

Abstract
Background
In a previous study, we found that a biomarker risk score (BRS) comprised of C‐reactive protein, fibrin‐degradation products, and heat shock protein‐70 predicts risk of myocardial infarction and death in coronary artery disease patients. We sought to: (1) validate the BRS in the independent BARI 2D (Bypass Angioplasty Revascularization Investigation 2 Diabetes) cohort, (2) investigate whether 1 year of intensive medical therapy is associated with improved BRS, and (3) elucidate whether an altered BRS parallels altered risk.
Methods and Results
Two thousand thirty‐two subjects with coronary artery disease were followed for 5.3±1.1 years for cardiovascular events. Biomarkers were measured at baseline and retested in 1304 subjects at 1 year. BRS was determined as the biomarker number above previously defined cut‐off values (C‐reactive protein >3 mg/L, heat shock protein‐70 >0.313 ng/mL, and fibrin‐degradation products >1 μg/mL). After adjustment for covariates, those with a BRS of 3 had a 4‐fold increased risk of all‐cause death and a 6.8‐fold increased risk of cardiac death compared with those with a BRS of 0 (95% CI, 2.9–16.0; P<0.0001). All individual biomarkers decreased by 1 year, with ≈80% of patients decreasing their BRS. BRS recalibrated at 1 year also predicted risk. Those with 1‐year BRS of 2 to 3 had a 4‐year mortality rate of 21.1% versus 7.4% for those with BRS of 0 to 1 (P<0.0001).
Conclusions
Our results validate the ability of the BRS to identify coronary artery disease patients at very high near‐term risk of myocardial infarction/death. After 1 year of intensive medical therapy, the BRS decreased significantly, and the reclassified BRS continued to track with risk. Our results suggest that repeated BRS measurements might be used to assess risk and recalibrate therapy.
Keywords: biomarker, coronary artery disease, major adverse cardiovascular event, risk score
Subject Categories: Biomarkers, Inflammation, Coronary Artery Disease
Introduction
Coronary artery disease (CAD) remains the predominant cause of death worldwide.1 Clinical tools, such as the Framingham risk score, predict risk of cardiovascular disease (CVD) and its adverse outcomes in community‐based populations free of known CVD, but fail to reliably predict risk in patients with established CAD.2, 3, 4 We have recently identified a pathway‐specific biomarker risk score (BRS) comprised of C‐reactive protein (CRP) representing inflammation, fibrin degradation products (FDP) representing the thrombosis pathway, and heat shock protein‐70 (HSP‐70) representing cell stress, that significantly predicts risk of MI and death in patients with suspected or established CAD and is independent of multiple clinical risk measures.5
BARI 2D (Bypass Angioplasty Revascularization Investigation 2 Diabetes) determined whether type II diabetics with complex CAD would benefit from early revascularization (coronary artery bypass grafting or percutaneous coronary intervention) and from insulin‐sensitizing (IS) medications. All recruited subjects received intensive medical therapy targeting individual cardiovascular risk factors with a goal for glycated hemoglobin (HbA1C) of <7%, blood pressure (BP) ≤130/80 mm Hg, low‐density lipoprotein level of <100 mg/dL, smoking cessation counseling, and other lifestyle modifications.
By analyzing samples drawn at baseline and after 1 year in the BARI 2D study, we aimed to: (1) validate the BRS in the BARI 2D cohort; (2) investigate whether 1 year of intensive medical therapy is associated with improved BRS; and (3) whether altered BRS is associated with altered risk.
Methods
Study Population and Design
Details of the study design were previously published.6 Briefly, 2368 patients with type II diabetes mellitus and CAD were enrolled from 49 clinical sites between 2001 and 2005 and randomized to 2 treatment strategies in a 2‐by‐2 factorial design. In the first strategy, patients were assigned to undergo prompt coronary revascularization (coronary artery bypass grafting or percutaneous coronary intervention) plus optimal medical therapy, or optimal medical therapy alone. In the second strategy, patients were assigned to undergo either IS or insulin‐providing therapy to achieve a target HbA1C level <7.0%. The study protocol was approved by the institutional review board at the University of Pittsburgh (Pittsburgh, PA), and all patients had provided written informed consent before enrollment in the BARI 2D study.
CAD was defined as ≥50% stenosis of a major epicardial coronary artery. Myocardial jeopardy index was used to assess CAD severity by calculating the percentage of myocardium jeopardized by significant lesions (≥50%) at a core laboratory.7, 8 Type II diabetes mellitus diagnosis was based on need for oral hypoglycemic or insulin therapy, or a confirmed elevated blood glucose level. Patients with left main disease, those with history of revascularization within the previous 12 months, or in need of urgent revascularization were excluded. Patients with elevated serum creatinine (≥2.0 mg/dL), HbA1C of ≥13%, or class III or IV heart failure were also excluded. Patients were followed monthly for 6 months and then every 3 months for a mean of 5.3 years.
Biomarker Measurements
Venous blood was drawn at baseline and at 1 year, and stored serum was available in a subset of 2032 subjects at baseline and 1304 subjects at 1‐year (Figure 1). Serum was stored at −70°C. Details of the biomarker assays were previously described5: serum high‐sensitivity CRP and FDP measurements were determined using a sandwich immunoassay by FirstMark, Inc (San Diego, CA). Serum HSP‐70 was measured by sandwich ELISA (R&D Systems, Minneapolis, MN) optimized by FirstMark. Minimum detectable CRP, FDP, and HSP‐70 were 0.1 mg/L, 0.06 μg/mL, and 0.313 ng/mL, respectively.
Figure 1.
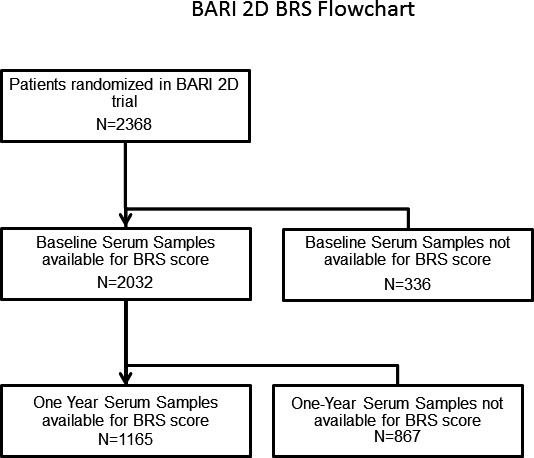
Flow chart demonstrates number of subjects at baseline and 1‐year time points with available biomarker samples. BARI 2D indicates Bypass Angioplasty Revascularization Investigation 2 Diabetes; BRS, biomarker risk score.
We used the same discriminatory cutoffs for each biomarker reported previously5 (9): Cut points for CRP, FDP, and HSP‐70 were 3 mg/L, 1.0 μg/mL, and 0.313 ng/mL, respectively.5 The BRS was computed by counting the number of biomarkers above their respective cutoffs.
Evaluation of Outcomes
The primary end point was all‐cause death; the principal secondary end point was a composite of death and myocardial infarction (MI).6 Other secondary end points were cardiac death and composite all‐cause death/MI/revascularization.
Diagnosis of MI was based on a doubling of cardiac enzymes and evidence of ischemia based on symptoms, ECG, or imaging. Periprocedural MI was defined as a 3‐ and a 10‐times increase in creatinine kinase MB after percutaneous coronary intervention and coronary artery bypass grafting, respectively. Silent MI was defined as a 2‐grade change of a Q‐wave on annual routine ECGs.6 Data on cause of death were adjudicated by personnel blinded to the group allocation, and cardiac death was defined as sudden death, fatal MI, congestive heart failure death, and presumed cardiac death. A subsequent revascularization procedure is defined as the first nonprotocol revascularization procedure for patients in the optimal medical treatment alone arm or a second revascularization procedure for patients in the prompt revascularization arm.
