

Abstract
Background
In cardiomyocytes from patients with hypertrophic cardiomyopathy, mechanical dysfunction and arrhythmogenicity are caused by mutation‐driven changes in myofilament function combined with excitation‐contraction (E‐C) coupling abnormalities related to adverse remodeling. Whether myofilament or E‐C coupling alterations are more relevant in disease development is unknown. Here, we aim to investigate whether the relative roles of myofilament dysfunction and E‐C coupling remodeling in determining the hypertrophic cardiomyopathy phenotype are mutation specific.
Methods and Results
Two hypertrophic cardiomyopathy mouse models carrying the R92Q and the E163R TNNT2 mutations were investigated. Echocardiography showed left ventricular hypertrophy, enhanced contractility, and diastolic dysfunction in both models; however, these phenotypes were more pronounced in the R92Q mice. Both E163R and R92Q trabeculae showed prolonged twitch relaxation and increased occurrence of premature beats. In E163R ventricular myofibrils or skinned trabeculae, relaxation following Ca2+ removal was prolonged; resting tension and resting ATPase were higher; and isometric ATPase at maximal Ca2+ activation, the energy cost of tension generation, and myofilament Ca2+ sensitivity were increased compared with that in wild‐type mice. No sarcomeric changes were observed in R92Q versus wild‐type mice, except for a large increase in myofilament Ca2+ sensitivity. In R92Q myocardium, we found a blunted response to inotropic interventions, slower decay of Ca2+ transients, reduced SERCA function, and increased Ca2+/calmodulin kinase II activity. Contrarily, secondary alterations of E‐C coupling and signaling were minimal in E163R myocardium.
Conclusions
In E163R models, mutation‐driven myofilament abnormalities directly cause myocardial dysfunction. In R92Q, diastolic dysfunction and arrhythmogenicity are mediated by profound cardiomyocyte signaling and E‐C coupling changes. Similar hypertrophic cardiomyopathy phenotypes can be generated through different pathways, implying different strategies for a precision medicine approach to treatment.
Keywords: excitation‐contraction coupling, hypertrophic cardiomyopathy, pathophysiology, sarcomere physiology, troponin T
Subject Categories: Cardiomyopathy, Pathophysiology, Contractile function, Calcium Cycling/Excitation-Contraction Coupling, Genetically Altered and Transgenic Models
Clinical Perspective
What is New?
Two hypertrophic cardiomyopathy (HCM) mouse models with R92Q and E163R TNNT2 mutations were characterized with echocardiographic and biophysical studies to assess the relative roles of myofilament dysfunction and excitation‐contraction coupling remodeling in HCM pathophysiology.
Both models exhibited diastolic dysfunction and increased arrhythmogenicity.
The E163R model showed severe myofilament abnormalities including altered sarcomere energetics, with preserved intracellular Ca2+ transients.
The R92Q model presented profound excitation‐contraction coupling alterations that slowed down Ca2+ transients and twitch contractions in the absence of myofilament energetic changes.
Increased calcium/calmodulin‐dependent protein kinase II activity and increased myofilament Ca2+ sensitivity were detected in both mutants, but the changes were much larger in the R92Q model.
What are the Clinical Implications?
The notion that cellular remodeling is mutation specific in HCM may be relevant for treatment and may explain why some patients with HCM may not respond to specific therapies.
Drugs targeting ion channels or Ca2+ fluxes, such as late Na‐current blockers or diltiazem, may be more effective in the presence of significant cardiomyocyte excitation‐contraction coupling remodeling (eg, R92Q mutation).
When HCM mutations have a direct impact on sarcomere energetics (eg, E163R), sarcomere‐targeting drugs may be preferred; trials of novel myosin inhibitors in patients are currently underway.
Disease mechanisms that are common to different HCM mutations (calcium/calmodulin‐dependent protein kinase II, myofilament Ca2+ sensitivity) should be considered as valid targets for future therapies.
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common Mendelian cardiac disease,1 defined as a disease of the sarcomere because of its association with myofilament gene mutations in most patients.2 HCM is a progressive disease with different clinical stages3 and a large spectrum of clinical phenotypes,4, 5 ranging from asymptomatic individuals with minimal echocardiographic anomalies6 to patients with extreme left ventricular (LV) remodeling and advanced heart failure.3 Despite the vast archipelago of clinical manifestations, LV hypertrophy (LVH), diastolic dysfunction, and an arrhythmic propensity are considered hallmarks of the disease and occur in most patients7, 8 independently of their genetic profile.
Biophysical experiments have demonstrated how HCM‐related mutations primarily alter sarcomere function, ie, crossbridge cycling kinetics9, 10 or the switched‐off state of the thin filament,11 leading to increased energy consumption during tension generation or diastole.12, 13, 14 In addition to altered sarcomere energetics, increased myofilament Ca2+ sensitivity has often been reported in human HCM myocardium9, 15 both as a direct effect of the sarcomeric protein mutation15, 16 and as a consequence of secondary myofilament posttranslational modifications.17 The complex and poorly known secondary remodeling process that occurs in HCM involves a number of myofilament, sarcoplasmic reticulum (SR), and sarcolemmal alterations,18, 19, 20 reflecting adaptive/maladaptive modifications in various signaling pathways.21, 22 We recently demonstrated that in myocardial samples from patients with severe HCM who underwent septal myectomy, changes of calcium/calmodulin‐dependent protein kinase II (CaMKII)–dependent signaling lead to prolonged action potentials, cellular arrhythmias, prolonged Ca2+ transients, and increased diastolic intracellular [Ca2+] and [Na+].22 Such abnormalities appeared unrelated to patients' individual mutations.22 However, the scarce availability of human samples does not allow us to properly define whether HCM‐related secondary remodeling is truly independent of the underlying genotype.
In an era of emerging therapies that use a precision medicine molecular approach, this notion may be directly relevant to future management of the disease. To investigate this complex issue, we used 2 HCM mouse models reflecting the human phenotype but caused by different cardiac troponin T (TnT) mutations. We assessed how primary changes in myofilament function concur with secondary abnormalities of cardiomyocyte excitation‐contraction (E‐C) coupling to determine contractile impairment and arrhythmogenicity in HCM.
Methods
Detailed methods are available in Data S1.
TnT Mutant Transgenic Mouse Lines
All experimental protocols were performed in agreement with current Italian and European regulations and were approved by the local institutional review board and the animal‐welfare committee of the Italian Ministry of Health. We used a total of sixty‐seven 6‐ to 8‐month‐old male C57BL/6N transgenic mice carrying the R92Q13, 23 or E163R24 mutation in the TNNT2 gene, as well as wild‐type (WT) littermates: 22 R92Q, 24 E163R, and 21 WT mice were used for the experiments described below. The mouse colonies were housed in the animal facility of the University of Florence and all experiments were conducted locally. The 2 transgenic lines were generated in laboratories as previously described.23, 24
Myocardial Mechanics From the Whole Heart to the Single Myofibril
Echocardiography was performed on isoflurane‐anesthetized mice as previously described25 to characterize LV morphology and systolic and diastolic function using B‐mode imaging and Doppler measurements of transmitral blood flow. LV intact trabeculae were dissected from explanted hearts26, 27 and used to record isometric force during electrical stimulation with different pacing protocols at baseline and following β‐adrenergic activation.18 Skinned trabeculae were used to obtain pCa‐tension curves as previously described.28 Sarcomere energetics was assessed in skinned trabeculae by simultaneous measurement of isometric force and ATPase activity with an enzyme‐coupled assay.12, 14 Single myofibrils were isolated from mouse ventricular samples and used for mechanical measurements using a fast solution switching technique to assess maximal Ca2+‐activated tension, resting tension, and the kinetics of active tension generation and relaxation.9, 29 Myofibril suspensions were also used to measure resting ATPase by assessing phosphate production with a rapid colorimetric method.
Assessment of Morphofunctional Myocardial and Cellular Remodeling
Single cardiomyocytes were isolated from excised hearts via enzymatic dissociation and used for intracellular Ca2+measurements using Ca‐sensitive fluorescent dyes18, 27 to evaluate the amplitude and kinetics of Ca2+ transients, diastolic [Ca2+], and the rate of spontaneous Ca2+ release, during stimulation with field electrodes.30 To assess T‐tubule density, myocytes were stained with membrane‐selective dyes and observed with a confocal microscope.18, 30 Fast‐frozen LV myocardial samples were processed to obtain total proteins, which were used for Western blot studies to assess expression and phosphorylation of CaMKII,22 phospholamban, troponin I, and nuclear factor of activated T cell, as well as the expression of SERCA. Formalin‐fixed LV slices were stained with Picrosirius red and used to assess intramyocardial fibrosis.30
Statistical Analysis
Data from myofibrils, cells, and muscles are expressed as mean±SEM (number of samples and animals are indicated in the respective figure legends). Statistical analysis was performed as previously described,22, 30 using SPSS version 23.0 (IBM) and STATA version 12.0 (StataCorp). In brief, all sets of variables were checked for normality (Shapiro‐Wilk test) and for homogeneity of variances among groups (Levene's test). The statistical tests used to calculate P values for each data set are indicated in the respective figure legends. For variables where a single measurement for each mouse is included (eg, echocardiography, Western blot), the 3 different groups were compared using 1‐way ANOVA with Tukey correction (for normally distributed homoscedastic data sets), Kruskal‐Wallis test with Dunn's multiple comparison test (for non‐Gaussian data sets), or Welch's ANOVA with Games‐Howell test (for heteroscedastic groups). For variables where measurements from an unequal number of different samples (eg, cells or trabeculae) from each mouse were included (eg, ion fluorescence and myofibrils isometric force data), we used linear mixed models to compare data groups, using the xtmixed function of the STATA 12.0 program (StataCorp). To account for intra‐subject correlation, we included mouse‐id as a random‐effect term in the model. The Tukey‐Kramer post hoc correction method was used to compute P values for all pairwise comparisons, using the pwcompare with mcompare(tukey) option in STATA. Overall, P<0.05 was considered statistically significant. The range of calculated P values for each comparison (0.05>P>0.01, 0.01>P>0.001, or P<0.001) is indicated in the respective figure panels using symbols: red symbols refer to R92Q versus WT comparisons, blue symbols to E163R versus WT comparisons, and purple symbols to R92Q versus E163R comparisons.
Results
Hallmarks of Human HCM Phenotype are Present in Both R92Q and E163R Mouse Hearts
Echocardiographic measurements were performed in anesthetized male mice from the 3 study groups (WT, R92Q, and E163R), using a standardized protocol.31 Average heart rate during echocardiographic measurement was similar in the 3 groups: 585±42 beats per minute in WT, 579±38 beats per minute in E163R, and 573±45 beats per minute in R92Q mice. Representative images of parasternal long‐axis views of the left ventricle are reported in Figure 1A. In both R92Q and E163R mice, septal thickness was significantly increased compared with WT mice (Figure 1B), highlighting the presence of asymmetric LVH in both HCM models. Short‐axis views at different levels of the left ventricle were used to estimate LV volumes (Figure 1C). In R92Q compared with WT and E163R mice, we found a modest reduction of end‐diastolic and end‐systolic LV volumes. This is consistent with more severe LVH in R92Q mice, as the increase of wall thickness diminishes LV cavity volume.
Figure 1.
