

Abstract
Background
Hypertrophic cardiomyopathy (HCM) is often caused by sarcomere gene mutations, resulting in left ventricular hypertrophy (LVH), myocardial fibrosis, and increased risk of sudden cardiac death and heart failure. Studies in mouse models of sarcomeric HCM demonstrated that early treatment with an angiotensin receptor blocker (ARB) reduced development of LVH and fibrosis. In contrast, prior human studies using ARBs for HCM have targeted heterogeneous adult cohorts with well-established disease. The VANISH trial is testing the safety and feasibility of disease-modifying therapy with an ARB in genotyped HCM patients with early disease.
Methods
A randomized, placebo-controlled, double-blind clinical trial is being conducted in sarcomere mutation carriers, 8 to 45 years old, with HCM and no/minimal symptoms, or those with early phenotypic manifestations but no LVH. Participants are randomly assigned to receive valsartan 80 to 320 mg daily (depending on age and weight) or placebo. The primary endpoint is a composite of 9 z-scores in domains representing myocardial injury/hemodynamic stress, cardiac morphology, and function. Total z-scores reflecting change from baseline to final visits will be compared between treatment groups. Secondary endpoints will assess the impact of treatment on mutation carriers without LVH, and analyze the influence of age, sex, and genotype.
Conclusions
The VANISH trial is testing a new strategy of disease modification for treating sarcomere mutation carriers with early HCM, and those at risk for its development. In addition, further insight into disease mechanisms, response to therapy, and phenotypic evolution will be gained.
Hypertrophic cardiomyopathy (HCM) is a primary myocardial disorder that affects up to 1 in 500 people.1 The disease has traditionally been defined as left ventricular hypertrophy (LVH) that occurs in the absence of identifiable triggers, such as pressure overload or infiltrative disease. At a histopathologic level, HCM is characterized by myocyte hypertrophy, disarray, and fibrosis. Most patients with HCM have normal longevity; however, effort intolerance, symptomatic heart failure, and atrial fibrillation may cause substantial morbidity, despite medical or invasive therapy.2–4 HCM can also have serious clinical consequences, including end-stage heart failure and sudden cardiac death. Notably, HCM is an important cause of sudden cardiac death in competitive athletes, adolescents, and young adults in the United States.5
Hypertrophic cardiomyopathy is often caused by mutations in genes encoding sarcomere proteins6 and is the most common monogenic heart disease. Pathogenic sarcomere mutations have been identified in up to 60% of patients with a family history of HCM.7 However, both the penetrance and phenotypic expression of sarcomere mutations are variable and highly age-dependent. Left ventricular wall thickness (LVWT), often normal during early childhood, typically increases in adolescence and young adulthood. Genetic testing allows at-risk sarcomere mutation carriers (G+) to be identified before diagnostic clinical features appear and thus before potentially irreversible changes to the myocardium are in place. Genetic testing also distinguishes sarcomeric HCM from other conditions that may similarly result in the relatively crude phenotype of “unexplained” LVH, but have different underlying causes and biology. Collectively, these factors provide remarkable opportunities to implement disease-modifying treatments in genetically susceptible individuals at a time when treatment may be most successful. Effective treatment would diminish disease progression, thereby reducing the morbidity and mortality associated with HCM.
Study rationale
Currently, HCM is treated only with therapy to palliate symptoms or with implantable cardioverter-defibrillators to prevent sudden cardiac death for patients considered to have increased risk. No treatments have been proven to alter the natural history of disease or to be beneficial for either sarcomere mutation carriers with normal LV wall thickness (denoted preclinical HCM) or for asymptomatic patients with HCM. With improved understanding of how sarcomere mutations cause HCM, more rational and mechanistic treatments can be developed, targeting early, disease-initiating pathways. The goal of these therapies would be to attenuate disease progression and, ultimately, disease emergence.8
Studies in mouse models of sarcomeric HCM have indicated the importance of transforming growth factor beta (TGF-β) activation in early disease pathogenesis.9 Although the mechanisms are not fully understood, numerous studies indicate that angiotensin II binding to the angiotensin type 1 receptor activates canonical and non-canonical TGF-β signaling pathways, stimulating pathways of collagen synthesis and inhibiting collagen degradation.10 Angiotensin receptor blockers (ARBs) can inhibit TGF-β activation.11
Two complementary strategies to reduce TGF-β activation decreased disease progression in a mouse model of sarcomeric HCM12 (Figure 1). Treatment with either TGF-β neutralizing antibodies or with the angiotensin II type 1 receptor blocker, losartan, were shown to attenuate the development of myocyte hypertrophy and fibrosis in treated animals compared with those receiving placebo. Notably, treatment was effective only when started at a young age, before LVH was established. In contrast, prior clinical studies using ARBs in patients with HCM targeted heterogeneous patient populations with well-established disease and have not shown consistent benefit.13–18
Figure 1.