Statistical Analysis
Means and SDs are presented for continuous normally distributed variables. One‐way ANOVA and Wilcoxon rank‐sum tests were used, as appropriate, to determine the differences between BRS groups. Kaplan–Meier cumulative incidence estimates were calculated 5 years after randomization and 4‐years after the 1‐year blood draw, and differences among BRS groups were detected using the log‐rank statistic. For the survival end points, patients without events were censored at the last time they were known to be alive. For the other outcomes, patients not experiencing the events were censored at the time of their last protocol follow‐up visit. Unadjusted and adjusted Cox proportional hazard models were used to estimate the relationship between the BRS and incident cardiovascular events. The adjusted models accounted for clinically relevant risk factors for CVD outcomes (age at baseline, sex, race, body mass index, history of hypertension, dyslipidemia, history of MI, history of coronary artery bypass grafting, smoking status, use of statins, aspirin, clopidogrel/ticlopidine, estimated glomerular filtration rate (calculated using the Modification of Diet in Renal Disease equation), myocardial jeopardy index, and left ventricular ejection fraction). Statistical interaction between BRS and each covariate was tested individually by adding an interaction term to the multivariable Cox model. Additionally, we analyzed the cardiac death end point using Fine–Gray competing risk models where noncardiac death events were handled as competing risks rather than censored values. Harrell's c‐statistics for discrimination, continuous net reclassification improvement, and integrated discrimination improvement metrics were calculated based on these multivariable Cox models. In addition, calibration was internally validated using 500 bootstrap samples for each of the multivariable Cox models and creating calibration plots based on the 4‐year predicted probability of an event. Landmark analyses censored individuals who did not survive or contribute a 1‐year serum sample at the 1‐year time point. Spearman correlations are presented for the 3 biomarkers. Values of P<0.05 from 2‐sided tests were considered to indicate statistical significance. SAS software (version 9.3; SAS Institute Inc, Cary, NC) was used for all analyses, except for the c‐statistics and calibration plots, where the Regression Modeling Strategies R package (http://biostat.mc.vanderbilt.edu/rms; January 1, 2017) was used.
Results
The study population of BARI 2D patients with a baseline serum sample was aged 62.4±8.9 years, 70% male, 66% white (Table 1). Patients with baseline serum samples were comparable with those who had missing samples (data not shown).
Table 1.
Baseline Characteristics
| Characteristic | Total (N=2032) | BRS=0 (N=372) | BRS=1 (N=918) | BRS=2 (N=617) | BRS=3 (N=125) | P Value |
|---|---|---|---|---|---|---|
| Treatment allocation | ||||||
| Early revascularization assignment, % (n) | 50.0 (1016) | 50.3 (187) | 48.9 (449) | 50.7 (313) | 53.6 (67) | 0.7468 |
| Insulin providing assignment, % (n) | 50.0 (1016) | 53.2 (198) | 48.7 (447) | 48.6 (300) | 56.8 (71) | 0.1750 |
| Randomization stratum, % (n) | ||||||
| PCI | 67.0 (1361) | 65.9 (245) | 67.3 (618) | 66.9 (413) | 68.0 (85) | 0.9566 |
| CABG | 33.0 (671) | 34.1 (127) | 32.7 (300) | 33.1 (204) | 32.0 (40) | |
| Demographics | ||||||
| Age at study entry, mean, SD | 62.4, 8.9 | 62.3, 8.6 | 61.9, 8.7 | 62.6, 9.2 | 64.8, 9.4 | 0.0076 |
| Male, % (n) | 69.6 (1414) | 82.8 (308) | 69.4 (637) | 65.2 (402) | 53.6 (67) | <0.0001 |
| Race/ethnicity, % (n) | <0.0001 | |||||
| White | 66.1 (1343) | 71.2 (265) | 67.9 (623) | 62.9 (388) | 53.6 (67) | |
| Black | 17.1 (348) | 9.4 (35) | 16.4 (151) | 20.4 (126) | 28.8 (36) | |
| Hispanic | 12.5 (254) | 11.0 (41) | 11.5 (106) | 13.9 (86) | 16.8 (21) | |
| Asian | 3.7 (76) | 7.8 (29) | 3.6 (33) | 2.1 (13) | 0.8 (1) | |
| Other | 0.5 (11) | 0.5 (2) | 0.5 (5) | 0.6 (4) | 0.0 (0) | |
| Clinical history | ||||||
| BMI, mean, SD | 31.7, 6.0 | 29.6, 4.5 | 32.1, 5.9 | 32.5, 6.4 | 31.8, 6.9 | <0.0001 |
| History of cigarettes smoking, % (n) | 66.7 (1355) | 67.7 (252) | 65.6 (602) | 67.9 (418) | 66.4 (83) | 0.7981 |
| Hypertension requiring tx, % (n) | 83.0 (1668) | 77.7 (285) | 83.4 (757) | 85.2 (520) | 85.5 (106) | 0.0166 |
| Hypercholesterolemia requiring tx, % (n) | 81.9 (1645) | 84.6 (312) | 81.0 (736) | 82.6 (502) | 77.2 (95) | 0.2340 |
| Hx triglycerides tx, % (n) | 32.5 (598) | 30.9 (105) | 36.7 (305) | 29.3 (163) | 22.1 (25) | 0.0015 |
| Classic angina class w/i 6 weeks, % (n) | 0.0080 | |||||
| Stable 1, 2 | 42.1 (856) | 42.5 (158) | 42.4 (389) | 42.4 (261) | 38.4 (48) | |
| Stable 3, 4 | 8.6 (174) | 5.6 (21) | 9.0 (83) | 10.1 (62) | 6.4 (8) | |
| Unstable | 9.6 (194) | 5.9 (22) | 9.7 (89) | 10.2 (63) | 16.0 (20) | |
| No angina | 39.7 (807) | 46.0 (171) | 38.9 (357) | 37.3 (230) | 39.2 (49) | |
| History of MI, % (n) | 31.4 (629) | 27.8 (101) | 29.5 (268) | 34.9 (211) | 39.2 (49) | 0.0139 |
| Sitting systolic BP, mean, SD | 131.9, 20.2 | 129.2, 20.5 | 131.9, 19.6 | 133.4, 20.5 | 132.6, 21.4 | 0.0206 |
| Sitting diastolic BP, mean, SD | 74.6, 11.4 | 73.7, 10.3 | 74.9, 11.5 | 75.3, 12.0 | 72.3, 10.2 | 0.0140 |
| Clinical labs | ||||||
| LDL mg/dL, mean, SD | 95.9, 33.3 | 91.3, 30.1 | 96.5, 33.0 | 97.3, 34.5 | 97.9, 38.4 | 0.0335 |
| HDL mg/dL, mean, SD | 38.2, 10.4 | 38.7, 9.9 | 37.9, 10.2 | 38.0, 10.1 | 39.7, 13.5 | 0.2351 |
| HbA1c %, mean, SD | 7.65, 1.61 | 7.29, 1.40 | 7.72, 1.63 | 7.78, 1.67 | 7.60, 1.62 | <0.0001 |
| GFR (MDRD algorithm), mean, SD | 79.1, 29.6 | 80.2, 20.9 | 79.6, 23.1 | 78.6, 40.7 | 75.3, 28.7 | 0.3945 |
| Angiographic | ||||||
| LVEF <50%, % (n) | 17.3 (341) | 16.6 (60) | 16.3 (145) | 17.6 (105) | 25.4 (31) | 0.0953 |
| LVEF not available, % (n) | 3.1 (63) | 3.0 (11) | 3.2 (29) | 3.2 (20) | 2.4 (3) | 0.9636 |
| Myocardial jeopardy, mean, SD | 44.4, 24.1 | 45.6, 24.1 | 43.4, 24.1 | 45.5, 24.8 | 43.3, 21.1 | 0.2540 |
| Medications | ||||||
| Statin, % (n) | 74.2 (1505) | 80.5 (298) | 73.1 (670) | 72.9 (450) | 69.6 (87) | 0.0169 |
| ACE inhibitor or ARB, % (n) | 77.4 (1570) | 78.4 (291) | 75.5 (692) | 78.6 (485) | 81.6 (102) | 0.2840 |
| Aspirin, % (n) | 88.2 (1789) | 91.1 (338) | 88.9 (814) | 86.4 (533) | 83.2 (104) | 0.0396 |
| Antiplatelet: ticlopodine/clopidogrel, % (n) | 17.8 (361) | 16.7 (62) | 18.0 (165) | 17.0 (105) | 23.2 (29) | 0.3796 |