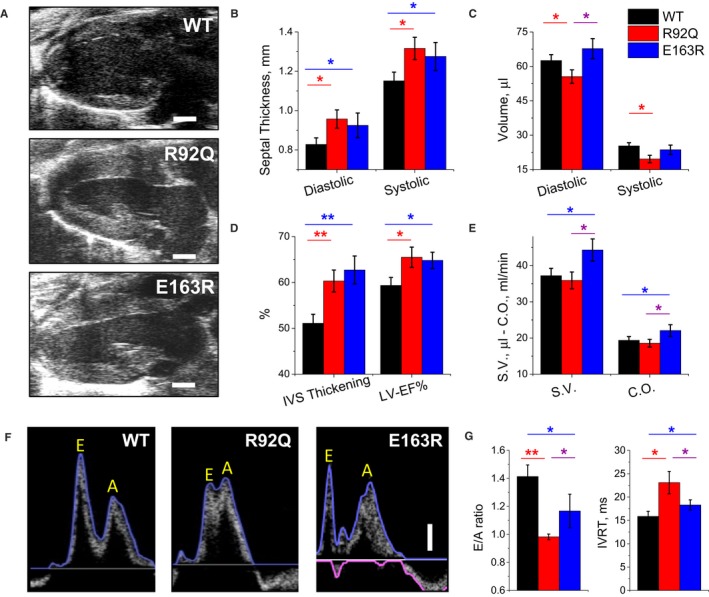
Echocardiographic measurements. A, Representative parasternal long‐axis views of the left ventricle at end diastole from wild‐type (WT), R92Q, and E163R mice. The horizontal scale bars equal 1 mm. B, Thickness of the interventricular septum (IVS) measured at end diastole (left) and at end systole (right) in WT, R92Q, and E163R mice. C, Left ventricular (LV) volumes are calculated using the Simpson technique at end diastole (left) and end systole (right) in mice from the 3 study groups. D, Systolic thickening of the IVS (left) and LV ejection fraction (LV‐EF%, right), are expressed as percentage of the diastolic values, measured in mice from the 3 cohorts. E, Stroke volume (SV) calculated from Simpson volumes and cardiac output (CO). F, Representative transmitral blood flow velocity curves recorded using pulsed‐wave Doppler echocardiography in apical views from WT, R92Q, and E163R mice. The vertical scale bar equals 100 mm/s. Early (passive, E) and late (atrial contraction, A) LV filling waves are indicated on the traces. G, Ratio between E and A waves (E/A ratio, left) and isovolumic LV relaxation time (IVRT, right) in mice from the 3 study cohorts. (B, E) Statistical tests: 1‐way ANOVA with Tukey correction. G, Statistical test: Welch's ANOVA with Games‐Howell test. (B through E and G) Means±SE from 10 mice per group. *=0.05>P>0.01; **=0.01>P>0.001.
In both R92Q and E163R mice compared with WT mice, we observed increased ejection fraction and septal fractional thickening during contraction (Figure 1D). In E163R mice, stroke volume and cardiac output were increased compared with that in WT mice (Figure 1E). In R92Q mice, instead, no differences were noted compared with that in WT mice, indicating that enhanced LV ejection fraction compensates for the reduced LV volumes.
Doppler studies of transmitral blood flow velocity (Figure 1F) were performed in 4‐chamber views to assess LV diastolic function. Reduced transmitral blood flow velocity during early diastole (ie, reduced E‐wave amplitude), paralleled by an increased proportion of LV diastolic filling during atrial contraction (ie, increased A‐wave amplitude), is widely recognized as an index of diastolic dysfunction. In both mutants, and more severely in R92Q, early LV filling was reduced and E/A ratio decreased (Figure 1G). Furthermore, in both R92Q and E163R compared with WT mice, we found prolonged isovolumic LV relaxation time, an additional index of diastolic dysfunction (Figure 1G). Taken together, these echocardiographic measurements indicate that 3 hallmarks of human HCM phenotype, ie, LVH, LV hypercontractility, and diastolic dysfunction are reproduced in both HCM models.
Twitch Relaxation is Prolonged and Spontaneous Activity Increased in Both R92Q and E163R Trabeculae
Isometric force was measured from intact left and right ventricular trabeculae or thin papillary muscles during field stimulation at 1 Hz, 30°C, and 2 mmol/L extracellular [Ca2+] (Figure 2). The amplitude of twitch contraction was similar in WT, R92Q, and E163R mice (Figure 2A and 2B). The baseline diastolic tension tended to be higher in E163R and R92Q compared with WT mice, although the difference was not significant (Figure 2B). Both E163R and R92Q trabeculae showed prolonged twitch duration compared with WT, caused by a significant prolongation of relaxation times (Figure 2A and 2C). This result is in line with the prolonged isovolumic LV relaxation time observed in vivo. The prolonged relaxation cannot be explained by changes in the myosin heavy chain isoform as myosin heavy chain‐α is the only significantly expressed isoform (>99%) in all 3 mouse lines.
Figure 2.
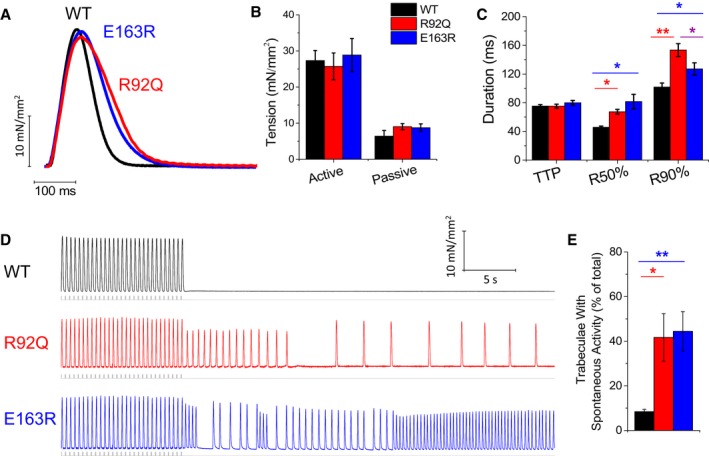
Steady‐state isometric twitches and occurrence of spontaneous contractions during stimulation pauses in intact trabeculae. A, Representative twitches elicited at 1 Hz in left ventricular trabeculae of wild‐type (WT), R92Q, and E163R mice. B, Passive and active twitch tension measured during 1‐Hz stimulation. C, Time from stimulus to peak contraction (TTP) and time from peak to 50% and 90% relaxation (R50% and R90%, respectively), measured in twitches at 1 Hz. D, Representative traces of the occurrence of spontaneous activity following a stimulation pause after 3‐Hz stimulation burst in the presence of isoproterenol 10−7 mol/L in intact trabeculae from the 3 study groups. At variance with WT, R92Q and E163R muscles display frequent spontaneous beats during pauses. The grey lines indicate electrical stimuli. E, Percentage of trabeculae that show spontaneous contractions following the protocol described in D. (B, C, E) Means±SE from 9 WT (6 mice), 11 R92Q (8 mice), and 9 E163R (6 mice) trabeculae. Statistical tests: linear mixed models with Tukey‐Kramer correction (corrected for heteroschedasticity in E). *=0.05>P>0.01; **=0.01>P>0.001.
We then analyzed the occurrence of spontaneous contractions in R92Q, E163R, and WT trabeculae to estimate the arrhythmogenic propensity associated with each mutation. In all mouse lines, the rate of spontaneous contractions was extremely low at baseline conditions (at 1‐Hz stimulation, <1% of preparations showed spontaneous contractions). A specific protocol to increase the probability of spontaneous events (Figure 2D) was applied, featuring the combination of isoproterenol 10−7 mol/L and a burst of high‐rate (3 Hz) stimuli followed by a 30‐second simulation pause. With this protocol, spontaneous activity occurred in more than 40% of E163R and R92Q trabeculae, while arrhythmic beats arose in only 10% of WT trabeculae (Figure 2E).
Responses to β‐Adrenergic Stimulation and Changes in Stimulus Interval Suggest Impaired SR Function in R92Q but not E163R Myocardium
Next, we studied the response of muscle contraction to steady‐state stimulation at different frequencies (0.2–6 Hz) at baseline (Figure 3) and in the presence of isoproterenol (10−7 mol/L) (Figure S1). While twitch amplitude at 1 Hz was the same in all experimental groups, the rate‐dependent increase of contractile force observed in WT and E163R trabeculae at frequencies above 2 Hz was significantly blunted in the R92Q trabeculae (Figure 3A and 2B). The increase of contractile force in response to isoproterenol was also significantly reduced in R92Q versus WT and E163R trabeculae (Figure S1). Mechanisms responsible of potentiating contraction in response to a high pacing rate or β‐adrenergic stimulation mainly rely on SR function32 The results suggest that SR function is impaired in R92Q but not E163R trabeculae. Relaxation times were prolonged in the 2 mutants at all investigated pacing rates, but rate adaptation of twitch duration (ie, acceleration of contraction kinetics with increase in stimulation frequency) was preserved in both R92Q and E163R models (Figure 3A through 3C). The positive lusitropic effect of β‐adrenergic stimulation was also preserved in both mutants (Figure S1).
Figure 3.
