

Abstract
Comprehensive understanding of a gene’s expression and regulation at the molecular level requires identification of all proteins interacting with the gene. HyCCAPP (Hybridization Capture of Chromatin Associated Proteins for Proteomics) is an approach that uses single-stranded DNA oligonucleotides to capture specific genomic sequences in cross-linked chromatin fragments and identify associated proteins by mass spectrometry. Previous studies have shown HyCCAPP to provide useful information on protein–DNA interactions, revealing the proteins associated with the GAL110 region in yeast. We present here a multiplexed version of HyCCAPP. Utilizing a toehold-mediated capture/release strategy, HyCCAPP is targeted to multiple genomic loci in parallel, and the protein binders at each locus are eluted in a programmable and selective fashion. Multiplexed HyCCAPP was applied to four genes (25S rDNA, ARX1, CTT1, and RPL30) in S. cerevisiae under normal and stressed conditions. Capture and release efficiencies and specificities were comparable to those obtained without multiplexing. Using mass spectrometry-based bottom-up proteomics, hundreds of proteins were discovered at each locus in each condition. Statistical analysis revealed 34–88 enriched proteins in each gene capture. Many of these proteins had expected functions, including DNA-related and ribosome biogenesis-associated activities. Multiplexed HyCCAPP provides a useful strategy for the identification of proteins interacting with specific chromatin regions.
Graphical abstract
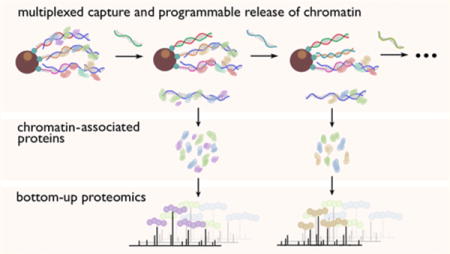
Numerous physiological functions in cells trace back to interactions between proteins and DNA at the molecular level. There are a variety of existing approaches to investigate these essential protein–DNA interactions. Both the protein-centric chromatin immunoprecipitation (ChIP)1 and DNA adenine methyltransferase identification (DamID)2 approaches map a specific protein’s binding loci in the genome, while both the DNA-centric PICh (Proteomics of Isolated Chromatin) strategy developed by Déjardin and Kingston3 and our group’s HyCCAPP (Hybridization Capture of Chromatin-Associated Proteins for Proteomics)4,5 reveal the proteins interacting with specific genomic regions. Our original HyCCAPP technique4 employed a single-stranded desthiobiotinylated capture oligonucleotide to hybridize with a specific sequence in formaldehyde-cross-linked chromatin fragments from a cell lysate. We then isolated hybridized fragments and associated proteins with streptavidin-conjugated magnetic particles and identified captured proteins using mass spectrometry-based bottom-up proteomics. However, this original HyCCAPP technique targeted only a single locus per experiment and required a large quantity of cells due to limitations in capture efficiency and mass spectrometer sensitivity. To alleviate these limitations, we sought to develop a multiplexed version of the HyCCAPP strategy, which would allow proteins to be identified from multiple loci in parallel.
We present here a multiplexed HyCCAPP technique that employs toehold-mediated DNA branch migration to allow the capture of multiple chromatin loci in parallel. The toehold-mediated capture and release strategy uses biotinylated toehold capture oligonucleotides and release oligonucleotides as illustrated in Figure 1 and has previously been shown to enable the selective release of targeted DNA sequence subsets from solid supports.6 Capture oligonucleotides for different targets are added to the cell lysate simultaneously, whereas release oligonucleotides, which recognize specific toehold sequences, are introduced sequentially to the bead suspension, releasing one target at a time (Figure 1).
Figure 1.
