

Abstract
Background
Gut microbial metabolites have been implicated as novel risk factors for cardiovascular events and premature death. The strength and consistency of associations between blood concentrations of the gut microbial metabolites, trimethylamine‐N‐oxide (TMAO) and its precursors, with major adverse cardiovascular events (MACE) or death have not been comprehensively assessed. We quantified associations of blood concentrations of TMAO and its precursors with risks of MACE and mortality.
Methods and Results
PubMed and Embase databases were searched up, and a total of 19 prospective studies from 16 publications (n=19 256, including 3315 incident cases) with quantitative estimates of the associations of TMAO with the development of MACE or death were included in our main analysis. Multivariate‐adjusted relative risks (RRs) were used when these were available. Elevated concentrations of TMAO were associated with a pooled RR of 1.62 (95% CI, 1.45, 1.80; P heterogeneity=0.2; I2=23.5%) for MACE compared with low TMAO levels, and 1 study of black participants influenced the heterogeneity of the association. After excluding the data of blacks, the RRs were not different according to body mass index, prevalence of diabetes mellitus, history of cardiovascular diseases, and kidney dysfunction. Furthermore, elevated TMAO concentrations were associated with a pooled RR of 1.63 (1.36, 1.95) for all‐cause mortality. Individuals with elevated concentrations of TMAO precursors (l‐carnitine, choline, or betaine) had an approximately 1.3 to 1.4 times higher risk for MACE compared to those with low concentrations.
Conclusions
Elevated concentrations of TMAO and its precursors were associated with increased risks of MACE and all‐cause mortality independently of traditional risk factors.
Keywords: major adverse cardiovascular events, meta‐analysis, risk, trimethylamine N‐oxide
Subject Categories: Cardiovascular Disease, Risk Factors, Epidemiology
Introduction
Gut microbiota has been recently implicated as a novel endocrine organ that plays an important role in regulation of host cardiometabolic function through modulating blood levels of bioactive metabolites.1, 2 Recent studies by Wang and Tang et al reported that circulating concentrations of trimethylamine N‐oxide (TMAO), a metabolite derived from the gut microbiota, were predictive of prevalent cardiovascular diseases (CVDs)1 and future cardiovascular events3 in clinical cohorts. TMAO is a small organic compound, mainly derived from choline (found in foods such as red meat, fish, poultry, and eggs), which is metabolized to produce trimethylamine (TMA) by microbiota1, 4 and then to TMAO by the hepatic enzyme, flavin monooxygenase 3.1, 5
An increasing number of studies have investigated the associations of circulating levels of TMAO and its metabolic precursors, such as l‐carnitine, choline, and betaine, with the risk of major adverse cardiovascular events (MACE) or all‐cause death.1, 3, 4, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26 Whereas these studies largely found that elevated blood concentrations of these TMAO‐related metabolites were associated with increased risks of the MACE outcomes and death, inconsistent associations have also been observed.10, 22 Differences in characteristics and disease status of participants across studies might affect TMAO levels14, 27, 28 and therefore partly explain such inconsistency. In addition, differential adjustment for potential confounders would add variance to the associations. No study has comprehensively analyzed the strength and consistency of associations of TMAO and its precursors with the risk of MACE.
We therefore performed a systematic review and meta‐analysis of prospective studies to quantify the association of blood concentrations of TMAO and its precursors, such as l‐carnitine, choline, or betaine, with the risks of MACE and death. We examined whether various clinical factors, including traditional cardiovascular risk factors, might modify the associations.
Methods
Data Sources and Search Strategy
We followed the standard criteria for conducting meta‐analyses of observational studies and reporting the results.29 The protocol of the present study was registered with the PROSPERO database: CRD42016052185. We searched MEDLINE (PubMed) and Embase up to December 6, 2016 for eligible studies with a search strategy that combined text word and Medical Subject Headings identifying reports relating blood concentrations of TMAO and the MACE. The following terms were used for the MEDLINE search: (trimethylamine n‐oxide [text] OR TMAO [text]) AND (atherosclerosis [text] OR death [text] OR mortality [text] OR stroke [text] OR heart failure [text] OR coronary [text], cardiovascular [text] OR Cerebrovascular Disorders [Mesh] OR Cardiovascular Diseases [Mesh]). Similar search terms were used for the Embase search. The reference lists of identified articles were also scanned to identify any other relevant studies. No language restrictions were applied. For TMAO precursors, eligible prospective studies were identified if they reported relative risks (RRs) and 95% CIs of categories of l‐carnitine, choline, or betaine for the outcome through the search of TMAO. We contacted authors4, 13, 18, 19, 24 to request additional data on number of incident cases, or RRs across categories of TMAO, l‐carnitine, choline, or betaine for the risk of MACE. The term MACE is a composite of clinical events that usually includes endpoints reflecting both “safety” and “effectiveness.”30 The “safety” definition includes death, myocardial infarction (MI), or stroke; and the “effectiveness” definition often includes target vessel revascularization, any repeat revascularization, or rehospitalization.30 There is no standard definition for MACE, and the definition varies by study, whereas most studies include MI and death to define MACE.30 In the current study, MACE was indicated by composites of MI, stroke, heart transplant, heart failure (HF), other ischemic cardiovascular events, or death (either cardiovascular or all‐cause death) as presented in Tables 1 and 2.
Table 1.
Characteristics of Identified Prospective Studies on TMAO Levels and Risk of MACE and Death
| Source | Country (Ethnicitya) | Study Population | Age, Mean, y | TMAO, Mean or Median, μmol/L | TMAO Ranges, μmol/L | Men, % | Total, N | Outcomes Assessed | Events, N | Follow‐up Duration, y |
|---|---|---|---|---|---|---|---|---|---|---|
| Tang et al (2013)3 | US | Patients undergoing elective coronary angiography | 63.0 | 3.7 | 2.4 to 6.2 (IQR) | 64.0 | 4007 | Death, MI, or stroke | 513 | Up to 3 years |
| Koeth et al (2013)4 | US | Patients undergoing elective cardiac evaluation, GeneBank study | 62 | 4.6 | ··· | 70 | 2595 | MACE (composite of death, MI, stroke, and revascularization) | 975 | 3 years |
| Lever et al (2014)7 | New Zealand (European ancestry, 83.0%) | Coronary Disease Cohort Study (CDCS), participants without diabetes mellitus | 68.0 | 4.8 | 3.0 to 29.1 (IQR) | 72.7 | 396 | All‐cause mortality | 62 | 4.96 years (median) |
| Acute MI | 65 | |||||||||
| Admission for HF | 62 | |||||||||
| Unstable angina | 56 | |||||||||
| All cardiovascular disease (CVD) events | 227 | |||||||||
| CDCS, participants with diabetes mellitus | 74.0 | 7.5 | 4.4 to 12.1 (IQR) | 73.4 | 79 | All‐cause mortality | 19 | 4.82 years (median) | ||
| Acute MI | 22 | |||||||||
| Admission for HF | 23 | |||||||||
| Unstable angina | 16 | |||||||||
| All CVD events | 56 | |||||||||
| Tang et al (2014)8 | US | Patients with stable heart failure (HF) undergoing cardiac evaluation | 66.0 | 5.0 | 3.0 to 8.5 (IQR) | 59.0 | 720 | All‐cause mortality | 207 | Up to 5 years |
| Wang et al (2014)9 | US | Patients undergoing elective diagnostic coronary angiography | 63 | 3.7 | 2.4 to 6.2 (IQR) | 64 | 3903 | MACE (death, MI, stroke) | 495 | 3 years |
| Kaysen et al (2015)10 | US (black, 28% whites, 69% of total) | Comprehensive Dialysis Study | 61.8 | 43.0 | 28 to 67 (IQR) | 55.3 | 235 | All‐cause mortality | 132 | 4 years (median) |
| 152 | CVD death or hospitalization | 48 | 2.5 years (median) | |||||||
