

Abstract
Background:
Limb-girdle muscular dystrophy (LGMD) is the most common adult-onset class of muscular dystrophies in India, but a majority of suspected LGMDs in India remain unclassified to the genetic subtype level. The next-generation sequencing (NGS)-based approaches have allowed molecular characterization and subtype diagnosis in a majority of these patients in India.
Materials and Methods:
(I) To select probable dysferlinopathy (LGMD2B) cases from other LGMD subtypes using two screening methods (i) to determine the status of dysferlin protein expression in blood (peripheral blood mononuclear cell) by monocyte assay (ii) using a predictive algorithm called automated LGMD diagnostic assistant (ALDA) to obtain possible LGMD subtypes based on clinical symptoms. (II) Identification of gene pathogenic variants by NGS for 34 genes associated with LGMD or LGMD like muscular dystrophies, in cases showing: absence of dysferlin protein by the monocyte assay and/or a typical dysferlinopathy phenotype, with medium to high predictive scores using the ALDA tool.
Results:
Out of the 125 patients screened by NGS, 96 were confirmed with two dysferlin variants, of which 84 were homozygous. Single dysferlin pathogenic variants were seen in 4 patients, whereas 25 showed no variants in the dysferlin gene.
Conclusion:
In this study, 98.2% of patients with absence of the dysferlin protein showed one or more variants in the dysferlin gene and hence has a high predictive significance in diagnosing dysferlinopathies. However, collection of blood samples from all over India for protein analysis is expensive. Our analysis shows that the use of the “ALDA tool” could be a cost-effective alternative method. Identification of dysferlin pathogenic variants by NGS is the ultimate method for diagnosing dysferlinopathies though follow-up with the monocyte assay can be useful to understand the phenotype in relation to the dysferlin protein expression and also be a useful biomarker for future clinical trials.
Keywords: Dysferlinopathy, limb-girdle muscular dystrophy, monocyte-assay, next-generation sequencing
INTRODUCTION
Limb-girdle muscular dystrophy (LGMD) is the most common adult-onset muscular dystrophy in India. LGMD subtype classification is generally based on immunocharacterization on muscle biopsies and clinical presentation with sarcoglycanopathies (LGMD2C-F) being the most common subtype identified in India and dysferlinopathy (also referred to as LGMD2B) being the second most common identified LGMD subtype.[1,2] However, in a vast population like India with limited diagnostic resources available to patients, a large group of LGMD patients remain unclassified.[1]
LGMD2B and Miyoshi myopathy (MM) are autosomal recessive forms of muscular dystrophy caused by pathogenic variants in the dysferlin gene (DYSF gene) located on chromosome 2p13.[3,4] The dysferlin gene comprises 55 exons spanning more than 150 kb, and encodes a 230 kDa transmembrane protein which is highly expressed in the muscle.[3,5] The dysferlin protein is localized at the sarcolemma of muscle cells and is known to be involved in membrane fusion events and appears necessary for calcium-dependent muscle membrane repair of the muscle following damage.[6] Disruption in muscle repair mechanism leads to dysferlin deficient muscle degeneration.[7]
LGMD2B and MM are initially characterized by different clinical presentations which include predominant weakness of muscles of the pelvic and shoulder girdle in the LGMD2B presentation[8] and distal weakness in the MM presentation.[9] Collectively, the clinical presentations caused by pathogenic variants in the DYSF gene and dysferlin protein deficiency are termed dysferlinopathy. The onset of dysferlinopathy is usually in late teens or adulthood with slow progression and marked elevation of CK levels. The exact prevalence of dysferlinopathy in India is yet to be elucidated, but clinical-pathological correlations have been documented by various groups.[2,10]
Screening of protein expression by immunohistochemistry (IHC) or immunoblot in muscle tissue has been a reliable method used for differential diagnosis of muscular dystrophies.[11] The discovery that the dysferlin protein is also expressed in blood monocytes and correlates well with the expression in skeletal muscle has led to the development of a blood-based diagnostic assay for dysferlinopathy.[12,13,14] The presence, absence, or reduced levels of dysferlin can be evaluated using an anti-Dysferlin antibody on protein lysate from monocytes isolated from the blood of subjects suspected of having LGMD[11,15] to determine the likelihood they have dysferlinopathy versus another form of LGMD. However, a definitive diagnosis of dysferlinopathy can only be achieved by gene sequencing and the identification of pathogenic variants in the DYSF gene.