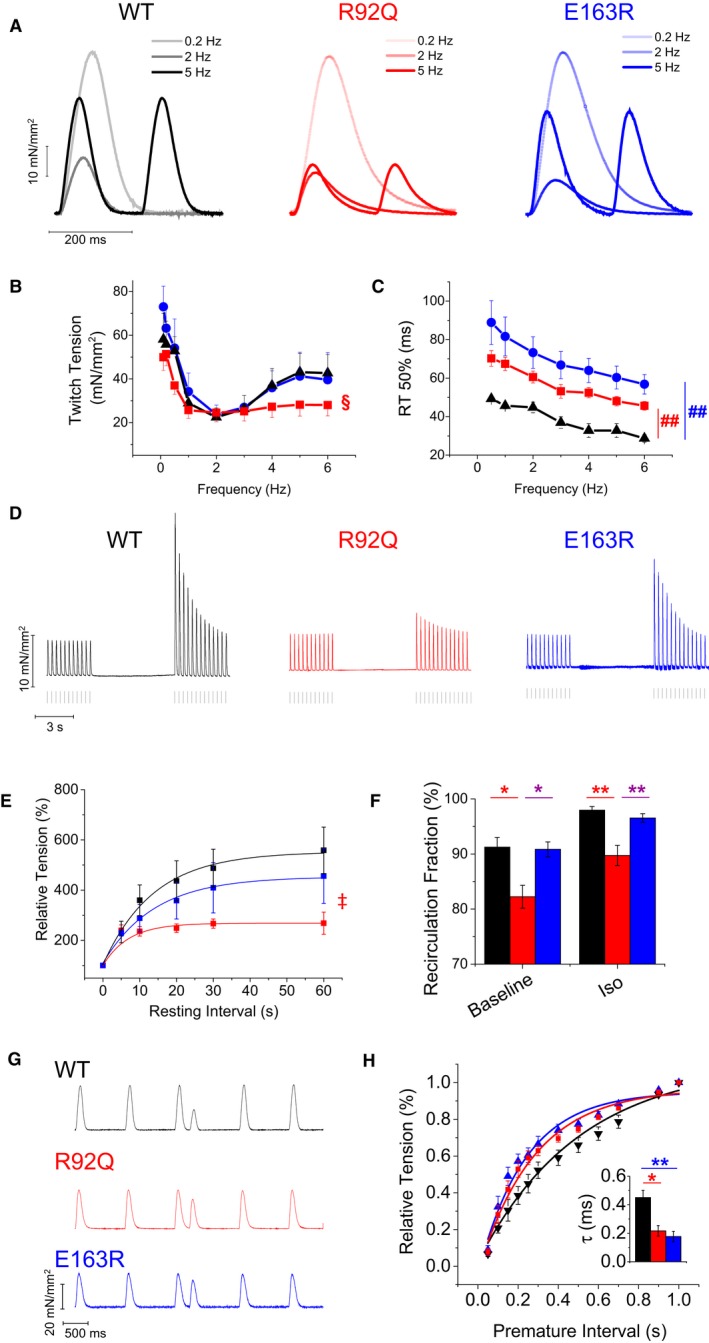
Steady‐state and short‐term interval force relationships. A, Representative superimposed force traces from trabeculae of wild‐type (WT), R92Q, and E163R mice, stimulated at 0.2 Hz, 2 Hz, and 5 Hz. B, Relationship between active twitch force and stimulation frequency (0.1–6 Hz) in trabeculae from the 3 groups of mice. §=0.05>P>0.01 (R92Q vsWT) for frequencies 0.1 to 0.5 Hz and 4 Hz and 0.01>P>0.001 for 5 to 6 Hz. C, Time from peak to 50% relaxation (R50%, right), measured in steady‐state twitches at different stimulation frequencies. ##=0.01>P>0.001 at all frequencies. D, Representative force traces of post‐rest potentiation in trabeculae from WT, R92Q, and E163R mice; steady‐state stimulation 3 Hz, pause duration 10 seconds. E, Average percentage increase of twitch force after different pause intervals from a basal frequency of 3 Hz in the 3 study groups. ‡=0.05>P>0.01 at resting intervals of 20 to 60 seconds. F, Ca2+ recirculation fraction estimated by the decline of potentiated beats following a period of high stimulation rate in trabeculae from WT, R92Q, and E163R mice at baseline and in the presence of isoproterenol 10−7 mol/L (Iso). G, Mechanical restitution protocol: representative traces from WT, R92Q, and E163R trabeculae: steady‐state stimulation 1 Hz, premature interval 250 ms. H, Restitution curves show the fractional recovery of force (percent of 1 Hz steady‐state peak force) in response to the premature stimulus plotted against the premature interval. Each curve was fitted by a single exponential to obtain the time constant of mechanical restitution (τ): mean τ values are reported in the inset. (B, C through F, H) Mean±SE from 9 WT (6 mice), 11 R92Q (8 mice), and 9 E193R (6 mice) trabeculae, Statistical tests: linear mixed models with Tukey‐Kramer correction (corrected for heteroschedasticity in C and F). * =0.05>P>0.01; **=0.01>P>0.001.
Stimulation pauses of variable duration were inserted into a steady‐state 1‐Hz series (Figure 3D) and the relative increase in amplitude of the first twitch after the pause was estimated (post‐rest potentiation) and plotted against the rest interval (Figure 3F). Maximum post‐rest potentiation was markedly lower in R92Q compared with WT and E163R trabeculae and was reached at shorter rest intervals. Pauses enhance twitch force mainly by allowing more Ca2+ to fill the SR during the prolonged diastolic period, thus favoring a larger Ca2+ release after the pause.33 Therefore, diminished post‐rest potentiation of R92Q myocardium suggests impaired SR Ca2+ reuptake.
The decay of post‐rest potentiation was studied to estimate SR Ca2+ recirculating fraction, a parameter that is generally assumed to reflect the relative contributions of SERCA and sarcolemma to cytosolic Ca2+ removal.34 In R92Q compared with WT or E163R trabeculae, SR Ca2+ recirculating fraction was significantly reduced both at baseline and in the presence of isoproterenol (Figure 3F), suggesting impaired SERCA function in the R92Q model.
Last, in intact trabeculae of the 3 mouse lines, we analyzed mechanical restitution by introducing a premature stimulus into a regular stimulus sequence at 1 Hz. The premature “extrasystolic” contraction was reduced in amplitude compared with steady‐state twitches (Figure 3G). The amplitude of the extrasystolic beat was plotted against the premature stimulus interval to obtain mechanical restitution curves (Figure 3F). In both R92Q and E163R versus WT trabeculae, the rate of mechanical restitution was faster (Figure 3F through G), suggestive of a shorter refractoriness of SR Ca2+ release27 and/or a lower Ca2+ threshold for myofilament activation.35
Ca2+ Handling, CaMKII Signaling, and Fibrosis in R92Q, WT, and E163R Myocardium
To further investigate the E‐C coupling process, intracellular Ca2+ measurements were performed in isolated, Fluoforte‐loaded cardiomyocytes during electrical field stimulation at 35°C. Representative traces are reported in Figure 4A and show considerable differences between the 2 mutants. At all stimulation frequencies tested, intracellular Ca2+ transient decay was markedly prolonged and its peak amplitude decreased in R92Q cardiomyocytes compared with both WT and E163R cardiomyocytes. Notably, WT and E163R cardiomyocytes exhibited the same Ca2+ transient amplitude and kinetics (Figure 4A through 4C). Diastolic [Ca2+]i was increased in both mutants but the largest change occurred in the R92Q model (Figure 4D).
Figure 4.
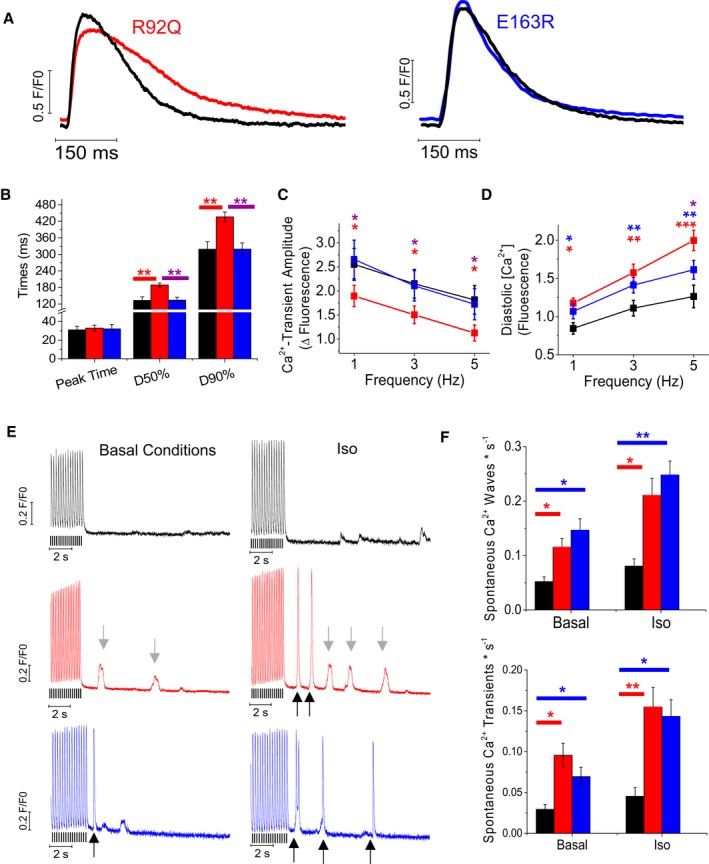
Intracellular Ca2+ measurements in intact ventricular cardiomyocytes. A, Representative superimposed Ca2+ transients elicited at 1 Hz in wild‐type (WT) and R92Q cardiomyocytes (left) and WT and E163R cells (right). B, Time from stimulus to peak (Peak Time), time from peak to 50% and 90% decay of Ca2+ transients (D50% and D90%, respectively) elicited at 1 Hz in cardiomyocytes from WT, R92Q, and E163R mouse hearts. C, Amplitude of Ca2+ transients in cardiomyocytes from the 3 study groups at different stimulation frequencies. D, Diastolic Ca2+ levels expressed as arbitrary units of fluorescence intensity during steady‐state stimulation at different frequencies in cells from mice of the 3 cohorts. (B through D) Mean±SE from 79 WT (6 mice), 91 R92Q (7 mice), and 109 E163R (8 mice) cardiomyocytes. *P<0.05. (E) Representative traces showing the stimulation pause protocol used to elicit spontaneous Ca2+ events in WT, R92Q, and E163R cardiomyocytes at basal conditions and in the presence of isoproterenol 10−7 mol/L (Iso). Notably, R92Q and E163R cardiomyocytes showed frequent Ca2+ waves (denoted by gray arrows) and premature Ca2+ transients (black arrows). The black lines below the traces indicate the times of stimulation. F, Frequency of spontaneous Ca2+ waves and spontaneous Ca2+ transients during stimulation pauses in WT, R92Q, and E163R cardiomyocytes at basal conditions and in the presence of Iso. Mean±SE from 88 WT (5 mice), 102 R92Q‐KET (7 mice), and 131 R92Q‐RAN (7 mice) cardiomyocytes. (B, D, F) Statistical tests: linear mixed models with Tukey‐Kramer correction (correction for heteroschedasticity was applied in F). *=0.05>P>0.01; **=0.01>P>0.001; ***=P<0.001.
To detect potential structural changes of T‐tubules, which may affect E‐C coupling18 we labelled intact cardiomyocytes with a membrane‐selective dye and imaged them with a confocal myocroscope. T‐tubule profile was quantified with dedicated software36 based on fast Fourier transform. The loss of T‐tubules, which was modest but statistically significant, was more pronounced in R92Q cardiomyocytes compared with E163R cardiomyocytes (Figure S2).
Next, we studied the occurrence of spontaneous Ca2+ waves and spontaneous Ca2+ transients after applying a specific stimulation protocol, ie, a burst of high‐rate stimuli followed by a stimulation pause (Figure 4E). The occurrence of spontaneous Ca2+ waves and spontaneous Ca2+ transients was significantly higher in R92Q and E163R versus WT models at baseline, and the difference increased after the administration of isoproterenol (10−7 mol/L).
To identify some of the potential mechanisms underlying the observed Ca2+ handling differences, fast‐frozen LV myocardial samples from WT, R92Q, and E163R animals were processed to obtain total proteins, which were used for Western blot studies aimed at identifying changes in CaMKII signaling and other molecular markers of cardiac remodelling, as previously described in human myocardium (Figure 5A).22, 37 Notably, CaMKII autophosphorylation, a marker of CaMKII activation, was increased in hearts from both R92Q and E163R compared with WT mice. Interestingly, the increase of CaMKII autophosphorylation was significantly more pronounced in R92Q compared with E163R hearts (Figure 5B). The total amount of SERCA protein was reduced in both R92Q and E163R hearts (Figure S3). Contrarily, phospholamban expression was significantly increased only in R92Q mice. Interestingly, the ratio between SERCA and phospholamban expression levels was markedly decreased in R92Q hearts. In E163R hearts, SERCA/phospholamban ratio was also reduced compared with WT, but was 3 times higher than that in R92Q. Finally, phospholamban phosphorylation at the protein kinase A site was decreased in both mutant lines, but the decrease was larger in R92Q hearts (Figure S3). The observed changes of the expression and phosphorylation of Ca2+‐handling proteins are in line with the marked reduction of the kinetics of Ca2+ transient decay observed in R92Q cardiomyocytes.
Figure 5.