| Tang et al (2015)11 | US | Patients with CKD who underwent elective diagnostic coronary angiography for cardiac evaluation | 70.0 | 7.9 | 5.2 to 12.4 (IQR) | 48.0 | 521 | All‐cause mortality | 174 | Up to 5 years |
| Patients without CKD who underwent elective diagnostic coronary angiography | 62.0 | 3.4 | 2.3 to 5.3 (IQR) | 66.0 | 3166 | All‐cause mortality | 292 | Up to 5 years | ||
| Tang et al (2015)12 | US | Patients with chronic systolic HF | 57.0 | 5.8 | 3.6 to 12.1 (IQR) | 75.0 | 112 | All‐cause mortality or cardiac transplantation | 40 | Up to 5 years |
| Troseid et al (2015)13 | Norway | Patients with stable HF | 57.0 | 9.2 in patients with dilated cardiomyopathy; 12.1 in patients with stable CAD | 1.2 to 124 (range) | 83.0 | 155 | All‐cause mortality or heart transplantation | 55 | 5.2 years (median) |
| Mueller et al (2015)14 | Austria | Patients who underwent coronary angiography | 63.0 | 1.74 | 0.382 to 3.48 (IQR) | 68.0 | 339 | Cardiac death, MI or stroke | 99 | Up to 8 years |
| Zhu et al (2016)15 | US | GeneBank Study,3 patients who underwent elective coronary angiography | ··· | ··· | ··· | ··· | 4007 | MI or stroke | ··· | Up to 3 years |
| Suzuki et al (2016)16 | UK | Patients with acute HF | 78.0 | 5.6 | 3.4 to 10.5 (IQR); 0.5 to 151.5 (range) | 61.0 | 972 | In‐hospital mortality | 72 | Up to 1 year |
| All‐cause mortality | 268 | Up to 1 year | ||||||||
| Death or rehospitalization because of HF | 384 | Up to 1 year | ||||||||
| Skagen et al (2016)17 | Norway | Patients with carotid artery atherosclerosis | 67.6 | 9.77 | 0.4 to 161.7 (range) | 68.6 | 259 | CVD mortality/non‐CVD mortality | 24/27 | 5.3 years (mean) |
| Missailidis et al (2016)18 | Sweden | Patients with CKD | 55.0 | 53.4 | 9.3, 170.0 (10th, 90th) | 65.0 | 179 | All‐cause mortality | 51 | Up to 5 years |
| Stubbs et al (2016)19 | US (whites, 90.9%; blacks, 7.3%; other, 1.8%) | Patients with CKD, Diabetes Genome Project | 69.7 | 6.9 | 4.8 to 10.9 (IQR); 0.63 to 163.03 (range) | 42.7 | 220 | All‐cause mortality | 36 | Up to 4 years |
| Kim et al (2016)20 | Canada (whites, 88.7%) | Patients with CKD, CanPREDDICT | 68.2 | 20.41 | 12.82 to 32.70 (IQR) | 62.5 | 2529 | Ischemic cardiovascular events | 264 | 3 years |
| Senthong et al (2016)21 | US | Patients with stable coronary artery disease | 63.0 | 3.8 | 2.5 to 6.5 (IQR) | 71.0 | 2235 | All‐cause mortality | 338 | Up to 5 years |
| Shafi et al (2017)22 | US (whites, 35%; black 65%) | Hemodialysis patients | 57.7 | 101.9 (whites: 98.4; blacks: 103.8) | 62 to 124 (whites: 63–120; blacks: 62–125) (IQR); (whites: 6.42–468; blacks: 2.25–682) (ranges) | 43.3 | 1232 (whites: 431; blacks: 801) | Cardiac death | 216 (whites: 96; blacks: 120) | 2.3 years |
| Sudden cardiac death | 124 (whites: 54; blacks: 70) | |||||||||
| 1148 (whites: 388; blacks: 760) | First cardiovascular event or any‐cause death | 626 (whites: 220; blacks: 406) | ||||||||
| 1232 (whites: 431; blacks: 801) | All‐cause mortality | 550 (whites: 217; blacks: 333) | ||||||||
| Robinson‐Cohen et al (2016)23 | US (whites, 64.5%; blacks, 25.5%; Asian/Pacific Islander 2.9%; American Indian/Native Alaskan 2.0%; Other 5.2%) | Patients with CKD, Seattle Kidney Study | 57.3 | 23.5 | >0 to >133 (ranges) | 69.0 | 339 | All‐cause mortality | 45 | 3.3 years (median) |
| Ottiger et al (2016)24 | Switzerland | Community‐acquired pneumonia patients | 72.0 | 3.0 | 1.7 to 5.4 (IQR) | 59.7 | 317 | All‐cause mortality | 143 | 6.1 years |
| Senthong et al (2016)25 | US | Patients with peripheral artery disease | 66 | 4.8 | 2.9 to 8 (IQR) | 66 | 821 | All‐cause mortality | 222 | 5 years |
| Tang et al (2017)26 | US | Patients with type 2 diabetes mellitus who underwent elective diagnostic coronary angiography | 64.4 | 4.4 | 2.8 to 7.7 (IQR) | 58 | 1216 | All‐cause mortality | 227 | 5 years |
| Major adverse cardiac events (death, nonfatal MI, and nonfatal stroke) | 209 | 3 years |
Details on ethnicity were provided if reported. CAD indicates coronary artery disease; CKD, chronic kidney disease; CVD, cardiovascular disease; HF, heart failure; IQR, interquartile range; MACE, major adverse cardiovascular events; MI, myocardial infarction; TMAO, trimethylamine N‐oxide.
Table 2.
RR of MACE According to TMAO Levels in Prospective Studies
| Source | Outcome | Comparison | Model | RR (95% CI) | Adjustment for Covariates |
|---|---|---|---|---|---|
| Tang et al (2013)3 | Death, MI, or stroke | Highest quartile (>6.18 μmol/L) vs lowest | Crude | 2.54 (1.96, 3.28) | NA |
| Multivariatea | 1.43 (1.05, 1.94) | Age, sex, smoking, diabetes mellitus, systolic blood pressure, HDL cholesterol, LDL cholesterol, triglycerides, log‐transformed hs‐CRP, log‐transformed eGFR, myeloperoxidase level, BMI, medication history, and angiographic extent of CAD | |||
| 1 SD | Crude | 1.40 (1.29, 1.51) | NA | ||
| Multivariate | 1.30 (1.20, 1.41) | Traditional risk factors and other baseline covariates | |||
| Koeth et al (2013)4 | MACE | Above median (>4.6 μmol/L) of TMAO and median of carnitine (46.8 μmol/L) vs below median of TMAO and carnitine | Crude | 2.5 (1.8, 3.4) | NA |
| Multivariate | 2.1 (1.5, 2.8) | Age, sex, history of diabetes mellitus, smoking, systolic blood pressure, LDL cholesterol, and HDL cholesterol | |||
| Lever et al (2014), participants without diabetes mellitus7 | All‐cause mortality | Highest quintile (>12.0 μmol/L) vs nonhighest quintile | Crude | 2.7 (1.6, 4.8) | NA |
| Admission for HF | Same as the above | Crudea | 1.9 (1.1, 3.4) | NA | |
| Lever et al (2014), participants with diabetes mellitus7 | All‐cause mortality | Highest quintile (>12.0 μmol/L) vs nonhighest quintile | Crude | 2.7 (1.1, 7.1) | NA |
| Acute MI | Same as the above | Crude | 4.0 (1.6, 9.8) | NA | |
| Admission for HF | Same as the above | Crude | 4.6 (2.0, 10.7) | NA | |
| Unstable angina | Same as the above | Crude | 9.1 (2.8, 29.7) | NA | |
| All CVD events | Same as the above | Crudea | 2.0 (1.1, 3.6) | NA | |
| Tang et al (2014)8 | All‐cause mortality | Highest quartile (≥8.51 μmol/L) vs lowest | Crude | 3.42 (2.24, 5.23) | NA |
| Multivariatea | 1.85 (1.14, 3.00) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, log‐transformed BNP, log‐transformed eGFR, and log‐transformed hs‐CRP | |||
| log‐transformed per 1 SD | Multivariate | 1.18 (1.06, 1.31) | Traditional cardiac risk factors | ||
| Wang et al (2014)9 | MACE | Above median (>3.7 μmol/L) of TMAO and median of choline (9.8 μmol/L) vs below median of TMAO and median of choline | Crude | 2.1 (1.7, 2.7) | NA |
| Multivariate | 1.6 (1.2, 2.0) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, and hs‐CRP | |||
| Above median (>3.7 μmol/L) of TMAO and median of betaine (41.1 μmol/L) vs below median of TMAO and median of betaine | Crude | 1.9 (1.5, 2.4) | NA | ||
| Multivariate | 1.6 (1.2, 2.0) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, and hs‐CRP | |||
| Kaysen et al (2015)10 | All‐cause mortality | Highest quartile (66.6–184 μmol/L) vs lowest | Crude | 0.61 (0.38, 0.97) | NA |
| Multivariate | 1.14 (0.67, 1.93) | Age, sex, race, BMI, diabetes mellitus, log‐transformed CRP, serum prealbumin, and serum albumin | |||
| Per log‐transformed | Crude | 0.84 (0.65, 1.09) | NA | ||
| Cardiovascular death or hospitalization | Highest quartile (>62 μmol/L) vs lowest | Crude | 0.71 (0.32, 1.59) | NA | |
| Highest quartile (66.6–184 μmol/L) vs lowest | Multivariatea | 0.92 (0.40, 2.10) | Race, diabetes mellitus, serum prealbumin | ||
| log‐transformed | Crude | 0.88 (0.57, 1.35) | NA | ||
| Tang et al (2015)11 | All‐cause mortality, CKD cohort | Highest quartile (≥12.4 μmol/L) vs lowest | Crude | 2.76 (1.74, 4.37) | NA |
| Multivariatea | 1.93 (1.13, 3.29) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, log‐transformed eGFR, and log‐transformed hs‐CRP | |||
| All‐cause mortality, non‐CKD cohort | Highest quartile (≥5.3 μmol/L) vs lowest | Crude | 2.21 (1.57, 3.12) | NA | |
| Multivariatea | 1.47 (1.02, 2.12) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, log‐transformed eGFR, and log‐transformed hs‐CRP | |||
| Tang et al (2015)12 | All‐cause mortality or cardiac transplantation | log‐transformed per 1 SD (0.99 μmol/L) | Crude | 1.48 (1.10, 1.96) | NA |
| Multivariate | 1.46 (1.03, 2.14) | Age, eGFR, mitral E/septal Ea, and NT‐proBNP | |||
