

Abstract
Objectives:
There are limited data regarding long-term follow-up and therapeutic outcomes in Sjogren's syndrome (SS)-associated peripheral neuropathy. In this study, we aim to study the clinical, electrophysiological spectrum and therapeutic responses among the different subtypes of SS-associated neuropathy. The predictors of suboptimal treatment response will be identified.
Methods:
The study included a retrospective cohort of patients with SS-associated neuropathy between January 2012 and November 2015. Baseline clinical, laboratory, electrophysiological data and details of treatment were noted. Therapeutic outcomes were assessed at follow-up and compared among the different subtypes. Prognostic predictors were determined using logistic regression analysis.
Results:
Fifty-four patients were included in the study. Sensory ataxic neuropathy (17, including 9 with sensory ganglionopathy) and radiculoneuropathy (11) were the main subtypes. Notable atypical presentations included acute neuropathies, pure motor neuropathies, and hypertrophic neuropathy. Concomitant autoimmune disorders were present in 24 (44.4%) patients. Most presentations were subacute-chronic (51, 94.4%). Minor salivary gland biopsy had a higher yield compared to serological markers (81.5 vs. 44.4%). Sensory ataxic neuropathy was associated with greater severity and autonomic dysfunction. Improvement was noted in 33 (61%) patients. Cranial neuropathy and radiculoneuropathy subtypes were associated with the best treatment responses. Chronicity, orthostatic hypotension, baseline severity, and marked axonopathy (nerve biopsy) were predictive of a suboptimal therapeutic response.
Conclusions:
The study highlights the heterogeneous spectrum, atypical presentations, and differential therapeutic responses. SS-associated neuropathy remains underdiagnosed. Early diagnosis and prompt initiation of immunotherapy before worsening axonal degeneration is paramount. SS-associated neuropathy need not necessarily be associated with a poor prognosis.
Keywords: Autoimmune, autonomic dysfunction, ganglionopathy, sensory ataxia, Sjogren's syndrome
INTRODUCTION
Sjogren's syndrome (SS) is a multisystem disorder with varied neurological manifestations. The reported prevalence of neuropathy in SS ranges from 5%–57%.[1,2] Nearly 36% can have isolated neurological involvement.[1,2] However, neurological symptoms can precede sicca symptoms by up to 6 years (47%).[1,2,3] SS-associated neuropathy remains an often underdiagnosed entity.
The spectrum of peripheral neuropathy is wide and includes sensory ataxic neuropathy, sensory neuropathy without sensory ataxia, multiple mononeuropathy, cranial neuropathy, and radiculoneuropathy.[4] There is scarce literature regarding the long-term outcomes, therapeutic responses, and predictive prognostic factors considering the heterogeneity and chronicity of this disorder. In this study, we assessed the clinical spectrum, electrophysiological features, and therapeutic responses in an Indian cohort with SS-associated neuropathy.
METHODS
The study included a retrospective analysis of a cohort of patients with SS-associated neuropathy admitted under neurology unit at a quaternary care teaching hospital in India during the period of January 2012–November 2015. Patients fulfilling the American–European Consensus Group criteria 2002 or American College of Rheumatology 2012 classification criteria were included in the study. Patients with other causes of neuropathy such as diabetes mellitus, paraproteinemias, and underlying malignancies were excluded from the study. The study variables obtained from our prospectively maintained electronic database included baseline demographic data, clinical presentation, duration of symptoms, presence of concomitant autoimmune disorders, serological profile, minor salivary gland biopsy, and cerebrospinal fluid (CSF) analysis findings. Ocular involvement was objectively documented using Schirmer's test (<5 mm in 5 min considered positive). Serological testing for anti-SSA and anti-SSB was done using commercial ELISA kits (value >20 Ru/ml considered positive). Histopathology of minor salivary gland showing inflammation (focus score >1, Chisholm-Mason Grade 3 or 4) was suggestive of SS [Figure 1].
Figure 1.