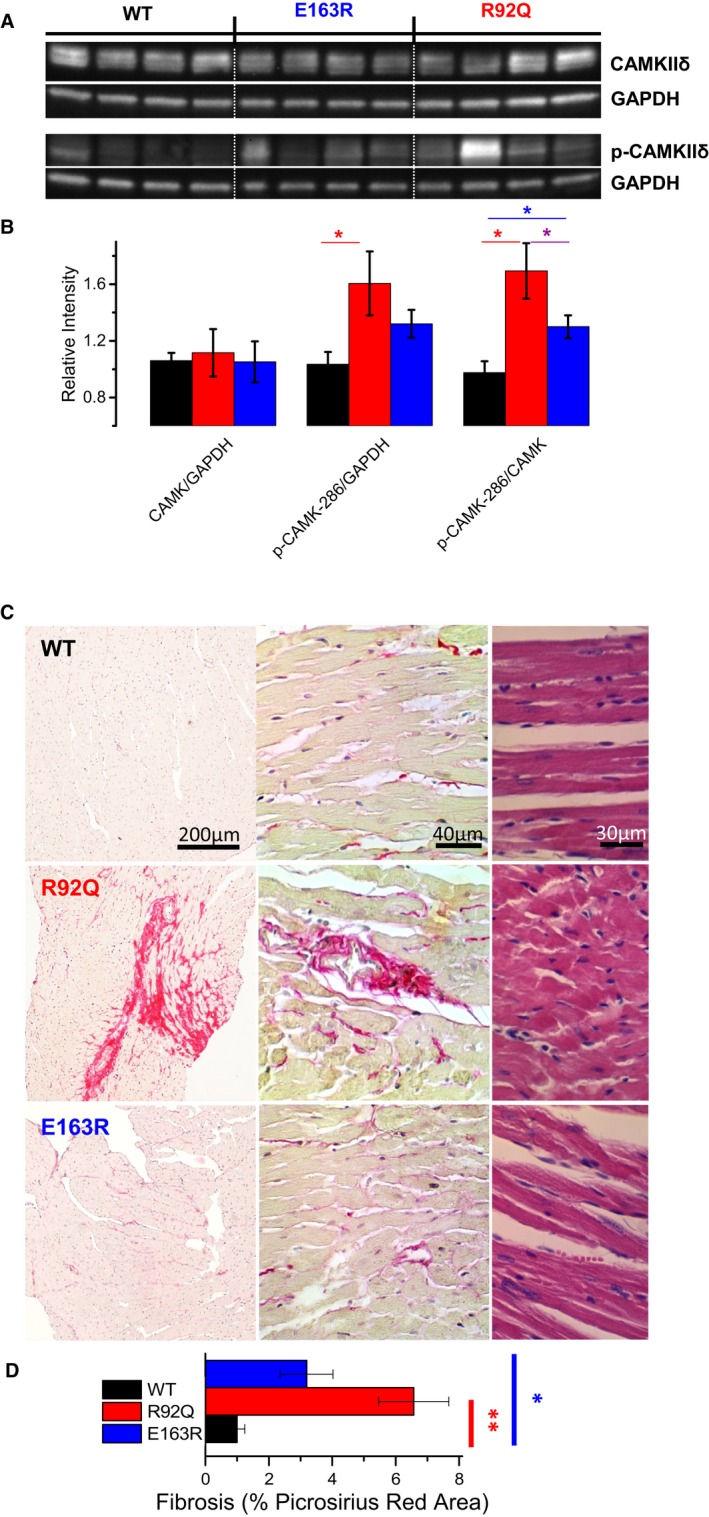
Molecular investigation of calcium/calmodulin‐dependent protein kinase II (CaMKII) signaling and myocardial fibrosis. A, Representative Western blots for total CaMKII, phospho‐CaMKII at tyrosine 287, and GAPDH. B, Average values in wild‐type (WT), R92Q, and E163R hearts (7 samples for each groups). The intensity of individual bands was quantified and normalized to that of GAPDH. Relative intensities of WT were set at 1. Statistical test: 1‐way ANOVA with Tukey correction. *=0.05>P>0.01. C, Representative histological sections of mouse left ventricular myocardium from WT, R92Q, and E163R mice. The left and central images show sections stained with hematoxylin (purple, marking cell nuclei) and Picrosirius red (pink) at different magnification; the bright pink strands denote collagen fibers. Sections shown on the right are stained with hematoxylin and eosin. D, Fraction of the myocardium occupied by collagen fibers, expressed as percentage of the whole section's area, measured in samples from mice of the 3 groups. Mean±SE from 9 mice per group. Five histological sections of the same size from each mouse were analyzed to calculate the extent of Picrosirius red–positive areas. Statistics: Kruskal–Wallis test with Dunn's multiple comparisons test. *=0.05>P>0.01; **=0.01>P>0.001.
Intramyocardial fibrosis is a pathological hallmark of HCM, both in human and in mouse models.30 We therefore explored disease‐related remodeling of extracellular matrix in the 2 transgenic lines by using Picrosirius red staining on LV tissue sections (Figure 5C). The amount of intramyocardial fibrosis, as identified by the extent of Picrosirius red–positive area in stained tissue sections, was increased in both R92Q and E163R hearts. However, the increase was more pronounced in R92Q versus E163R myocardium (Figure 5D).
We conclude that Ca2+ handling alterations in R92Q mice (driven, at least in part, by altered CaMKII signaling) may account for prolonged relaxation in trabeculae and diastolic dysfunction observed in vivo. In E163R mice, the extent of secondary myocardial remodeling, although qualitatively similar to that of R92Q mice, is much more limited. Indeed, the phenotype of E163R hearts is determined by different patho‐mechanisms.
Sarcomere Mechanical and Kinetic Properties are Altered in E163R but Preserved in R92Q Myofibrils
Ventricular myofibrils from R92Q, E163R, and WT hearts, mounted for force recording at 15°C and optimum myofilament overlap, were maximally calcium‐activated (pCa 4.5) and fully relaxed (pCa 8.0) by fast solution switching, as previously described.9 Figure 6A through 6C shows representative traces of activation‐relaxation protocols, while average data for the 3 myofibril groups are reported in Figure 6D through 6F. R92Q myofibrils showed no changes compared with WT. Specifically, maximal isometric tension, resting tension, the rates of tension generation and tension redevelopment, and all kinetic parameters of tension relaxation were the same in R92Q and WT myofibrils (Figure 6). E163R myofibrils, instead, showed a number of significant changes compared with both WT and R92Q. While maximal isometric tension was preserved (Figure 6A), resting tension was significantly increased and tension activation and tension redevelopment were faster, indicative of faster crossbridge turnover in the E163R sarcomeres (Figure 6B, 6D, and 6E). Significant changes were also found in the kinetics of tension relaxation of E163R myofibrils (Figure 6C and 6F). As previously described,34, 35 the time course of tension relaxation upon Ca2+ removal in all myofibril groups was biphasic with an early slow relaxation phase, which occurs while sarcomeres are isometric, followed by a fast exponential phase, starting with the “give” of few sarcomeres and dominated by intersarcomere dynamics. The rate of the early phase (slow k REL), predominantly reflecting the apparent rate with which attached crossbridges leave force‐generating states,34, 35 was significantly faster in the E163R myofibrils, indicating that this mutation may increase the energy cost of active tension generation10 (Figure 6F). In spite of the acceleration in kinetics of the slow isometric relaxation phase, the overall duration of the force relaxation transient was prolonged in E163R myofibrils compared with both WT and R92Q myofibrils. Indeed, the duration of the slow tension decay was significantly longer while the rate of the fast exponential phase (fast k REL) was significantly slower in E163R myofibrils (Figure 6F).
Figure 6.
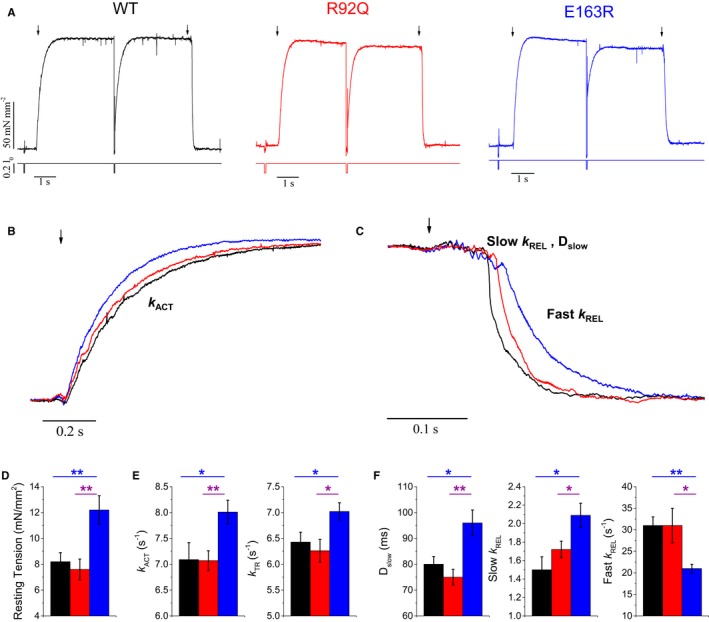
Mechanical measurements on single myofibrils. A, Tension generation and relaxation: representative tension responses of wild‐type (WT), E163R, and R92Q myofibrils, maximally activated (pCa 4.5) and fully relaxed (pCa 8) by fast solution switching technique at 15°C, [MgATP] 5 mmol/L; [Pi] <5 μmol/L. The timing of solution switch is indicated by black arrows. (B through C) Superimposed representative tension curves from single myofibrils isolated from WT, R92Q, and E163R hearts, highlighting the kinetics of force generation (B) and relaxation (C). Of note, relaxation is biphasic, with an initial linear phase (slow) followed by an exponential decay of force (fast phase). D, Resting tension in WT, R92Q, and E163R myofibrils. E, Kinetic parameters of tension generation in WT, R92Q, and E163R myofibrils: time constant of tension activation (k ACT, left) and of tension redevelopment upon myofibril relengthening after forced slacking (k TR, right). F, Kinetic parameters of myofibril relaxation: duration of the slow linear phase (Dslow, left) and time constant of the slow phase of relaxation (Slow k REL, center) and of the fast exponential phase (Fast k REL, right). (D through F) Mean±SE from 32 WT, 34 R92Q, and 36 E163R myofibrils, 3 mice per group. Statistical tests: linear mixed models with Tukey‐Kramer correction. *=0.05>P>0.01; **=0.01>P>0.001.
Prolonged tension relaxation and increased diastolic tension suggest that diastolic dysfunction of E163R hearts is primarily related to sarcomere dysfunction. Mutation‐driven impairment of the blocked state of thin filaments, by allowing recruitment of force‐generating crossbridges in the absence of Ca2+, may be responsible for both the relaxation and diastolic tension changes found in the E163R myofibrils. This mechanism can also lead to increased sarcomere energy consumption at rest.