| Beta‐blocker, % (n) | 73.1 (1484) | 73.3 (272) | 73.9 (677) | 72.3 (446) | 71.2 (89) | 0.8614 |
| Revascularization history | ||||||
| Previous PCI, % (n) | 19.9 (405) | 19.6 (73) | 19.9 (183) | 19.8 (122) | 21.6 (27) | 0.9692 |
| Previous stent, % (n) | 13.7 (278) | 11.0 (41) | 15.0 (138) | 13.0 (80) | 15.2 (19) | 0.2465 |
| Previous CABG, % (n) | 6.1 (124) | 4.3 (16) | 6.0 (55) | 6.8 (42) | 8.8 (11) | 0.2358 |
| Biomarkers | ||||||
| CRP mg/L, mean, SD | 5.84, 3.64 | 1.65, 0.80 | 5.93, 3.49 | 7.74, 2.88 | 8.30, 2.54 | <0.0001 |
| HSP‐70 ng/mL, mean, SD | 0.65, 0.96 | 0.31, 0.00 | 0.46, 0.58 | 1.01, 1.35 | 1.23, 1.43 | <0.0001 |
| FDP μg/mL, mean, SD | 0.98, 1.75 | 0.45, 0.21 | 0.65, 1.01 | 1.49, 2.47 | 2.53, 2.63 | <0.0001 |
ACE indicates angiotensin‐converting enzyme; ARB, angiotensin receptor blocker; BMI, body mass index; BP, blood pressure; BRS, biomarker risk score; CABG, coronary artery bypass grafting; CRP, C‐reactive protein; FDP, fibrin degradation products; GFR, glomerular filtration rate; HbA1c, glycated hemoglobin; HDL, high‐density lipoprotein; HSP‐70, heat shock protein‐70; Hx, history; LDL, low‐density lipoprotein; LVEF, left ventricular ejection fraction; MDRD, Modification of Diet in Renal Disease; MI, myocardial infarction; PCI, percutaneous coronary intervention; tx, treatment; w/I, within.
Participants were separated based on the serum levels of CRP, HSP‐70, and FDP at the baseline visit into 4 BRS categories as described above. Thus, 18.3%, 45.2%, 30.3%, and 6.2% of patients had a BRS of 0, 1, 2, and 3, respectively, based on the number of biomarkers above the cut‐off threshold value. Those with a higher BRS were older, more likely to be female, obese, hypertensive, and more likely to have severe angina, a higher HbA1C, higher low‐density lipoprotein level, a history of MI, and less likely to be white or to be on statins and aspirin (Table 1). There were no differences in the myocardial jeopardy index or left ventricular ejection fraction between categories of the BRS. Importantly, a similar proportion of subjects in each BRS category were randomized to either the revascularization versus medical therapy or insulin‐providing versus IS treatments.
Relationship Between the Individual Biomarkers and Incident CVD Events
There were modest, but significant, correlations among the 3 individual biomarkers (Table 2). All 3 biomarkers (continuous variables) were associated with incident adverse events in both unadjusted and multivariable models adjusted for aforementioned covariates (Table 3). Elevated levels (above cut points) of CRP, HSP‐70, and FDP were all associated with increased risk of all‐cause death, the composite of all‐cause death and MI, cardiac death, and composite of all‐cause death/MI/revascularization in unadjusted and adjusted models (Table 3).
Table 2.
Relationship Between Individual Biomarkers in 2032 Subjects and MJI (n=2030): Spearman Correlation Coefficients and P Values Are Presented
| HSP‐70 | FDP | MJI | |
|---|---|---|---|
| CRP | |||
| R | 0.075 | 0.223 | −0.0433 |
| P value | 0.0007 | <0.0001 | 0.051 |
| HSP‐70 | |||
| R | 0.133 | 0.0523 | |
| P value | <0.0001 | 0.0184 | |
| FDP | |||
| R | −0.0312 | ||
| P value | 0.159 | ||
CRP indicates C‐reactive protein; FDP, fibrin degradation products; HSP‐70, heat shock protein‐70; MJI, myocardial jeopardy index; R, correlation coefficient.
Table 3.
Association of Individual Biomarkers and the BRS With Adverse Events
| All Patients (2032) | Unadjusted | Adjusted | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Value | HR | 95% CI | P Value | |
| All cause death (n=274) | ||||||
| Model 1 | ||||||
| CRP ≥3.0 mg/L | 1.62 | 1.21, 2.15 | 0.0011 | 1.84 | 1.37, 2.48 | <0.0001 |
| HSP‐70 >0.313 ng/mL | 1.42 | 1.12, 1.82 | 0.0045 | 1.29 | 1.00, 1.65 | 0.0470 |
| FDP ≥1.0 μg/mL | 2.05 | 1.58, 2.66 | <0.0001 | 1.63 | 1.24, 2.14 | 0.0005 |
| Model 2 | ||||||
| BRS per 1 unit increase | 1.67 | 1.44, 1.93 | <0.0001 | 1.54 | 1.33, 1.78 | <0.0001 |
| Model 3 | ||||||
| 1 Positive biomarker vs 0 | 1.72 | 1.13, 2.60 | 0.011 | 1.75 | 1.15, 2.67 | 0.0093 |
| 2 Positive biomarkers vs 0 | 2.68 | 1.76, 4.08 | <0.0001 | 2.49 | 1.62, 3.82 | <0.0001 |
| 3 Positive biomarkers vs 0 | 4.98 | 3.01, 8.24 | <0.0001 | 3.99 | 2.37, 6.73 | <0.0001 |
| Cardiac death (n=114) | ||||||
| Model 1 | ||||||
| CRP ≥3.0 mg/L | 1.93 | 1.20, 3.11 | 0.0067 | 2.12 | 1.30, 3.46 | 0.0028 |
| HSP‐70 >0.313 ng/mL | 1.71 | 1.18, 2.48 | 0.0047 | 1.49 | 1.02, 2.18 | 0.0409 |
| FDP ≥1.0 μg/mL | 2.24 | 1.51, 3.33 | <0.0001 | 1.82 | 1.20, 2.76 | 0.0050 |
| Model 2 | ||||||
| BRS per 1 unit increase | 1.94 | 1.55, 2.43 | <0.0001 | 1.75 | 1.39, 2.20 | <0.0001 |
| Model 3 | ||||||
| 1 Positive biomarker vs 0 | 2.27 | 1.07, 4.82 | 0.034 | 2.35 | 1.10, 5.04 | 0.0277 |
| 2 Positive biomarkers vs 0 | 3.71 | 1.74, 7.88 | 0.0007 | 3.42 | 1.59, 7.35 | 0.0016 |
| 3 Positive biomarkers vs 0 | 8.80 | 3.85, 20.14 | <0.0001 | 6.82 | 2.90, 16.02 | <0.0001 |
| death/MI (n=443) | ||||||
| Model 1 | ||||||
| CRP ≥3.0 mg/L | 1.47 | 1.18, 1.83 | 0.0007 | 1.57 | 1.25, 1.99 | 0.0001 |
| HSP‐70 >0.313 ng/mL | 1.33 | 1.10, 1.61 | 0.0036 | 1.23 | 1.01, 1.50 | 0.0364 |
| FDP ≥1.0 μg/mL | 1.90 | 1.54, 2.35 | <0.0001 | 1.73 | 1.38, 2.15 | <0.0001 |
| Model 2 | ||||||
| BRS per 1 unit increase | 1.54 | 1.37, 1.72 | <0.0001 | 1.47 | 1.31, 1.65 | <0.0001 |
| Model 3 | ||||||
| 1 Positive biomarker vs 0 | 1.68 | 1.22, 2.30 | 0.0013 | 1.77 | 1.28, 2.45 | 0.0005 |
| 2 Positive biomarkers vs 0 | 2.41 | 1.74, 2.32 | <0.0001 | 2.38 | 1.71, 3.31 | <0.0001 |
| 3 Positive biomarkers vs 0 | 3.94 | 2.64, 5.89 | <0.0001 | 3.53 | 2.32, 5.34 | <0.0001 |
| death/MI/revascularization (n=848) | ||||||
| Model 1 | ||||||
| CRP ≥3.0 mg/L | 1.42 | 1.21, 1.65 | <0.0001 | 1.41 | 1.20, 1.66 | <0.0001 |
| HSP‐70 >0.313 ng/mL | 1.18 | 1.03, 1.36 | 0.0167 | 1.18 | 1.02, 1.35 | 0.0221 |
| FDP ≥1.0 μg/mL | 1.32 | 1.12, 1.56 | 0.0012 | 1.26 | 1.06, 1.50 | 0.0099 |
| Model 2 | ||||||
| BRS per 1 unit increase | 1.30 | 1.20, 1.41 | <0.0001 | 1.27 | 1.17, 1.38 | <0.0001 |
| Model 3 | ||||||
| 1 Positive biomarker vs 0 | 1.40 | 1.14, 1.72 | 0.0014 | 1.38 | 1.12, 1.70 | 0.0029 |
| 2 Positive biomarkers vs 0 | 1.80 | 1.46, 2.23 | <0.0001 | 1.75 | 1.40, 2.18 | <0.0001 |
| 3 Positive biomarkers vs 0 | 2.11 | 1.56, 2.86 | <0.0001 | 1.97 | 1.44, 2.69 | <0.0001 |
BRS indicates biomarker risk score; CRP, C‐reactive protein; FDP, fibrin degradation products; HR, hazard ratio; HSP‐70, heat shock protein‐70; MI, myocardial infarction.