| Troseid et al (2015)13 | All‐cause mortality or heart transplantation | Highest tertile vs remaining groups | Crude | 2.24 (1.28, 3.92) | NA |
| Multivariate | 1.79 (0.90, 1.79) | HF severity, age, hypertension, type 2 diabetes mellitus, HF etiology, eGFR, CRP, and NT‐proBNP | |||
| Highest tertile (12.8–124 μmol/L) vs lowest | Crudea | 1.76 (0.93, 3.33)b | NA | ||
| Mueller et al (2015)14 | Cardiac death, MI, or stroke | 1 μmol/L | Multivariate | 1.01 (0.984, 1.04) | Age, sex, smoking, metabolic syndrome, HbA1c, CRP, eGFR |
| Zhu et al (2016)15 | MI or stroke | Highest quartile (6.18–312 μmol/L) vs lowest | Multivariate | 1.64 (1.12, 2.39) | Age, sex, BMI, systolic blood pressure, LDL cholesterol, HDL cholesterol, triglycerides, estimated creatinine clearance, smoking, diabetes mellitus, medication use, and history of CVD |
| Suzuki et al (2016)16 | Death or rehospitalization because of HF | Highest tertile (8.2–151.5 μmol/L) vs lowest | Multivariatea | 2.12 (1.54, 2.93) | Age, sex, history of HF, IHD, hypertension, diabetes mellitus, HF severity, current smoking, edema, atrial fibrillation, systolic blood pressure, heart rate, Hb, respiratory rate, sodium, urea, eGFR, NT‐proBNP |
| log‐transformed per 1 SD | Crude | 1.33 (1.20, 1.46) | NA | ||
| Multivariate | 1.18 (1.05, 1.33) | Age, sex, history of HF, IHD, hypertension, diabetes mellitus, HF severity, current smoking, edema, atrial fibrillation, systolic blood pressure, heart rate, Hb, respiratory rate, sodium, urea, NT‐proBNP | |||
| All‐cause mortality | log‐transformed per 1 SD | Crude | 1.35 (1.21, 1.51) | NA | |
| Multivariate | 1.16 (1.01, 1.33) | Age, sex, history of HF, IHD, hypertension, diabetes mellitus, HF severity, current smoking, edema, atrial fibrillation, systolic blood pressure, heart rate, Hb, respiratory rate, sodium, urea, and NT‐proBNP | |||
| Skagen et al (2016)17 | Cardiovascular (MI or stroke) death | log‐transformed per 1 SD | Crude | 1.81 (1.29, 2.53) | NA |
| Multivariate | 1.38 (0.91, 2.08) | Age and eGFR | |||
| Missailidis et al (2016)18 | All‐cause mortality | Top 2 tertiles (32.2 or more μmol/L) vs lowest | Crude | 6.29 (2.67, 14.8) | NA |
| Multivariate | 6.68 (2.33, 19.1) | Age, sex, diabetes mellitus, and hs‐CRP | |||
| Multivariatea | 4.32 (1.32, 14.2) | Age, sex, diabetes mellitus, and hs‐CRP and GFR | |||
| Stubbs et al (2016)19 | All‐cause mortality | Highest tertile (9.26–163.03 μmol/L) vs lowest | Crudea | 1.95 (0.91, 4.17) | NA |
| 10 μmol/L | Crude | 1.19 (1.10, 1.29) | NA | ||
| Multivariate | 1.26 (1.13, 1.40) | Age, sex, race, smoking, BMI, hypertension, diabetes mellitus, eGFR, CKD stage, triglycerides, cholesterol, history of percutaneous intervention, histories of coronary artery bypass grafting, MI, cerebrovascular accident, peripheral vascular disease and congestive HF | |||
| Kim et al (2016)20 | Ischemic cardiovascular events | Highest quartile (>32.67 μmol/L) vs lowest | Crude | 2.33 (1.63, 3.33) | NA |
| Multivariatea | 1.37 (0.91, 2.06) | Age, sex, race, diabetes mellitus, cardiovascular comorbidities at baseline, systolic blood pressure, total cholesterol, HDL cholesterol, smoking, medication use, eGFR, ln‐transformed ACR and Hb | |||
| Natural log‐transformed per 1 SD | Crude | 1.45 (1.28, 1.64) | NA | ||
| Multivariate | 1.24 (1.07, 1.43) | Age, sex, race, diabetes mellitus, cardiovascular comorbidities at baseline, systolic blood pressure, total cholesterol, HDL cholesterol, smoking, medication use, eGFR, ln‐transformed ACR and Hb | |||
| Senthong et al (2016)21 | All‐cause mortality | Highest quartile (median, 9.7 μmol/L) vs lowest | Crude | 3.90 (2.78, 5.48) | NA |
| Multivariatea | 1.71 (1.11, 2.61) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, medication use, number of stenotic vessels, log‐transformed hs‐CRP, log‐transformed myeloperoxidase, log‐transformed eGFR, and log‐transformed BNP | |||
| Shafi et al (2017)22 | Cardiac death, white | Highest quintile (135–468 μmol/L) vs lowest quintile | Crude | 1.82 (1.23, 2.69) | NA |
| Multivariatea | 1.78 (1.12, 2.82) | Age, sex, index of coexisting disease severity score, cause of end‐stage renal disease, BMI, systolic blood pressure, albumin, and relative volume removed on dialysis and residual kidney function | |||
| Sudden cardiac death, white | Same as the above | Crude | 2.98 (1.38, 6.44) | NA | |
| Multivariate | 2.76 (1.22, 6.24) | Same | |||
| First cardiovascular event or any‐cause death, white | Same as the above | Crude | 1.53 (1.01, 2.32) | NA | |
| Multivariate | 1.45 (0.99, 2.15) | Same | |||
| Any‐cause mortality, white | Same as the above | Crude | 1.40 (0.98, 2.00) | NA | |
| Multivariate | 1.50 (1.03, 2.18) | Same | |||
| Cardiac death, black | Highest quintile (135–682 μmol/L) vs lowest quintile | Crude | 0.73 (0.49, 1.09) | NA | |
| Multivariatea | 0.78 (0.51, 1.18) | Same | |||
| Sudden cardiac death, black | Same as the above | Crude | 0.73 (0.48, 1.11) | NA | |
| Multivariate | 0.80 (0.51, 1.25) | Same | |||
| First cardiovascular event or any‐cause death, black | Same as the above | Crude | 0.99 (0.70, 1.40) | NA | |
| Multivariate | 1.03 (0.80, 1.31) | Same | |||
| Any‐cause mortality, black | Same as the above | Crude | 0.85 (0.65, 1.11) | NA | |
| Multivariate | 0.89 (0.66, 1.20) | Same | |||
| Robinson‐Cohen et al (2016)23 | All‐cause mortality | Highest tertile (1.71 μg/mL) vs lowest tertile | Multivariatea | 1.25 (0.48, 3.28) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, log‐transformed CRP, log‐transformed eGFR |
| Ottiger et al (2016)24 | All‐cause mortality | Highest quartile (median, 8.9 μmol/L) vs lowest quartile | Crude | 3.5 (2.1, 5.8)b | NA |
| Multivariatea | 1.9 (1.1, 3.3)b | CAD, congestive heart failure, cerebrovascular disease, peripheral artery occlusive disease, diabetes mellitus, CKD, neoplastic disease and chronic obstructive pulmonary disease | |||
| log‐transformed | Crude | 2.3 (1.7, 3.3) | NA | ||
| Multivariate | 1.6 (1.01, 2.4) | CAD, congestive heart failure, cerebrovascular disease, peripheral artery occlusive disease, diabetes mellitus, CKD, neoplastic disease, and chronic obstructive pulmonary disease | |||
| Senthong et al (2016)25 | All‐cause mortality | Highest quartile (≥8.01 μmol/L) vs lowest quartile | Crude | 2.69 (1.82, 3.97) | NA |
| Multivariatea | 1.59 (1.03, 2.45) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, log‐transformed hs‐CRP, and log‐transformed eGFR | |||
| Multivariate | 1.88 (1.21, 2.92) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, history of CAD, statin use, apolipoprotein A1, apolipoprotein B, log‐transformed myeloperoxidase, and log‐transformed hs‐CRP | |||
| log‐transformed per 1 SD | Crude | 1.53 (1.35, 1.74) | NA | ||
| Multivariate | 1.26 (1.03, 1.53) | Age, sex, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, diabetes mellitus, log‐transformed hs‐CRP, and log‐transformed eGFR | |||
| Tang et al (2017)26 | All‐cause mortality | Highest tertile (≥6.3 μg/mL) vs lowest tertile | Crude | 3.63 (2.53, 5.21) | NA |
| Multivariate | 1.85 (1.21, 2.84) | Age, sex, history of CVD, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, hs‐CRP, log‐transformed HbA1c, log‐transformed eGFR, log‐transformed BMI, and history of HF | |||
| MACE | Highest tertile (≥6.3 μg/mL) vs lowest tertile | Crude | 3.03 (2.08, 4.42) | NA | |
| Multivariatea | 1.94 (1.23, 3.05) | Age, sex, history of CVD, systolic blood pressure, LDL cholesterol, HDL cholesterol, smoking, hs‐CRP, log‐transformed HbA1c, log‐transformed eGFR, log‐transformed BMI, and history of HF |
ACR indicates albumin‐to‐creatinine ratio; BMI, body mass index; BNP, B‐type natriuretic peptide; CAD, coronary artery disease; CKD, chronic kidney disease; CVD, cardiovascular disease; eGFR, estimated glomerular filtration rate; Hb, hemoglobin; HbA1c, glycosylated hemoglobin; HDL, high‐density lipoprotein; HF, heart failure; hs‐CRP, high‐sensitivity C‐reactive protein; IHD, ischemic heart disease; LDL, low‐density lipoprotein; MACE, major adverse cardiovascular events; MI, myocardial infarction; NA, not applicable; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; RR, relative risk; TMAO, trimethylamine N‐oxide.