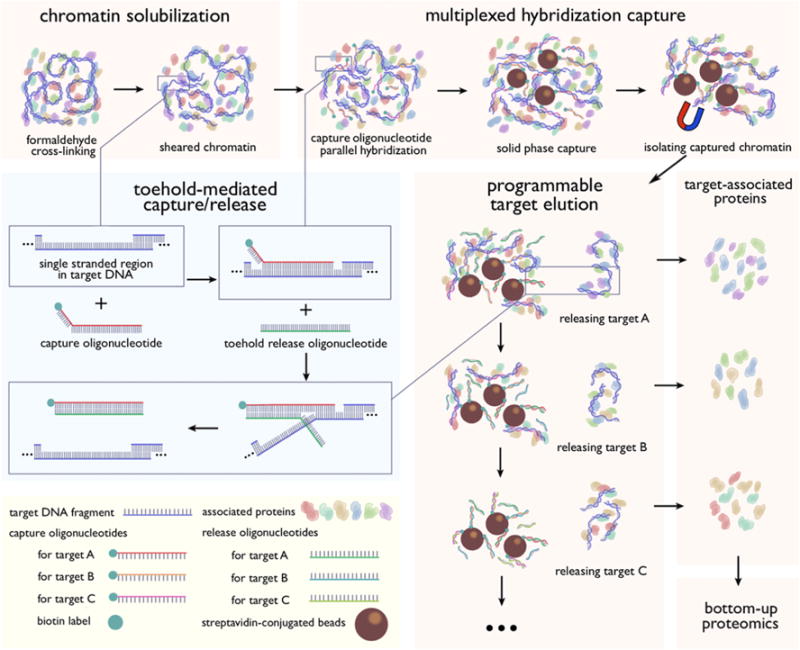
Schematic diagram for multiplexed HyCCAPP with toehold-mediated capture/release strategy. Capture oligonucleotides contain a 30-nucleotide sequence, complementary to target DNAs, and an 8 to 9-nucleotide toehold at the 3′ end. Complete sequences of all oligonucleotides are provided in the SI Tables S-1 and S-2. The 3′ end is biotinylated to enable solid-phase capture with streptavidin magnetic beads. Release oligonucleotides are single-stranded oligonucleotides 38 to 39 nucleotides in length whose sequence is completely complementary to their capture oligonucleotide counterparts. Since the toehold sequence does not hybridize with the targeted genomic sequence, the release oligonucleotides are thermodynamically favored to hybridize with the capture oligonucleotides when introduced, displacing the previously bound targets. Sequential addition of different release oligonucleotides permits the programmable and stepwise elution of captured chromatin regions.
In our previous proof-of-principle study of multiplexed toehold release, the specific capture and release of chromatin regions corresponding to three S. cerevisiae genomic loci was demonstrated at a scale of 109 cells,6 suitable for validation of the results at a nucleic acid level but insufficient for proteomic analysis of the associated proteins. The DNA results indicated satisfactory capture specificity, with an average target/nontarget ratio of 290. In the present work the strategy has been scaled up to 1011 cells, a level adequate for proteomic analysis. We targeted one multicopy and three single-copy regions in S. cerevisiae, the 25S rDNA, ARX1, RPL30, and CTT1 genes. The first three genes play important roles in ribosome biogenesis, while CTT1 encodes cytosolic catalase that is involved in the yeast stress response.7–10 The functionalities of these genes can be reflected by the sets of proteins identified using multiplexed HyCCAPP, validating this as a powerful technology to profile protein–DNA interactions at multiple genomic loci of interest from a single cell lysate preparation.