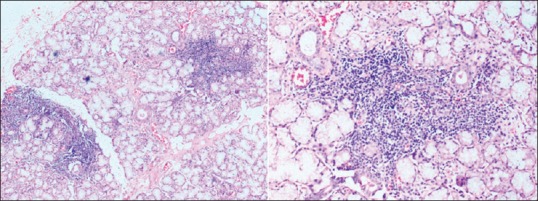
Hematoxylin- and eosin-stained sections of a minor salivary gland showing seromucinous glands with two foci of lymphoid infiltrate each composed of >50 cells (×50 and ×100 magnification, respectively)
The electrophysiological variables including motor and sensory amplitudes, conduction velocities, and distal latencies were noted. The type of neuropathy was classified into sensory ataxic neuropathy, painful sensory neuropathy without sensory ataxia, multiple mononeuropathy, radiculoneuropathy, isolated cranial neuropathy, pure autonomic neuropathy, and pure motor neuropathy based on the clinical profile and electrophysiological findings. A diagnosis of sensory neuronopathy was made in patients with ataxia in the upper or lower limbs, asymmetric distribution, sensory loss not restricted to the lower limbs, abnormal sensory action potentials in the upper limbs, and <2 nerves with abnormal motor conduction study in the lower limbs.[5]
The severity of neuropathy was documented using a modified version of total neuropathy score (TNSr). Quantitative sensory testing was not used in the TNSr. The severity of involvement was graded according to the TNSr score as 1–9 (mild), 10–19 (moderate), and >20 (severe). All the patients received immunotherapy, and the details of treatment received were noted. The therapeutic response at the time of last follow-up was classified as improvement, stable disease, or worsening. Improvement included objective clinical improvement and/or improvement in nerve conduction parameters. Participants were considered to have a suboptimal therapeutic response when they did not have improvement with treatment (this included the groups with both stable disease and worsening).
The statistical analysis was performed using SPSS (Statistical Package for the Social Science) version 16.0 (Chicago, IL). Data were presented as mean + standard deviation (SD). Descriptive analysis was performed using Chi-square test for categorical variables and ANOVA test for continuous variables. Wilcoxon signed-rank test was used when the data were not normally distributed. Statistical significance was taken to be at the two-tailed 0.05 level. The predictors of suboptimal therapeutic response were determined using logistic regression analysis.
RESULTS
Patient characteristics
A total of 54 patients were included in the study. There were 37 females (68.5%) and 17 males (31.5%). Mean age of presentation was 49.28 years (SD 14.07). The mean duration of illness at the time of presentation was 24.57 months (SD 12). Majority of the patients (49/54) were diagnosed to have SS during evaluation for the neuropathy.
Antinuclear antibody was positive in twenty patients (37%). Positive SS-A was present in 17 (31.5%) and positive SS-B in 7 (12.9%) patients. Minor salivary gland biopsy showed Grade 3 (22 patients) and Grade 4 (22 patients) inflammation in 44 patients (81.5%). Concomitant autoimmune disorders were present in 24 (44.4%) patients. These included autoimmune thyroiditis (14), rheumatoid arthritis (3), systemic lupus erythematosus (3), scleroderma (1), lichen planus (1), ulcerative colitis (1), and IgG4-associated pancreatitis. CSF protein was elevated in 26 patients (48.1%). Seven patients had secondary SS.
Classification of neuropathy and electrophysiological features
Sensory ataxic neuropathy was the most common clinical presentation in our cohort with 17 patients (31.5%). This group also included nine patients who had clinical and electrophysiological features of a sensory neuronopathy. The other presentations included painful sensory neuropathy without ataxia 8 (14.8%), mononeuritis multiplex 6 (11.1%), radiculoneuropathy 11 (20.4%), isolated cranial neuropathy 9 (16.7%), and pure autonomic neuropathy 1 (1.8%). In addition, two patients (3.7%) had pure motor neuropathy. In the cranial neuropathy group, majority had trigeminal neuropathy (six patients), and other cranial nerves included facial nerve (five patients), glossopharyngeal and vagus nerves (four patients), oculomotor nerve (two patients), and vestibulocochlear nerve (one patient).