Data used for main analysis using 19 data points.
Data from authors.
Study Selection, Data Extraction, and Quality Assessment
We included studies in this meta‐analysis if they satisfied the following criteria: the study design was prospective, the exposure of interest was blood concentrations of TMAO, the outcome was MACE or death, and the investigators reported RRs with 95% CIs for quantitative categories of TMAO levels. Also, studies with available data on an RR and 95% CIs per 1 μmol/L or per 1 SD log‐transformed TMAO for the MACE or death were included to perform the dose‐response analysis. We excluded reviews, editorials, comments, and nonhuman studies. Studies of other exposures and other disease outcomes, case‐control studies, and cross‐sectional studies that examined associations of TMAO and markers of atherosclerosis were also excluded. Two reviewers (Y.H. and W.M.) independently extracted data on study design and population characteristics and compared results to ensure consistency. Inconsistencies in data were resolved by a third reviewer (L.Q.). In detail, we extracted the following data from each study: the first author's last name, publication year, country or region where the study was conducted, ethnicity, number of participants and cases, follow‐up time, mean or median TMAO concentrations among total study participants or among the highest category for estimating the risk, ranges or interquartile ranges (IQRs) of TMAO, and other characteristics of the study. The original articles used tertiles, quartiles, or quintiles as categories for TMAO levels. We extracted number of cases/N, exposure levels, RRs and 95% CIs for categories of TMAO, and covariates in adjusted models. Quality assessment was performed using to the Newcastle–Ottawa quality assessment scale.31 We also extracted RRs and 95% CIs across categories of TMAO precursors (l‐carnitine, choline, or betaine) for the outcome.
Main Analysis and Secondary Analyses
We performed a main analysis to estimate a pooled RR of MACE associated with high concentrations of TMAO in comparison with low concentrations. Also, we calculated a pooled RR of elevated TMAO concentrations for all‐cause mortality alone. The high TMAO group was indicated by the highest tertile,13, 16, 19, 23, 26 the top 2 tertiles18 (because a RR of the top 2 tertiles vs the lowest tertile was presented in the original study), the highest quartile,1 or the highest quintile.7, 22 Results stratified by disease status at baseline or ethnicity were treated as 2 separate data points.7, 11, 22 We performed a sensitivity analysis to check whether use of the publications from the same study group would not alter our results. For studies reporting RRs for multiple outcomes (such as cardiovascular outcomes and all‐cause mortality),7, 10, 22, 26 we used RRs for the vascular outcomes in our main analysis, whereas we performed a sensitivity analysis when we used RRs for all‐cause mortality. A pooled RR for MACE without including all‐cause mortality was also calculated. We explored heterogeneity of the main results according to study characteristics such as length of follow‐up, mean (median) age, proportion of smokers, mean or median TMAO levels among the total participants, mean body mass index (BMI), degree of kidney dysfunction, prevalence of diabetes mellitus, or past cardiovascular histories at a baseline examination.
For the analysis of TMAO precursors and risk of MACE, we calculated a pooled RR of elevated concentrations of betaine (highest in comparison with the lowest)7, 9, 13, 18, 26 and that of elevated concentrations of either l‐carnitine or choline.4, 9, 13, 18, 26 Because both choline and l‐carnitine are metabolized by intestinal bacteria to induce TMA, and then further metabolized to TMAO,1, 4 therefore l‐carnitine and choline were combined in our analysis.
Statistical Analysis
We used unadjusted or multivariable‐adjusted RRs and 95% CIs that were reported in the original articles and calculated log‐RRs and log‐standard error for performing our analysis. If studies reported several multivariate‐adjusted RRs, we used the effect estimate that was most fully adjusted for potential confounders, except for 2 studies for which we could not calculate the SE of its logarithm in the final model.13, 20 Heterogeneity was assessed by the Cochran Q test and I2 statistic; low, moderate, and high I2 values were considered to be 25%, 50%, and 75%, respectively.32, 33 We used a fixed‐effect model, using the method of Mantel and Haenszel when there was no significant heterogeneity, and a random effect model when heterogeneity was significant.34 The cutpoint of P value 0.05 was used for assessing significance of heterogeneity. We assessed publication bias by using Begg's and Egger's tests and visual inspection of the funnel plot if 10 or more studies were available.35
For the dose‐response analysis of TMAO concentrations and risk of MACE, we used RRs and 95% CIs per 1 μmol/L or per 1 SD log‐transformed TMAO for the outcome. We also estimated dose‐response associations based on data for categories of TMAO levels on median dose, number of cases and participants, and effect estimates with corresponding SEs using the generalized least‐squares for trend estimation method of Greenland and Longnecker.36 If medians for categories of TMAO levels were not reported, approximate medians were estimated using the midpoint of the lower and upper bounds. For categories without upper limit, median values were defined as 1.5 times the lower limit of the category. An original study reported data separately for whites and blacks; a nonlinear association of TMAO and cardiovascular events was shown in blacks.22 Therefore, we only included data in whites in our dose‐response analysis. Stratified analysis was performed using meta‐regression. All analyses were conducted with Stata software (version 14; StataCorp LP, College Station, TX).
Results
Literature Flow
We identified 24 potentially eligible articles, including 2 cross‐sectional studies and 22 prospective studies (Figure 1). We also reviewed details of the cross‐sectional studies (Tables S1 and S2), whereas these were not included in the meta‐analysis. The 22 publications were enrolled from the United States (14 publications), Canada (1 publication), New Zealand, (1 publication), UK (1 publication), Switzerland (1 publication), Sweden (1 publication), Austria (1 publication), and Norway (2 publications). The mean or median value of TMAO ranged from 1.74 to 103.8 μmol/L across the 22 prospective studies listed in Table 1. One study14 introduced a newly developed method for the measurement of TMAO showing the lowest value of TMAO. Of the 22 studies, 2 only investigated combinations of TMAO and choline, betaine,9 or l‐carnitine4; thus we did not include the 2 studies4, 9 in the analysis for TMAO, although these studies were eligible for the analysis of TMAO precursors and risk of MACE. For publications from the same study group, we treated as different data if the number of study participants and outcome measurements were not completely the same. An exclusion of publications from the same group did not alter our results. Two studies3, 15 included the same number of total study participants from the same cohort so that we only used data from the first publication.3 We observed fundamentally similar results when we used data from the other study.15 Subsequently, the remaining 19 articles published from 2013 to 2017 assessed the RRs of TMAO (either as a categorical or continuous variable) and risk of MACE. All of the studies included clinical cohorts, including patients at higher cardiovascular risk, that is, enriched for baseline heart disease,3, 8, 12, 13, 14, 15, 16, 17, 21 kidney disease,10, 11, 18, 19, 20, 22, 23 peripheral artery disease,25 or diabetes mellitus.7, 26 Of the 19 publications, a total of 16 publications, including 19 data points, were eligible for our main analysis to calculate a pooled RR of elevated TMAO levels as compared with low levels3, 7, 8, 10, 11, 13, 16, 18, 19, 20, 21, 22, 23, 24, 25, 26 (Table 1 and Figure 1).
Figure 1.
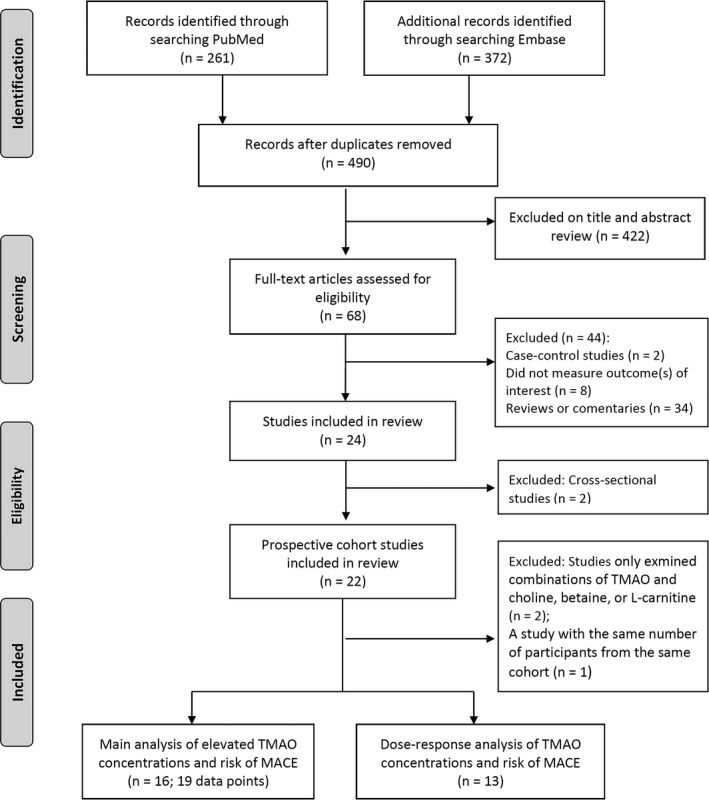
Selection of studies for meta‐analysis. MACE indicates major adverse cardiovascular events; TMAO, trimethylamine N‐oxide.
The RRs for MACE according to TMAO levels in their original studies are summarized in Table 2. Among the prospective studies, 5 studies10, 13, 19, 20, 23 did not find a significantly elevated RR in the highest category of TMAO levels as compared with the lowest category, in either unadjusted or multivariate‐adjusted models among total participants. One study showed a significant association of high TMAO levels and elevated risk of outcomes in whites but not in blacks.22 Also, another 3 studies did not show a significantly elevated RR when TMAO was analyzed as a continuous variable in model.10, 14, 17 In most original studies, RRs were carefully adjusted with various traditional cardiovascular risk factors as shown in Table 2. Of the total 22 publications, multivariate‐adjusted RRs were available for 21 (95%) publications; of the 19 data points used for the main analysis, 79% (n=15) were multivariate‐adjusted data.