Screening the entire dysferlin gene of 55 exons by Sangers sequencing or using a next-generation sequencing (NGS) panel test for pathogenic variants can be very expensive and time-consuming. Therefore, a method to delineate the patients most likely to have a dysferlinopathy before sequencing was followed. Two screening methods were employed: (i) evaluation of the expression of dysferlin protein by immunoblot and/or (ii) the use of ALDA, which stands for automated LGMD diagnostic assistant. The ALDA tool was developed by the staff of the Jain Foundation and uses a Bayesian algorithm to predict the most likely subtypes of LGMD a patient has based on their clinical, biochemical, and histopathological findings.[16] The ALDA tool assesses both the LGMD2B and Miyoshi myopathy clinical presentations of dysferlinopathy. While the monocyte assay has been shown to be accurate for provisional diagnosis, it can be laborious and not a cost-effective option for the continual prediction of likely dysferlinopathy cases over the long-term. Therefore, the use of the ALDA tool has become the method we currently use for predicting the most likely India cases to have dysferlinopathy.
In this paper, we show the results of our analysis of patients determined to likely have a dysferlinopathy based on the dysferlin monocyte assay and/or ALDA. From this analysis, we show that these methods are highly efficient in predicting individuals who will show dysferlin pathogenic variants on sequencing.
MATERIALS AND METHODS
Patient sample collection
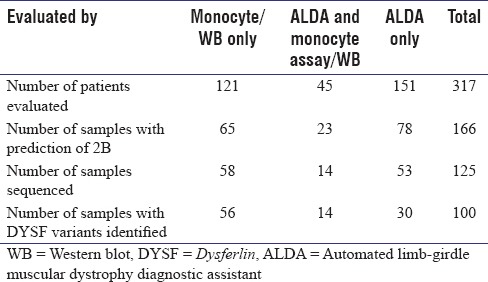
Blood samples from a total of 166 patients who were predicted to have a dysferlinopathy (LGMD2B or MM) based on clinical, histopathological, and immunohistochemical evaluation, unclassified LGMD patients who had self-registered on the Jain Foundation website (121 patients) or patients who were determined to be probable dysferlinopathy patients (LGMD2B or MM) based on the ALDA tool (45 patients) were collected from all parts of India for blood monocyte assay screening [Table 1]. After the blood samples were collected, protein lysate was prepared, and DNA extraction was carried out and stored at −80°C and −20°C, respectively
In addition, a total of 151 patients were screened using the ALDA tool alone for the likelihood of dysferlinopathy and for the 78 that were determined to be probable dysferlinopathy (LGMD2B or MM), blood samples were collected for DNA extraction for direct NGS sequencing [Table 1].
Table 1.
Total patient sample analysis, using monocyte assay/western blot and automated limb-girdle muscular dystrophy diagnostic assistant for screening, followed by sequencing
Clinical presentation
The clinical features largely conformed to the literature. The age at onset of the cohort of 244 patients was between 15 and 26 years. All patients presented with lower limb weakness. The disease typically started with calf weakness and atrophy of legs in the majority of patients (70%) and in some patients (30%), the initial muscle weakness was of limb girdle with the onset of lower limb proximal muscle weakness. In a few patients (64), the first symptom was progressive weakness of the tibialis anterior muscle with foot drop, which later progressed to involve the gastrocnemius and the proximal muscles. The majority of the patients presented with inability to stand on toes (72%). The serum CK of the cohort ranged from 500 to 3000 IU/L.
Dysferlin protein analysis by blood monocyte assay
A modified and simplified blood protein assay involving protein analysis directly in lysates from peripheral blood mononuclear cells (PBMCs) was used for dysferlin protein screening. In brief, PBMCs were isolated from 2 ml of whole blood by Ficoll-Hypaque gradient separation. The cell pellet was lysed by M-PER in the presence of protease inhibitor to prepare protein lysate. Protein quantification was carried out using the Bradford method.[17] Protein analysis was performed using SDS-PAGE electrophoresis using 4%–15% gradient gels. Monoclonal dysferlin antibody (NCL Hamlet) and GAPDH antibody were used at 1:3000 and 1:80,000 dilution, respectively, to detect dysferlin and the housekeeping protein GAPDH. For complete details of protein extraction and western blotting protocol please refer to our recent report.[18]
Automated limb-girdle muscular dystrophy diagnostic assistant tool
A total of 196 patients who were suspected of having a dysferlinopathy based on their clinical presentation or unclassified LGMD patients were evaluated using the LGMD subtyping tool, ALDA, developed by the Jain Foundation. ALDA includes prediction of all LGMD types as well as other muscular dystrophies having similar clinical presentations [Table 1].