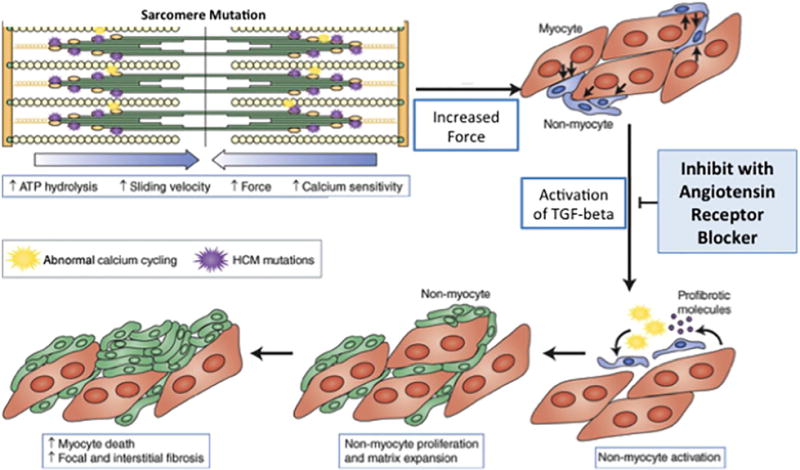
Proposed model of how sarcomere mutations may lead to hypertrophic cardiomyopathy and the potential impact of angiotensin receptor blockade. Studies using mouse models of hypertrophic cardiomyopathy (HCM) have suggested that sarcomere mutations increase force generation and calcium sensitivity, and activate profibrotic pathways early in disease pathogenesis, before cardiac hypertrophy develops. These abnormalities activate numerous cellular pathways, including transforming growth factor-beta (TGF-β) signaling pathways, culminating in the development of myocardial fibrosis and hypertrophy. In mouse models of HCM, inhibiting TGF-β activation, using either neutralizing antibody or losartan, attenuated the development of left ventricular hypertrophy and fibrosis if given early in the prehypertrophic phase of disease (modified from Teekakirikul P, et al. JCI; 2010 (12)).
In a randomized clinical trial, the safety and efficacy of valsartan, an angiotensin II, type I-selective angiotensin receptor blocker, is being evaluated in modifying the course of HCM. The target population is an at-risk population with early sarcomeric disease identified through clinical evaluation and genetic testing. The trial is testing the hypothesis that valsartan will have beneficial effects on reducing adverse changes in cardiac structure, function, and serum biomarker concentrations, indicating attenuated progression of HCM. Here, we summarize the study protocol of this trial. The authors are solely responsible for the design and conduct of this study, all study analyses, the drafting and editing of the manuscript, and its final contents.
Trial design
The VANISH study is a phase II multicenter, randomized, placebo-controlled, double-blind clinical trial. Two cohorts of sarcomere mutation carriers will be enrolled: one with early disease (primary analysis cohort) and one at a preclinical stage with increased risk for developing HCM (exploratory cohort). Each cohort will have both a treatment and a placebo group.
The primary analysis cohort consists of sarcomere mutation carriers 8 to 45 years old with at least borderline LVH who are asymptomatic or mildly symptomatic (New York Heart Association (NYHA) class I–II). Patients in this cohort are anticipated to have early disease, given their relatively young age and minimal symptom burden. The primary analysis will be performed on this cohort (target enrollment of 150 subjects) because disease pathways are more likely to be already activated and these patients may be more responsive to therapy.
The exploratory cohort consists of mutation carriers 10 to 25 years old who have normal LV wall thickness (no diagnosis with HCM) but some evidence of phenotypic impact, such as impaired relaxation (reduced tissue Doppler E′ velocity) or electrocardiographic abnormalities (Q waves or ST changes). Evidence of phenotypic impact was included as an eligibility criterion for this cohort because disease progression during the study period is not expected to be substantial in mutation carriers without any discernible abnormalities at enrollment. No enrollment target is specified for the exploratory cohort.
Patients in each cohort are randomly assigned to receive either valsartan or placebo for 2 years (Figure 2). Study procedures are identical for the 2 cohorts. The trial is being conducted in the HCMNet collaborative network (Table I) and is registered at ClinicalTrials.gov (NCT01912534). An independent Data Safety Monitoring Board will oversee the conduct of the trial. This trial is funded by the National Institutes of Health (NIH, 1P50HL112349). Valsartan and matching placebo tablets have been donated by Novartis Pharmaceuticals Corporation. Novartis had no part in developing this protocol and will not be involved in data analysis or publication decisions.
Figure 2.
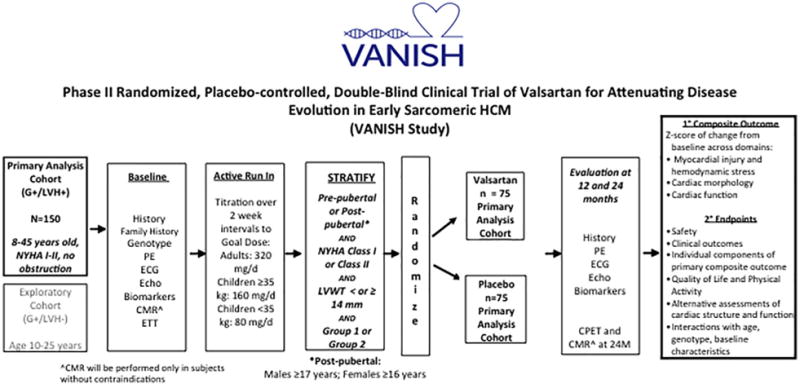
VANISH trial schema. This schematic illustrates the overall structure of the trial from patient selection to completion of 2 years of blinded therapy, followed by primary and secondary analyses.