The temporal profile of evolution was classified as acute (within 4 weeks), subacute (4 weeks to 6 months), and chronic (more than 6 months). Subacute presentation was the most frequent seen in 27 patients (50%). A chronic presentation was noted in 24 patients (44.4%). Interestingly, an acute presentation was noted in 3 patients. Autonomic dysfunction was present in 35 patients (64.8%). The mean duration of symptoms of autonomic dysfunction was 9.13 months (SD 8.93). The major autonomic manifestations were orthostatic hypotension (ten patients, 18.5%), cardiovascular involvement (five patients, 9.2%), gastrointestinal (19 patients, 35.1%), genitourinary (17 patients, 31.5%), skin involvement (22 patients, 40.7%), and secretomotor (38 patients, 70.4%). Three patients had a history of syncope.
Nerve conduction studies were done in all patients. Excluding the ten patients who had isolated cranial or autonomic neuropathy, nerve conduction studies showed axonal features in 32 (59.2%), predominant demyelinating features with conduction blocks in 4 (7.4%), and mixed axonal and demyelinating features in 8 (14.8%) patients. Sympathetic skin response was absent in 27 (50%) patients. Nerve biopsy was done in 32 patients. Nerve fiber loss (loss of myelin sheath and axons) was classified as mild (10, 31.2%), moderate (8, 25%), and marked (14, 43.7%). Features of vasculitis (transmural inflammation, perivascular inflammation with Wallerian degeneration/asymmetric myelin loss) were seen in 11 of these patients. The mean duration of illness was 12.19 (SD 15.37) months in the group with vasculitis compared to 30.64 (SD 32.08) in the group without vasculitis (P = 0.03).
Treatment and follow-up
All the patients received immunotherapy. Steroids were given in all patients. Mycophenolate mofetil was used as steroid-sparing agent in 26 patients (48.1%). The other immunosuppressants used included cyclophosphamide in 25 (46.3%), azathioprine in 3 (5.5%), intravenous immunoglobulin in 4 (7.4%), and plasma exchange and rituximab in one patient each. Follow-up data were available in 50/54 patients. The mean duration of follow-up was 13.8 months (SD 9.6). During follow-up, improvement was noted in 33 (61.1%), stable disease in 12 (22.2%), and worsening in 3 (5.5%) patients. On subgroup analysis, the improvement in each of the clinical subtypes was as follows: sensory ataxic neuropathy (41.1%), painful sensory neuropathy without sensory ataxia (62.8%), mononeuritis multiplex (33.3%), radiculoneuropathy (81.8%), and isolated cranial neuropathy (100%). Nerve conduction studies were repeated at follow-up in 36 patients, and there was evidence of electrophysiological improvement in 22 (61.1%) patients.
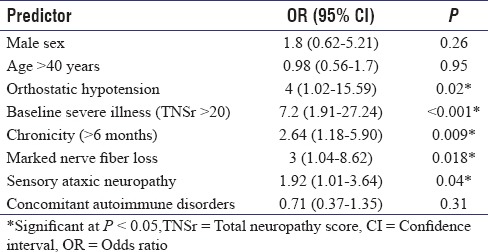
The comparison of clinical profile, electrophysiological findings, and therapeutic response among the main subtypes has been provided in Table 1. Sensory ataxic neuropathy was associated with greater baseline severity of illness and presence of autonomic dysfunction. Vasculitis on nerve biopsy was associated with mononeuritis multiplex and radiculoneuropathy. The comparison of pre- and post-treatment severity using TNSr has been depicted in Figure 2. The predictors of suboptimal therapeutic response are shown in Table 2. On multivariate logistic regression analysis, chronicity of illness (>6 months), presence of orthostatic hypotension, baseline severity (TNSr >20), and marked nerve fiber loss on the nerve biopsy remained predictive of a suboptimal therapeutic response.
Table 1.