The tool results give two parameters, “probability” and “concordance.” The probability percentage indicates the three most likely diagnosis based on the patient's symptoms and the concordance score indicates how well the patient's symptoms correspond to the predicted LGMD subtypes, based on what has been cited in the medical literature. Cases in which ALDA indicated LGMD2B as one of the top three predictions with medium to high concordance were selected for sequencing if the clinical data were provided by a physician on clinical examination. In cases where the clinical data were obtained from the patient directly, lower concordance value was also considered probable dysferlinopathy cases and selected for DNA analysis.
Dysferlin sequencing analysis for molecular confirmation
DNA was extracted from whole blood using the Qiagen genomic DNA extraction (Puregen Blood core) Kit according to the manufacturer's instructions. Sanger sequencing analysis of the 55 coding exons of the DYSF gene was performed for 15 patients initially at Emory genetics laboratory in their CLIA approved diagnostic laboratory using standard sequencing techniques.[15] The DNA analysis for the remaining 110 patients was performed using a NGS panel that included 34 genes associated with LGMDs or associated muscular dystrophies [Figure 1].
Figure 1.

Next generation sequencing limb-girdle muscular dystrophy panel containing the following 34 genes
For the NGS sequencing, the coding and flanking intronic regions up to 50 bp were enriched using the IDT technology and were sequenced using the Illumina HiSeq 2500 system. Direct sequencing of the captured regions was performed using NGS at Emory Genetics Laboratory. The patient's gene sequences were then compared to a standard reference sequence (DYSF NM_003494.3). Sequence variations were classified as pathogenic, likely pathogenic, benign, likely benign, or variants of uncertain significance (VOUS).[19,20]
Target sequencing, for recently identified pathogenic mutation in DYSF Intron50 region was also carried out using PCR of the appropriate intron 50 region, followed by Sanger sequencing.
RESULTS
Patients evaluated
Blood-based screening for dysferlin protein expression by immunoblot analysis using peripheral blood mononuclear cells
Blood samples were collected and protein lysate from the PBMCs isolated from the blood monocytes was run for a total of 166 clinically suspected cases of dysferlinopathy and unclassified LGMD that were collected over a 3-year period from all parts of India including from individuals and referring Neurologists from Mumbai, NIMHANS (Bengaluru) and NIZAMS (Hyderabad) [Table 1]. Protein lysate from control as well as patient monocytes was analyzed for absence and presence of dysferlin protein expression as shown in Figure 2.
Figure 2.
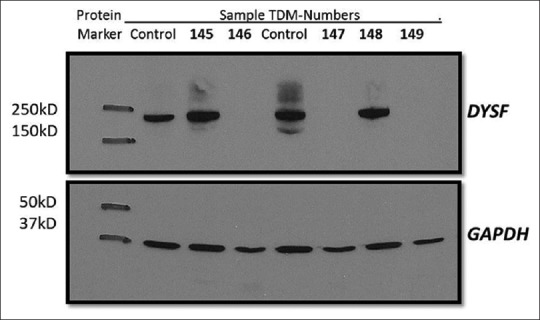
Western blot showing dysferlin protein expression. Immunoblotting analysis of dysferlin protein expression. Protein lysate from control as well as patient monocytes analyzed for dysferlin expression using NCL-hamlet antibody specific for 235 kDa dysferlin protein. Lane 2 and 5 control lysate shows an intense band. Lane 4, 6 and 8 shows absence of dysferlin protein in TDM-146 (JF tool results 97.34% probability with high concordance), TDM-147 (JF tool results 97.28% probability with high concordance), TDM-149 (JF tool results 64.9% probability with medium concordance) respectively. Lane 3 and 7 shows presence of dysferlin protein in TDM-145 (JF tool result 52.2% probability with low concordance) and TDM-148 (JF tool results 95.11% probability with high concordance). GAPDH used as a loading control (37 kDa)
Out of the 166 samples analyzed by the monocyte assay, absence of dysferlin protein was seen in 88 patients, and 78 showed no loss of dysferlin protein on immunoblotting.
With the development of ALDA, clinical data obtained for these patients were run on the tool. WB was done on 45 patients, after running them on ALDA where LGMD2B was predicted as one of the probabilities, 51% showed the absence of DYSF protein [Table 1].