TMAO Levels With the Risk of MACE and All‐Cause Mortality
Our meta‐analysis of elevated TMAO levels and the risk of MACE enrolled 19 256 participants and 3315 incident cases from the 19 data points. The pooled RR of elevated TMAO levels for the development of MACE as compared to low TMAO levels using the fixed‐effect model was 1.62 (95% CI, 1.45, 1.80; P<0.001; P heterogeneity=0.2; I2=23.5%; Figure 2A). The pooled RR using the random‐effect model was 1.62 (95% CI, 1.43 1.85). The Begg and Egger regression tests showed no substantial publication bias (P>0.1 for both tests). Of the 19 data points, the study in blacks by Shafi et al22 affected the heterogeneity; removal of the data in blacks22 resulted in a pooled RR of 1.70 (95% CI, 1.52, 1.91; P<0.001; P heterogeneity=0.9; I2=0%; fixed‐effect model; Figure S1A). In results of a sensitivity analysis omitting 1 study at a time and calculating the pooled RRs for the remainder of 18 studies (the data in blacks were not included), the pooled RRs ranged from 1.65 (95% CI, 1.47, 1.86) to 1.75 (95% CI, 1.55, 1.98), and no other study was identified as an influential outlier across the 18 studies (P heterogeneity ranged 0.8–0.9; I2 was consistently 0%). When we performed an analysis further omitting data from the same group, the pooled RR of high TMAO levels for the development of MACE was 1.73 (95% CI, 1.50, 2.00; P heterogeneity=0.3; I2=15.4%). Of the 18 data points, 4 were unadjusted and 14 were multivariate adjusted in the original studies; when we only included multivariate‐adjusted data, the pooled RR was 1.68 (95% CI, 1.49, 1.89; P<0.001; P heterogeneity=0.7; I2=0%). Because we primarily used RRs for the cardiovascular outcomes when there were multiple outcomes presented,7, 10, 22, 26 we also performed a sensitivity analysis for the 18 data when we replaced RRs for all‐cause mortality in these publications7, 10, 22, 26; the pooled RR was 1.69 (95% CI, 1.52, 1.89; P<0.001; P heterogeneity=0.6; I2=0%). In results of further sensitivity analysis removing 12 studies that included RRs for any death or all‐cause mortality, a pooled RR of elevated levels of TMAO for MACE (MACE: CVD events, admission for HF, cardiovascular death/hospitalization, ischemic cardiovascular events, cardiac death, or MACE) was 1.66 (95% CI, 1.35, 2.05; P heterogeneity= 0.6; I2=0%).
Figure 2.
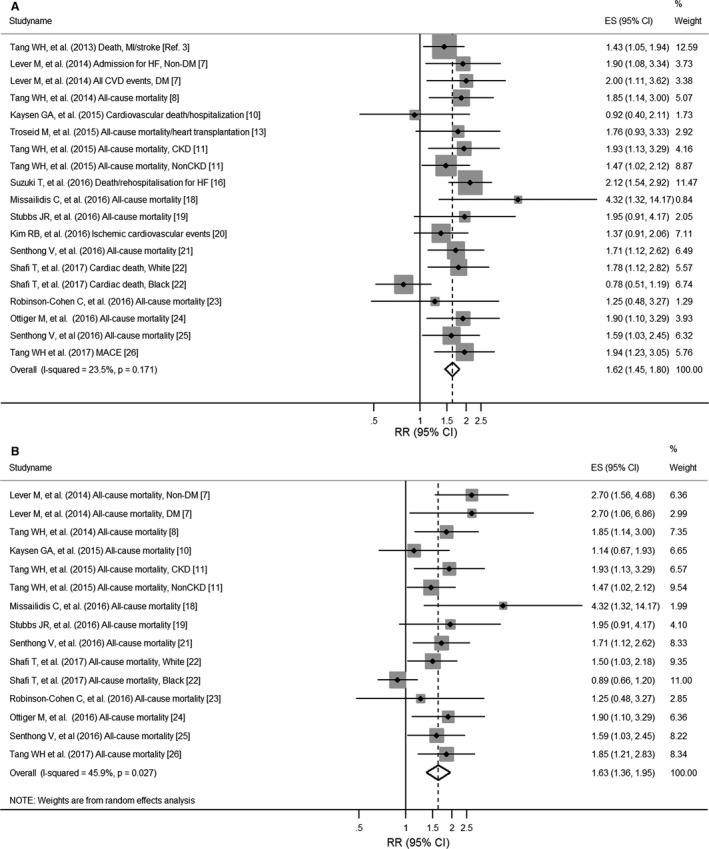
Pooled relative risks of high trimethylamine N‐oxide (TMAO) levels for the risk of major adverse clinical events/death (A) and all‐cause mortality (B). Dashed lines represent the overall effect, and gray boxes represent weight. ES indicates effect size; RR, relative risk.
For calculating the risk for only all‐cause mortality, 15 data from 12 articles7, 8, 10, 11, 18, 19, 21, 22, 23, 24, 25, 26 were available, including 2498 deaths among 11 676 participants. Overall, elevated concentrations of TMAO were significantly associated with an increased risk of all‐cause mortality (pooled RR, 1.63; 95% CI, 1.36, 1.95; P heterogeneity=0.027; I2=45.9% using random‐effect model; Figure 2B). Again, after removal of the data in blacks, there was no heterogeneity across the studies (pooled RR, 1.72; 95% CI, 1.50, 1.97; P heterogeneity=0.7; I2=0%; Figure S1B).
Stratified Analyses
We then investigated potential sources of heterogeneity in stratified analyses using the metaregression analysis based on the 18 data (4 were unadjusted and 14 were multivariate adjusted) that were used for the main analysis without the data in blacks (Table 3). In the stratified analysis using a cut‐off value of median/mean TMAO value of 5.0 μmol/L (lower 39% of the total 18 studies), there was no heterogeneity between <5.0 or ≥5.0 μmol/L of TMAO concentrations at baseline. In addition, there were 5 studies with extremely high mean/median TMAO values (>20 μmol/L). We also performed a stratified analysis using a cut‐off value of 8.0 μmol/L. We observed similarly increased risks of MACE regardless of studies with <8.0 (range, 3.0–7.9) or ≥8.0 (range, 20.41–98.4) μmol/L mean/median TMAO levels. Again, there was no heterogeneity between low or high TMAO concentrations at baseline. The association between elevated TMAO levels and the risk of MACE was consistently observed across strata of age, BMI at baseline, smoking, degree of kidney function, prevalence of CVD or diabetes mellitus at a baseline examination, the follow‐up duration, and whether kidney function markers, lipids, blood pressure, or C‐reactive protein (CRP) levels were controlled for in models. Associations appeared to be stronger among studies with participants who had generally lower BMI, or lower prevalence of smoking, although such differences did not attain statistical significance.
Table 3.
RRs of High TMAO for Major Cardiovascular Events According to Study Characteristics
| Characteristics | N of Total Studies [Adjusted data] | RR (95% CI) | P for Interaction |
|---|---|---|---|
| Age, y | |||
| <65 | 9 [8] | 1.59 (1.34, 1.90) | 0.3 |
| ≥65 | 9 [6] | 1.82 (1.53, 2.16) | |
| Body mass index | |||
| <27.0 kg/m2 | 5 [3] | 1.91 (1.42, 2.58) | 0.2 |
| ≥27.0 kg/m2 | 7 [5] | 1.51 (1.22, 1.87) | |
| Smoking habit, yes | |||
| <30% | 4 [3] | 2.12 (1.58, 2.84) | 0.1 |
| ≥30% | 7 [6] | 1.61 (1.34, 1.94) | |
| TMAO levels at baseline (average TMAO) | |||
| (a) <5.0 μmol/L (4.0 μmol/L) | 7 [6] | 1.62 (1.37, 1.92) | 0.4 |
| ≥5.0 μmol/L (25.4 μmol/L) | 11 [8] | 1.79 (1.51, 2.14) | |
| (b) <8.0 μmol/L (5.3 μmol/L) | 13 [9] | 1.75 (1.53, 2.00) | 0.3 |
| ≥8.0 μmol/L (47.7 μmol/L) | 5 [5] | 1.51 (1.13, 2.02) | |
| Kidney function | |||
| High (eGFR ≥60 mL/min per 1.73 m2) | 10 [7] | 1.67 (1.43, 1.94) | 0.6 |
| Low (eGFR <60 mL/min per 1.73 m2) | 8 [7] | 1.77 (1.45, 2.17) | |
| Controlling for eGFR or other renal function markers, or albuminuria in models | |||
| No | 5 [1] | 1.74 (1.26, 2.38) | 0.9 |
| Yes | 13 [13] | 1.70 (1.49, 1.94) | |
| Controlling for serum cholesterol levels or use of cholesterol lowering medications in models | |||
| No | 9 [5] | 1.92 (1.57, 2.34) | 0.1 |
| Yes | 9 [9] | 1.58 (1.36, 1.85) | |
| Controlling for hs‐CRP or CRP in models | |||
| No | 9 [5] | 1.78 (1.49, 2.14) | 0.5 |
| Yes | 9 [9] | 1.64 (1.39, 1.94) | |
| Controlling for blood pressure measurements, hypertension, or use of antihypertensive medications in models | |||
| No | 8 [4] | 1.83 (1.44, 2.32) | 0.5 |
| Yes | 10 [10] | 1.66 (1.44, 1.92) | |
| Prevalence of diabetes mellitus | |||
| <40% | 8 [6] | 1.73 (1.47, 2.04) | 0.8 |
| ≥40% | 10 [8] | 1.68 (1.40, 2.01) | |
| Prevalence of individuals with cardiovascular disease histories at baseline | |||
| <40% | 6 [4] | 1.61 (1.22, 2.13) | 0.7 |
| ≥40% | 9 [9] | 1.72 (1.48, 1.99) | |
| Follow‐up time | |||
| <5 years | 10 [7] | 1.69 (1.44, 1.98) | 0.9 |
| ≥5 years | 8 [7] | 1.73 (1.43, 2.09) | |
CRP indicates C‐reactive protein; eGFR, estimated glomerular filtration rate. RR, relative risks; TMAO, trimethylamine N‐oxide.