Table I.
Participating sites for the VANISH trial
| Trial site | Principal investigator | Location |
|---|---|---|
| Brigham and Women’s Hospital | Carolyn Ho, MD (Trial Principal Investigator) | Boston, MA |
| University of Michigan | Sharlene Day, MD; Mark Russell, MD |
Ann Arbor, MI |
| Cleveland Clinic Foundation | Harry Lever, MD; Kenneth Zahka, MD |
Cleveland, OH |
| Boston Children’s Hospital | Steven Colan, MD; Renee Margossian, MD |
Boston, MA |
| Yale University (virtual site) | Kevin Hall, MD | New Haven, CT |
| Vanderbilt University | Jason Becker, MD | Nashville, TN |
| Cincinnati Children’s Hospital Medical Center | John Lynn Jeffries, MD | Cincinnati, OH |
| University of Chicago | Amit Patel, MD | Chicago, IL |
| Northwestern University (virtual site) | Lubna Choudhury, MD | |
| Johns Hopkins University | Anne Murphy, MD | Baltimore, MD |
| Washington University School of Medicine | Charles Canter, MD; Richard Bach, MD |
St. Louis, MO |
| University of Colorado, Denver | Matthew Taylor, MD; Luisa Mestroni, MD |
Denver, CO |
| Stanford University | Matthew Wheeler, MD | Palo Alto, CA |
| Toronto Hospital for Sick Children | Lee Benson, MD | Toronto, ON Canada |
| Hospital of the University of Pennsylvania | Anjali Owens, MD | Philadelphia, PA |
| Children’s Hospital of Philadelphia | Joseph Rossano, MD; Kim Lin, MD |
Philadelphia, PA |
| Lurie Children’s Hospital of Chicago | Elfriede Pahl, MD | Chicago, IL |
| Penn State Health, Hershey Medical Center | Eric Popjes, MD | Hershey, PA |
| Massachusetts General Hospital | Michael Fifer, MD | Boston, MA |
| INCOR (Instituto do Coração) | Alexandre Pereira, MD | Sao Paolo, Brazil |
| Rigshospitalet | Henning Bundgaard, MD; Anna Axelsson Raja, MD |
Copenhagen, Denmark |
Patient selection and eligibility criteria
All patients must have a pathogenic or likely pathogenic HCM sarcomere mutation and provide informed consent. Variant pathogenicity is determined using standard criteria accounting for segregation, conservation, literature review, review of publicly available databases,7,19,20 and very low frequency in appropriate ethnically-matched control populations (Exome Aggregation Consortium [ExAC], Cambridge, MA [URL: http://exac.broadinstitute.org]).21 A panel with expertise in genotyping, led by the principal investigator, reviews each questionable variant and approves or denies eligibility by consensus. Inclusion and exclusion criteria are listed in Table II.
Table II.
Eligibility criteria for the VANISH trial
| Inclusion Criteria | Exclusion Criteria |
|---|---|
Primary analysis cohort
Exploratory analysis cohort
|
|
Eligibility criteria were selected to attempt to target therapy to individuals that are most likely to respond (in the treatment group) or to demonstrate phenotypic progression (in the placebo group). The age limits for each cohort also considered the delayed penetrance of sarcomere mutations. Mutation carriers typically develop clinical manifestations during or after adolescence, although disease expression is variable and may be incomplete. Patients older than 45 years are excluded from the primary cohort out of concern that prolonged exposure to disease may have resulted in irreversible changes that will not respond to disease-modifying therapy. Children under 10 years of age are excluded from the exploratory cohort given the low likelihood that phenotypic progression will occur in very young children during the trial. Similarly, patients older than 25 years are excluded from the exploratory cohort out of concern that sarcomere mutation carriers who have not expressed a more pronounced phenotype by this age may have substantially delayed penetrance or be non-penetrant mutation carriers and thus unlikely to progress during the trial.
Treatment and active run-in period
High doses of ARBs are generally believed necessary to inhibit TGF-β, although the threshold for efficacy is unknown. Therefore, the maximum FDA approved dose of valsartan was chosen as the target dose (adults, 320 mg daily; children <17 years old weighing ≥35 kg, 160 mg daily; children <17 years old weighing <35 kg, 80 mg daily). To ensure that patients can be maintained on the target dose, eligible patients first enter a 2- to 6-week active run-in period during which the dose is titrated up to the target dose. Only those who tolerate the target dose are randomly assigned and maintained on blinded therapy for 2 years.