Comparison of clinical, laboratory findings and therapeutic responses among the subtypes of Sjogren's syndrome-associated neuropathy
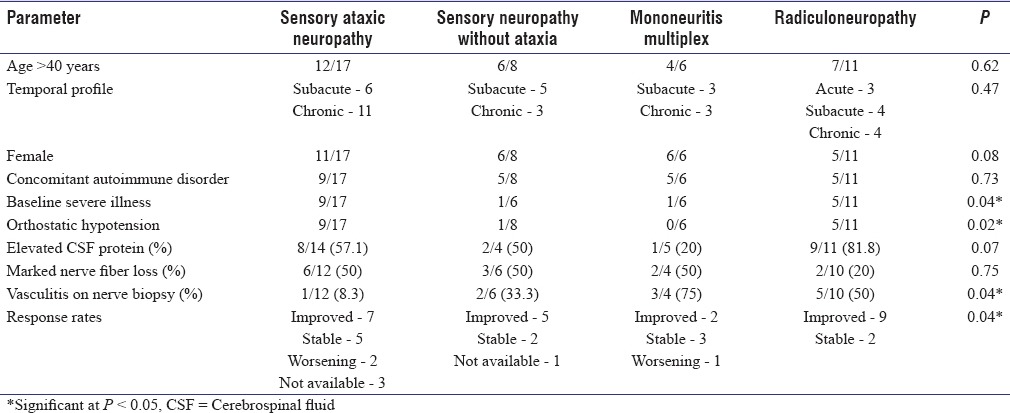
Figure 2.
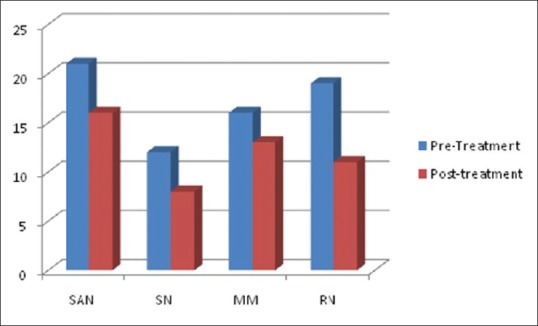
Comparison of the mean total neuropathy score (pre- and post-treatment) among the subtypes of Sjogren's syndrome-associated neuropathy. There was statistically significant difference in the mean total neuropathy score (pre- and post-treatment) in the radiculoneuropathy (19, 11, P = 0.002), sensory ataxic neuropathy (21, 16, P = 0.02), and the painful sensory neuropathy (12, 8, P = 0.04) groups. There was also modest improvement in the mononeuritis multiplex group (16, 13, P = 0.07)
Table 2.
Predictors of suboptimal therapeutic response in the cohort
DISCUSSION
This study comprising a cohort of patients with SS-associated neuropathy had some interesting observations. The classification of neuropathies was done based on the observations from prominent studies done earlier in SS-associated neuropathies.[4] Some of the salient observations from this cohort included (a) the atypical clinical presentations, (b) higher yield of minor salivary gland biopsy in the diagnosis compared to standard serological tests, (c) high proportion of concomitant autoimmune disorders, and (d) differential treatment responses among the various subtypes. The treatment responses emphasize the importance of early and aggressive immunotherapy in the eventual therapeutic outcome.
Clinical presentations
The most common clinical presentation in our cohort was sensory ataxic neuropathy. In addition, we also had a substantial number of patients with isolated cranial neuropathy [Figure 3a] and radiculoneuropathy. There were three cases with acute presentation mimicking a Guillain-Barre syndrome (GBS). The nerve conductions in all these patients showed features of a demyelinating polyneuropathy. All the patients improved on immunotherapy. Ganglioside antibodies were negative. All the patients continued to have sicca symptoms and arthralgias on follow-up. One patient had recurrence of lower limb paresthesias on tapering of immunotherapy at 6-month follow-up, the symptoms improved following re-initiation of steroids. There is limited literature including a case report of a fulminant acute motor axonal neuropathy in SS.[6] It is important to recognize this subset of patients early as they will need continuation of treatment unlike the prototype GBS which has a monophasic course. A comprehensive immunological testing for other autoimmune disorders may be relevant, especially in patients with atypical “GBS.”
Figure 3.