Correlation between the absence of dysferlin protein expression and pathogenic variants found in the dysferlin gene
Of the 88 patients showing the absence of dysferlin protein, 75 patients were selected for sequencing (15 by Sanger sequencing and 60 by NGS) [Table 1]. The other 13 patients were not sequenced due to insufficient blood/DNA, and patient follow up to obtain more sample was not possible. Of the 75 samples sequenced, the sequencing was successful for 72 and the remaining 3 failed due to low-quality sequencing data and are being repeated.
Of the 72 samples successfully sequenced, 97% of the patients showing the absence of dysferlin protein by the monocyte assay showed two variants in the dysferlin gene. One patient showed the presence of a single pathogenic dysferlin mutation and one patient showed 2 variants in the CAPN3 gene. These results indicate how accurate the absence of dysferlin protein by the blood monocyte assay is in predicting who has a dysferlinopathy. Therefore, the blood monocyte assay method is a robust and accurate method to detect likely dysferlinopathy patients.
Performance of automated limb-girdle muscular dystrophy diagnostic assistant in limb-girdle muscular dystrophy subtype prediction
A total of 196 patients of suspected cases of dysferlinopathy (LGMD2B) based on the physician's assessment of the clinical presentation and protein expression evaluated by an immunohistochemical method on muscle biopsies as well as unclassified LGMDs were evaluated using the ALDA tool.[16] The ALDA results showed 101 patients with a probability of LGMD2B and the rest were predicted to have other LGMD subtypes or LGMD-like muscular dystrophies. NGS was carried out on 67 of the 101 predicted dysferlinopathy patients showing concordance between high to medium. A few patients with medium low to low concordance were also sequenced [Figure 3]. The remaining 34 were not sequenced due to low concordance scores. Of the 67 samples sequenced, 75%–70% of patients showing high to medium high concordance showed one or more variants in dysferlin gene, indicating that higher the concordance, the more likely is the probability of finding dysferlin variants [Figure 3].
Figure 3.
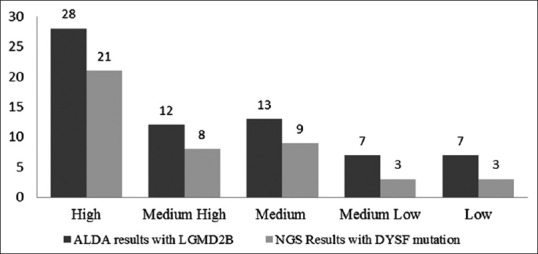
Limb-girdle muscular dystrophy prediction tool and next generation sequencing result for 67 patients: Correlation between automated limb-girdle muscular dystrophy diagnostic assistant scores and dysferlin gene variants. Higher the concordance for limb-girdle muscular dystrophy 2B, the more likely is the probability of finding dysferlin variants
Analysis of the variant data obtained on sequencing
Molecular confirmation and identification of causative pathogenic variants in DYSF gene were performed on 125 patients selected based on monocyte and/or ALDA results. Sanger sequencing was performed on DYSF gene (exons and flanking exon/intron boundaries up to 50 bp) only in the first 15 patients, and for the other 110 patients, sequencing was done by NGS for a panel of 34 genes [Figure 1]. Two predicted causative DYSF pathogenic variants were detected in 90 patients, VOUS were seen in 6 patients. These 6 patients with VOUS were confirmed to be dysferlinopathies due to the absence of DYSF protein on monocyte assay. A single predicted causative DYSF variant was detected in 4 patients, and no causative pathogenic variants of the DYSF gene were identified in 25 patients. Of the 25 patients without causative DYSF mutation, 5 had causative variants in other genes (i.e. GNE, CAPN3) that could explain their phenotype, whereas 11 did not have variants that could clearly diagnose their disease. A novel pathogenic variant in intron 50 of DYSF (c.5668-824C>T) was recently identified by Dr. R.Y. Brown from University of Massachusetts Medical School USA (unpublished data) and was not evaluated by the NGS panels. Therefore, individuals with single or no DYSF variants identified by the other sequencing methods were evaluated for the presence of this intron 50 DYSF pathogenic variant. The intron50 DYSF pathogenic variant (c.5668-824C>T) was detected in 7 patients with 5 cases showing it in the homozygous state and in 2 cases the heterozygous state in combination with an already identified DYSF causative variant. This brings the total number of individuals with 2 identified causative DYSF variants to 77% of those sequenced. The analysis performed for this study did not include the assessment for large deletions or duplications. Therefore, some of the remaining individuals could have these types of mutations in DYSF. Moreover, the cases predicted as LGMD2B on ALDA but with no DYSF variants on NGS sequencing could have a type of LGMD that is very similar in clinical presentation to LGMD2B but is caused by mutations in a gene yet to be identified.