Energetic Changes are Only Observed in E163R Skinned Trabeculae, While Myofilament Ca2+ Sensitivity is Enhanced in Both Mutants
Direct demonstration of the specific impact of E163R on sarcomere energetics at rest and during contraction was obtained by simultaneously measuring isometric tension and ATPase activity in demembranated ventricular trabeculae at 20°C. Representative recordings are reported in Figure 7A. Maximal Ca2+‐activated tension was the same in all groups of preparations (Figure 7B), in line with what observed in myofibrils. Resting tension tended to be higher in E163R trabeculae compared with WT and R92Q, although, at variance with myofibrils, this difference did not reach statistical significance (Figure 7B). Importantly, in E163R skinned trabeculae, both resting and maximal Ca2+‐activated ATPase were increased compared with WT and R92Q (Figure 7C). An increase in resting ATPase was confirmed in E163R trabeculae by measuring steady‐state ATP hydrolysis from myofibril suspensions in relaxing solution (Figure S4). The ratio between maximal Ca2+‐activated ATPase activity and active tension, representing the energetic cost of tension generation, was markedly higher in E163R compared with both WT and R92Q trabeculae (Figure 7D). The result was confirmed by measuring tension cost from the slope of the ATPase/tension plot obtained in skinned trabeculae by simultaneously measuring isometric ATPase and tension at different levels of Ca2+ activation (Figure S5).
Figure 7.
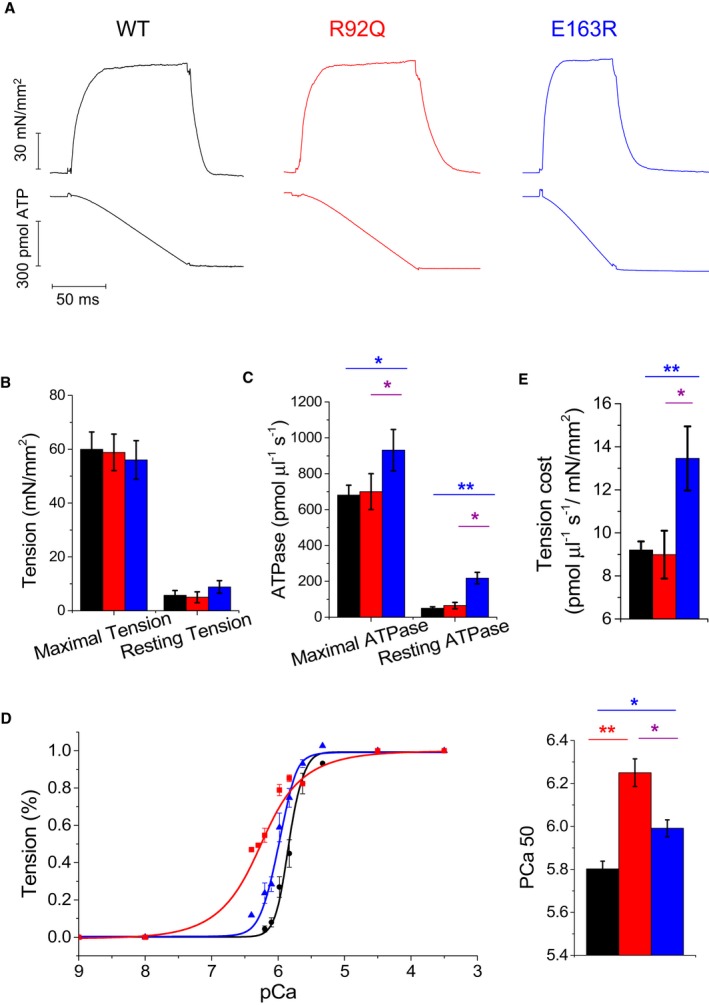
Isometric tension and ATPase measurements in skinned ventricular trabeculae. A, Representative tension (top) and ATPase activity (bottom) traces from wild‐type (WT), R92Q, and E163R trabeculae that were maximally Ca2+‐activated (pCa 4.5) at 20°C. (B through D) Mean±SE of maximal active tension (P 0) and resting tension (B), maximal Ca2+‐activated ATPase and resting ATPase activities (C), tension cost estimated as the ratio between maximal Ca2+‐activated ATPase and maximal active tension (D) in WT (n=16), R92Q (n=12), and E163R (n=15) trabeculae. (E) Tension‐pCa curves (left) and mean values of pCa at half‐maximal activation (pCa50, right) from WT (n=10), R92Q (n=7), and E163R (n=9) trabeculae. Statistical tests: linear mixed models with Tukey‐Kramer correction. *=0.05>P>0.01; **=0.01>P>0.001.
Mean pCa‐active tension curves obtained from skinned trabeculae showed that myofilament Ca2+ sensitivity was significantly increased in both R92Q and E163R compared with WT preparations (Figure 7E). The change was milder in the E163R trabeculae and much more marked in the R92Q preparations that showed the highest pCa50 value.
Discussion
In HCM, functional alterations at the sarcomere level9, 14 are associated with secondary modifications in E‐C coupling that are responsible for the proarrhythmogenic phenotype and contribute to alterations of cardiac mechanical function.22 The relative contribution of primary, mutation‐driven, sarcomeric changes versus adverse cardiomyocyte remodeling to the development of HCM phenotype is unclear, and, in the present work, we suggest that it varies depending on the underlying mutation. Previous work on human samples17, 22 described a number of “disease‐specific” aspects of HCM‐related myocardial remodeling, different from those observed in heart failure or secondary hypertrophy and without a major impact of the specific patient genotype. Here, we characterized in vivo heart phenotype and in vitro biophysical changes in sarcomere function and E‐C coupling in 2 HCM mouse models carrying different mutations in cardiac TnT and found that HCM pathophysiology is “mutation specific” rather than “disease specific” (Figure 8).
Figure 8.
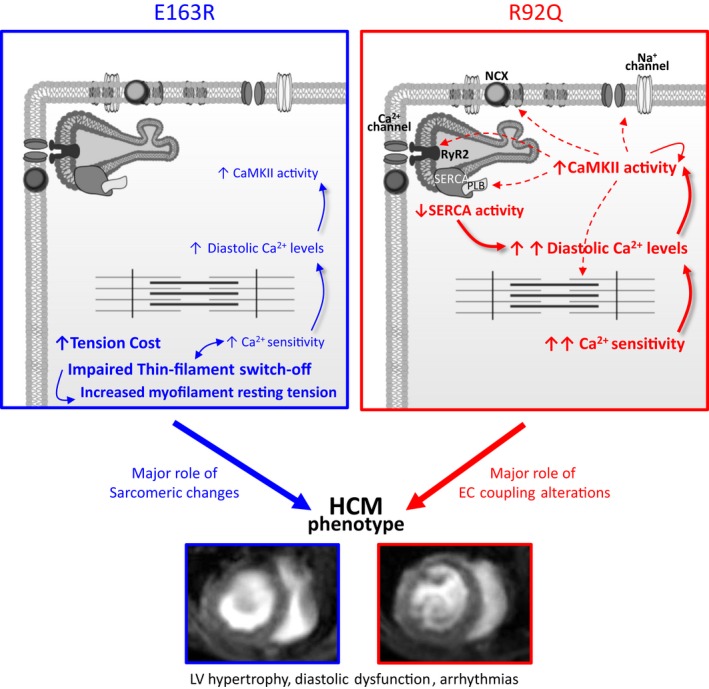
Different mutation‐specific mechanisms lead to similar phenotypes. Scheme depicting the proposed mutation‐specific molecular and cellular mechanisms leading to hypertrophic cardiomyopathy (HCM)–related cardiac structural and functional abnormalities in mice carrying the R92Q or E163 mutations. The figure includes 2 short‐axis magnetic resonance scans (mid‐left ventricular [LV] level) at end diastole from mice of the 2 strains, obtained with a 7T magnetic resonance setup. CaMKII indictes calcium/calmodulin‐dependent protein kinase II; E‐C, excitation‐contraction.
Similar Phenotypes Through Different Pathogenic Pathways
Findings from echocardiographic and Doppler measurements showed that several hallmarks of human HCM phenotype (increased septal thickness, increased ejection fraction, diastolic dysfunction) are present in both HCM mouse models, with the phenotype more pronounced in the R92Q mice (Figure 1). Mechanical experiments in intact trabeculae demonstrated that (in the absence of a switch in myosin isoforms) isometric twitch relaxation was markedly prolonged and resting tension tended to increase in both HCM mice compared with WT mice (Figure 2A through C), in line with the diastolic dysfunction observed in vivo (Figure 1F and 1G). Compared with WT, both HCM models exhibited a significantly higher frequency of spontaneous activity during pauses, following a period of high‐frequency stimulation in the presence of isoproterenol (Figure 2D and 2E). Previous telemetry evaluations in the R92Q models highlighted increased occurrence of both atrial and ventricular arrhythmias,38 while no evaluation of the arrhythmic burden was performed on living E163R mice.
In spite of the similarities between the 2 mouse models, further experiments investigating the inotropic reserve of intact trabeculae indicated that the function of the Ca2+‐handling machinery was substantially different (Figure 3 and Figure S1). While E163R trabeculae behaved like WT, R92Q trabeculae exhibited a blunted response to changes in stimulation frequency and to other inotropic interventions, as previously shown in Langendorff‐perfused working hearts.39 Experiments in isolated cardiomyocytes confirmed that E‐C coupling abnormalities were mostly present in the R92Q mouse model (Figure 4). As previously described,38 R92Q cardiomyocytes exhibited smaller and slower Ca2+ transients compared with WT, providing a reasonable explanation for diastolic dysfunction in vivo. Differently, in E163R cardiomyocytes, the kinetics of Ca2+ transients were normal (Figure 4) and no major E‐C coupling changes were found that could explain prolonged twitch duration in trabeculae. Experiments performed in single myofibrils and skinned ventricular trabeculae showed that E163R uniquely alters the switched‐off state of the thin filament in the absence of Ca2+ (Figures 6, 7 and Figure S4), thus directly impairing the mechanics and energetics of cardiac relaxation and diastole. In addition, simultaneous measurements of isometric tension and ATPase demonstrated that the energy cost of tension generation was increased in E163R but unchanged in R92Q trabeculae (Figure 7 and Figure S5), the latter in line with previous observations.28 Despite these differences, some similarities were observed. Myofilament Ca2+ sensitivity in skinned trabeculae was increased in both HCM mutant lines compared with WT, although the change was much greater in the R92Q model (Figure 7). Intramyocardial fibrosis was increased in both mouse lines as compared with WT, but the extent was much larger in R92Q hearts (Figure 4). In cardiomyocytes, diastolic Ca2+ levels and CaMKII autophosphorylation were abnormally elevated in both mutant lines, with greater changes occurring in the R92Q model (Figure 4). Increased myofilament Ca2+ sensitivity and cytosolic Ca2+ overload may be plausibly linked to the increased rate of arrhythmias observed with both mutations. Overall, these results indicate that similar HCM phenotypes can be generated through different pathogenic pathways (see scheme in Figure 8). In the E163R model, mutation‐driven abnormalities in myofilament function largely account for cardiac dysfunction. In the R92Q model, instead, profound E‐C coupling changes related to aggressive cardiomyocyte remodeling seem to drive the cardiac HCM phenotype (Figure 8).
In addition to the comprehensive biophysical characterization in 6‐month‐old mice, data obtained at different ages, including 3 months and 12 months, are reported in Figure S6. Studies of 3‐month‐old mutant mice demonstrated that both sarcomere and E‐C coupling changes are already present, although in R92Q mice, some of the intracellular calcium alterations (eg, increased diastolic Ca2+ levels, prolonged Ca2+ transient duration) are more severe in older animals, while no progression was observed in the E163R model.
