεR
High affinity IgE receptor
- FcγRIIIA
IgG receptor III A
- L-NAME
L-NG-Nitroarginine Methyl Ester
- MCPT-1
mast cell protease-1
- NOS3
nitric oxide synthase type III = endothelial nitric oxide synthase
- NO
nitric oxide
- OVX
ovariectomy/ovariectomized
- PSA
passive systemic anaphylaxis
- PAEC
pulmonary artery endothelial cells
- P-eNOS
Ser1177-phosphorylated eNOS
- PAF
platelet activating factor
- WT
wild type
References
- 1.Gonzalez-Perez A, Aponte Z, Vidaurre CF, Rodriguez LA. Anaphylaxis epidemiology in patients with and patients without asthma: a United Kingdom database review. J Allergy Clin Immunol. 2010;125:1098–104 e1. doi: 10.1016/j.jaci.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Webb LM, Lieberman P. Anaphylaxis: a review of 601 cases. Ann Allergy Asthma Immunol. 2006;97:39–43. doi: 10.1016/S1081-1206(10)61367-1. [DOI] [PubMed] [Google Scholar]
- 3.Liew WK, Williamson E, Tang ML. Anaphylaxis fatalities and admissions in Australia. J Allergy Clin Immunol. 2009;123:434–42. doi: 10.1016/j.jaci.2008.10.049. [DOI] [PubMed] [Google Scholar]
- 4.Ross MP, Ferguson M, Street D, Klontz K, Schroeder T, Luccioli S. Analysis of food-allergic and anaphylactic events in the National Electronic Injury Surveillance System. J Allergy Clin Immunol. 2008;121:166–71. doi: 10.1016/j.jaci.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 5.International Collaborative Study of Severe A. Risk of anaphylaxis in a hospital population in relation to the use of various drugs: an international study. Pharmacoepidemiol Drug Saf. 2003;12:195–202. doi: 10.1002/pds.822. [DOI] [PubMed] [Google Scholar]
- 6.Kvedariene V, Martins P, Rouanet L, Demoly P. Diagnosis of iodinated contrast media hypersensitivity: results of a 6-year period. Clin Exp Allergy. 2006;36:1072–77. doi: 10.1111/j.1365-2222.2006.02532.x. [DOI] [PubMed] [Google Scholar]
- 7.Khan DA, Yocum MW. Clinical course of idiopathic anaphylaxis. Ann Allergy. 1994;73:370–74. [PubMed] [Google Scholar]
- 8.Bauer CS, Kampitak T, Messieh ML, Kelly KJ, Vadas P. Heterogeneity in presentation and treatment of catamenial anaphylaxis. Ann Allergy Asthma Immunol. 2013;111:107–11. doi: 10.1016/j.anai.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Lieberman P. Catamenial anaphylaxis. J Allergy Clin Immunol Pract. 2014;2:358–59. doi: 10.1016/j.jaip.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Meggs WJ, Pescovitz OH, Metcalfe D, Loriaux DL, Cutler G, Jr, Kaliner M. Progesterone sensitivity as a cause of recurrent anaphylaxis. N Engl J Med. 1984;311:1236–38. doi: 10.1056/NEJM198411083111907. [DOI] [PubMed] [Google Scholar]
- 11.Gilfillan AM, Austin S, Metcalfe DD. Mast cell biology: introduction and overview. Adv Exp Med Biol. 2011;716:2–12. doi: 10.1007/978-1-4419-9533-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finkelman FD. Anaphylaxis: lessons from mouse models. J Allergy Clin Immunol. 2007;120:506–15. doi: 10.1016/j.jaci.2007.07.033. [DOI] [PubMed] [Google Scholar]
- 13.Finkelman FD, Rothenberg ME, Brandt EB, Morris SC, Strait RT. Molecular mechanisms of anaphylaxis: lessons from studies with murine models. J Allergy Clin Immunol. 2005;115:449–57. doi: 10.1016/j.jaci.2004.12.1125. [DOI] [PubMed] [Google Scholar]
- 14.Jonsson F, Daeron M. Mast cells and company. Front Immunol. 2012;3:16. doi: 10.3389/fimmu.2012.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A, Teuber SS, Gershwin ME. Why do people die of anaphylaxis? A clinical review Clin Dev Immunol. 2005;12:281–87. doi: 10.1080/17402520500376277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olivera A, Eisner C, Kitamura Y, Dillahunt S, Allende L, Tuymetova G, et al. Sphingosine kinase 1 and sphingosine-1-phosphate receptor 2 are vital to recovery from anaphylactic shock in mice. J Clin Invest. 2010;120:1429–40. doi: 10.1172/JCI40659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan KJ. Biochemistry of aromatase: significance to female reproductive physiology. Cancer Res. 1982;42:3342s–44s. [PubMed] [Google Scholar]