Stratification and random assignment
After baseline data collection and successfully completing active run-in, eligible patients are randomly assigned in a 1:1 ratio to receive to valsartan or placebo (Figure 3). Subjects determined to be ineligible and those who do not tolerate active run-in will be followed in a registry with information gathered from clinical evaluation. Because group assignment, age, pubertal status, and baseline NYHA class are anticipated to influence outcomes and phenotypic progression, the randomization scheme is stratified by each of these factors. This stratification effectively creates 2 parallel trials: one comprised the primary analysis cohort of patients with early disease and the other comprised the exploratory cohort of patients at elevated risk for disease expression. Patients with borderline LVH (LV wall thickness 12–14 mm or z-score of 4–6) may be in the early stages of phenotypic evolution and may be expected to either have a more dynamic course or be more responsive to treatment than those with much more established and severe LVH. Therefore, the primary analysis cohort will also be stratified by LV wall thickness (<14 vs ≥14 mm or z-score of 6).
Figure 3.
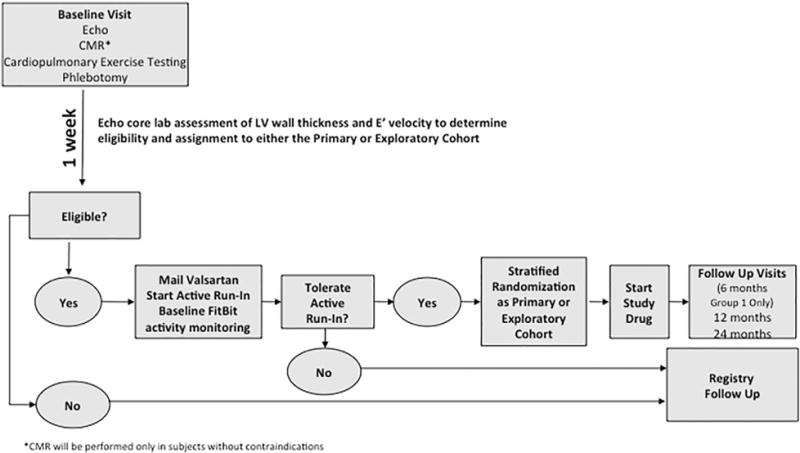
Summary of the flow from enrollment to randomization in the VANISH Trial. Participants will undergo baseline studies and await rapid echocardiographic core laboratory evaluation to confirm eligibility and to determine whether they will be stratified to the primary or exploratory cohort (based on measurement of maximal left ventricular wall thickness, LV thickness to dimension ratio, and E′ velocity). Eligible participants will then enter into active run-in and undergo up titration of valsartan to target dose over the course of 2 to 6 weeks. Those who tolerate active run-in will undergo stratified randomization and begin 24-months of blinded treatment with study medication. Ineligible subjects will be followed in a registry.
The randomization scheme is stratified with a block size of 4 and with cells defined by NYHA class, pubertal status, and entry LV wall thickness (stratification factors shown in the Supplemental Figure). The Data Coordinating Center (DCC) generates the randomization lists for each stratum using SAS programming and enters these lists into the data management system (eCOS). When a participant’s eligibility and stratification for randomization is determined, eCOS will assign the participant the next treatment from the list for that stratum. After assignment, a participant will remain on the same Treatment Allocation Code for the duration of the study. Only the DCC has access to the randomization lists; participants and investigators are blinded to the study treatment.
Follow-up visits and procedures
Family history is assessed at baseline and updated at subsequent visits (Table III). Three-generation pedigrees are constructed to capture family structure and history of HCM and related complications. Physical examination, collection of blood samples for safety assessment and for biomarker analysis, 12-lead electrocardiograms, and trans-thoracic echocardiography are obtained at baseline, year 1, and year 2. In addition, these visits will assess quality of life (using validated Pediatric Quality of Life Inventory forms22 appropriate for the patient’s age) and physical activity using the abbreviated version of the Physical Activity and Physical Fitness survey of the National Health and Nutrition Examination survey (NHANES Physical Activity and Cardiovascular Fitness Data Tutorial. http://www.cdc.gov/nchs/tutorials/PhysicalActivity/SurveyOrientation/DataOverview/ [Accessed July 18, 2016]). Physical activity is also being assessed with FitBit activity monitors (accelerometer/pedometer) worn by participants for the duration of the trial.
Table III.
Schedule of trial visits and procedures
| Active Run-In* | Follow-Up† | ||||||
|---|---|---|---|---|---|---|---|
| Visit Number | 1 | 2 Titration |
3 Titration to Target Dose |
4 On Target Dose |
5 F/U |
6 F/U |
7 F/U |
| Time Point | Screening/Baseline | 2 weeks (from drug initiation‡) |
4 weeks (from drug initiation‡) |
6 weeks (from drug initiation‡) |
6 Months (Group 1 subjects only) |
1 Year (from drug initiation) |
2 Years (from drug initiation) |
| Physical Exam (* BP only) | X | X* | X* | X | X | X | X |
| Cardiac MRI | X | X | |||||
| Echocardiography | X | X | X | ||||
| Cardiopulmonary Exercise Test | X | X | |||||
| ECG | X | X | X | ||||
| Assess for Adverse Events | X | X | X | X | X | X | X |
| Blood for Safety | X | X | X | X | |||
| Pregnancy testing | X | X | X | ||||
| Blood for DNA Banking | X | ||||||
| Fasting Blood for Biomarkers | X | X | X | ||||
| Quality Of Life/Physical activity Questionnaire | X | X | X | ||||
| Dose Adjustment | X | X | |||||
F/U, Follow up; BP, blood pressure; MRI, magnetic resonance imaging; ECG, electrocardiogram.