Homozygosity for the identified variants was detected in 84% of the patients [Figure 4] which indicates the high probability of marriages between closely related individuals, endogamy and consanguineous marriages. No recurring DYSF pathogenic variants, except for the 7 cases of the novel intron 50 mutation were identified. For the cases with DYSF variants, variant classification showed missense pathogenic variants in 36.37% of patients, followed by nonsense pathogenic variants at 25.25% and splice site and frameshift pathogenic variants both at around 19% each [Figure 5]. In 22 out of the 100 patients in which DYSF variants were found to be the cause of the disease, other gene variants were also detected.
Figure 4.

Next generation sequencing results of 100 samples showing dysferlin pathogenic variants: 100 patients showing variants in the dysferlin gene. 90 with two pathogenic variants (80 homozygous and 10 heterozygous); 6 with variants of uncertain significance (4 homozygous and 2 heterozygous); 4 with single pathogenic variant
Figure 5.
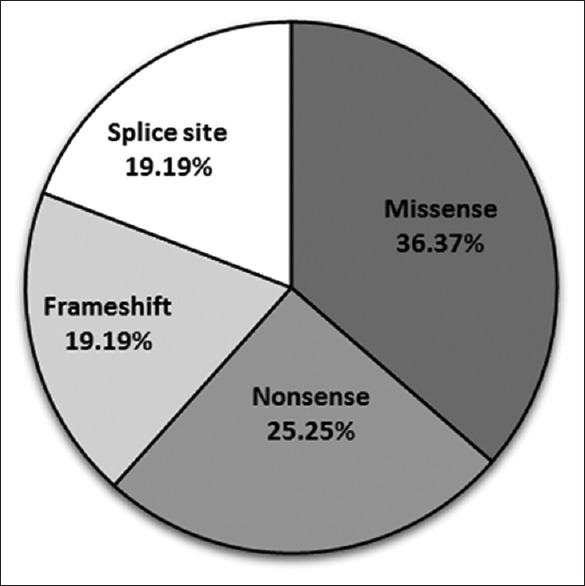
Variant classification of dysferlin gene pathogenic variants
DISCUSSION
Most of the laboratories in India currently depend on the expression of dysferlin protein seen in muscle biopsy specimen by IHC methods or by WB using muscle tissue protein for confirming a clinical diagnosis of dysferlinopathy. Different IHC patterns of dysferlin expression in muscle biopsies from patients of various LGMDs have been described by many authors, but only a few have compared with the expression seen in peripheral blood monocyte.[2,12,14] dysferlin protein levels in the blood have been shown to correlate with what is seen in the muscle, and mutant proteins known to result in the secondary reduction in dysferlin are not expressed in the blood. Therefore, as shown in this study and by others,[15,18] the dysferlin blood monocyte assay can be a reliable screening method to delineate primary dysferlinopathies from other LGMD subtypes.
In addition to the high specificity of this blood-based assay for identifying dysferlinopathy, this method is less invasive as it requires only a blood draw versus a muscle biopsy. This not only provides a significant cost saving benefit, especially in India but also makes the collection of the sample a much more approachable option than collecting muscle biopsies from affected individuals from various centers across the country. The monocyte assay can also be a useful biomarker for future clinical trials.
However, from some parts of India and from other countries, it is difficult to get the necessary sample to do the monocyte assay as the blood needs to be kept cold and arrive at the laboratory within 24 hrs. of the draw. Therefore, a tool that would accurately identify individuals with dysferlinopathy based on clinical symptoms could be useful in recruiting more patients from both within and outside of India as we would then only need to collect blood for the isolation of DNA which does not require the blood being kept cold or collect already isolated DNA for sequencing confirmation. The Jain Foundation developed such a tool, called ALDA, that uses clinical symptoms to predict the most likely form of LGMD or related muscular dystrophy a person may have. In this study, a cohort of patients was subjected to analysis by the ALDA tool for subtype prediction of LGMD. Our analysis showed that causative pathogenic variants in the dysferlin gene were found in 57% of those predicted to have a dysferlinopathy based solely on ALDA. The efficacy of ALDA prediction for dysferlinopathy phenotype was directly correlated with the quality of the clinical information received. Cases, in which the clinical history came directly from the patient or where limited clinical information was available, were less predictive. A small cohort of individuals was analyzed by both the monocyte assay and ALDA. A good correlation between the ALDA tool prediction and the monocyte assay was observed. Any discordance between the two was likely due to the lack of detailed clinical findings which is known to limit ALDA's prediction capability.