Relationship Between the BRS and Incident CVD Events
There was a significant association between the BRS and all incident CVD events in both unadjusted and adjusted models (Table 3). Each unit increase in the BRS corresponded to a 54% increase in risk of future all‐cause death and a 75% increase in risk of cardiac death after adjustment for the aforementioned variables (P<0.0001). Figure 2 demonstrates Kaplan–Meier survival curves for the outcomes of death, cardiac death, death/MI, and death/MI/revascularization. Compared with those with a BRS of 0, adjusted hazard ratios for incident cardiac death for patients with a BRS of 1, 2, and 3 were 2.4, 3.4, and 6.8, for all‐cause death were 1.75, 2.49, and 3.99, and for death/MI were 1.77, 2.38, and 3.53. Five‐year Kaplan–Meier event probabilities for all‐cause death for a BRS of 0, 1, 2, and 3 were 6.2%, 10.4%, 15.8%, and 26.4%, respectively (P<0.0001; Figure 2). Treating the noncardiac deaths as competing risks rather than censored events, the Fine–Gray models for cardiac death yielded comparable results to the Cox model results shown in Table 3.
Figure 2.
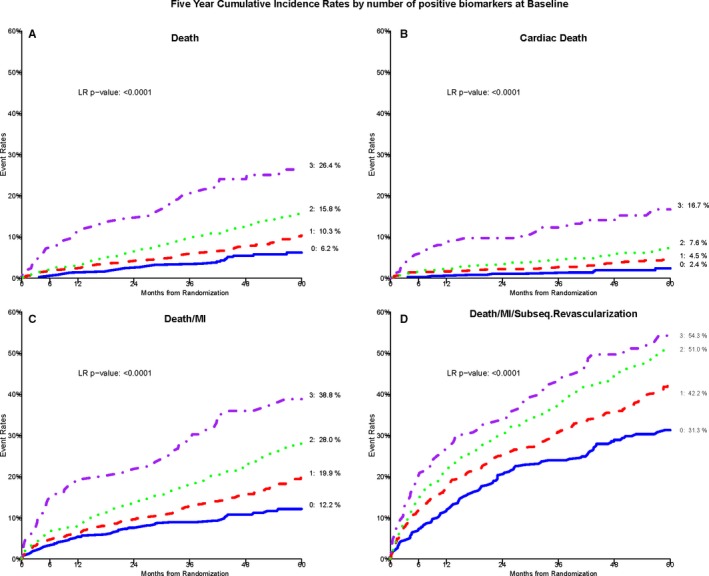
Kaplan–Meier cumulative incidence curves by BRS score. A through D, Demonstrate this association with all‐cause death, cardiac death, composite death/MI, and death/MI/revascularization, respectively, in those with BRS of 0, 1, 2, and 3. BRS, biomarker risk score; LR, likelihood ratio; MI, myocardial infarction; Subseq., subsequent.
Sensitivity Analysis
To evaluate whether the association of the BRS with adverse outcomes was modified by demographic and clinical risk factor covariates or the assigned treatment strategies, interaction analyses were performed and revealed no significant effect modification (Figure 3). Importantly, there were no interactions between randomization assignments or the assigned strata with the death/MI outcome.
Figure 3.
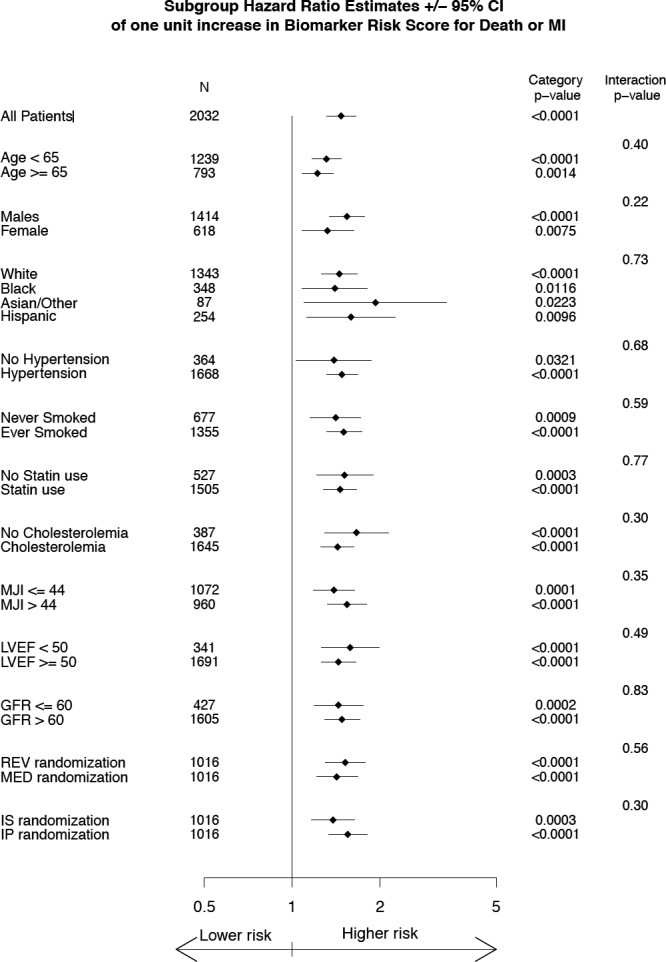
Sensitivity analysis of the biomarker risk score in association with death/MI outcome with respect to individual covariates. GFR indicates glomerular filtration rate; IP, insulin‐providing; IS, insulin‐sensitizing; LVEF, left ventricular ejection fraction; MED, medical therapy; MI, myocardial infarction; MJI, myocardial jeopardy index.