EXPERIMENTAL SECTION
Materials
Yeast extract peptone dextrose (YPD) (Y1375), 37% formaldehyde (F8775), protease inhibitors for fungi (P8215), capture and release oligonucleotides (SI Tables S-1 and S-2), trichloroacetic acid (TCA) (T0699), urea (U5378), deoxycholic acid (D2510), DL-dithiothreitol solution (DTT) (646563), iodoacetamide (I6125), trifluoroacetic acid (TFA) (T6508), ethyl acetate (270989), acetone (270725), and TWEEN 20 (P7949) were purchased from Sigma-Aldrich Co. (St. Louis, MO). The 20% sodium dodecyl sulfate (SDS) (161-0418) was purchased from Bio-Rad (Hercules, CA). Phosphate buffered saline (PBS) (P0191), 5 M Tris pH = 8 (T5581), 1 M Tris pH = 7 (T1070), 5 M sodium chloride (NaCl) (S0250), 500 mM ethylenediaminetetraacetic acid (EDTA) (E0306), and 0.2 M ammonium bicarbonate buffer (A2012) were purchased from Teknova (Hollister, CA). RNaseA (12091-039) was purchased from Life Technologies (Carlsbad, CA). qPCR probes and primers (SI Table S-3) were ordered from Integrated DNA Technologies (Coralville, IA). qPCR probes master solutions (0470749001) was purchased from Roche (Indianapolis, IN). Sera-Mag streptavidin particles (30152104010350) and formic acid (M1116701000) were purchased from Thermo Fisher Scientific (Waltham, MA). DNaseI (M0303S) was purchased from New England BioLabs (Ipswich, MA). Trypsin (V5111) was purchased from Promega (Madison, WI). Acetonitrile (ACN) (AH015-4) was purchased from Honeywell (Morris Plains, NJ).
Cell Lysate Preparation and Chromatin Solubilization
Cell culture and lysis procedures of S. cerevisiae strain Y1788 were similar to those previously described.4 Briefly, yeast cells were grown in four flasks of 1.5 L YPD media to an OD600 of about 2.0. For stressed conditions, 232.5 mL of YPD containing 5 M NaCl sterile solution was added into each flask to give a salt concentration of 0.7 M. The salt-stressed culture continued to shake for 30 min, allowing biological changes to take place. Both normal and stressed cultures were cross-linked at room temperature (RT) with 3% formaldehyde for 30 min and quenched with 664 mM Tris for 10 min at RT. Cells were pelleted and washed with PBS. Pellets from 6 L of culture were resuspended in 180 mL of lysis buffer (200 mM NaCl, 20 mM EDTA, 50 mM Tris pH 7) containing 200× diluted protease inhibitors DMSO cocktail solution and were lysed with a Constant Systems TS series cell disruptor at 30 kpsi.
SDS was added to the cell lysate to a 1% final concentration, and cell lysate was incubated in a 65 °C water bath for 5 min. Lysate was then sonicated in an ice water bath with a MisoniX Ultrasonic Processor S4000 at 20 V for 3 min with 4 s on/4 s off intervals in 50 mL aliquots (four rounds to process about 200 mL cell lysate). Sonicated lysate was centrifuged at 4 °C at 8 000g for 12 min. Supernatant was diluted 5-fold in lysis buffer containing protease inhibitors to lower the SDS concentration to 0.2%. The total volume is about 1 L at this point and was split in half into two 1 L Erlenmeyer flasks. A volume of 1.5 mL of 20 mg/mL RNaseA was added to each flask. The lysate was then shaken at 150 rpm at 37 °C for 1 h and centrifuged at 23 °C at 15 000g for 15 min.