With the evaluation of the performance, diagnostic specificity, and sensitivity of ALDA, clinical diagnosis can become more straightforward and affordable. The clinicians seeing the patients can use this freely available online tool in real time while examining the patient and asking the questions put forth by ALDA tool. This could allow the physician to come to a diagnosis or at least could narrow down the possibilities to a small subset of potential diagnoses for further genetic confirmation. While the specificity and sensitivity of the tool for other LGMD subtypes has not been evaluated in this study, the Jain Foundation is doing a larger analysis and has so far determined that some of the more common LGMDs or LGMD-like muscular dystrophies such as LGMD2I, Bethlem, and Becker show predictive abilities similar to what has been seen in this study for dysferlinopathy (personal communication).
Dysferlinopathy or LGMD2B and LGMD2A are the two most common subtypes of LGMDs worldwide and in India as well, and therefore, the ability to distinguish at least these two from each other and from other LGMD subtypes by itself can be very useful and significantly helpful. Two founder pathogenic variants in the Calpain 3 gene were identified in a cohort of patients with LGMD2A from unrelated families of the “Agrawal” community sharing common Indian ancestry has been reported.[15] Two sibling belonging to the “Agrawal” community with an LGMD phenotype, who did not show the founder mutation when screened for CAPN3, were then included for the LGMD2B study. When their clinical details were run on the ALDA tool, they revealed a high probability score of more than 90% with high concordance for LGMD2B. The monocyte assay was done for both these siblings and showed the absence of dysferlin protein expression. On NGS, two pathogenic variants in the dysferlin gene were identified.
A wide range of dysferlin pathogenic variants, including missense, nonsense, frameshift, deletion/insertion, splice-site mutation, and large deletions have been identified.[4,21,22,23,24,25,26] Limited mutation data on the dysferlin gene are available for Indian patients so far, except, one reported case of an Indian family with a 22 year of follow-up, one heterozygous mutation in Exon 54, c.6124C>T was identified in the dysferlin gene.[27] This study shows a similar pattern of pathogenic variant types in the Indian populations as has been seen in other populations. Given the number of variants identified and the high frequency of missense variants, the use of a tool such as the Universal Mutation Database for dysferlin (UMD-DYSF), a Locus-Specific Database has been developed by Blandin et al.[28] which provides an updated compilation of mutational data and relevant interactive tools for the analysis of DYSF sequence variants is very helpful for diagnostic and research purposes.
Other important findings of this study are the lack of recurrent pathogenic variants and the high degree of homozygosity in the Indian cohort. Founder pathogenic variants in dysferlinopathy patients have been reported in native Canadian, Libyan, Jewish, Spanish, Italian, and Caucasian Jewish population.[29,30,31,32] Frequencies of such common and founder pathogenic variants in different Indian subpopulations and their significance in clinical genetics have been reviewed elsewhere.[15] However, in the present study, no founder-mutation effect in any of the specific ethnic groups was seen. This means that the entire dysferlin gene should be evaluated in each patient to identify the causative pathogenic variants instead of just evaluating for a single founder mutation. This makes the genetic analysis for dysferlinopathy more costly and highlights the benefit of narrowing down the patient pool using the above-described tools before embarking on DNA sequencing. The high frequency of homozygous pathogenic variants (91.66%) seen in our patients is likely because in the Indian population the majority of people marry within the same ethnic groups and consanguineous marriages are also highly prevalent. This highlights the need for careful genetic counselling of the families involved, and hence they understand the recurrence risks and the possible screening methods to be undertaken to mitigate these risks.
CONCLUSION
Given the diversity of ethnicity and genetic background of the Indian population, the use of the non-invasive dysferlin blood monocyte test and the ALDA tool serves as a guide to clinicians in delineating dysferlinopathies from other overlapping LGMD phenotypes. Having a cohort of highly probable dysferlinopathy patients identified based on these tools before initiating DNA sequencing makes the genetic confirmation of these individuals much more reliable and cost effective. This in turn could lead to larger numbers of dysferlinopathy patients being identified and a better understanding and management of dysferlinopathy in the Indian population.