Discrimination and Calibration Analyses
Addition of the BRS to a baseline model consisting of the aforementioned demographic and clinical variables was associated with significant improvements in the c‐statistic for all outcome measures, including all‐cause death (area under the curve [AUC], 0.700–0.715; Δ AUC=0.015; P=0.044), all‐cause death/MI (AUC, 0.634–0.655; Δ AUC=0.021; P=0.002), all‐cause death/MI/revascularization (AUC, 0.574–0.589; Δ AUC=0.015; P=0.015), and for cardiac death (AUC, 0.740–0.760; Δ AUC=0.020; P=0.053) with a trend toward significance. Similarly, the addition of the BRS to the baseline model was associated with improvements in both net reclassification improvement and integrated discrimination improvement for each outcome measure (Table 4). The continuous net reclassification improvement for the composite outcome of all‐cause death/MI was 15%, which corresponded to 76% and 61% improvements in reclassification of risk in event‐ and nonevent groups, respectively. Calibration plots from each of the 4 models (corresponding to the adjusted model 3 for each outcome in Table 3) indicate that the risk models are fairly well calibrated (Figure 4).
Table 4.
Discrimination Metrics for the BRS in Relation to Primary and Secondary Outcome Measures
| Discrimination Indices | |
|---|---|
| Death | |
| Events correctly reclassified | 0.69 |
| Nonevents correctly reclassified | −0.54 |
| NRI | 0.151 |
| IDI | 0.037 |
| Cardiac death | |
| Events correctly reclassified | 0.84 |
| Nonevents correctly reclassified | −0.63 |
| NRI | 0.224 |
| IDI | 0.014 |
| Death/MI | |
| Events correctly reclassified | 0.76 |
| Nonevents correctly reclassified | −0.61 |
| NRI | 0.151 |
| IDI | 0.0339 |
| Death/MI/revascularization | |
| Events correctly reclassified | 0.68 |
| Nonevents correctly reclassified | −0.54 |
| NRI | 0.133 |
| IDI | 0.0091 |
BRS indicates biomarker risk score; IDI, integrated discrimination improvement; MI, myocardial infarction; NRI, net reclassification improvement.
Figure 4.
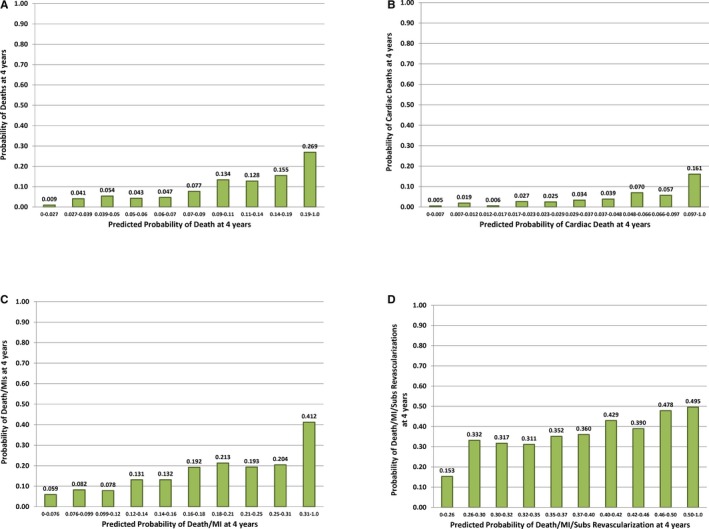
Calibration plots for the multivariable Cox Models for the predicted probability of an event 4 years after randomization. The calibration plots correspond to the multivariable adjusted Cox model 3 shown in Table 3 for all‐cause death (A), cardiac death (B), death/MI (C), and death/MI/revascularization (D). Each bar represents ≈10% of the patients rank ordered based on their predicted probability of an event from the Cox model (ie, deciles of risk). The height of the bar is the observed proportion of patients who had an event by 4 years. MI indicates myocardial infarction.
Change in Biomarker Levels at 1 Year
Biomarker measurements were available in 1304 subjects at 1 year largely attributed to a protocol‐related delay in storing follow‐up samples. Minor differences were noted between patients with 1‐year serum samples and those who had missing samples (Table 5). There were significant reductions in all 3 biomarker levels at 1 year compared with baseline (Δ CRP=−1.2±3.5 mg/L, Δ HSP‐70=−0.36±1.2 ng/mL, and Δ FDP=−0.24±1.45 μg/mL; P=0.0001 for all). There were also substantial improvements in cardiovascular risk factors between baseline and 1 year (Δ systolic BP=−5.1±21.9 mm Hg, Δ diastolic BP=−2.6±11.6 mm Hg, Δ low‐density lipoprotein=−12.4±37.5 mg/dL, Δ high‐density lipoprotein=2.5±8.2 mg/dL, Δ HbA1c=−0.6±1.5%, and Δ glomerular filtration rate=−3.4±30.3). There were significant correlations between the change in HbA1c, low‐density lipoprotein, and high‐density lipoprotein levels and the change in CRP level (r=0.14, P=0.0001; r=0.07, P=0.01; and r=−0.18, P=0.0001, respectively). Similarly, there were significant correlations between the change in HSP‐70 and the change in systolic and diastolic BPs (r=0.09, P=0.002 and r=0.07, P=0.008; Table 6). Importantly, the significant reduction in the biomarkers levels at 1 year occurred irrespective of the randomization strategy for revascularization or insulin‐producing or IS assignment (data not shown).
Table 5.
Baseline Characteristics by Availability of 1‐Year Biomarker Information
| Characteristic | Total With Baseline BRS Data (N=2032) | Baseline and 1‐Y BRS Data (N=1165) | Baseline but No 1‐Y BRS Data (N=867) | P Value |
|---|---|---|---|---|
| Study design | ||||
| Early revascularization assignment, % (n) | 50.0 (1016) | 48.9 (570) | 51.4 (446) | 0.2622 |
| Insulin providing assignment, % (n) | 50.0 (1016) | 49.4 (575) | 50.9 (441) | 0.5011 |
| Randomization stratum, % (n) | ||||
| PCI | 67.0 (1361) | 64.2 (748) | 70.7 (613) | 0.0021 |
| CABG | 33.0 (671) | 35.8 (417) | 29.3 (254) | |
| Demographics | ||||
| Age at study entry, mean, SD | 62.4, 8.9 | 62.2, 8.8 | 62.6, 9.0 | 0.2374 |
| Male, % (n) | 69.6 (1414) | 69.0 (804) | 70.4 (610) | 0.5146 |
| Race/ethnicity, % (n) | ||||
| White | 66.1 (1343) | 67.1 (782) | 64.7 (561) | 0.1291 |
| Black | 17.1 (348) | 16.0 (186) | 18.7 (162) | |
| Hispanic | 12.5 (254) | 13.0 (151) | 11.9 (103) | |
| Asian | 3.7 (76) | 3.7 (43) | 3.8 (33) | |
| Other | 0.5 (11) | 0.3 (3) | 0.9 (8) | |
| Clinical history | ||||
| BMI, mean, SD | 31.7, 6.0 | 31.4, 5.8 | 32.2, 6.2 | 0.0045 |
| History of cigarettes smoking, % (n) | 66.7 (1355) | 65.2 (759) | 68.9 (596) | 0.0760 |
| Hypertension requiring tx, % (n) | 83.0 (1668) | 83.5 (965) | 82.4 (703) | 0.5306 |
| Hypercholesterolemia requiring tx, % (n) | 81.9 (1645) | 83.4 (962) | 79.9 (683) | 0.0453 |
| Hx triglycerides tx, % (n) | 32.5 (598) | 33.1 (357) | 31.6 (241) | 0.5016 |
| Classic angina class w/i 6 weeks, % (n) | ||||
| Stable 1, 2 | 42.1 (856) | 46.0 (536) | 37.0 (320) | <0.0001 |
| Stable 3, 4 | 8.6 (174) | 9.6 (112) | 7.2 (62) | |
| Unstable | 9.6 (194) | 7.4 (86) | 12.5 (108) | |
| No angina | 39.7 (807) | 37.0 (431) | 43.4 (376) | |