Case illustrations. (a) a 34-year-old woman with bilateral trigeminal neuropathy, postcontrast scan showing enhancement of the trigeminal nerve on the left side and nerve thickening on the right side, (b) a 32-year-old woman with features of radiculoneuropathy and hypertrophied nerve roots (c) a 25-year-old male with intestinal pseudo-obstruction on barium meal follow-through, (d) a 25-year-old male with beat-to-beat recordings showing orthostatic hypotension with tachycardia (d) and resolution of these autonomic disturbances (e) with treatment
There was a higher prevalence of radiculoneuropathy in this cohort compared to previous studies. Two of the patients also had hypertrophic neuropathy [Figure 3b] which is rarely described in literature. The association of SS and chronic inflammatory demyelinating polyneuropathy (CIDP) has also been reported.[7,8] The high proportion of concomitant autoimmune disorders also needs mention. This association is likely to be an epiphenomenon and attributable to an immune dysregulation with antigens triggering a cross-immune response. The autoimmunity associated with SS may induce a “CIDP-like” inflammatory polyneuropathy. Studies have also revealed that HLA class II markers in addition to conferring a genetic susceptibility to SS also support autoantibody production and epitope spreading.[9] It was noted that the prevalence of concomitant autoimmune disorders neither varied among the different subgroups nor did it influence the eventual therapeutic outcome.
There were two patients with a pure motor neuropathy mimicking an anterior horn cell disorder. Such presentations are extremely rare.[10,11] Both the patients had asymmetric “foot onset weakness” with lower motor neuron (LMN) signs. On evaluation, one patient had elevated CSF protein (107 mg%) and nerve-muscle biopsy which showed loss of myelin sheath, axons, myelin ovoid formation and perivascular infiltrates of lymphocytes, and plasma cells around endomysial vessels. The other patient had a long-standing inflammatory arthritis with a recent onset motor neuropathy and positive ganglioside antibody (weak-positive IgM anti-GM3). There were no conduction blocks on the nerve conduction studies in the second patient. After an initial period of stabilization with immunotherapy (steroids and cyclophosphamide), both of them showed gradual worsening of LMN-type weakness which remained confined to the initially involved limbs (follow-up of 13 and 15 months, respectively). There was no clinical or electrophysiological involvement of the previously unaffected segments (including bulbar/respiratory) on serial follow-up. The initial improvement and absence of relentless progression during follow-up made the possibility of other LMN syndromes such as progressive muscular atrophy (PMA) less likely. However, a long-term follow-up is essential in this subset of patients considering the phenotypic heterogeneity of LMN syndromes which include PMA and multifocal motor neuropathy. In addition, there were two patients with lower cranial neuropathy mimicking a “bulbar-onset motor neuron disease.” Both these patients had a good response to immunotherapy. Upper motor neuron signs have also been reported in SS secondary to involvement of the central nervous system. This emphasizes that SS in a given clinical setting should be considered in the differential diagnosis of motor neuron disease.[12,13,14]
There were a high proportion of patients with autonomic dysfunction in this cohort. Autonomic dysfunction was associated with the sensory ataxic neuropathy and radiculoneuropathy subtypes [Table 1]. The specific domains of autonomic dysfunction and the presence or absence of subclinical autonomic dysfunction need to be estimated by quantitative autonomic function tests using beat-to-beat monitoring. A representative case illustration showing intestinal pseudo-obstruction [Figure 3c] and orthostatic hypotension improving posttreatment has been illustrated [Figure 3d and e]. The autonomic dysfunction in SS has been associated with the presence of alpha 3 nicotinic receptor antibody.[4] The role of a holistic treatment regimen addressing these autonomic symptoms need not be overemphasized.