May be performed locally
Quarterly telephone communication will also occur during follow up to assess for adverse events and compliance.
Study drug initiation will occur after eligibility is confirmed.
Cardiopulmonary exercise testing and cardiac magnetic resonance imaging (CMR; if not contraindicated) are performed at baseline and year 2. CMR will obtain information adjunctive to echocardiography on cardiac structure and function. In addition, to assess myocardial tissue characteristics, replacement fibrosis will be estimated by quantifying late gadolinium enhancement, and T1 mapping will estimate effective extracellular volume. All imaging and exercise testing studies are obtained with standardized protocols, and personnel, blinded to treatment assignment but not to the purpose of the trial, will analyze echocardiographic, CMR, exercise testing, electrocardiographic, and biomarker data at core laboratories (Supplemental Table).
Blood for biomarker analysis is processed within 60 minutes of phlebotomy and stored at −80°C. Validated, commercial immunoassays will examine multiple potential biomarkers of disease progression by monitoring myocardial injury (including high-sensitivity cardiac troponin I and T), hemodynamic stress (including natriuretic peptides, and soluble ST2), and collagen metabolism.23 Assays for all samples will be run at the end of the trial. Additional assays may be included as guided by state-of-the-art knowledge available at the time of data analysis. Remaining samples will be banked for future study. In addition, blood for safety laboratory assessments will be drawn at the end of the run-in period and yearly to detect renal dysfunction and hyperkalemia. Doses will be reduced on confirmation that the serum potassium concentration is greater than 5 mmol/L, or that the estimated glomerular filtration rate (eGFR) decreased by 25% or more, as determined by the modified Schwartz formula for children24 or the CKD-EPI equation for adults,25 or for eGFR <75/1.73 m2. Laboratory studies will be repeated within 1 week, and treatment will be stopped if values do not improve despite dose reduction.
Trial timeline and preliminary baseline characteristics of randomized subjects
Enrollment is estimated to require 32 months and began in April 2014. As of September 2016, 139 patients have been enrolled, including 114 in the primary analysis cohort (76% of target enrollment) (Table IV). The baseline characteristics of the randomized subjects to date demonstrate recruitment of intended population. Participants will receive study medication for 24 months and trial completion is anticipated in 2019. All participating sites have received institutional review or ethics board approval and all participants have provided informed consent.
Table IV.
Demographic characteristics of VANISH patients as of September 27, 2016
| Variable | Overall, N = 139 | Primary cohort, N = 114 | Exploratory cohort, N = 25 |
|---|---|---|---|
| Age at baseline, mean (SD), years | 20.3 (8.9) | 21.1 (9.5) | 16.6 (4.5) |
| Sex, n (%) | |||
| Female | 60 (43) | 51 (45) | 9 (36) |
| Male | 79 (57) | 63 (55) | 16 (64) |
| Race, n (%) | |||
| White | 134 (96) | 109 (96) | 25 (100) |
| Black | 0 (0) | ||
| Other | 5 (4) | 5 (4) | |
| Maximal LV Wall Thickness Z-Score, mean (SD) | 6.0 (4.1) | 6.8 (4.2) | 2.0 (0.7) |
| E″ lateral Z-Score, mean (SD) | −1.6 (1.4) | −1.8 (1.3) | −0.4 (1.0) |
| LVEF (%), mean (SD), years | 65 (7) | 67 (7) | 63 (4) |
| Peak VO2, mean (SD), mL/kg/min | 33.8 (9.8) | 32.8 (10.0) | 37.3 (8.3) |
| NYHA class, n (%) | |||
| I | 134 (96) | 109 (96) | 25 (100) |
| II | 5 (4) | 5 (4) | |
| Gene, n (%) | |||
| MYBPC3 | 67 (49) | 55 (50) | 12 (48) |
| MYH7 | 47 (35) | 37 (33) | 10 (40) |
| TNNT2 | 9 (7) | 9 (8) | 0 |
| TNNI3 | 5 (4) | 3 (3) | 0 |
LV, Left ventricle; EF, ejection fraction; VO2, volume of oxygen consumption; NYHA, New York Heart Association; MYBPC3, myosin binding protein C; MYH7, myosin heavy chain; TNNT2, cardiac troponin T; TNNI3, cardiac troponin I.
Statistical analysis plan
Developing the statistical analysis plan for the VANISH trial faced the following challenges: (1) the effect of valsartan in modifying disease is unknown; (2) objective clinical outcomes (such as death or major adverse clinical events) are unlikely to occur during the trial because participants are young and relatively healthy; (3) factors that drive disease progression and outcomes in HCM are unknown; (4) surrogate outcomes that are potentially responsive to therapy or associated with a strong and consistent treatment effects have not yet been identified for early HCM; and (5) with the modest sample size in the VANISH trial, the power to detect a small but clinically important difference in any single outcome variable may be insufficient.