| History of MI, % (n) | 31.4 (629) | 33.3 (383) | 28.9 (246) | 0.0347 |
| Sitting systolic BP average, mean, SD | 131.9, 20.2 | 132.6, 20.9 | 131.0, 19.1 | 0.0821 |
| Sitting diastolic BP average, mean, SD | 74.6, 11.4 | 75.5, 11.6 | 73.5, 11.0 | 0.0002 |
| Clinical labs | ||||
| Core: LDL mg/dL, mean, SD | 95.9, 33.3 | 96.7, 34.9 | 94.7, 31.2 | 0.1892 |
| Core: HDL mg/dL, mean, SD | 38.2, 10.4 | 37.6, 9.6 | 39.0, 11.3 | 0.0025 |
| Core: HbA1c %, mean, SD | 7.65, 1.61 | 7.74, 1.68 | 7.53, 1.51 | 0.0026 |
| GFR (MDRD algorithm), mean, SD | 79.1, 29.6 | 80.5, 33.8 | 77.2, 22.6 | 0.0086 |
| Angiographic | ||||
| LVEF <50%, % (n) | 17.3 (341) | 13.9 (158) | 22.0 (183) | <0.0001 |
| LVEF not available, % (n) | 3.1 (63) | 2.2 (26) | 4.3 (37) | 0.0088 |
| Myocardial jeopardy, mean, SD | 44.4, 24.1 | 43.9, 24.3 | 45.0, 23.9 | 0.3042 |
| Medications | ||||
| Statin, % (n) | 74.2 (1505) | 75.5 (878) | 72.5 (627) | 0.1256 |
| ACE or ARB, % (n) | 77.4 (1570) | 76.9 (893) | 78.1 (677) | 0.5107 |
| Aspirin, % (n) | 88.2 (1789) | 89.3 (1039) | 86.6 (750) | 0.0594 |
| Antiplatelet: ticlopodine/clopidogrel, % (n) | 17.8 (361) | 16.2 (188) | 20.0 (173) | 0.0250 |
| Beta‐blocker, % (n) | 73.1 (1484) | 72.7 (845) | 73.8 (639) | 0.5698 |
| Revascularization history | ||||
| Previous PCI, % (n) | 19.9 (405) | 20.3 (237) | 19.4 (168) | 0.5897 |
| Previous stent, % (n) | 13.7 (278) | 15.1 (176) | 11.8 (102) | 0.0309 |
| Previous CABG, % (n) | 6.1 (124) | 6.8 (79) | 5.2 (45) | 0.1384 |
ACE indicates angiotensin‐converting enzyme; ARB, angiotensin receptor blocker; BMI, body mass index; BP, blood pressure; BRS, biomarker risk score; CABG, coronary artery bypass grafting; GFR, glomerular filtration rate; HbA1c, glycated hemoglobin; HDL, high‐density lipoprotein; Hx, history; LDL, low‐density lipoprotein; LVEF, left ventricular ejection fraction; MDRD, Modification of Diet in Renal Disease; MI, myocardial infarction; PCI, percutaneous coronary intervention; tx, treatment; w/I, within.
Table 6.
Spearman Correlation Among Change in Biomarkers (1 Year‐Baseline) and Change in Traditional Cardiac Risk Factors (1 Year‐Baseline)
| R P Value | ΔCRP (1 Y‐Base) (n=1163) | ΔHSP70 (1 Y‐Base) (n=1165) | ΔFDP (1 Y‐Base) (n=1165) |
|---|---|---|---|
| ΔSBP (1 y‐base) | −0.0055 | 0.092 | −0.081 |
| 0.85 | 0.002 | 0.006 | |
| ΔDBP (1 y‐base) | 0.0115 | 0.078 | −0.048 |
| 0.70 | 0.0081 | 0.10 | |
| ΔLDL (1 y‐base) | 0.072 | 0.0094 | −0.008 |
| 0.019 | 0.76 | 0.79 | |
| ΔHDL (1 y‐base) | −0.183 | 0.080 | −0.049 |
| 0.0001 | 0.0066 | 0.096 | |
| ΔHbA1c (1 y‐base) | 0.14 | −0.007 | −0.052 |
| 0.0001 | 0.82 | 0.077 | |
| ΔeGFR (1 y‐base) | −0.011 | −0.025 | 0.054 |
| 0.72 | 0.40 | 0.063 |
In each box, the top number is the correlation coefficient and the bottom number is the P value. CRP indicates C‐reactive protein; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate; FDP, fibrin degradation products; HbA1c, glycated hemoglobin; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; SBP, systolic blood pressure.
Change in the BRS at 1 Year Compared to Baseline
Among patients with a BRS of 2 and 3 at baseline, the BRS fell in 81% and 68% of patients, respectively, in the first year of the study to levels of 0 or 1. A change from low levels to higher levels was less common; only 6% and 13% of patients with a baseline BRS of 0 and 1, respectively, increased to a score of 2 at 1 year (Table 7). The change in the BRS from baseline to 1 year was not associated with the revascularization versus medical therapy randomization, but its association with glycemic control randomization showed statistically significant difference (Tables 8 and 9).
Table 7.
Change in the BRS at 1 Year Compared to Baseline
| Total (N=1165) | Biomarker Risk Score at Baseline | P Value | ||||
|---|---|---|---|---|---|---|
| 0 (N=189) | 1 (N=487) | 2 (N=410) | 3 (N=79) | |||
| BRS at 1 y (n) | ||||||
| 0 | 38.6 (450) | 67.7 (128) | 41.1 (200) | 24.4 (100) | 27.8 (22) | <0.0001 |
| 1 | 45.9 (535) | 26.5 (50) | 45.8 (223) | 56.1 (230) | 40.5 (32) | |
| 2 | 14.0 (163) | 5.8 (11) | 11.7 (57) | 17.6 (72) | 29.1 (23) | |
| 3 | 1.5 (17) | 0.0 (0) | 1.4 (7) | 2.0 (8) | 2.5 (2) | |
Rows represent biomarker risk score at 1 year whereas columns represent biomarker risk score at baseline. Second column represents percentage (number) of subjects at 1 year in different categories of the biomarker risk score. Columns 3 to 6 represent the percentage (number) of subjects at baseline who have the same biomarker risk score as 1 year. BRS indicates biomarker risk score.
Table 8.
Change in BRS Comparing 1 Year to Baseline Between Medical Therapy and Revascularization Randomization Arms
| Characteristic | Total (N=1165) | MED (N=595) | REV (N=570) | P Value |
|---|---|---|---|---|
| Change in BRS score 1 y‐baseline, mean, SD | −0.542, 0.938 | −0.511, 0.929 | −0.575, 0.946 | 0.2405 |
BRS indicates biomarker risk score; med, medical therapy.
Table 9.
Change in BRS Comparing 1 Year to Baseline Between IS and IP Randomization Arms
| Characteristic | Total (N=1165) | IP (N=575) | IS (N=590) | P Value |
|---|---|---|---|---|
| Change in BRS score 1 y‐baseline, mean, SD | −0.542, 0.938 | −0.437, 0.949 | −0.646, 0.915 | 0.0001 |
BRS indicates biomarker risk score; IP, insulin‐providing; IS, insulin‐sensitizing.
Association Between the BRS at 1 Year and CVD Events
The reclassified BRS derived at 1 year after randomization among the 1304 survivors significantly predicted risk of all‐cause mortality. Those who had a BRS of 2 to 3 at 1 year had a 4‐year mortality rate of 21.1% compared with a 7.7% in those with a BRS of 1 and 7.1% with a BRS of 0 (P<0.001). Similarly, the 4‐year death/MI event rate was 30.3% in those with a BRS of 2 to 3 at 1 year as compared with 14.4% for those with a BRS of 1 and 14.1% with a BRS of 0 (P<0.0001; Figure 5).
Figure 5.