Time to diagnosis and utility of minor salivary gland biopsy
The high number of patients diagnosed to have SS after the onset of neuropathy as seen in this cohort is consistent with previous reports in literature. The neurological involvement can precede the classical sicca symptoms in 40-93%, and this could attribute to the underdiagnosis of SS in peripheral neuropathies.[1,2,3] These observations could be biased due to the fact that majority of the patients were recruited from the neurology outpatient services. The wide range and heterogeneity can be explained by the specialty clinic (neurology, rheumatology, and internal medicine), from which these patients are getting recruited into the study as well as the different inclusion criteria used. The mean age of onset was 49.28 years. Neuropathy associated with SS usually presents in older patients compared to those without neurological involvement.[1,4]
The standard serological tests (anti-SS-A, SS-B) were less sensitive compared to minor salivary gland biopsy in the diagnosis of SS in this cohort. This is a very important observation. Minor salivary gland biopsy can be inconclusive in patients with prior steroid use and chronic disease with atrophic changes. Procedures such as salivary scintigraphy, unstimulated whole salivary flow estimation, parotid sialography, and ocular staining are not routinely available in clinical practice in resource crunch situations. A positive minor salivary gland biopsy (Grade 3, 4 inflammation) can potentially obviate the need for these procedures and help in establishing the diagnosis of SS in a given clinical setting. The lack of specific biomarkers in patients with neuropathy does not exclude SS. There could also be a role for rescreening for the commonly described autoantibodies during follow-up visits as seroconversion has been reported.[2] The specificity of other autoantibodies such as anti-alpha-fodrin antibodies in patients with SS-associated neuropathy is also being studied.[4] These could be potential biomarkers in the near future.
Therapeutic responses
The differential therapeutic responses among the various subtypes are explained by the various pathophysiological processes involved.[1,2,3,4] The sensory neuronopathies and trigeminal neuropathy are attributable to a ganglioneuronitis whereas multiple mononeuropathy is associated with a vasculitic process. Sensory neuronopathy in SS has been reported to be chronic and debilitating.[15] Progression has been reported in spite of treatment. Axonal degeneration warrants treatment as early as possible. As the disease progresses, axonal neuropathy induces central chromatolysis of the neuronal cell body, and this induces a central-peripheral distal axonopathy of a dying back type. Use of an aggressive immunotherapy induction regimen before significant axonopathy occurs might help in limiting disease progression.
In our cohort, we noted that a significant proportion of patients had improvement or stabilization of disease with potent immunotherapy. The responses were excellent in the isolated cranial neuropathy and radiculoneuropathy group with the majority showing a significant improvement. A sizeable proportion in the sensory ataxic neuropathy group also showed improvement and stabilization of disease [Table 1 and Figure 2]. The suboptimal response in the sensory ataxic neuropathy group could be attributable to the fact that 9/17 (52.9%) patients had features of sensory neuronopathy suggesting significant axonal degeneration. The poor outcome predictors in the cohort included chronicity (presentations >6 months), baseline clinical severity (TNSr >20), and marked axonopathy before initiation of treatment.
The limitations of the study included a relatively small number of patients when individually considering the size of the various subtypes, their differential clinical profile and therapeutic responses. The treatment regimen chosen was not randomized in the different groups as all patients received treatment based on the subtype of neuropathy and baseline clinical severity. Hence, therapeutic regimens could not be compared. All these data were limited by the retrospective nature of our study and the fact that the treatment-free outcome of SN is not well known.