Multiplexed Chromatin Capture
To remove interfering endogenous biotinylated molecules naturally present in yeast cells, lysate was pretreated with streptavidin-conjugated particles. Four mL of Sera-Mag streptavidin particles were washed using wash buffer (200 mM NaCl, 0.2% SDS, 50 mM Tris pH 8) and then resuspended in 5 mL of wash buffer. A volume of 2.5 mL of the particle suspension was added to each flask, followed by shaking at 150 rpm at 23 °C for 1 h. The particles were then removed with DynaMag-50 magnets (Life Technologies) from 50 mL aliquots of lysate. A 200 μL aliquot of lysate was employed to measure input target sequence concentration by qPCR. In total, 80 pmol of each single-copy locus (ARX1, CTT1, PRL30) capture oligonucleotides (15 = 3 × 5 oligonucleotides), and 400 pmol of the 25S locus oligonucleotide were added to the lysate. The sample was split in half again and shaken at 150 rpm at 37 °C for 3 h. Four mL of the streptavidin particles was washed and resuspended in 5 mL of wash buffer. A volume of 2.5 mL of the washed particle solution was added to each flask followed by shaking at 150 rpm at 23 °C for 1 h. The particles were then magnetically isolated in 50 mL aliquots. A volume of 4.8 mL of the lysate was saved as the uncaptured lysate control in four 1.7 mL tubes, and the remainder was discarded. Collected particles were washed in 100 mL of wash buffer 4 times. Each wash was done at RT with gentle rotation for 5 min. Particles were then concentrated into 6 mL (1.5 mL in four 1.7 mL low retention tubes) and washed for 5 min and then 1 h. Release oligonucleotide solutions, one for each locus, were prepared by adding 8 nmol of each release oligonucleotide, 5 total oligonucleotides per single-copy locus (ARX1 CTT1, RPL30), or 40 nmol for the multicopy 25S locus to 4.8 mL of wash buffer. A volume of 1.2 mL of the first release oligonucleotide solution (25S) was added to each tube of particles, and the tube was rotated for 15 min at room temperature. The particles were magnetically isolated, and the solution from each tube was transferred to a new 1.7 mL tube. Thus, a total volume of 4.8 mL of released solution was collected in four tubes. Particles were washed twice with 1.5 mL wash buffer prior to the addition of the subsequent release oligonucleotide solution, in the order of CTT1, ARX1, and RPL30. Three biological replicates were performed for both normal and salt-stressed conditions. To quantify DNA capture efficiencies and specificity, qPCR Analysis was conducted as previously described.4
Mass Spectrometry
Proteins in the released samples were precipitated with TCA. Protein pellets can be stored at −80 °C at this step. Pellets then went through DNase treatment and enhanced filter-aided sample preparation (eFASP) to eliminate contaminants such as extra release oligonucleotides, salts, and lipids. Proteins were digested on filters during eFASP, which was able to recover most peptides at this low protein level.11 The samples were further desalted using C18 solid-phase extraction tips (Agilent Technologies). See the Supporting Information for details of these sample preparation procedures. Samples and control were analyzed by HPLC–ESI-MS/MS. The microcolumn employed for HPLC was packed with 20 cm of 1.7 μm-diameter/130 Å pore size C18 beads. Full-mass profile scans were performed on an LTQ Velos Orbitrap mass spectrometer (Thermo Fisher Scientific) with parameters as described previously.4 Two technical replicates were performed on each biological replicate.
Data Analysis
The 60 resultant raw data files ([4 loci samples + 1 uncaptured lysate control] × 2 technical replicates × 3 biological replicates × 2 conditions) were searched by MaxQuant against the UniProt S. cerevisiae proteome database allowing a 1% false discovery rate. Detailed search parameter settings can be found in the Supporting Information. The identified proteins and their abundance values (LFQ, for Label Free Quantification)12 were imported into Microsoft Excel. LFQ values were normalized across biological replicates, and the average was calculated. Proteins were given percentile rank values in each sample based on replicate average LFQ. Differences in percentile rank were calculated by subtracting capture rank from the uncaptured lysate rank. Proteins with a rank difference above 10% in nonstressed experiments and 5% for stressed experiments were considered enriched. Enriched proteins were examined in terms of their molecular functions. In addition, Perseus software13 was used to highlight significantly enriched proteins and to visualize quantification results in a heat map.
RESULTS AND DISCUSSION
Multiplexed DNA Capture
In this multiplexed HyCCAPP study, five capture oligonucleotides were designed for each single-copy gene (ARX1, CTT1, and RPL30). The target sequences were spread across each gene region, separated by several hundred bases to maximize coverage. For the multicopy 25S rDNA targets, a single capture oligonucleotide was designed to complement an antisense sequence near the 3′ end (SI Figure S-1). Each capture oligonucleotide contains a 5′ biotin moiety, an 8–9 nucleotide toehold region, and a 30-nucleotide sequence for hybridization. Capture oligonucleotides within a target set share a unique toehold sequence (SI Tables S-1 and S-2).