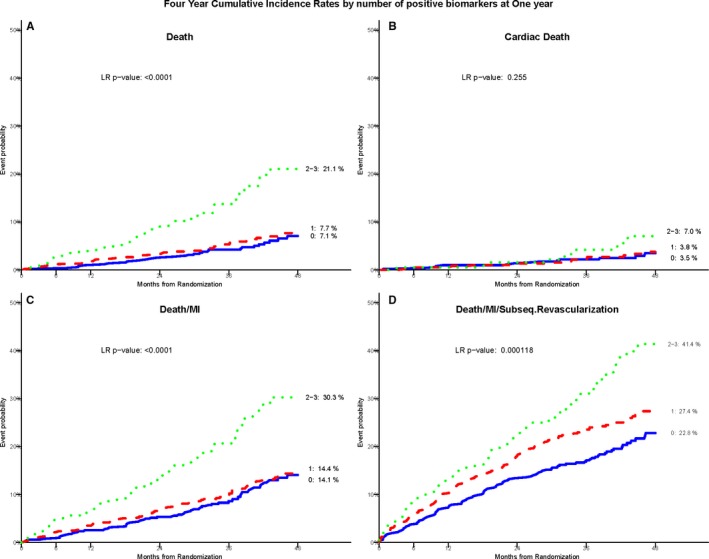
Kaplan–Meier cumulative incidence curves by the biomarker risk score measured in survivors at 1‐year postrandomization. A through D, Demonstrate this association with all‐cause death, cardiac death, composite death/MI, and death/MI/revascularization, respectively. LR indicates likelihood ratio; MI, myocardial infarction.
Association of the Change in BRS at 1 Year With Risk of Incident Outcomes
Among patients with a baseline BRS of 2 to 3, 10.1% suffered death or MI at 1 year. In the survivors at 1 year, those who had a lowering of their BRS from 2 to 3 to 0 to 1 had significantly lower 4‐year death/MI rate compared with those whose risk score remained unchanged (19% versus 34.2%; P=0.0025; Figure 6).
Figure 6.
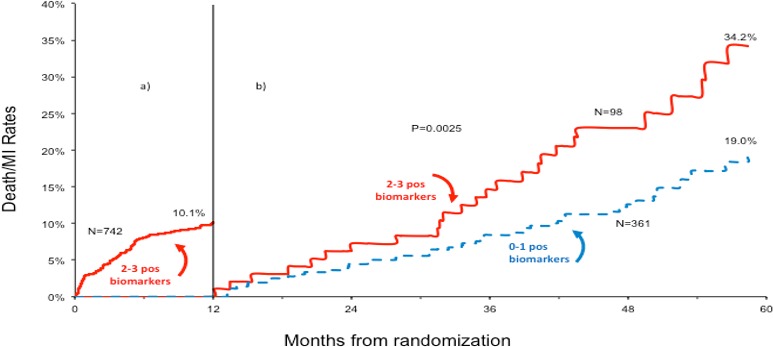
Kaplan–Meier death/MI cumulative incidence curve in those with the biomarker risk score of 2 to 3 at baseline. A, Demonstrates the death/MI event curve during the first year of follow‐up. B, Demonstrates event rates from years 2 to 5 in subjects in whom the BRS decreased to 0 or 1 compared with those in whom the BRS remained high at 2 or 3. MI indicates myocardial infarction.
Discussion
In patients with diabetes mellitus and stable CAD enrolled in the BARI 2D study, a BRS comprised of markers of inflammation (CRP), thrombosis (FDP), and cell stress (HSP‐70) identifies a subgroup of CAD patients at very high near‐term risk of incident MI and death. The BRS was additive to the clinical, demographic, and angiographic features. Thus, when BRS was added to the baseline model, it was associated with significant improvement in c‐statistic and risk reclassification metrics. These results confirm those previously obtained in a totally independent cohort of CAD patients.5
Additionally, we found that following aggressive medical and lifestyle intervention, all biomarkers and the BRS were modifiable, such that 78% of subjects with a BRS of 2 or 3 demonstrated a decrease to 0 to 1 after 1 year. These reductions were associated with improvements in CVD risk factors, including the lipid profile, BP, and HbA1C, but not associated with either assignment to revascularization or insulin‐providing compared to IS strategies.
Intriguingly, the decline in the BRS was associated with improved future prognosis. Thus, the observed 4‐year death/MI rates were 45% lower in those who started with a high BRS and had a reduction in their BRS, compared with those in whom the BRS remained high. This suggests that repeat measurements of the BRS accurately reflect time‐related alteration of risk and might be used to calibrate therapy. Furthermore, the almost one quarter of subjects with low BRS have a very low risk of adverse CVD events and a very low probability of increasing their BRS over the first year of participation in the BARI 2D study. The findings of this investigation lend major support to the importance of a strategy for identifying the “vulnerable patient.”9
Vulnerability of atherosclerotic plaques involves activation of several pathways that synergistically promote phenotypic changes that precipitate plaque rupture and consequent acute coronary events. Traditional risk models, such as the Framingham risk score, fail to predict risk in those with established CVD.4 We found that the aggregate of 3 biomarkers—CRP, FDP, and HSP‐70—each individually representing different biologic pathways (but not exclusively), independently predict risk of incident MI/death in CAD patients both at baseline and after 1 year.5 Although studies have identified high‐risk subgroups using single biomarkers, they have failed to identify patients with the extremely high or low near‐term risk as can be achieved using our aggregate biomarker strategy.10, 11
Elevated CRP levels, reflecting activation of the inflammatory pathway, has been shown to predict incident CVD events.10, 11 HSP‐70, a marker of cell stress, is an intracellular protein within the family of heat shock proteins that aid in the cellular response to acute stress.12 However, its relationship with CAD has been controversial.13 Whereas lower levels are associated with long‐term development of CAD,13 higher levels are associated with higher risk of plaque rupture and incident future outcomes.5 The association of HSP‐70 with future outcomes we found in this study is novel and validates our previous findings.5 Ongoing fibrin/fibrinogen degradation increases FDP levels, which measure D‐dimer, fragments D and E, and additional intermediate products of fibrin degradation. Elevation of FDP is indicative of a prothrombotic milieu and predicts incident CVD events in patients with peripheral vascular disease.14
A previous BARI 2D analysis demonstrated reductions in CRP, fibrinogen, and fibrinopeptide‐A after 1 year.15 Other studies have also demonstrated that inflammatory markers are modifiable with aggressive medical therapy, including statin regimens,16, 17 and that the magnitude of benefit from statin therapy is associated with the magnitude of reduction of CRP levels.18, 19, 20, 21, 22 In the BARI 2D trial, baseline levels of CRP, d‐dimer, but not fibrinogen, predicted risk of death.15 The present study extends the previous observations for CRP by incorporating FDP and HSP‐70 measurements and, most important, by calculating an aggregate of these 3 biomarkers into a BRS.
Limitations
We cannot definitely conclude that the improved 1‐year BRS was caused by the aggressive medical and lifestyle interventions in the BARI 2D study. Also, we were able to measure biomarkers on fewer subjects after 1 year because of missing sample collection and thus cannot be certain that patient selection factors played a role in our results.
Conclusion
An aggregate risk score based on serum levels of CRP, FDP, and HSP‐70 significantly predicts the risk of death and MI in patients with diabetes mellitus and stable CAD and improves risk reclassification. Importantly, the BRS is modifiable and is significantly reduced with aggressive medical and lifestyle intervention. A decline in the BRS is associated with a significant improvement in risk, whereas a persistent elevation is associated with persistent high risk. Thus, serial measurements of the BRS may be used by physicians to titrate therapy and provide continuing information on changing risk in an individual patient, thereby permitting individualized tailoring of therapeutic strategies on an ongoing basis.