Therefore, rather than trying to detect a large effect of valsartan on a single outcome measure, our analysis focuses on identifying a series of composite outcome measures that are relevant to the pathophysiology of HCM, reflecting 3 domains of cardiac structure, function, and myocardial injury or stress (Table V). We postulate that consistent moderate effects across the component measures in these domains will indicate an important treatment response, and will be more sensitive than monitoring any single component in isolation. Individual variables captured in these domains will measure LV wall thickness, LV cavity size, diastolic function, longitudinal systolic function, and serum biomarkers of myocardial stress and injury. These variables (increased LV wall thickness, decreased cavity size, diastolic and longitudinal systolic dysfunction, and increased biomarkers) have been shown to reflect early phenotypic expression of sarcomere mutations.26–28 As such, they are anticipated to be clinically relevant surrogate outcomes to monitor treatment response and disease progression. Values for each of the component outcomes from each domain will be provided by the appropriate core laboratories and then summed to create a single composite outcome z-score.29 We hypothesize that valsartan treatment will improve the primary composite z-score, indicating an important treatment response. We believe that this approach of assessing non-redundant variables that monitor relevant aspects of cardiac structure and function provides the highest likelihood of detecting any apparent signal of treatment effect and/or disease progression. This strategy has been used successfully in other phase II heart failure trials.30,31
Table V.
Outcome domains and components for the primary composite endpoint
| Domain | Component outcomes |
|---|---|
| Serum biomarkers of myocardial injury and stress | High-sensitivity cardiac troponin NTproBNP |
| Cardiac morphology | CMR LV mass* CMR LA volume* CMR end-diastolic LV volume* CMR end-systolic LV volume* Echocardiographic maximum LV wall thickness z-score |
| Cardiac function | Echocardiographic E′ z-score Echocardiographic S′ z-score |
NTproBNP, Amino terminal propeptide of B-type natriuretic peptide; CMR, cardiac magnetic resonance imaging; LA, left atrium; LV, left ventricle.
CMR metrics will be indexed to body surface area; echocardiographic z-scores to substitute if CMR imaging unavailable.
Analysis will be performed as intention-to-treat after all patients have completed 2 years of treatment. The primary efficacy analyses will be based on an average change in the composite z-score across the 3 domains listed above. For each patient and each component, the change between baseline and 24 months will be calculated. Using the anticipated distribution of data, relative change (percent change from baseline) will be used to evaluate serum biomarker concentrations, and absolute change will be used for all other components. Higher composite z-scores reflect relatively greater improvement in phenotype in one or more components of the composite outcome.
Sample size considerations
Power calculations for the composite z-score were based on the work of Sun et al,29 who considered a composite z-score of 6 measures commonly used in heart failure trials. Simulations were used to calculate the power for a composite z-score of the 3 domains described above, with the same correlation of 0.12 between component measures and assuming that the z-score for each domain would show a standardized beneficial effect of valsartan of between 0.22 and 0.25 z-score standard deviations. Given these assumptions, the target enrollment for VANISH is 150 patients in the primary cohort (75 per treatment arm), which provides an estimated 76% to 88% power to detect standardized effect sizes of 0.22 to 0.25 z-score SDs.
Primary safety analyses
To determine whether valsartan is safe, well-tolerated, and does not reduce the quality of life of young, asymptomatic patients, we will compare the incidence of adverse drug reactions, frequency of treatment discontinuation, and responses to the Pediatric Quality of Life Inventory forms22 between the valsartan and placebo groups.
Secondary analyses
A variety of complementary and biologically relevant metrics will be analyzed to characterize more broadly the impact of valsartan on disease pathology. Improvement in, stability, or attenuation of progression in metrics of interest will be assessed, including the 9 components of the composite outcome, other measures of LV and left atrial size, new measures of LV systolic and diastolic function, measures of metabolic exercise testing, and new serum biomarkers.
Several features are anticipated to affect treatment response and the natural history of the disease. Therefore, pre-specified subgroup analyses will consider phenotypic status (primary analysis versus exploratory cohorts), sex, underlying genotype (MYBPC3, MYH7, TNNI3, and TNNT2), pubertal status, growth by height velocity during the trial, and baseline LV wall thickness. Due to the diverse geography of the participating sites, the influence of participating site and ethnicity will also be explored in secondary analyses.
The effect of valsartan on the exploratory cohort in isolation will be analyzed as described for the primary analysis cohort. In addition, the effect of valsartan on development of LVH and clinically overt HCM will be analyzed. A Clinical Events Committee comprised of adult and pediatric cardiologists will adjudicate clinical outcomes (hospitalization or unscheduled clinical visits, development of atrial or ventricular arrhythmias, need for procedures, etc) for evaluation in secondary analyses. Quality of life and physical activity metrics will also be analyzed.