Financial support and sponsorship
Jain Foundation, USA.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
Special thanks are due to Dr. Sasikumar K. Menon, Assistant Director, TDM Labs Mumbai for providing the infrastructural facilities and Ms. Mugdha Devalkar, Research Fellow, for her assistance in the project work. The study was entirely funded by the Jain Foundation, Seattle, USA.
REFERENCES
- 1.Khadilkar SV, Singh RK, Agarwal P, Krahn M, Levy N. Twenty-two year follow-up of an Indian family with dysferlinopathy-clinical, immunocytochemical, Western blotting and genetic features. Neurol India. 2008;56:388–90. doi: 10.4103/0028-3886.43459. [DOI] [PubMed] [Google Scholar]
- 2.Nalini A, Gayathri N. Dysferlinopathy: A clinical and histopathological study of 28 patients from India. Neurol India. 2008;56:379–85. doi: 10.4103/0028-3886.40964. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–6. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen K, Bassez G, Bernard R, Krahn M, Labelle V, Figarella-Branger D, et al. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum Mutat. 2005;26:165. doi: 10.1002/humu.9355. [DOI] [PubMed] [Google Scholar]
- 5.Matsuda C, Aoki M, Hayashi YK, Ho MF, Arahata K, Brown RH., Jr Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology. 1999;53:1119–22. doi: 10.1212/wnl.53.5.1119. [DOI] [PubMed] [Google Scholar]
- 6.Doherty KR, McNally EM. Repairing the tears: Dysferlin in muscle membrane repair. Trends Mol Med. 2003;9:327–30. doi: 10.1016/s1471-4914(03)00136-9. [DOI] [PubMed] [Google Scholar]
- 7.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–72. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 8.Urtizberea JA, Bassez G, Leturcq F, Nguyen K, Krahn M, Levy N. Dysferlinopathies. Neurol India. 2008;56:289–97. doi: 10.4103/0028-3886.43447. [DOI] [PubMed] [Google Scholar]
- 9.Miyoshi K, Kawai H, Iwasa M, Kusaka K, Nishino H. Autosomal recessive distal muscular dystrophy as a new type of progressive muscular dystrophy. Seventeen cases in eight families including an autopsied case. Brain. 1986;109(Pt 1):31–54. doi: 10.1093/brain/109.1.31. [DOI] [PubMed] [Google Scholar]
- 10.Khadilkar SV, Singh RK, Kulkarni KS, Chitale AR. A study of clinical and laboratory features of 14 Indian patients with dysferlinopathy. J Clin Neuromuscul Dis. 2004;6:1–8. doi: 10.1097/01.cnd.0000134859.41385.6e. [DOI] [PubMed] [Google Scholar]
- 11.Anderson L. Multiplex Western blot analysis of muscular dystrophy proteins. Methods Mol Med. 1999;43:369–86. doi: 10.1016/S0002-9440(10)65354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho M, Gallardo E, McKenna-Yasek D, De Luna N, Illa I, Brown RH., Jr A novel, blood-based diagnostic assay for limb girdle muscular dystrophy 2B and Miyoshi myopathy. Ann Neurol. 2002;51:129–33. doi: 10.1002/ana.10080. [DOI] [PubMed] [Google Scholar]
- 13.De Luna N, Freixas A, Gallano P, Caselles L, Rojas-García R, Paradas C, et al. Dysferlin expression in monocytes: A source of mRNA for mutation analysis. Neuromuscul Disord. 2007;17:69–76. doi: 10.1016/j.nmd.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Gallardo E, de Luna N, Diaz-Manera J, Rojas-García R, Gonzalez-Quereda L, Flix B, et al. Comparison of dysferlin expression in human skeletal muscle with that in monocytes for the diagnosis of dysferlin myopathy. PLoS One. 2011;6:e29061. doi: 10.1371/journal.pone.0029061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ankala A, Kohn JN, Dastur R, Gaitonde P, Khadilkar SV, Hegde MR. Ancestral founder mutations in calpain-3 in the Indian Agarwal community: Historical, clinical, and molecular perspective. Muscle Nerve. 2013;47:931–7. doi: 10.1002/mus.23763. [DOI] [PubMed] [Google Scholar]
- 16.LGMD Subtyping tool Version 2.0 Jain Foundation. 2013. Available from: http://www.jain-foundation.org/lgmd-subtyping-diagnosis-tool .