Primary Sarcomeric Changes Directly Impair Energetics and Diastolic Function in E163R Hearts
While all mechanical and kinetic parameters of maximally activated R92Q myofibrils were the same as those of WT myofibrils, in E163R myofibrils, kinetics of force generation (tension activation and tension redevelopment) and isometric relaxation (slow k REL) were significantly faster compared with both WT and R92Q, preparations suggesting that the mutation primarily affects crossbridge kinetics and energetics. According to a simple crossbridge model, these results indicate that a modest (but significant) increase in g app (the apparent rate with which crossbridges leave their force‐generating state under isometric conditions) in E163R mice accounts for both faster crossbridge turnover and increased tension cost, without significantly affecting maximal tension generation.9 As to the impact of E163R on g app, pioneer studies reviewed by Gordon et al40 reported that HCM‐associated cardiac TnT mutations may cause changes in regulated acto‐S1 ATPase and unloaded thin filament sliding speed. This implies that cardiac TnT can modulate strongly bound crossbridge detachment rate in addition to its ability to control the attachment of crossbridges to the thin filament. However, the potential molecular mechanism of this effect of the E163R mutation remains unclear.
In spite of the faster rate of the slow isometric phase of relaxation following Ca2+ removal, overall relaxation from maximal activation was prolonged in E163R myofibrils (slower fast k REL and prolonged duration of the early isometric phase), while resting tension was significantly higher compared with WT and R92Q myofibrils (Figure 6). This behavior, together with the increase in resting tension, suggests that in E163R sarcomeres, the thin filament may not turn off properly upon Ca2+ removal and may be unable to fully maintain its switched‐off state during diastole, in analogy with previously described HCM‐related tropomyosin mutations.41, 42, 43 The modest increase in myofilament Ca2+ sensitivity in E163R myocardium is in line with previous findings44 and may be a consequence of the impaired thin filament blocked state that allows recruitment of some force‐generating crossbridges at low Ca2+.24, 45, 46 We excluded that the increase of myofilament Ca2+ sensitivity depended on a reduction of troponin I phosphorylation at the protein kinase A site (Figure S3), although we have not explored other possible posttranslational modifications of sarcomere proteins (eg, MyPBC3). The observed increase of resting ATPase in E163R skinned trabeculae and myofibrils is likely a direct consequence of residual thin filament activation at low [Ca2+]. Residual thin filament activation following Ca2+ removal may promote formation of new crossbridges,41, 42, 43 contributing to slow down relaxation, and increase diastolic tension in E163R myocardium. These sarcomeric abnormalities are likely to be the basis of the prolonged twitch contraction and the impaired diastolic function observed in vivo (Figure 8).
While these considerations suggest that altered sarcomere function is the main pathogenic element underlying impaired relaxation in E163R myocardium, a contribution of other mechanisms cannot be excluded. For instance, previous work24 described considerable degrees of myofibril disarray and Z‐line misalignment in E163R cardiomyocytes. This is consistent with the modest structural disorganization of T‐tubules observed in E163R myocytes and may contribute to impair cardiomyocyte relaxation properties.26
Moreover, the expression of mutant TnT protein in the R92Q line is 67%,13, 23 while it is 50% in the E163R line.24 Such a difference is unlikely to have accounted for the observed divergences in myofilament energetics and myofibril relaxation between the 2 lines; however, we cannot exclude that the higher expression of mutant protein in R92Q mice affects some features of myocardial dysfunction in mutant mice (eg, myofilament Ca2+ sensitivity) and acknowledge this issue as a limitation of our work.
Secondary E‐C Coupling Changes are the Major Cause for Contractile Dysfunction in R92Q
Diastolic dysfunction evaluated in vivo through echocardiography was more severe in R92Q compared with E163R mice. Mechanical measurements in intact trabeculae confirmed that twitch contraction was prolonged. In addition, despite preserved baseline systolic contraction, inotropic reserve was reduced in R92Q, at variance with E163R preparations (Figure 2 and 3). Reduced contractile reserve is a feature of adverse remodeling and disease progression in human HCM, previously observed in transgenic mice carrying different TnT mutations in the same locus.47 Approximately 20% of HCM patients showed an insufficient blood pressure response to exercise and 10% displayed overt systolic dysfunction (ejection fraction <50%).3 Both features are validated predictors of adverse outcome and sudden cardiac death in patients with HCM.48
At variance with E163R cells, R92Q cardiomyocytes display severe prolongation of Ca2+ transient decay, which can account for prolonged twitch duration and contribute to the diastolic dysfunction observed in vivo (Figure 8). A number of mechanisms may contribute to this aspect. First, we confirmed47 that the reduced SERCA activity underlies the slower kinetics of Ca2+ transients in R92Q cells and is caused by a lower SERCA expression paralleled by increased phospholamban expression and phosphorylation levels (Figure S3). The reduction of SR Ca2+ recirculation fraction observed in R92Q trabeculae further confirms the reduction of SERCA activity.26 Interestingly, cardiomyocytes isolated from septal samples of patients with obstructive HCM undergoing myectomy consistently displayed slower Ca2+ transients, associated with reduced SERCA expression.22 Furthermore, slower Ca2+ transient kinetics and reduction in SERCA expression have been observed in the majority of animal models with experimental LVH and LV failure and in cardiomyocytes from patients with terminal heart failure.49 Thus, it does not represent a disease‐specific feature. Similarly, as in human HCM,50 we describe a certain degree of T‐tubule disruption in R92Q cardiomyocytes, in line with our previous data on Δ160 cardiac TnT mutant mouse model.18 The extent of T‐tubule structural remodeling in R92Q cardiomyocytes, however, is much less pronounced compared with models of secondary hypertrophy and failure51 and is likely to play only a minor role in the slowing of Ca2+ transient and contraction kinetics. In parallel with a large number of HCM mutations in thin and thick filament proteins, we observed a large increase in myofilament Ca2+ sensitivity in R92Q skinned trabeculae, likely related to higher Ca2+ binding affinity of troponin complexes incorporating the mutant protein.46 As a consequence, myofilament Ca2+ dissociation is slower and may contribute to prolong the last phase of Ca2+ transient decay, resulting in increased end‐diastolic [Ca2+].35 Increased myofilament Ca2+ sensitivity in combination with reduced SERCA activity can explain the elevated diastolic [Ca2+]i and excessive increase in baseline [Ca2+]i at high stimulation frequencies, which were particularly prominent in R92Q myocytes. In E163R myocardium, we found a slight reduction of SERCA expression and a minimal increase of phospholamban expression and phosphorylation compared with WT myocardium (Figure S3). Such changes were much less pronounced with respect to R92Q hearts and are unlikely to determine a significant slowing of SERCA Ca2+‐reuptake rate, as indicated by the preserved Ca2+ transient decay rate in E163R myocytes. Therefore, increased myofilament Ca2+ sensitivity could be the sole cause of the increase of diastolic [Ca2+]i observed in E163R myocardium.35
As previously shown in human HCM myocardium,22 a sustained increase in diastolic [Ca2+]i is associated with enhanced activation of CaMKII, as confirmed by the higher degree of CaMKII autophosphorylation observed in R92Q myocardium. Increased CaMKII activity plays a central role in driving cellular and extracellular remodeling in a number of genetic and acquired cardiac diseases,52 including HCM,22 and may play a leading role in R92Q mice. In line with the lesser degree of cellular remodeling and the less prominent increase of diastolic [Ca2+]i in E163R versus R92Q myocytes (Figure 4), the increase of CaMKII autophosphorylation was less pronounced in E163R myocardium as compared with R92Q. CaMKII activates transcription factors that drive increased myocardial fibrosis in cardiac diseases53 In line with that, fibrosis was more pronounced in hearts from R92Q mice compared with E163R mice. Of note, both mouse lines well reproduce the increased CaMKII activity, recently observed in myectomy samples from HCM patients22, 37 and described in humans as a hallmark of myofilament‐positive HCM.
Other pathways of hypertrophic remodeling, including the nuclear factor of activated T cell pathway, do not appear to be changed in either of the 2 mutant lines (Figure S3), consistent with studies on human samples.37
Cellular Mechanisms of Arrhythmias in R92Q and E163R Myocardium: Open Questions
Increased arrhythmogenesis in hearts from patient and animal models with HCM is the result of pathological changes that occur both at the cell and tissue level. The latter include intramyocardial fibrosis, myocyte disarray, and regional hypertrophy, which all promote the establishment of reentrant arrhythmias. In the present work, we found that the mechanism underlying the increased rate of premature beats during prolonged pauses and β‐adrenergic stimulation in trabeculae was likely an increased rate of spontaneous Ca2+ waves and Ca2+ transients in cardiomyocytes, providing a trigger for propagated contractions. In recent work on a mouse model of catecholaminergic polymorphic ventricular tachycardia,27 we showed that Ca2+‐dependent cellular arrhythmias depend on the increased open probability of the ryanodine receptor (RyR), and that increased RyR open probability alone causes faster mechanical restitution. Indeed, in both R92Q and E163R mutants, and previously in the Δ160 TnT mouse line,18 we observed a faster rate of mechanical restitution. This suggests that in TnT mutants, Ca2+ waves are mediated by an increased RyR open probability during diastole. The increase in diastolic cytosolic [Ca2+]i, observed in cells from both TnT mouse lines, is the most direct explanation for the observed instability of RyR channels, as the main trigger for RyR opening is [Ca2+] at the dyad level. However, in our experimental conditions, maximal diastolic [Ca2+]i is observed at high pacing rates, while Ca2+ waves are more frequent during prolonged pauses. An alternative explanation has been provided by Knollman et al,35 who found that in HCM‐linked TnT mutations, high myofilament Ca2+ sensitivity leads to increased cytosolic Ca2+‐buffering capacity. The excessive amount of Ca2+ that binds to the myofilaments is slowly released during pauses and accumulates in the SR, determining SR Ca2+ overload, thus enhancing RyR open probability through luminal control.54 This may represent the main trigger for Ca2+ waves and delayed afterdepolarizations during pauses. The proposed mechanism occurs only in the presence of a mutation that increases the binding rate of Ca2+ to the troponin complex. This is likely the case for R92Q mutation. Whether the E163R mutation affects Ca2+ binding to myofilaments and intracellular Ca2+ buffering remains to be determined.
Implications for Human Disease
Our data support the view that different mutations, even within the same gene, can generate cardiac dysfunction with markedly different mechanisms. While diastolic dysfunction and arrhythmogenesis are common consequences of all HCM mutations, a complex structural and functional remodeling of cardiomyocytes may occur only with certain variants, such as R92Q, and may not be present in others, such as E163R. Interestingly, mutations at the R92 site of TNNT2 are regarded as severe, with a high risk of lethal arrhythmias during adolescence and early adulthood,55, 56 while mutations at the E163 site lead to milder forms of HCM.57 The notion that cellular remodeling is mutation specific may be relevant for treatment. Drugs targeting membrane ion channels, such as late Na‐current blockers22 or diltiazem58 may be more effective in the presence of significant cardiomyocyte E‐C coupling remodeling. We previously showed that in R92Q cardiomyocytes, the late Na+‐current blocker ranolazine (10 μmol/L) significantly hastened Ca2+ transient kinetics and reduced diastolic Ca2+ and the rate of spontaneous beats and Ca2+ waves.30 Additionally, lifelong treatment with ranolazine prevented most of the aspects of cardiac remodeling and dysfunction in R92Q mice.30 Contrarily, recent unpublished experiments show that the acute administration of ranolazine is unable to exert similar effects on E163R cardiomyocytes.