Dose‐Response Analysis
We further investigated whether there was a dose‐response relationship between TMAO concentrations and the risk for MACE (Table 4). The pooled unadjusted RR was 1.05 (95% CI, 1.03, 1.07) per 1‐μmol/L increment in TMAO concentrations. Although the RR was attenuated, it was significantly elevated with a pooled adjusted RR of 1.02 (1.01, 1.03) using data in multivariate‐adjusted models. One study14 introduced a newly developed method for the measurement of TMAO, and their IQRs of TMAO were lower than other studies, so that we calculated a RR without data from the study. Results of the analysis showed a similar pooled adjusted RR per 1‐μmol/L increment in TMAO of 1.02 (1.01, 1.04). We also analyzed the dose‐response relationship on log‐transformed TMAO levels; and the pooled RR per 1‐SD increment of log‐transformed TMAO was 1.43 (95% CI, 1.34, 1.52) in the unadjusted model or 1.21 (95% CI, 1.14, 1.29) in the adjusted model, respectively.
Table 4.
Pooled RRs Per 1 μmol/L or 1 SD Log‐Transformed Increment of TMAO for Major Adverse Cardiovascular Events
| Variables | N | Study ID (Reference) | RR (95% CI) | I2 | Heterogeneity P Value |
|---|---|---|---|---|---|
| 1 μmol/L increment of TMAO, unadjusted | 9 | 11, 13, 19, 20, 22, 24, 25, 26 | 1.05 (1.03, 1.07) | 95.0% | <0.001 |
| 1 μmol/L increment of TMAO, adjusted | 9 | 11, 14, 19, 20, 22, 24, 25, 26 | 1.02 (1.01, 1.03) | 81.7% | <0.001 |
| 1 μmol/L increment of TMAO, adjusteda | 8 | 11, 19, 20, 22, 24, 25, 26 | 1.02 (1.01, 1.04) | 83.9% | <0.001 |
| 1 SD increment of log‐transformed TMAO, unadjusted | 5 | 12, 16, 17, 20, 25 | 1.43 (1.34, 1.52) | 22.7% | 0.3 |
| 1 SD increment of log‐transformed TMAO, adjusted | 6 | 8, 12, 16, 17, 20, 25 | 1.21 (1.14, 1.29) | 0% | 0.8 |
Without a study of reference.14 RR indicates relative risks; TMAO, trimethylamine N‐oxide.
Associations of TMAO Precursors With the Risk of MACE
Finally, we conducted analyses on the associations between circulating levels of TMAO precursors and the risk and MACE using data from 6 publications4, 7, 9, 13, 18, 26 (Figure 3). For the association between elevated l‐carnitine or choline concentrations and the risk of MACE, the summary RR was 1.26 (95% CI, 1.10, 1.44; P heterogeneity=0.9; I2=0%; Figure 3A). For the association of elevated betaine concentrations with the risk of MACE, the pooled RR was 1.43 (95% CI, 1.19, 1.73; P heterogeneity=0.4; I2=6.4%; Figure 3B).
Figure 3.
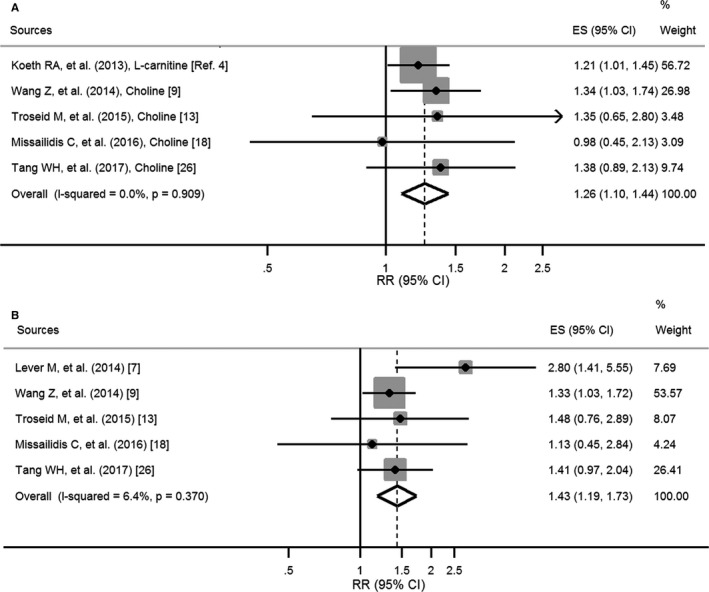
Pooled relative risks of elevated concentrations of l‐carnitine or choline (A) and betaine (B) for major adverse cardiovascular events/death. Dashed lines represent the overall effect, and gray boxes represent weight. ES indicates effect size; RR, relative risk.
Discussion
Our meta‐analysis of data from prospective studies provides quantitative pooled estimates of the associations of circulating TMAO level with the incidence of MACE and all‐cause death. As compared to participants with low TMAO levels, those with high levels had a 62% increased risk for the development of MACE and a 63% increased risk for all‐cause death. In addition, elevated concentrations of TMAO were associated with 1.7‐fold increased risks for MACE and all‐cause mortality compared with low TMAO levels when we estimated the RRs without using data in blacks. The associations did not significantly differ according to the past histories of CVD, prevalence of diabetes mellitus, kidney dysfunction, follow‐up duration, or TMAO levels at baseline. However, no prospective cohort studies were available in the population at general risk. In addition, we found that the association between blood TMAO levels and development of MACE was dose dependent. Moreover, our quantitative estimates for the precursors of TMAO and the risk of MACE/death revealed that individuals with elevated concentrations of l‐carnitine, choline, or betaine had approximately of 1.3 to 1.4 times higher risk for MACE compared to those with low concentrations. Our results indicated that the relations of elevated TMAO and its precursors with MACE and all‐cause death were independent of conventional risk factors, such as kidney dysfunction, diabetes mellitus, and obesity.
Strengths and Limitations
Our study has several strengths. The included original studies were all prospective and therefore our analysis minimized the likelihood of reverse causation. We carefully assessed the influence of several potential confounders and traditional risk factors for CVDs. In addition, our sensitivity analyses indicated that the results were not affected by varying definitions of outcomes or elevated TMAO levels. Several limitations warrant consideration. First, no data were available among general populations for our analysis. The included studies have been conducted in clinical cohorts including patients with pre‐existing cardiovascular risk, and thus we cannot determine whether results will be similar in lower‐risk populations. Second, the definition of elevated TMAO levels was difference across individual studies. Third, similar to other observational studies, we could not exclude the possibility of residual confounding attributed to unmeasured factors. Fourth, the gut microbiota are affected by environmental factors37, 38 such as dietary intakes,39, 40, 41 which may, in turn, influence blood levels of TMAO and its precursors.4 However, detailed assessment of dietary intakes was not available in all the included studies; this limited our abilities to investigate the potential roles of dietary factors in the associations of gut microbiota metabolites and the outcomes. Fifth, most of the study participants were from Europe and the United States and included mainly (approximately of 65–90% of total study participants) whites,10, 19, 20, 23 so that our results might not be applicable to other race/ethnic groups such as Asians and Africans. Furthermore, TMAO levels were measured at 1 time point, which might not capture the long‐term levels of gut microbiota metabolites. Finally, further studies, especially clinical trials to lower TMAO levels, are warranted to inform causality.
Association Between Our Results and Other Studies
The associations of TMAO and the risk of CVDs have been reviewed previously42, 43, 44, 45; however, these studies did not aim to perform a meta‐analysis to quantify the risks of TMAO and its precursors for MACE. We observed the highly consistent positive associations between TMAO levels and the outcomes across studies, even among studies with low (<5.0 μmol/L) concentrations of TMAO at baseline, particularly after excluding the data among blacks. As previously reported that the associations of TMAO and risk of cardiovascular events in hemodialysis patients differed by race,22 the inclusion of data in black hemodialysis patients influenced the heterogeneity of results in our study. It has also been reported that morality risk may differ between black and white dialysis patients.46 Nonetheless, median values of TMAO concentrations in healthy individuals were reported as ranges of 3 to 6 μmol/L,8, 17, 18, 19 and the study by Shafi et al22 consisted of hemodialysis patients with the substantially high mean vale of TMAO (101.9 μmol/L among total participants). It may not be generalizable to other groups of blacks from the general population, and further studies are warranted on the ethnic difference in the associations of TMAO and the risk of MACE.