- 18.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–65. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 19.Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer. 2004;90(Suppl 1):S2–6. doi: 10.1038/sj.bjc.6601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slater JE, Kaliner M. Effects of sex hormones on basophil histamine release in recurrent idiopathic anaphylaxis. J Allergy Clin Immunol. 1987;80:285–90. doi: 10.1016/0091-6749(87)90033-9. [DOI] [PubMed] [Google Scholar]
- 21.Zaitsu M, Narita S, Lambert KC, Grady JJ, Estes DM, Curran EM, et al. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol Immunol. 2007;44:1977–85. doi: 10.1016/j.molimm.2006.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen F, Woudwyk M, Teles A, Woidacki K, Taran F, Costa S, et al. Estradiol and progesterone regulate the migration of mast cells from the periphery to the uterus and induce their maturation and degranulation. PLoS One. 2010;5:e14409. doi: 10.1371/journal.pone.0014409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dombrowicz D, Flamand V, Miyajima I, Ravetch JV, Galli SJ, Kinet JP. Absence of Fc epsilonRI alpha chain results in upregulation of Fc gammaRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between Fc epsilonRI and Fc gammaRIII for limiting amounts of FcR beta and gamma chains. J Clin Invest. 1997;99:915–25. doi: 10.1172/JCI119256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strait RT, Morris SC, Yang M, Qu XW, Finkelman FD. Pathways of anaphylaxis in the mouse. J Allergy Clin Immunol. 2002;109:658–68. doi: 10.1067/mai.2002.123302. [DOI] [PubMed] [Google Scholar]
- 25.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119:1871–79. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–09. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cauwels A, Janssen B, Buys E, Sips P, Brouckaert P. Anaphylactic shock depends on PI3K and eNOS-derived NO. J Clin Invest. 2006;116:2244–51. doi: 10.1172/JCI25426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi T, Yamada K, Esaki T, Kuzuya M, Satake S, Ishikawa T, et al. Estrogen increases endothelial nitric oxide by a receptor-mediated system. Biochem Biophys Res Commun. 1995;214:847–55. doi: 10.1006/bbrc.1995.2364. [DOI] [PubMed] [Google Scholar]
- 29.Hishikawa K, Nakaki T, Marumo T, Suzuki H, Kato R, Saruta T. Up-regulation of nitric oxide synthase by estradiol in human aortic endothelial cells. FEBS Lett. 1995;360:291–93. doi: 10.1016/0014-5793(95)00124-r. [DOI] [PubMed] [Google Scholar]
- 30.Kleinert H, Wallerath T, Euchenhofer C, Ihrig-Biedert I, Li H, Forstermann U. Estrogens increase transcription of the human endothelial NO synthase gene: analysis of the transcription factors involved. Hypertension. 1998;31:582–88. doi: 10.1161/01.hyp.31.2.582. [DOI] [PubMed] [Google Scholar]
- 31.Cui H, Okamoto Y, Yoshioka K, Du W, Takuwa N, Zhang W, et al. Sphingosine-1-phosphate receptor 2 protects against anaphylactic shock through suppression of endothelial nitric oxide synthase in mice. J Allergy Clin Immunol. 2013;132:1205–14 e9. doi: 10.1016/j.jaci.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 32.Makabe-Kobayashi Y, Hori Y, Adachi T, Ishigaki-Suzuki S, Kikuchi Y, Kagaya Y, et al. The control effect of histamine on body temperature and respiratory function in IgE-dependent systemic anaphylaxis. J Allergy Clin Immunol. 2002;110:298–303. doi: 10.1067/mai.2002.125977. [DOI] [PubMed] [Google Scholar]
- 33.Yang WW, Krukoff TL. Nitric oxide regulates body temperature, neuronal activation and interleukin-1 beta gene expression in the hypothalamic paraventricular nucleus in response to immune stress. Neuropharmacology. 2000;39:2075–89. doi: 10.1016/s0028-3908(00)00054-x. [DOI] [PubMed] [Google Scholar]