Sources of Funding
Ghasemzadeh's research fellowship in preventive cardiology was supported by the Katz Family Foundation Preventive Cardiology Grant, Atlanta, GA. Sample measurements were conducted by FirstMark, Division of GenWay Biotech Inc (San Diego, CA). This study was funded by GenWay Biotech Inc. The Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) is funded by the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases (U01 HL061744, U01 HL061746, U01 HL061748, U01 HL063804, and R21 HL121495). BARI 2D receives significant supplemental funding provided by GlaxoSmithKline (Collegeville, PA), Lantheus Medical Imaging, Inc (formerly Bristol‐Myers Squibb Medical Imaging, Inc; North Billerica, MA), Astellas Pharma US, Inc (Deerfield, IL), Merck & Co, Inc (Whitehouse Station, NJ), Abbott Laboratories, Inc (Abbott Park, IL), and Pfizer Inc (New York, NY). Generous support is given by Abbott Laboratories Ltd, MediSense Products (Mississauga, Ontario, Canada), Bayer Diagnostics (Tarrytown, NY), Becton, Dickinson and Company (Franklin Lakes, NJ), J. R. Carlson Labs (Arlington Heights, IL), Centocor, Inc (Malvern, PA), Eli Lilly and Company (Indianapolis, IN), LipoScience, Inc (Raleigh, NC), Merck Sante (Lyon, France), Novartis Pharmaceuticals Corporation (East Hanover, NJ), and Novo Nordisk, Inc (Princeton, NJ).
Disclosures
Epstein and Quyyumi are equity holders in Firstmark, GenWay Biotech and receive consulting fees. Sikora is an equity holder in Firstmark, GenWay Biotech. Brooks reports significant research grants from Firstmark and Genway Biotech, Inc. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2017;6:e003587 DOI: 10.1161/JAHA.116.003587.)28673897
References
- 1. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker‐Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez‐Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O'Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez‐Ruiz F, Perico N, Phillips D, Pierce K, Pope CA III, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De Leon FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui‐Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilson PW, D'Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. [DOI] [PubMed] [Google Scholar]
- 3. Pencina MJ, D'Agostino RB Sr, Larson MG, Massaro JM, Vasan RS. Predicting the 30‐year risk of cardiovascular disease: the Framingham Heart Study. Circulation. 2009;119:3078–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shlipak MG, Ix JH, Bibbins‐Domingo K, Lin F, Whooley MA. Biomarkers to predict recurrent cardiovascular disease: the Heart and Soul Study. Am J Med. 2008;121:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eapen DJ, Manocha P, Patel RS, Hammadah M, Veledar E, Wassel C, Nanjundappa RA, Sikora S, Malayter D, Wilson PW, Sperling L, Quyyumi AA, Epstein SE. Aggregate risk score based on markers of inflammation, cell stress, and coagulation is an independent predictor of adverse cardiovascular outcomes. J Am Coll Cardiol. 2013;62:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Group BDS , Frye RL, August P, Brooks MM, Hardison RM, Kelsey SF, MacGregor JM, Orchard TJ, Chaitman BR, Genuth SM, Goldberg SH, Hlatky MA, Jones TL, Molitch ME, Nesto RW, Sako EY, Sobel BE. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360:2503–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alderman EL, Kip KE, Whitlow PL, Bashore T, Fortin D, Bourassa MG, Lesperance J, Schwartz L, Stadius M; Bypass Angioplasty Revascularization I . Native coronary disease progression exceeds failed revascularization as cause of angina after five years in the Bypass Angioplasty Revascularization Investigation (BARI). J Am Coll Cardiol. 2004;44:766–774. [DOI] [PubMed] [Google Scholar]
- 8. Beohar N, Davidson CJ, Massaro EM, Srinivas VS, Sansing VV, Zonszein J, Davis AM, Helmy T, Lopes NH, Thomas SB, Brooks MM. The impact of race/ethnicity on baseline characteristics and the burden of coronary atherosclerosis in the Bypass Angioplasty Revascularization Investigation 2 Diabetes trial. Am Heart J. 2011;161:755–763. [DOI] [PubMed] [Google Scholar]
- 9. Arbab‐Zadeh A, Fuster V. The myth of the “Vulnerable Plaque”: transitioning from a focus on individual lesions to atherosclerotic disease burden for coronary artery disease risk assessment. J Am Coll Cardiol. 2015;65:846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sabatine MS, Morrow DA, Jablonski KA, Rice MM, Warnica JW, Domanski MJ, Hsia J, Gersh BJ, Rifai N, Ridker PM, Pfeffer MA, Braunwald E; Investigators P . Prognostic significance of the Centers for Disease Control/American Heart Association high‐sensitivity C‐reactive protein cut points for cardiovascular and other outcomes in patients with stable coronary artery disease. Circulation. 2007;115:1528–1536. [DOI] [PubMed] [Google Scholar]
- 11. Hemingway H, Philipson P, Chen R, Fitzpatrick NK, Damant J, Shipley M, Abrams KR, Moreno S, McAllister KS, Palmer S, Kaski JC, Timmis AD, Hingorani AD. Evaluating the quality of research into a single prognostic biomarker: a systematic review and meta‐analysis of 83 studies of C‐reactive protein in stable coronary artery disease. PLoS Med. 2010;7:e1000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu Q. Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:1547–1559. [DOI] [PubMed] [Google Scholar]
- 13. Zhu J, Quyyumi AA, Wu H, Csako G, Rott D, Zalles‐Ganley A, Ogunmakinwa J, Halcox J, Epstein SE. Increased serum levels of heat shock protein 70 are associated with low risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2003;23:1055–1059. [DOI] [PubMed] [Google Scholar]
- 14. Fowkes FG, Lowe GD, Housley E, Rattray A, Rumley A, Elton RA, MacGregor IR, Dawes J. Cross‐linked fibrin degradation products, progression of peripheral arterial disease, and risk of coronary heart disease. Lancet. 1993;342:84–86. [DOI] [PubMed] [Google Scholar]
- 15. Sobel BE, Hardison RM, Genuth S, Brooks MM, McBane RD III, Schneider DJ, Pratley RE, Huber K, Wolk R, Krishnaswami A, Frye RL; Investigators BD . Profibrinolytic, antithrombotic, and antiinflammatory effects of an insulin‐sensitizing strategy in patients in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Circulation. 2011;124:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long‐term effects of pravastatin on plasma concentration of C‐reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1999;100:230–235. [DOI] [PubMed] [Google Scholar]
- 17. Albert MA, Danielson E, Rifai N, Ridker PM; Investigators P . Effect of statin therapy on C‐reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA. 2001;286:64–70. [DOI] [PubMed] [Google Scholar]
- 18. Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, Gotto AM Jr; Air Force/Texas Coronary Atherosclerosis Prevention Study I . Measurement of C‐reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344:1959–1965. [DOI] [PubMed] [Google Scholar]
- 19. Ridker PM, Rifai N, Pfeffer MA, Sacks FM, Moye LA, Goldman S, Flaker GC, Braunwald E. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1998;98:839–844. [DOI] [PubMed] [Google Scholar]
- 20. Morrow DA, de Lemos JA, Sabatine MS, Wiviott SD, Blazing MA, Shui A, Rifai N, Califf RM, Braunwald E. Clinical relevance of C‐reactive protein during follow‐up of patients with acute coronary syndromes in the Aggrastat‐to‐Zocor Trial. Circulation. 2006;114:281–288. [DOI] [PubMed] [Google Scholar]
- 21. Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, Pfeffer MA, Braunwald E; Pravastatin or Atorvastatin E and Infection Therapy‐Thrombolysis in Myocardial Infarction I . C‐reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28. [DOI] [PubMed] [Google Scholar]
- 22. Ridker PM, Morrow DA, Rose LM, Rifai N, Cannon CP, Braunwald E. Relative efficacy of atorvastatin 80 mg and pravastatin 40 mg in achieving the dual goals of low‐density lipoprotein cholesterol <70 mg/dl and C‐reactive protein <2 mg/l: an analysis of the PROVE‐IT TIMI‐22 trial. J Am Coll Cardiol. 2005;45:1644–1648. [DOI] [PubMed] [Google Scholar]