Small-scale experiments (between 1/10th to 1/100th of the material obtained using the protocol detailed in the Experimental Section) were conducted to test the efficiency and specificity of the multiplexed capture. DNA from the released samples and the uncaptured lysate sample were analyzed with qPCR assays designed to detect the four targeted gene loci (SI Figure S-1). Efficiency of DNA capture was determined by the amount of the target DNA sequence in the sample compared to that in the lysate and was generally comparable to that obtained in standard (not multiplexed) HyCCAPP4 (0.22–1.11%) (SI Figure S-2). Although capture oligonucleotides were hybridized to the chromatin at the same time, release oligonucleotides were added sequentially to specifically release each targeted DNA–protein complex. The release process using the toehold-mediated strategy took 15 min per target, representing a significant increase in speed compared to the former 2-h release with free biotin.4 Capture specificity was estimated by qPCR for each single copy locus as the ratio of target DNA captured to the average capture of the off-target DNAs and ranged from 35 to 75-fold. The order of target release does not drastically affect efficiency and specificity (SI Figure S-3).
Associated Proteins
Mass spectrometric analysis was performed on the specifically captured and released chromatin samples as detailed in the Experimental Section. Hundreds of proteins were identified from each of the released samples obtained under normal and stressed conditions for three biological replicates (Table 1). These proteins, like those generally obtained in affinity purification experiments, include both true binders and background contaminants. We employed several bioinformatics tools to assist in the identification of true binders. Analysis of normal condition data with Perseus revealed significantly enriched proteins in capture samples compared to uncaptured lysate (SI Figure S-4). Gene ontology (GO) analysis using FunSpec14 showed these proteins to be highly enriched in GO terms such as DNA-directed RNA polymerase activity (p-value = 4.859 × 10−7), transcription of nuclear rRNA (1.035 × 10−6), and DNA-directed RNA polymerase I (3.74 × 10−9), as one might expect for chromatin-associated proteins. On the other hand, the proteins that were present at higher levels in lysate than in capture samples are associated with GO terms such as catalytic activity, metabolic process, mitochondrion, and cytoplasm. To identify additional proteins enriched at the targeted sequences, a method based upon changes in rank abundance in capture samples compared to lysate was employed.4 All proteins were ranked based upon their MaxQuant LFQ abundance in the respective samples/lysate and were assigned a percentile rank value between 0% and 100%. Proteins whose changes in percentile rank (delta-percentile-rank) between lysate and capture samples were greater than 10% (normal conditions) or 5% (stressed conditions) were selected for further analysis. This process revealed 161 total proteins (SI Table S-4), 69–88 of which were capture-enriched in normal yeast growth conditions and 34–48 of which were capture-enriched in stressed conditions (Table 1 and SI Table S-5). Enriched GO terms for the pooled set of proteins included DNA-directed RNA polymerase activity (p-value = 3.371 × 10−10), nucleotide binding (4.857 × 10−9), RNA polymerase I complex (6.137 × 10−12), nucleolus (1.84 × 10−5), and RNA polymerase II core complex (1.667 × 10−4). Nucleotide polymerases were prominent among the captured proteins. These included several protein subunits of RNA polymerase I complex (Pol I), namely, Rpa12, Rpa135, Rpa190, Rpa34, Rpa43, and Rpa49. Pol I is involved in rRNA synthesis, playing an important role in eukaryotic ribosome biogenesis.15 Proteins Rpb2 and Rpb10, which are subunits of RNA polymerase II, the enzyme responsible for mRNA synthesis, were enriched at the protein-coding ARX1, CTT1, and RPL30 loci. Interestingly, the method also captured several kinases that are known to bind RNA Pol II and are associated with genes during transcription, including Hog1, Hrr25, Cdc28, and Rck2.16,17
Table 1.