The same can be said for drugs that act on extracellular remodeling, such as sartans or statins, which have shown promising results in transgenic HCM animal models with selected “severe” mutations59, 60 but failed to show convincing results in patients who were not selected based on genotype.61, 62 Conversely, in the presence of sarcomeric mutations leading to cardiac dysfunction through direct mechanisms, sarcomere‐targeting drugs may be preferred, such as novel myosin inhibitors, currently in the pipeline for the treatment of HCM.63
Conclusions
Based on the results of the present investigation, we envision a future where the treatment of every patient will be decided based on his/her specific mutation, driven by studies of the specific mutation‐linked mechanisms of disease. Although we understand that producing animal models as a predictive tool is not practical, cardiomyocytes differentiated from patient‐specific induced pluripotent stem cells may be a feasible tool to assess mutation‐specific pathomechanisms and predict the efficacy of drugs in individual patients.19, 64
Sources of Funding
This work was supported by Telethon Italy (GGP13162 and GGP16191), the European Commission (STREP Project 241577 “BIG HEART,” 7th European Framework Program), the Italian Ministry of Health (GR‐2011‐02350583, RF‐2013‐02356787 and NET‐2011‐02347173), Regione Toscana (FAS‐Salute 2014, ToRSADE project), and the National Institues of Health (projects HL075619 and HL62426).
Disclosures
None.
Supporting information
Data S1. Supplemental Methods
Figure S1. Steady‐state contractions with isoproterenol. A, Steady‐state 1‐Hz twitch tension at basal conditions and in the presence of isoproterenol 10 to 7 mol/L. B, Time to peak and time from peak to 50% relaxation (R50%, right), measured in steady‐state twitches at different stimulation frequencies at basal conditions and in the presence of isoproterenol 10−7 mol/L.
Figure S2. Alterations of T‐tubules. A, Representative confocal images from isolated left ventricular (LV) cardiomyocytes stained with di‐3‐aneppdhq from wild‐type (WT; right), R92Q (middle), and E163R (right) hearts. The horizontal bar equals 10 μm. B, T‐tubule power, as calculated using the TTorg ImageJ plugin, in cardiomyocytes from the 3 study groups. C, Mean T‐tubule period (equivalent to sarcomere length) in resting LV cardiomyocytes from the 3 cohorts of mice. (B through C) Means±SE from 68 WT (4 mice), 142 R92Q (6 mice), and 69 E163R (4 mice) cardiomyocytes. Statistics: 1‐way ANOVA with Tukey correction. *P<0.05.
Figure S3. Additional protein studies. A, Representative Western blots for total SERCA, phospholamban (PLB), phosphorylated PLB at Serine 16 (P‐PLB(S16)), and GAPDH. B, Average relative intensity values of SERCA, PLB, SERCA/PLB, and p‐PLB(S16)/PLB ratios in wild‐type (WT), R92Q, and E163R hearts (8 samples for each group). C, Representative Western blots for total cardiac troponin I (cTnI), phosphorylated cTnI (P‐cTnI), total nuclear factor of activated T cell (NFATc1), phosphorylated NFATc1 (P‐NFATc1), and GAPDH. D, Average relative intensity values of cTnI, P‐cTnI, NFATc1, and P‐NFATc1 in WT, R92Q, and E163R hearts (5 samples for each group). (B through D) The intensity of individual SERCA bands was quantified and normalized to that of GAPDH. Relative intensities of WT were set at 1. Statistics: 1‐way ANOVA with Tukey correction. *P<0.05. **P<0.01.
Figure S4. Effect of the E163R mutation on the relationship between steady‐state isometric force and ATPase activity. A, TC was determined from the slope of the relationship between ATPase activity and active tension measured at various pCa. ATPase vs active tension values were obtained from 10 wild‐type and 8 E163R trabeculae. B, Mean slopes of the force‐ATPase relations in the 2 groups.
Figure S5. Steady‐state ATP hydrolysis from myofibril suspensions in relaxing solution. A, Representative ATPase activity for wild‐type (WT; black square) and E163R (red square) myofibrils measured in relaxing condition using malachite green phosphate assay and normalized by myosin head concentration. Each point is the mean±SEM of 2 measurements from the same batch of myofibrils. B, Average slope of regression lines (s−1) for WT (n=6 hearts) and E163R (n=2 hearts) myofibrils.
Figure S6. Intracellular Ca2+ handling in transgenic mice of different ages. A, Representative superimposed Ca transients elicited at 1 Hz from myocytes of 3‐month‐old wild‐type (WT) and E163R mice (left), as well as WT and R92Q mice (right). B, Amplitude of Ca transients at 1 Hz (left), 3 Hz (center), and 5 Hz (right) in cells from WT, E163R, and R92Q mice of differente ages (3 months, 6 months, and 12 months). C, Diastolic [Ca] during steady‐state stimulation at 1 Hz (left), 3 Hz (center), and 5 Hz (right) in cells from WT, E163R, and R92Q mice of differente ages (3 months, 6 months, and 12 months). D, Kinetics of Ca transients elicited at 1 Hz in cells from mice of different ages. Time to peak (TTP) on the left, time to 50% decay (center), and time from peak to 90% decay (right). E, Representative Ca fluorescence traces of the protocol used to elicit spontaneous Ca‐release events from 3‐month‐old mice: R92Q above and E163R below. F, Frequency of spontaneous Ca‐release events during pauses in mice of different ages: Ca waves above and Ca transients below. (B through D and F) 3 Months: means±SEM from 38 WT (2 mice), 44 E163R (3 mice), and 39 R92Q (2 mice) cells; 6 months: 65 WT (3 mice), 65 E163R (4 mice), and 55 R92Q (3 mice) cells; 12 months: 55 WT (3 mice), 41 E163R (2 mice), and 53 R92Q (3 mice) cells. Statistics: linear mixed models Tukey correction.
(J Am Heart Assoc. 2017;6:e005407 DOI: 10.1161/JAHA.116.005407.)28735292
References
- 1. Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254. [DOI] [PubMed] [Google Scholar]
- 2. Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck‐Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res. 2015;105:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Patterns of disease progression in hypertrophic cardiomyopathy: an individualized approach to clinical staging. Circ Heart Fail. 2012;5:535–546. [DOI] [PubMed] [Google Scholar]
- 4. Olivotto I, Girolami F, Nistri S, Rossi A, Rega L, Garbini F, Grifoni C, Cecchi F, Yacoub MH. The many faces of hypertrophic cardiomyopathy: from developmental biology to clinical practice. Cardiovasc Transl Res. 2009;2:349–367. [DOI] [PubMed] [Google Scholar]
- 5. Olivotto I, d'Amati G, Basso C, Van Rossum A, Patten M, Emdin M, Pinto Y, Tomberli B, Camici PG, Michels M. Defining phenotypes and disease progression in sarcomeric cardiomyopathies: contemporary role of clinical investigations. Cardiovasc Res 2015;105:409–423. [DOI] [PubMed] [Google Scholar]
- 6. Maron MS, Rowin EJ, Olivotto I, Casey SA, Arretini A, Tomberli B, Garberich RF, Link MS, Chan RH, Lesser JR, Maron BJ. Contemporary natural history and management of nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2016;67:1399–1409. [DOI] [PubMed] [Google Scholar]
- 7. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. [DOI] [PubMed] [Google Scholar]
- 8. Poggesi C, Ho CY. Muscle dysfunction in hypertrophic cardiomyopathy: what is needed to move to translation? J Muscle Res Cell Motil. 2014;35:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belus A, Piroddi N, Scellini B, Tesi C, Amati GD, Girolami F, Yacoub M, Cecchi F, Olivotto I, Poggesi C. The familial hypertrophic cardiomyopathy‐associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol. 2008;586:3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferrantini C, Belus A, Piroddi N, Scellini B, Tesi C, Poggesi C. Mechanical and energetic consequences of HCM‐causing mutations. Cardiovasc Transl Res. 2009;2:441–451. [DOI] [PubMed] [Google Scholar]
- 11. Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, Maier LS, Ashrafian H, Huke S, van der Velden J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res. 2015;105:457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Witjas‐Paalberends ER, Ferrara C, Scellini B, Piroddi N, Montag J, Tesi C, Stienen GJ, Michels M, Ho CY, Kraft T, Poggesi C, van der Velden J. Faster cross‐bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J Physiol. 2014;592:3257–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Javadpour MM, Tardiff JC, Pinz I, Ingwall JS. Decreased energetics in murine hearts bearing the R92Q mutation in cardiac troponin T. J Clin Investig. 2003;112:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Witjas‐Paalberends ER, Guclu A, Germans T, Knaapen P, Harms HJ, Vermeer AM, Christiaans I, Wilde AA, Dos Remedios C, Lammertsma AA, van Rossum AC, Stienen GJ, van Slegtenhorst M, Schinkel AF, Michels M, Ho CY, Poggesi C, van der Velden J. Gene‐specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc Res. 2014;103:248–257. [DOI] [PubMed] [Google Scholar]
- 15. Hoskins AC, Jacques A, Bardswell SC, McKenna WJ, Tsang V, dos Remedios CG, Ehler E, Adams K, Jalilzadeh S, Avkiran M, Watkins H, Redwood C, Marston SB, Kentish JC. Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2010;49:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in troponin that cause HCM, DCM and RCM: what can we learn about thin filament function? J Mol Cell Cardiol. 2010;48:882–892. [DOI] [PubMed] [Google Scholar]
- 17. Sequeira V, Wijnker PJ, Nijenkamp LL, Kuster DW, Najafi A, Witjas‐Paalberends ER, Regan JA, Boontje N, Ten Cate FJ, Germans T, Carrier L, Sadayappan S, van Slegtenhorst MA, Zaremba R, Foster DB, Murphy AM, Poggesi C, Dos Remedios C, Stienen GJ, Ho CY, Michels M, van der Velden J. Perturbed length‐dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ Res. 2013;112:1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crocini C, Ferrantini C, Scardigli M, Coppini R, Mazzoni L, Lazzeri E, Pioner JM, Scellini B, Guo A, Song LS, Yan P, Loew LM, Tardiff J, Tesi C, Vanzi F, Cerbai E, Pavone FS, Sacconi L, Poggesi C. Novel insights on the relationship between T‐tubular defects and contractile dysfunction in a mouse model of hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2016;91:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lan F, Lee AS, Liang P, Sanchez‐Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT, Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient‐specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haim TE, Dowell C, Diamanti T, Scheuer J, Tardiff JC. Independent fhc‐related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J Mol Cell Cardiol. 2007;42:1098–1110. [DOI] [PubMed] [Google Scholar]
- 21. Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E, Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–584. [DOI] [PubMed] [Google Scholar]