- 17.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 18.Ankala A, Nallamilli B, Rufibach L, Hwang E, Hegde M. Diagnostic overview of blood-based dysferlin protein assay for dysferlinopathies. Muscle Nerve. 2014;50:333–9. doi: 10.1002/mus.24195. [DOI] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ankala A, da Silva C, Gualandi F, Ferlini A, Bean LJ, Collins C, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015;77:206–14. doi: 10.1002/ana.24303. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen K, Bassez G, Krahn M, Bernard R, Laforêt P, Labelle V, et al. Phenotypic study in 40 patients with dysferlin gene mutations: High frequency of atypical phenotypes. Arch Neurol. 2007;64:1176–82. doi: 10.1001/archneur.64.8.1176. [DOI] [PubMed] [Google Scholar]
- 22.Krahn M, Béroud C, Labelle V, Nguyen K, Bernard R, Bassez G, et al. Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum Mutat. 2009;30:E345–75. doi: 10.1002/humu.20910. [DOI] [PubMed] [Google Scholar]
- 23.Pramono ZA, Tan CL, Seah IA, See JS, Kam SY, Lai PS, et al. Identification and characterisation of human dysferlin transcript variants: Implications for dysferlin mutational screening and isoforms. Hum Genet. 2009;125:413–20. doi: 10.1007/s00439-009-0632-y. [DOI] [PubMed] [Google Scholar]
- 24.Tagawa K, Ogawa M, Kawabe K, Yamanaka G, Matsumura T, Goto K, et al. Protein and gene analyses of dysferlinopathy in a large group of Japanese muscular dystrophy patients. J Neurol Sci. 2003;211:23–8. doi: 10.1016/s0022-510x(03)00041-8. [DOI] [PubMed] [Google Scholar]
- 25.Aoki M, Liu J, Richard I, Bashir R, Britton S, Keers SM, et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology. 2001;57:271–8. doi: 10.1212/wnl.57.2.271. [DOI] [PubMed] [Google Scholar]
- 26.Klinge L, Aboumousa A, Eagle M, Hudson J, Sarkozy A, Vita G, et al. New aspects on patients affected by dysferlin deficient muscular dystrophy. J Neurol Neurosurg Psychiatry. 2010;81:946–53. doi: 10.1136/jnnp.2009.178038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khadilkar SV, Singh RK. Limb girdle muscular dystrophies in India. Neurol India. 2008;56:281–8. doi: 10.4103/0028-3886.43446. [DOI] [PubMed] [Google Scholar]
- 28.Blandin G, Beroud C, Labelle V, Nguyen K, Wein N, Hamroun D, et al. UMD-DYSF, a novel locus specific database for the compilation and interactive analysis of mutations in the dysferlin gene. Hum Mutat. 2012;33:E2317–31. doi: 10.1002/humu.22015. [DOI] [PubMed] [Google Scholar]
- 29.Argov Z, Sadeh M, Mazor K, Soffer D, Kahana E, Eisenberg I, et al. Muscular dystrophy due to dysferlin deficiency in Libyan Jews. Clinical and genetic features. Brain. 2000;123(Pt 6):1229–37. doi: 10.1093/brain/123.6.1229. [DOI] [PubMed] [Google Scholar]
- 30.Cagliani R, Fortunato F, Giorda R, Rodolico C, Bonaglia MC, Sironi M, et al. Molecular analysis of LGMD-2B and MM patients: Identification of novel DYSF mutations and possible founder effect in the Italian population. Neuromuscul Disord. 2003;13:788–95. doi: 10.1016/s0960-8966(03)00133-0. [DOI] [PubMed] [Google Scholar]
- 31.Vilchez JJ, Gallano P, Gallardo E, Lasa A, Rojas-García R, Freixas A, et al. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the Spanish population. Arch Neurol. 2005;62:1256–9. doi: 10.1001/archneur.62.8.1256. [DOI] [PubMed] [Google Scholar]
- 32.Leshinsky-Silver E, Argov Z, Rozenboim L, Cohen S, Tzofi Z, Cohen Y, et al. Dysferlinopathy in the Jews of the Caucasus: A frequent mutation in the dysferlin gene. Neuromuscul Disord. 2007;17:950–4. doi: 10.1016/j.nmd.2007.07.010. [DOI] [PubMed] [Google Scholar]