Protein Data Summarya
| normal | stressed | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| proteins in uncaptured lysate | 1014 | 1503 | ||||||
|
| ||||||||
| 25S rDNA | ARX1 | CTT1 | RPL30 | 25S rDNA | ARX1 | CTT1 | RPL30 | |
| total proteins identified | 639 | 657 | 635 | 650 | 517 | 667 | 530 | 509 |
| enriched proteins | 75 | 69 | 72 | 88 | 34 | 48 | 38 | 39 |
| enriched at locus | 24 | 17 | 11 | 26 | 13 | 25 | 22 | 18 |
| proteins with expected GO term keywords | ||||||||
| nucleus | 30 | 25 | 31 | 37 | 13 | 18 | 11 | 12 |
| nucleotide binding | 21 | 21 | 29 | 31 | 14 | 19 | 11 | 13 |
| DNA | 14 | 14 | 13 | 15 | 9 | 9 | 10 | 11 |
| nucleolus | 11 | 9 | 10 | 10 | 7 | 7 | 5 | 7 |
| rRNA | 10 | 9 | 9 | 10 | 5 | 7 | 7 | 8 |
| RNA polymerase | 7 | 9 | 9 | 9 | 5 | 6 | 7 | 9 |
| ribosome biogenesis | 5 | 3 | 5 | 6 | 2 | 4 | 0 | 1 |
| ribosomal export | 3 | 2 | 3 | 3 | 0 | 0 | 0 | 0 |
| ribosomal proteins | 5 | 5 | 6 | 4 | 2 | 3 | 2 | 2 |
| stress response | 0 | 0 | 0 | 1 | 0 | 3 | 0 | 0 |
Bottom-up proteomics identified around 600 proteins from each capture sample. Delta-percentile-rank analysis revealed between 34 and 88 enriched proteins, between 11 and 26 of which were enriched at specific locus (SI Table S-5). Many of the enriched proteins have GO terms associated with DNA, nucleus, and other chromatin-related functions. Complete lists of all proteins identified are provided in Supporting Information Table S-6.
A heat map was generated showing abundance levels for the 161 enriched proteins (Figure 2). While overall, similar patterns of protein abundance and their changes in response to stress were evident for each of the four loci, there are a number of interesting examples of significant abundance differences for particular subgroups or individual proteins. For example, DNA polymerase ϕ (Pol5) was found to be enriched at the 25S rDNA locus under normal conditions but was not seen at the other loci nor under stressed conditions. This is consistent with Pol5’s known function in transcription regulation at the rDNA loci.18 The nucleolar proteins Rlp7, Nog1, and Nip7 were also specifically enriched in the 25S rDNA capture. These proteins are involved in pre-rRNA processing and modification which occur cotranscriptionally at the DNA loci,19 and it is thus reasonable for them to be found associated with the region. Rlp7 is required for 25S rDNA precursor processing and binds to the 5′ end of the 25S pre-rRNA.20 This binding site is close to the 3′ end of the rDNA template strand, which is where the capture oligonucleotide was designed to hybridize and thus a region that is likely to be captured with high efficiency. Decreases were also observed in the levels of the Pol I protein at the 25S rDNA locus and of Pol II proteins at the ARX1 and RPL30 loci under stressed conditions. While this was not surprising for these transcription activities, which are known to decrease under stressed conditions,21 it was unexpected for Pol I to be observed at non-rDNA loci, as it only transcribes rDNA. It was also unexpected that no increase of Pol II enrichment was observed at the CTT1 locus, as this gene has been reported to be up-regulated during stress.10 Another interesting observation was the 50% increase in the relative abundance (percentile rank) of DNA topoisomerase 1 (Top1) at the 25S rDNA locus in response to salt stress (SI Table S-4). This is consistent with its role in rDNA transcription silencing, which was shown in a TOP1 knockout study.22
Figure 2.