CONCLUSIONS
SS-associated neuropathy has a heterogeneous spectrum, and atypical presentations are not uncommon. There is considerable overlap with concomitant autoimmune disorders. Diagnosis is often delayed as the neuropathy precedes the systemic manifestations. Minor salivary gland biopsy has a superior diagnostic yield compared to currently available serological markers. The diagnosis of SS-associated neuropathy is not associated with a poor prognosis. Early diagnosis and prompt initiation of immunotherapy before worsening axonal degeneration is paramount in the management. There is a need for prospective randomized trials with large sample size and long-term follow-up to help in identifying the optimal therapeutic regimen.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Tobón GJ, Pers JO, Devauchelle-Pensec V, Youinou P. Neurological disorders in primary Sjögren's syndrome. Autoimmune Dis 2012. 2012 doi: 10.1155/2012/645967. 645967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delalande S, de Seze J, Fauchais AL, Hachulla E, Stojkovic T, Ferriby D, et al. Neurologic manifestations in primary Sjögren syndrome: A study of 82 patients. Medicine (Baltimore) 2004;83:280–91. doi: 10.1097/01.md.0000141099.53742.16. [DOI] [PubMed] [Google Scholar]
- 3.Lafitte C, Amoura Z, Cacoub P, Pradat-Diehl P, Picq C, Salachas F, et al. Neurological complications of primary Sjögren's syndrome. J Neurol. 2001;248:577–84. doi: 10.1007/s004150170135. [DOI] [PubMed] [Google Scholar]
- 4.Mori K, Iijima M, Koike H, Hattori N, Tanaka F, Watanabe H, et al. The wide spectrum of clinical manifestations in Sjögren's syndrome-associated neuropathy. Brain. 2005;128(Pt 11):2518–34. doi: 10.1093/brain/awh605. [DOI] [PubMed] [Google Scholar]
- 5.Camdessanché JP, Jousserand G, Ferraud K, Vial C, Petiot P, Honnorat J, et al. The pattern and diagnostic criteria of sensory neuronopathy: A case-control study. Brain. 2009;132(Pt 7):1723–33. doi: 10.1093/brain/awp136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awad A, Mathew S, Katirji B. Acute motor axonal neuropathy in association with Sjögren syndrome. Muscle Nerve. 2010;42:828–30. doi: 10.1002/mus.21830. [DOI] [PubMed] [Google Scholar]
- 7.Lin WS, Hsu YD. Sjögren's syndrome with chronic inflammatory demyelinating polyneuropathy. Neurol India. 2011;59:476–8. doi: 10.4103/0028-3886.82754. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Li X. Chronic inflammatory demyelinating polyneuropathy with Sjögren's syndrome. Case Study Case Rep. 2016;6:19–26. [Google Scholar]
- 9.Gottenberg JE, Busson M, Loiseau P, Cohen-Solal J, Lepage V, Charron D, et al. In primary Sjögren's syndrome, HLA class II is associated exclusively with autoantibody production and spreading of the autoimmune response. Arthritis Rheum. 2003;48:2240–5. doi: 10.1002/art.11103. [DOI] [PubMed] [Google Scholar]
- 10.Mochizuki H, Kamakura K, Masaki T, Hirata A, Nakamura R, Motoyoshi K. Motor dominant neuropathy in Sjögren's syndrome: Report of two cases. Intern Med. 2002;41:142–6. doi: 10.2169/internalmedicine.41.142. [DOI] [PubMed] [Google Scholar]
- 11.Saitoh BY, Hayashi S, Kamada T, Murai H, Omoto M, Kira J. A case of chronic progressive motor-dominant multiple mononeuritis associated with primary Sjögren's syndrome. Rinsho Shinkeigaku. 2015;55:753–8. doi: 10.5692/clinicalneurol.cn-000748. [DOI] [PubMed] [Google Scholar]
- 12.Yamashita K, Kobayashi S, Yamaguchi S, Okada K, Arimoto S, Fujihara S, et al. A case of bulbar palsy associated with Sjögren syndrome. Nihon Naika Gakkai Zasshi. 1988;77:1280–1. doi: 10.2169/naika.77.1280. [DOI] [PubMed] [Google Scholar]
- 13.Attout H, Rahmeh F, Ziegler F. de Gorgerot-Sjogren syndrome simulating amyotrophic lateral sclerosis. Rev Med Interne. 2000;21:708–10. doi: 10.1016/s0248-8663(00)80030-2. [DOI] [PubMed] [Google Scholar]
- 14.Dar WR, Latief M, Sofi NU, Dar I, Parry M, Qayoom O. J Gen Practice. 2015;3:4. http://dx.doi.org/10.4172/2329-9126.1000210. [Google Scholar]
- 15.Pereira PR, Viala K, Maisonobe T, Haroche J, Mathian A, Hié M, et al. Sjögren sensory neuronopathy (Sjögren ganglionopathy): Long-term outcome and treatment response in a series of 13 cases. Medicine (Baltimore) 2016;95:e3632. doi: 10.1097/MD.0000000000003632. [DOI] [PMC free article] [PubMed] [Google Scholar]