Several studies10, 11, 19, 28 specifically considered the role of kidney dysfunction in the associations between TMAO and the risk of clinical adverse outcomes. The burden of CVD is high among patients with chronic kidney disease (CKD) or end‐stage renal disease,47 and the circulating TMAO is predominantly excreted by the kidneys.42 Nonetheless, we did not observe a statistically significant difference in the associations according to kidney dysfunction. Emerging evidence has shown associations of gut microbiota with obesity.48, 49, 50, 51 We found a stronger association in the subgroup with lower BMI and speculated that populations with lower BMI were less likely to be affected by diseases and might have less confounding factors, and effects of dietary differences influencing TMAO levels might be more apparent in the low‐BMI group. However, the difference between low‐ and high‐BMI groups was not statistically significant. Whether overweight or obesity accounted for the association of TMAO with the risk of MACE should be further investigated.
Additionally, we found that the pooled risk of elevated TMAO was stronger than those of its precursors, including betaine and choline or l‐carnitine, for the risk of MACE or all‐cause mortality. In our analyses, the associations of these correlated gut microbiota metabolites could not be mutually adjusted; therefore, we could not determine whether the TMAO or its precursors were independently associated with the outcomes. A study4 showed that plasma l‐carnitine concentrations predicted MACE independent of traditional cardiovascular risk factors, but the significant association disappeared after adjustment for plasma TMAO concentration. In addition, it was noted that elevated concentrations of l‐carnitine were only predictive of MACE risk among individuals with higher TMAO levels.4 In another study, it was found that only TMAO predicted the risk of MACE when choline, betaine, and TMAO were simultaneously included in the multivariate‐adjusted model, and choline and betaine predicted risk of MACE only among participants with an above median value of TMAO (>3.7 μmol/L) concentrations.9
Potential Mechanisms
In our recent study, we found that high intake of phosphatidylcholine, which could lead to a higher production of TMAO, was significantly associated with an increased risk of all‐cause and CVD‐specific mortality.52 Dietary choline and l‐carnitine are metabolized by intestinal bacteria to produce TMA, which is, in turn, absorbed into the bloodstream and oxidized to TMAO by enzyme flavin monooxygenase 3 in the liver.1, 4, 5 Koeth et al showed that dietary supplementation of mice with choline or l‐carnitine4 increased TMAO levels and enhanced the development of atherosclerosis.1 Flavin monooxygenase 3 is reported to be a key integrator of hepatic cholesterol and lipid metabolism and inflammation.53 TMAO was found to modulate cholesterol and sterol metabolism that would, at least partly, contribute to the increasing risk of CVDs.4 Higher TMAO levels were associated with the presence of increased atherosclerotic burden and complexity among patients with coronary artery disease (CAD).54 A recent study has shown that TMAO directly interacts with platelets altering calcium signaling, fostering platelet hyper‐reactivity and a prothrombotic phenotype in vivo.15 Similar, TMAO acutely induces aortic endothelial cell inflammatory gene profile, suggesting another potential pathway by which TMAO contributes to CVD.55 Betaine is a metabolite of choline,1, 56 and dietary betaine administration induced production of TMAO in animals.9 l‐carnitine in red meat can also be transformed to gamma‐butyrobetaine by gut bacteria before being converted to TMA and TMAO.2, 5 Whether betaine, choline, or l‐carnitine have independent effects on MACE and all‐cause mortality and whether other mechanisms are involved need further investigation.
Conclusion
Our meta‐analysis of published prospective studies indicates that higher circulating levels of gut microbiota metabolites, including TMAO and its precursors, are associated with an increased risk of MACE, regardless of conventional risk factors. Further studies are needed to investigate these associations in general‐risk populations, as well as the causality of the associations.
Sources of Funding
The study was supported by grants from the National Heart, Lung, and Blood Institute (HL071981, HL034594, and HL126024), the National Institute of Diabetes and Digestive and Kidney Diseases (DK091718, DK100383, and DK078616), the Boston Obesity Nutrition Research Center (DK46200), and United States–Israel Binational Science Foundation Grant 2011036. Qi was a recipient of the American Heart Association Scientist Development Award (0730094N). Heianza was a recipient of a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science.
Disclosures
None.
Supporting information
Table S1. Characteristics of the Identified Studies of Trimethylamine N‐Oxide (TMAO) Levels and the Prevalent Cardiovascular Diseases (Cross‐Sectional Studies)
Table S2. Relative Risks (RRs) of Major Cardiovascular Events According to Trimethylamine N‐Oxide (TMAO) Levels in Cross‐Sectional Studies
Figure S1. Pooled relative risks of high trimethylamine N‐oxide (TMAO) levels for therisk of major adverse clinical events/death (A) and all‐cause mortality (B) using 18 data points.3‐18
Acknowledgments
We thank Drs Stanley L. Hazen, Yuping Wu, Jason Stubbs, Marius Trøseid, Catharina Missailidis, Manuel Ottiger, and Philipp Schuetz, for providing data and clarifying aspects of their studies.
(J Am Heart Assoc. 2017;6:e004947 DOI: 10.1161/JAHA.116.004947.)28663251
Note
References
- 1. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koeth RA, Levison BS, Culley MK, Buffa JA, Wang Z, Gregory JC, Org E, Wu Y, Li L, Smith JD, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. gamma‐Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L‐carnitine to TMAO. Cell Metab. 2014;20:799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, Didonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WHW, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL, Lusis AJ. Trimethylamine‐N‐oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mente A, Chalcraft K, Ak H, Davis AD, Lonn E, Miller R, Potter MA, Yusuf S, Anand SS, McQueen MJ. The relationship between trimethylamine‐N‐oxide and prevalent cardiovascular disease in a multiethnic population living in Canada. Can J Cardiol. 2015;31:1189–1194. [DOI] [PubMed] [Google Scholar]
- 7. Lever M, George PM, Slow S, Bellamy D, Young JM, Ho M, McEntyre CJ, Elmslie JL, Atkinson W, Molyneux SL, Troughton RW, Frampton CM, Richards AM, Chambers ST. Betaine and trimethylamine‐N‐oxide as predictors of cardiovascular outcomes show different patterns in diabetes mellitus: an observational study. PLoS One. 2014;9:e114969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang WH, Wang Z, Fan Y, Levison B, Hazen JE, Donahue LM, Wu Y, Hazen SL. Prognostic value of elevated levels of intestinal microbe‐generated metabolite trimethylamine‐N‐oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 2014;64:1908–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. Prognostic value of choline and betaine depends on intestinal microbiota‐generated metabolite trimethylamine‐N‐oxide. Eur Heart J. 2014;35:904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaysen GA, Johansen KL, Chertow GM, Dalrymple LS, Kornak J, Grimes B, Dwyer T, Chassy AW, Fiehn O. Associations of trimethylamine N‐oxide with nutritional and inflammatory biomarkers and cardiovascular outcomes in patients new to dialysis. J Ren Nutr. 2015;25:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa‐Boyle B, Li XS, Levison BS, Hazen SL. Gut microbiota‐dependent trimethylamine N‐oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang WH, Wang Z, Shrestha K, Borowski AG, Wu Y, Troughton RW, Klein AL, Hazen SL. Intestinal microbiota‐dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J Card Fail. 2015;21:91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Troseid M, Ueland T, Hov JR, Svardal A, Gregersen I, Dahl CP, Aakhus S, Gude E, Bjorndal B, Halvorsen B, Karlsen TH, Aukrust P, Gullestad L, Berge RK, Yndestad A. Microbiota‐dependent metabolite trimethylamine‐N‐oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. 2015;277:717–726. [DOI] [PubMed] [Google Scholar]
- 14. Mueller DM, Allenspach M, Othman A, Saely CH, Muendlein A, Vonbank A, Drexel H, von Eckardstein A. Plasma levels of trimethylamine‐N‐oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis. 2015;243:638–644. [DOI] [PubMed] [Google Scholar]
- 15. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, Sartor RB, McIntyre TM, Silverstein RL, Tang WH, DiDonato JA, Brown JM, Lusis AJ, Hazen SL. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suzuki T, Heaney LM, Bhandari SS, Jones DJ, Ng LL. Trimethylamine N‐oxide and prognosis in acute heart failure. Heart. 2016;102:841–848. [DOI] [PubMed] [Google Scholar]
- 17. Skagen K, Troseid M, Ueland T, Holm S, Abbas A, Gregersen I, Kummen M, Bjerkeli V, Reier‐Nilsen F, Russell D, Svardal A, Karlsen TH, Aukrust P, Berge RK, Hov JE, Halvorsen B, Skjelland M. The carnitine‐butyrobetaine‐trimethylamine‐N‐oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis. 2016;247:64–69. [DOI] [PubMed] [Google Scholar]