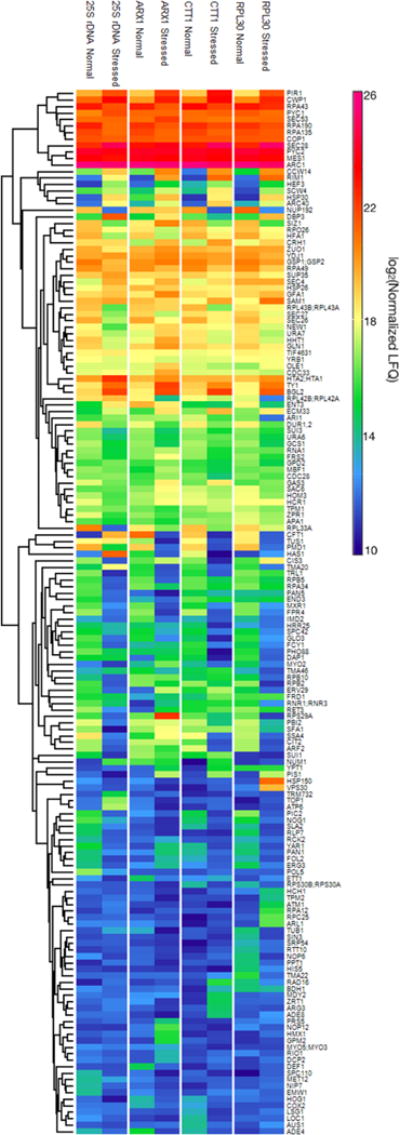
Heat map of enriched proteins. As described in the main text, 161 proteins were identified as enriched at one or more of the four loci. The corresponding protein abundance levels are shown here for each of the four loci and two conditions (normal vs stressed) investigated. Details of the heat map generation are provided in the Supporting Information, along with an alternative arrangement of the figure (SI Figure S-5) to facilitate comparison of results from the different loci.
Proteins with no known DNA-related functions were also observed, such as biotin carboxylases, actin binding proteins, and vesicle transporters, which were at higher abundance under stressed conditions. The reasons for the presence of these proteins are not clear. Proteins Pyc1, Pyc2, and Hfa1 contain biotin carboxylase domains whose cofactor is biotin. It is possible that they are associated with biotin groups that bind to the streptavidin beads employed in HyCCAPP. Ribosome synthesis is decreased in response to stress, and vesicular trafficking is known to be involved in the regulation of that process, providing a possible link between these pathways.23 This might explain finding vesicle transport proteins such as Arc40, Cop1, Sec4, especially under stressed conditions. Related proteins were also reported to be associated with chromatin in the previous yeast HyCCAPP study.5
CONCLUSIONS
We have described here a multiplexed version of the HyCCAPP strategy for sequence-specific capture of targeted genomic regions and identification of associated proteins. Multiplexed HyCCAPP allows multiple regions to be captured and analyzed in parallel, increasing the throughput and decreasing the cost and cell number requirements of the process. The toehold release strategy provides results comparable to those obtained in the original HyCCAPP procedure but enables multiplex analysis. The ability of multiplexed HyCCAPP to identify chromatin-associated proteins in a locus-specific manner has great potential to provide insight into the molecular mechanisms that control gene regulation, although as is generally true for interactome studies, follow-up studies and validation of putative interactors are required for confirmation.
Supplementary Material
Acknowledgments
We thank Professor Ronald T. Raines for use of the cell disruptor and Professor Eric R. Strieter for use of the sonicator. We thank August R. Schultz and Michael C. Chemello for videotaping the HyCCAPP procedure. This work was supported by the NIH Center of Excellence in Genomic Sciences Grant 1 P50 HG004952.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b01784.
Oligonucleotide and qPCR probe sequences; supplementary experimental methods; raw data files; figures showing targeted genomic sites, capture efficiencies, specificities, and enriched proteins from Perseus analysis; tables of enriched proteins from percentile-rank analysis; an alternatively arranged heat map of enriched proteins (PDF)
Complete list of identified proteins and their raw LFQ values obtained using MaxQuant (XLSX)
ORCID
Yunxiang Dai: 0000-0001-7999-798X
Notes
The authors declare no competing financial interest.