- 34.Brown AF, McKinnon D, Chu K. Emergency department anaphylaxis: A review of 142 patients in a single year. J Allergy Clin Immunol. 2001;108:861–66. doi: 10.1067/mai.2001.119028. [DOI] [PubMed] [Google Scholar]
- 35.De Marco R, Locatelli F, Sunyer J, Burney P. Differences in incidence of reported asthma related to age in men and women. A retrospective analysis of the data of the European Respiratory Health Survey. Am J Respir Crit Care Med. 2000;162:68–74. doi: 10.1164/ajrccm.162.1.9907008. [DOI] [PubMed] [Google Scholar]
- 36.Poulos LM, Waters AM, Correll PK, Loblay RH, Marks GB. Trends in hospitalizations for anaphylaxis, angioedema, and urticaria in Australia, 1993–1994 to 2004–2005. J Allergy Clin Immunol. 2007;120:878–84. doi: 10.1016/j.jaci.2007.07.040. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi T, Adachi Y, Hasegawa K, Morimoto M. Less sensitivity for late airway inflammation in males than females in BALB/c mice. Scand J Immunol. 2003;57:562–67. doi: 10.1046/j.1365-3083.2003.01269.x. [DOI] [PubMed] [Google Scholar]
- 38.Ober C, Loisel DA, Gilad Y. Sex-specific genetic architecture of human disease. Nat Rev Genet. 2008;9:911–22. doi: 10.1038/nrg2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–42. [PubMed] [Google Scholar]
- 40.Loscalzo J, Welch G. Nitric oxide and its role in the cardiovascular system. Prog Cardiovasc Dis. 1995;38:87–104. doi: 10.1016/s0033-0620(05)80001-5. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Forstermann U. Nitric oxide in the pathogenesis of vascular disease. J Pathol. 2000;190:244–54. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 42.Yang S, Bae L, Zhang L. Estrogen increases eNOS and NOx release in human coronary artery endothelium. J Cardiovasc Pharmacol. 2000;36:242–47. doi: 10.1097/00005344-200008000-00015. [DOI] [PubMed] [Google Scholar]
- 43.Teede HJ. Sex hormones and the cardiovascular system: effects on arterial function in women. Clin Exp Pharmacol Physiol. 2007;34:672–76. doi: 10.1111/j.1440-1681.2007.04658.x. [DOI] [PubMed] [Google Scholar]
- 44.Rosselli M, Imthurm B, Macas E, Keller PJ, Dubey RK. Circulating nitrite/nitrate levels increase with follicular development: indirect evidence for estradiol mediated NO release. Biochem Biophys Res Commun. 1994;202:1543–52. doi: 10.1006/bbrc.1994.2107. [DOI] [PubMed] [Google Scholar]
- 45.Caliman IF, Lamas AZ, Dalpiaz PL, Medeiros AR, Abreu GR, Figueiredo SG, et al. Endothelial relaxation mechanisms and oxidative stress are restored by atorvastatin therapy in ovariectomized rats. PLoS One. 2013;8:e80892. doi: 10.1371/journal.pone.0080892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reckelhoff JF, Hennington BS, Moore AG, Blanchard EJ, Cameron J. Gender differences in the renal nitric oxide (NO) system: dissociation between expression of endothelial NO synthase and renal hemodynamic response to NO synthase inhibition. Am J Hypertens. 1998;11:97–104. doi: 10.1016/s0895-7061(97)00360-9. [DOI] [PubMed] [Google Scholar]
- 47.SGAK Brown, Lieberman SF, Anaphylaxis PL. In: Middleton’s Allergy Principles and Practive. Adkinson NFB BS, Burks AW, Busse WW, Holgate ST, Lemanske RF, O’Hehir RE, editors. Philadelphia, PA: Elsevier Saunders; 2013. pp. 1237–59. [Google Scholar]
- 48.Cotter G, Kaluski E, Milo O, Blatt A, Salah A, Hendler A, et al. LINCS: L-NAME (a NO synthase inhibitor) in the treatment of refractory cardiogenic shock: a prospective randomized study. Eur Heart J. 2003;24:1287–95. doi: 10.1016/s0195-668x(03)00193-3. [DOI] [PubMed] [Google Scholar]
- 49.Evora PR, Simon MR. Role of nitric oxide production in anaphylaxis and its relevance for the treatment of anaphylactic hypotension with methylene blue. Ann Allergy Asthma Immunol. 2007;99:306–13. doi: 10.1016/S1081-1206(10)60545-5. [DOI] [PubMed] [Google Scholar]
- 50.Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev. 2008;60:210–41. doi: 10.1124/pr.107.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.