- 18. Missailidis C, Hallqvist J, Qureshi AR, Barany P, Heimburger O, Lindholm B, Stenvinkel P, Bergman P. Serum trimethylamine‐N‐oxide is strongly related to renal function and predicts outcome in chronic kidney disease. PLoS One. 2016;11:e0141738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stubbs JR, House JA, Ocque AJ, Zhang S, Johnson C, Kimber C, Schmidt K, Gupta A, Wetmore JB, Nolin TD, Spertus JA, Yu AS. Serum trimethylamine‐N‐oxide is elevated in CKD and correlates with coronary atherosclerosis burden. J Am Soc Nephrol. 2016;27:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim RB, Morse BL, Djurdjev O, Tang M, Muirhead N, Barrett B, Holmes DT, Madore F, Clase CM, Rigatto C, Levin A, Can PI. Advanced chronic kidney disease populations have elevated trimethylamine N‐oxide levels associated with increased cardiovascular events. Kidney Int. 2016;89:1144–1152. [DOI] [PubMed] [Google Scholar]
- 21. Senthong V, Wang Z, Li XS, Fan Y, Wu Y, Tang WH, Hazen SL. Intestinal microbiota‐generated metabolite trimethylamine‐N‐oxide and 5‐year mortality risk in stable coronary artery disease: the contributory role of intestinal microbiota in a COURAGE‐like patient cohort. J Am Heart Assoc. 2016;5:e002816 DOI: 10.1161/JAHA.115.002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shafi T, Powe NR, Meyer TW, Hwang S, Hai X, Melamed ML, Banerjee T, Coresh J, Hostetter TH. Trimethylamine N‐oxide and cardiovascular events in hemodialysis patients. J Am Soc Nephrol. 2017;28:321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson‐Cohen C, Newitt R, Shen DD, Rettie AE, Kestenbaum BR, Himmelfarb J, Yeung CK. Association of FMO3 variants and trimethylamine N‐oxide concentration, disease progression, and mortality in CKD patients. PLoS One. 2016;11:e0161074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ottiger M, Nickler M, Steuer C, Odermatt J, Huber A, Christ‐Crain M, Henzen C, Hoess C, Thomann R, Zimmerli W, Mueller B, Schuetz P. Trimethylamine‐N‐oxide (TMAO) predicts fatal outcomes in community‐acquired pneumonia patients without evident coronary artery disease. Eur J Intern Med. 2016;36:67–73. [DOI] [PubMed] [Google Scholar]
- 25. Senthong V, Wang Z, Fan Y, Wu Y, Hazen SL, Tang WH. Trimethylamine N‐oxide and mortality risk in patients with peripheral artery disease. J Am Heart Assoc. 2016;5:e004237 DOI: 10.1161/JAHA.116.004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang WH, Wang Z, Li XS, Fan Y, Li DS, Wu Y, Hazen SL. Increased trimethylamine N‐oxide portends high mortality risk independent of glycemic control in patients with type 2 diabetes mellitus. Clin Chem. 2017;63:297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dambrova M, Latkovskis G, Kuka J, Strele I, Konrade I, Grinberga S, Hartmane D, Pugovics O, Erglis A, Liepinsh E. Diabetes is associated with higher trimethylamine N‐oxide plasma levels. Exp Clin Endocrinol Diabetes. 2016;124:251–256. [DOI] [PubMed] [Google Scholar]
- 28. Bain MA, Faull R, Fornasini G, Milne RW, Evans AM. Accumulation of trimethylamine and trimethylamine‐N‐oxide in end‐stage renal disease patients undergoing haemodialysis. Nephrol Dial Transplant. 2006;21:1300–1304. [DOI] [PubMed] [Google Scholar]
- 29. Stroup DF, Berlin JA, Morton SC, Olkin I, Williamson GD, Rennie D, Moher D, Becker BJ, Sipe TA, Thacker SB. Meta‐analysis of observational studies in epidemiology: a proposal for reporting. Meta‐analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA. 2000;283:2008–2012. [DOI] [PubMed] [Google Scholar]
- 30. Kip KE, Hollabaugh K, Marroquin OC, Williams DO. The problem with composite end points in cardiovascular studies: the story of major adverse cardiac events and percutaneous coronary intervention. J Am Coll Cardiol. 2008;51:701–707. [DOI] [PubMed] [Google Scholar]
- 31. Wells GA, Shea B, O'Connell D, Peterson J, Welch V, Losos M, Tugwell P. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. 2014 Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp. Accessed May 7, 2016.
- 32. Higgins JP. Commentary: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2008;37:1158–1160. [DOI] [PubMed] [Google Scholar]
- 33. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ. 2003;327:557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lau J, Ioannidis JP, Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med. 1997;127:820–826. [DOI] [PubMed] [Google Scholar]
- 35. Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta‐analysis: power of statistical tests and prevalence in the literature. J Clin Epidemiol. 2000;53:1119–1129. [DOI] [PubMed] [Google Scholar]
- 36. Greenland S, Longnecker MP. Methods for trend estimation from summarized dose‐response data, with applications to meta‐analysis. Am J Epidemiol. 1992;135:1301–1309. [DOI] [PubMed] [Google Scholar]
- 37. Scalbert A, Brennan L, Manach C, Andres‐Lacueva C, Dragsted LO, Draper J, Rappaport SM, van der Hooft JJ, Wishart DS. The food metabolome: a window over dietary exposure. Am J Clin Nutr. 2014;99:1286–1308. [DOI] [PubMed] [Google Scholar]
- 38. Wong JM. Gut microbiota and cardiometabolic outcomes: influence of dietary patterns and their associated components. Am J Clin Nutr. 2014;100(suppl 1):369S–377S. [DOI] [PubMed] [Google Scholar]
- 39. Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, Harris HM, Coakley M, Lakshminarayanan B, O'Sullivan O, Fitzgerald GF, Deane J, O'Connor M, Harnedy N, O'Connor K, O'Mahony D, van Sinderen D, Wallace M, Brennan L, Stanton C, Marchesi JR, Fitzgerald AP, Shanahan F, Hill C, Ross RP, O'Toole PW. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. [DOI] [PubMed] [Google Scholar]
- 40. Kong LC, Holmes BA, Cotillard A, Habi‐Rachedi F, Brazeilles R, Gougis S, Gausseres N, Cani PD, Fellahi S, Bastard JP, Kennedy SP, Dore J, Ehrlich SD, Zucker JD, Rizkalla SW, Clement K. Dietary patterns differently associate with inflammation and gut microbiota in overweight and obese subjects. PLoS One. 2014;9:e109434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, Almeida M, Quinquis B, Levenez F, Galleron N, Gougis S, Rizkalla S, Batto JM, Renault P; Consortium ANRM , Dore J, Zucker JD, Clement K, Ehrlich SD. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585–588. [DOI] [PubMed] [Google Scholar]
- 42. Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. 2014;124:4204–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu TX, Niu HT, Zhang SY. Intestinal microbiota metabolism and atherosclerosis. Chin Med J. 2015;128:2805–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tang WH, Hazen SL. Microbiome, trimethylamine N‐oxide, and cardiometabolic disease. Transl Res. 2017;179:108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wilson A, McLean C, Kim RB. Trimethylamine‐N‐oxide: a link between the gut microbiome, bile acid metabolism, and atherosclerosis. Curr Opin Lipidol. 2016;27:148–154. [DOI] [PubMed] [Google Scholar]
- 46. Kucirka LM, Grams ME, Lessler J, Hall EC, James N, Massie AB, Montgomery RA, Segev DL. Association of race and age with survival among patients undergoing dialysis. JAMA. 2011;306:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kahn MR, Robbins MJ, Kim MC, Fuster V. Management of cardiovascular disease in patients with kidney disease. Nat Rev Cardiol. 2013;10:261–273. [DOI] [PubMed] [Google Scholar]
- 48. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. [DOI] [PubMed] [Google Scholar]
- 50. Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G, Vieira‐Silva S, Wang J, Imhann F, Brandsma E, Jankipersadsing SA, Joossens M, Cenit MC, Deelen P, Swertz MA, Weersma RK, Feskens EJ, Netea MG, Gevers D, Jonkers D, Franke L, Aulchenko YS, Huttenhower C, Raes J, Hofker MH, Xavier RJ, Wijmenga C, Fu J. Population‐based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352:565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sonnenburg JL, Backhed F. Diet‐microbiota interactions as moderators of human metabolism. Nature. 2016;535:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zheng Y, Li Y, Rimm EB, Hu FB, Albert CM, Rexrode KM, Manson JE, Qi L. Dietary phosphatidylcholine and risk of all‐cause and cardiovascular‐specific mortality among US women and men. Am J Clin Nutr. 2016;104:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, Marshall S, McDaniel A, Schugar RC, Wang Z, Sacks J, Rong X, Vallim TA, Chou J, Ivanova PT, Myers DS, Brown HA, Lee RG, Crooke RM, Graham MJ, Liu X, Parini P, Tontonoz P, Lusis AJ, Hazen SL, Temel RE, Brown JM. The TMAO‐generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 2015;10:326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Senthong V, Li XS, Hudec T, Coughlin J, Wu Y, Levison B, Wang Z, Hazen SL, Tang WH. Plasma trimethylamine N‐oxide, a gut microbe‐generated phosphatidylcholine metabolite, is associated with atherosclerotic burden. J Am Coll Cardiol. 2016;67:2620–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, Lusis AJ, Shih DM. Trimethylamine N‐oxide promotes vascular inflammation through signaling of mitogen‐activated protein kinase and nuclear factor‐kappaB. J Am Heart Assoc. 2016;5:e002767 DOI: 10.1161/JAHA.115.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lever M, Slow S. The clinical significance of betaine, an osmolyte with a key role in methyl group metabolism. Clin Biochem. 2010;43:732–744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of the Identified Studies of Trimethylamine N‐Oxide (TMAO) Levels and the Prevalent Cardiovascular Diseases (Cross‐Sectional Studies)
Table S2. Relative Risks (RRs) of Major Cardiovascular Events According to Trimethylamine N‐Oxide (TMAO) Levels in Cross‐Sectional Studies
Figure S1. Pooled relative risks of high trimethylamine N‐oxide (TMAO) levels for therisk of major adverse clinical events/death (A) and all‐cause mortality (B) using 18 data points.3‐18