When the trial was first designed, the primary analysis cohort had more restrictive eligibility requirements, including an upper age of 30 years and a maximum LV wall thickness of ≤20 mm (z-score ≤14). Owing to difficulties identifying a sufficiently large cohort of individuals meeting entrance criteria for trial enrollment (which began in April 2014), the eligibility criteria were modified and are as shown in Table II. Thus, the impact of treatment using the original eligibility criteria will be assessed with sensitivity analyses. Furthermore, up to 5 members of the same family are permitted to participate. Although family membership will be accounted for in the statistical analysis, the impact of relatedness will be assessed with sensitivity analyses by including only the first family member enrolled.
In addition to continuous outcome variables, each participant will be scored as a “success” or “failure” based on improvement in any of the 9 component z-scores at 24 months. The proportion of successes and failures will be compared between treatment groups in secondary analyses using Fisher’s exact test and adjusted for confounding with logistic regression.
Discussion
Determining the genetic basis of inherited heart disease provides the remarkable opportunity to define the specific cause of disease, to identify at-risk individuals before clinical diagnosis, and to develop new therapies intended to diminish or prevent clinical expression. Based on the knowledge and mechanistic insight gained from basic science discovery, clinical trials can now be developed to explore whether pharmacologic therapy may counteract the phenotypic expression of an inherited gene mutation. The VANISH trial will investigate this new paradigm for genetic cardiomyopathy by testing the safety and efficacy of using valsartan as disease-modifying therapy for patients anticipated to be in the early stages of sarcomeric HCM.
Several trials have investigated the use of ARBs in regressing disease in adults with established HCM.13–18 Results have been inconsistent and no strong signals of benefit have been detected. Notably, these studies have had several key limitations. All but the study by Axelsson et al18 included fewer than 30 patients. The patient cohorts were heterogeneous and may not have targeted the most responsive patient population. For example, prior studies targeted older adults and did not incorporate genotyping into eligibility criteria. In contrast, all preclinical work supporting the use of ARB therapy used young animals with sarcomere mutations in the pre-hypertrophic stage of disease. Therefore, the patients enrolled in prior studies included many whose disease may have been irreversibly entrenched and not responsive to disease-modifying treatment.
The VANISH trial was designed to address the limitations and the unanswered questions associated with earlier trials of ARB therapy in HCM. Particular attention was paid to capture patients anticipated to be most responsive to disease-modifying therapy. Younger patients with less advanced disease are targeted for because they may be at a more plastic and modifiable period in disease evolution. Furthermore, patients are required to have genetically-confirmed sarcomeric HCM to focus study on a cohort that most closely replicates the fundamental basis for the trial.
Limitations of the trial
Few clinical trials have tested disease-modifying therapies in genetic heart disease and important challenges are present. The underlying pathophysiology of HCM typically evolves over years to decades, requiring large numbers of patients to be followed over long periods; an approach beyond the scope of this Phase II trial of patients with a relatively rare disease. The young, healthy patients in this trial are anticipated to have low event rates, making it difficult to detect treatment response. As such, this trial will rely on a composite of 9 surrogate endpoints believed to reflect the pathophysiology of HCM and important aspects of cardiac structure and function, but whose relationship to clinical disease has not yet been firmly established. Choosing the target dose of valsartan for this trial was difficult because this strategy has not been previously tested, and because it is not possible to accurately translate doses used in mouse models to those that will be required to affect change in humans.
Conclusions
By helping to pioneer disease-modifying therapy, the VANISH trial has the potential to advance the care of patients and families with inherited cardiomyopathies. Experience gained with patient selection and with the surrogate outcomes used in this Phase II trial will yield valuable information to inform the development of future clinical trials. Analogous data from the placebo groups will further understanding of factors that influence disease progression and clinical outcomes in patients with sarcomeric HCM. This knowledge will help target therapy to responsive individuals at highest risk who may receive the greatest benefit from new strategies for disease modification and prevention. As such, VANISH can serve as a paradigm for learning how to harness genetic insights to better understand pathogenesis and advance clinical practice. By fostering discovery of new ways to disrupt disease progression, a precision approach of gene-based diagnosis and management can be brought closer to reality.
Supplementary Material
Acknowledgments
Eugene Braunwald, MD; Harold Dietz, MD; Steven Lipshultz, MD; Calum MacRae, MD; William McKenna, MD; John McMurray, MD; John Orav, PhD; Scott Solomon, MD.
Clinical Events Committee:
Chair: Akshay Desai, MD.
Neal Lakdawala, MD; Elizabeth Blume, MD.
Funding: The National Institutes of Health (1P50HL112349 to CYH) funded this study but is not involved with study design or analysis.
Study medication was provided as a donation from Novartis Pharmaceuticals Corporation. Novartis plays no role in the design, conduct, or reporting of the study.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ahj.2017.02.008.
Footnotes
RCT# NCT01912534.
Conflict of Interest: NONE.