References
- 1.Solomon MJ, Larsen PL, Varshavsky A. Cell. 1988;53:937–947. doi: 10.1016/s0092-8674(88)90469-2. [DOI] [PubMed] [Google Scholar]
- 2.van Steensel B, Henikoff S. Nat Biotechnol. 2000;18:424–428. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- 3.Dejardin J, Kingston RE. Cell. 2009;136:175–186. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennedy-Darling J, Guillen-Ahlers H, Shortreed MR, Scalf M, Frey BL, Kendziorski C, Olivier M, Gasch AP, Smith LM. J Proteome Res. 2014;13:3810–3825. doi: 10.1021/pr5004938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guillen-Ahlers H, Rao PK, Levenstein ME, Kennedy-Darling J, Perumalla DS, Jadhav AY, Glenn JP, Ludwig-Kubinski A, Drigalenko E, Montoya MJ, Goring HH, Anderson CD, Scalf M, Gildersleeve HI, Cole R, Greene AM, Oduro AK, Lazarova K, Cesnik AJ, Barfknecht J, Cirillo LA, Gasch AP, Shortreed MR, Smith LM, Olivier M. Genomics. 2016;107:267–273. doi: 10.1016/j.ygeno.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy-Darling J, Holden MT, Shortreed MR, Smith LM. ChemBioChem. 2014;15:2353–2356. doi: 10.1002/cbic.201402343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geerlings TH, Vos JC, Raue HA. RNA. 2000;6:1698–1703. doi: 10.1017/s1355838200001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hung NJ, Lo KY, Patel SS, Helmke K, Johnson AW. Mol Biol Cell. 2008;19:735–744. doi: 10.1091/mbc.E07-09-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bragulat M, Meyer M, Macias S, Camats M, Labrador M, Vilardell J. RNA. 2010;16:2033–2041. doi: 10.1261/rna.2366310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berry DB, Guan Q, Hose J, Haroon S, Gebbia M, Heisler LE, Nislow C, Giaever G, Gasch AP. PLoS Genet. 2011;7:e1002353. doi: 10.1371/journal.pgen.1002353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erde J, Loo RR, Loo JA. J Proteome Res. 2014;13:1885–1895. doi: 10.1021/pr4010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. Nat Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 14.Robinson MD, Grigull J, Mohammad N, Hughes TR. BMC Bioinf. 2002;3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell J, Zomerdijk JC. Biochem Soc Symp. 2006;73:203–216. doi: 10.1042/bss0730203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chasman D, Ho YH, Berry DB, Nemec CM, MacGilvray ME, Hose J, Merrill AE, Lee MV, Will JL, Coon JJ, Ansari AZ, Craven M, Gasch AP. Mol Syst Biol. 2014;10:759. doi: 10.15252/msb.20145120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pokholok DK, Zeitlinger J, Hannett NM, Reynolds DB, Young RA. Science. 2006;313:533–536. doi: 10.1126/science.1127677. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu K, Kawasaki Y, Hiraga S, Tawaramoto M, Nakashima N, Sugino A. Proc Natl Acad Sci U S A. 2002;99:9133–9138. doi: 10.1073/pnas.142277999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kos M, Tollervey D. Mol Cell. 2010;37:809–820. doi: 10.1016/j.molcel.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunbar DA, Dragon F, Lee SJ, Baserga SJ. Proc Natl Acad Sci U S A. 2000;97:13027–13032. doi: 10.1073/pnas.97.24.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Alfonso A, Di Felice F, Carlini V, Wright CM, Hertz MI, Bjornsti MA, Camilloni G. J Mol Biol. 2016;428:4905–4916. doi: 10.1016/j.jmb.2016.10.032. [DOI] [PubMed] [Google Scholar]
- 23.Kingsbury JM, Cardenas ME. G3: Genes, Genomes, Genet. 2016;6:641–652. doi: 10.1534/g3.115.023911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.