References
- 1.Maron BJ, Gardin JM, Flack JM, et al. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults-Echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation. 1995;92:785–9. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Casey SA, Hauser RG, et al. Clinical course of hypertrophic cardiomyopathy with survival to advanced age. J Am Coll Cardiol. 2003;42:882–8. doi: 10.1016/s0735-1097(03)00855-6. [DOI] [PubMed] [Google Scholar]
- 3.Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687–713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 4.Gersh BJ, Maron BJ, Bonow RO, et al. ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;2011 doi: 10.1016/j.jacc.2011.10.825. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064–75. doi: 10.1056/NEJMra022783. [DOI] [PubMed] [Google Scholar]
- 6.Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res. 2011;108:743–50. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–8. doi: 10.1038/gim.2014.205. [DOI] [PubMed] [Google Scholar]
- 8.Ho CY, Lakdawala NK, Cirino AL, et al. Diltiazem Treatment for Pre-Clinical Hypertrophic Cardiomyopathy Sarcomere Mutation Carriers: A Pilot Randomized Trial to Modify Disease Expression. JACC Heart failure. 2014 doi: 10.1016/j.jchf.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim JB, Porreca GJ, Song L, et al. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science. 2007;316:1481–4. doi: 10.1126/science.1137325. [DOI] [PubMed] [Google Scholar]
- 10.Lopez B, Gonzalez A, Diez J. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation. 2010;121:1645–54. doi: 10.1161/CIRCULATIONAHA.109.912774. [DOI] [PubMed] [Google Scholar]
- 11.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–21. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teekakirikul P, Eminaga S, Toka O, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–9. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Araujo AQ, Arteaga E, Ianni BM, et al. Effect of Losartan on left ventricular diastolic function in patients with nonobstructive hypertrophic cardiomyopathy. Am J Cardiol. 2005;96:1563–7. doi: 10.1016/j.amjcard.2005.07.065. [DOI] [PubMed] [Google Scholar]
- 14.Kawano H, Toda G, Nakamizo R, et al. Valsartan decreases type I collagen synthesis in patients with hypertrophic cardiomyopathy. Circ J. 2005;69:1244–8. doi: 10.1253/circj.69.1244. [DOI] [PubMed] [Google Scholar]
- 15.Penicka M, Gregor P, Kerekes R, et al. The effects of candesartan on left ventricular hypertrophy and function in nonobstructive hypertrophic cardiomyopathy: a pilot, randomized study. J Mol Diagn. 2009;11:35–41. doi: 10.2353/jmoldx.2009.080082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimada YJ, Passeri JJ, Baggish AL, et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart failure. 2013;1:480–7. doi: 10.1016/j.jchf.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamazaki T, Suzuki J, Shimamoto R, et al. A new therapeutic strategy for hypertrophic nonobstructive cardiomyopathy in humans. A randomized and prospective study with an Angiotensin II receptor blocker. Int Heart J. 2007;48:715–24. doi: 10.1536/ihj.48.715. [DOI] [PubMed] [Google Scholar]
- 18.Axelsson A, Iversen K, Vejlstrup N, et al. Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2015;3:123–31. doi: 10.1016/S2213-8587(14)70241-4. [DOI] [PubMed] [Google Scholar]
- 19.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karczewski KJ, Weisburd B, Thomas B, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varni JW, Burwinkle TM, Seid M. The PedsQL as a pediatric patient-reported outcome: reliability and validity of the PedsQL Measurement Model in 25,000 children. Expert Rev Pharmacoecon Outcomes Res. 2005;5:705–19. doi: 10.1586/14737167.5.6.705. [DOI] [PubMed] [Google Scholar]
- 23.Ibrahim NE, Gaggin HK, Konstam MA, et al. Established and Emerging Roles of Biomarkers in Heart Failure Clinical Trials. Circ Heart Fail. 2016;9 doi: 10.1161/CIRCHEARTFAILURE.115.002528. [DOI] [PubMed] [Google Scholar]
- 24.Chavers BM, Rheault MN, Foley RN. Kidney function reference values in US adolescents: National Health And Nutrition Examination Survey 1999–2008. Clin J Am Soc Nephrol. 2011;6:1956–62. doi: 10.2215/CJN.10311110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–12. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho CY, Abbasi SA, Neilan TG, et al. T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ Cardiovasc Imaging. 2013;6:415–22. doi: 10.1161/CIRCIMAGING.112.000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic Strain Imaging to Assess Early and Late Consequences of Sarcomere Mutations in Hypertrophic Cardiomyopathy. Circ Cardiovasc Genet. 2009;2:314–21. doi: 10.1161/CIRCGENETICS.109.862128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho CY, Lopez B, Coelho-Filho OR, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010;363:552–63. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun H, Davison BA, Cotter G, et al. Evaluating treatment efficacy by multiple end points in phase II acute heart failure clinical trials: analyzing data using a global method. Circ Heart Fail. 2012;5:742–9. doi: 10.1161/CIRCHEARTFAILURE.112.969154. [DOI] [PubMed] [Google Scholar]
- 30.Greenberg B, Yaroshinsky A, Zsebo KM, et al. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b) JACC Heart Fail. 2014;2:84–92. doi: 10.1016/j.jchf.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 31.Greenberg B, Butler J, Felker GM, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–86. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.