- 23. Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, Moore RL, Robbins J, Leinwand LA. Cardiac troponin T mutations result in allele‐specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J Clin Investig. 1999;104:469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moore RK, Abdullah S, Tardiff JC. Allosteric effects of cardiac troponin TNT1 mutations on actomyosin binding: a novel pathogenic mechanism for hypertrophic cardiomyopathy. Arch Biochem Biophys. 2014;552–553:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pistner A, Belmonte S, Coulthard T, Blaxall B. Murine echocardiography and ultrasound imaging. J Vis Exp. 2010;42:pii:2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferrantini C, Coppini R, Sacconi L, Tosi B, Zhang ML, Wang GL, de Vries E, Hoppenbrouwers E, Pavone F, Cerbai E, Tesi C, Poggesi C, ter Keurs HE. Impact of detubulation on force and kinetics of cardiac muscle contraction. J General Physiol. 2014;143:783–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferrantini C, Coppini R, Scellini B, Ferrara C, Pioner JM, Mazzoni L, Priori S, Cerbai E, Tesi C, Poggesi C. R4496c RYR2 mutation impairs atrial and ventricular contractility. J General Physiol. 2016;147:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chandra M, Rundell VL, Tardiff JC, Leinwand LA, De Tombe PP, Solaro RJ. Ca(2+) activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am J Physiol Heart Circ Physiol. 2001;280:H705–713. [DOI] [PubMed] [Google Scholar]
- 29. Belus A, Piroddi N, Ferrantini C, Tesi C, Cazorla O, Toniolo L, Drost M, Mearini G, Carrier L, Rossi A, Mugelli A, Cerbai E, van der Velden J, Poggesi C. Effects of chronic atrial fibrillation on active and passive force generation in human atrial myofibrils. Circ Res. 2010;107:144–152. [DOI] [PubMed] [Google Scholar]
- 30. Coppini R, Mazzoni L, Ferrantini C, Gentile F, Pioner JM, Laurino T, Santini L, Bargelli V, Rotellini M, Bartolucci G, Crocini C, Sacconi L, Tesi C, Belardinelli L, Tardiff J, Mugelli A, Olivotto I, Cerbai E, Poggesi C. Ranolazine prevents phenotype development in a mouse model of hypertrophic cardiomyopathy. Circ Heart Fail. 2017;10(3). pii: e003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Merx MW, Gorressen S, van de Sandt AM, Cortese‐Krott MM, Ohlig J, Stern M, Rassaf T, Godecke A, Gladwin MT, Kelm M. Depletion of circulating blood nos3 increases severity of myocardial infarction and left ventricular dysfunction. Basic Res Cardiol. 2014;109:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Briston SJ, Dibb KM, Solaro RJ, Eisner DA, Trafford AW. Balanced changes in Ca buffering by SERCA and troponin contribute to Ca handling during beta‐adrenergic stimulation in cardiac myocytes. Cardiovasc Res. 2014;104:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maier LS, Bers DM, Pieske B. Differences in Ca(2+)‐handling and sarcoplasmic reticulum Ca(2+)‐content in isolated rat and rabbit myocardium. J Mol Cell Cardiol. 2000;32:2249–2258. [DOI] [PubMed] [Google Scholar]
- 34. Roe AT, Frisk M, Louch WE. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr Pharm Des. 2015;21:431–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause‐dependent Ca‐triggered arrhythmia. Circ Res. 2012;111:170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo A, Song LS. AutoTT: automated detection and analysis of T‐tubule architecture in cardiomyocytes. Biophys J. 2014;106:2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Helms AS, Alvarado FJ, Yob J, Tang VT, Pagani F, Russell MW, Valdivia HH, Day SM. Genotype‐dependent and ‐independent calcium signaling dysregulation in human hypertrophic cardiomyopathy. Circulation. 2016;134:1738–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jimenez J, Tardiff JC. Abnormal heart rate regulation in murine hearts with familial hypertrophic cardiomyopathy‐related cardiac troponin T mutations. Am J Physiol Heart Circ Physiol. 2011;300:H627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He H, Hoyer K, Tao H, Rice R, Jimenez J, Tardiff JC, Ingwall JS. Myosin‐driven rescue of contractile reserve and energetics in mouse hearts bearing familial hypertrophic cardiomyopathy‐associated mutant troponin T is mutation‐specific. J Physiol. 2012;590:5371–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. [DOI] [PubMed] [Google Scholar]
- 41. Bai F, Weis A, Takeda AK, Chase PB, Kawai M. Enhanced active cross‐bridges during diastole: molecular pathogenesis of tropomyosin's HCM mutations. Biophys J. 2011;100:1014–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janco M, Kalyva A, Scellini B, Piroddi N, Tesi C, Poggesi C, Geeves MA. Alpha‐tropomyosin with a D175N or E180G mutation in only one chain differs from tropomyosin with mutations in both chains. Biochemistry. 2012;51:9880–9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ly S, Lehrer SS. Long‐range effects of familial hypertrophic cardiomyopathy mutations E180G and D175N on the properties of tropomyosin. Biochemistry. 2012;51:6413–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J Biol Chem. 2004;279:14488–14495. [DOI] [PubMed] [Google Scholar]
- 45. Manning EP, Guinto PJ, Tardiff JC. Correlation of molecular and functional effects of mutations in cardiac troponin T linked to familial hypertrophic cardiomyopathy: an integrative in silico/in vitro approach. J Biol Chem. 2012;287:14515–14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Manning EP, Tardiff JC, Schwartz SD. Molecular effects of familial hypertrophic cardiomyopathy‐related mutations in the TNT1 domain of cTnT. J Mol Biol. 2012;421:54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ertz‐Berger BR, He H, Dowell C, Factor SM, Haim TE, Nunez S, Schwartz SD, Ingwall JS, Tardiff JC. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc Natl Acad Sci USA. 2005;102:18219–18224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Authors/Task Force Members ; Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 49. Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of T‐tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci USA. 2009;106:6854–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T‐tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Swaminathan PD, Purohit A, Hund TJ, Anderson ME. Calmodulin‐dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kreusser MM, Backs J. Integrated mechanisms of camkii‐dependent ventricular remodeling. Front Pharmacol. 2014;5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, Tian X, Jones PP, O'Mara ML, Liu Y, Mi T, Zhang L, Bolstad J, Semeniuk L, Cheng H, Zhang J, Chen J, Tieleman DP, Gillis AM, Duff HJ, Fill M, Song LS, Chen SR. The ryanodine receptor store‐sensing gate controls Ca2+ waves and Ca2+‐triggered arrhythmias. Nat Med. 2014;20:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, Seidman CE. Mutations in the genes for cardiac troponin T and alpha‐tropomyosin in hypertrophic cardiomyopathy. New Engl J Med. 1995;332:1058–1064. [DOI] [PubMed] [Google Scholar]
- 56. Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H. Sudden death due to troponin T mutations. J Am Coll Cardiol. 1997;29:549–555. [DOI] [PubMed] [Google Scholar]
- 57. Koga Y, Toshima H, Kimura A, Harada H, Koyanagi T, Nishi H, Nakata M, Imaizumi T. Clinical manifestations of hypertrophic cardiomyopathy with mutations in the cardiac beta‐myosin heavy chain gene or cardiac troponin T gene. J Cardiac Fail. 1996;2:S97–103. [DOI] [PubMed] [Google Scholar]
- 58. Ho CY, Lakdawala NK, Cirino AL, Lipshultz SE, Sparks E, Abbasi SA, Kwong RY, Antman EM, Semsarian C, Gonzalez A, Lopez B, Diez J, Orav EJ, Colan SD, Seidman CE. Diltiazem treatment for pre‐clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail. 2015;3:180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, Quinones MA, Zoghbi WA, Entman ML, Roberts R, Marian AJ. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lim DS, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, Roberts R, Marian AJ. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nagueh SF, Lombardi R, Tan Y, Wang J, Willerson JT, Marian AJ. Atorvastatin and cardiac hypertrophy and function in hypertrophic cardiomyopathy: a pilot study. Eur J Clin Invest. 2010;40:976–983. [DOI] [PubMed] [Google Scholar]
- 62. Axelsson A, Iversen K, Vejlstrup N, Ho C, Norsk J, Langhoff L, Ahtarovski K, Corell P, Havndrup O, Jensen M, Bundgaard H. Efficacy and safety of the angiotensin ii receptor blocker losartan for hypertrophic cardiomyopathy: the inherit randomised, double‐blind, placebo‐controlled trial. Lancet Diabetes Endocrinol. 2015;3:123–131. [DOI] [PubMed] [Google Scholar]
- 63. Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG, Seidman CE. A small‐molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351:617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pioner JM, Racca AW, Klaiman JM, Yang KC, Guan X, Pabon L, Muskheli V, Zaunbrecher R, Macadangdang J, Jeong MY, Mack DL, Childers MK, Kim DH, Tesi C, Poggesi C, Murry CE, Regnier M. Isolation and mechanical measurements of myofibrils from human induced pluripotent stem cell‐derived cardiomyocytes. Stem Cell Reports. 2016;6:885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods
Figure S1. Steady‐state contractions with isoproterenol. A, Steady‐state 1‐Hz twitch tension at basal conditions and in the presence of isoproterenol 10 to 7 mol/L. B, Time to peak and time from peak to 50% relaxation (R50%, right), measured in steady‐state twitches at different stimulation frequencies at basal conditions and in the presence of isoproterenol 10−7 mol/L.
Figure S2. Alterations of T‐tubules. A, Representative confocal images from isolated left ventricular (LV) cardiomyocytes stained with di‐3‐aneppdhq from wild‐type (WT; right), R92Q (middle), and E163R (right) hearts. The horizontal bar equals 10 μm. B, T‐tubule power, as calculated using the TTorg ImageJ plugin, in cardiomyocytes from the 3 study groups. C, Mean T‐tubule period (equivalent to sarcomere length) in resting LV cardiomyocytes from the 3 cohorts of mice. (B through C) Means±SE from 68 WT (4 mice), 142 R92Q (6 mice), and 69 E163R (4 mice) cardiomyocytes. Statistics: 1‐way ANOVA with Tukey correction. *P<0.05.
Figure S3. Additional protein studies. A, Representative Western blots for total SERCA, phospholamban (PLB), phosphorylated PLB at Serine 16 (P‐PLB(S16)), and GAPDH. B, Average relative intensity values of SERCA, PLB, SERCA/PLB, and p‐PLB(S16)/PLB ratios in wild‐type (WT), R92Q, and E163R hearts (8 samples for each group). C, Representative Western blots for total cardiac troponin I (cTnI), phosphorylated cTnI (P‐cTnI), total nuclear factor of activated T cell (NFATc1), phosphorylated NFATc1 (P‐NFATc1), and GAPDH. D, Average relative intensity values of cTnI, P‐cTnI, NFATc1, and P‐NFATc1 in WT, R92Q, and E163R hearts (5 samples for each group). (B through D) The intensity of individual SERCA bands was quantified and normalized to that of GAPDH. Relative intensities of WT were set at 1. Statistics: 1‐way ANOVA with Tukey correction. *P<0.05. **P<0.01.
Figure S4. Effect of the E163R mutation on the relationship between steady‐state isometric force and ATPase activity. A, TC was determined from the slope of the relationship between ATPase activity and active tension measured at various pCa. ATPase vs active tension values were obtained from 10 wild‐type and 8 E163R trabeculae. B, Mean slopes of the force‐ATPase relations in the 2 groups.
Figure S5. Steady‐state ATP hydrolysis from myofibril suspensions in relaxing solution. A, Representative ATPase activity for wild‐type (WT; black square) and E163R (red square) myofibrils measured in relaxing condition using malachite green phosphate assay and normalized by myosin head concentration. Each point is the mean±SEM of 2 measurements from the same batch of myofibrils. B, Average slope of regression lines (s−1) for WT (n=6 hearts) and E163R (n=2 hearts) myofibrils.
Figure S6. Intracellular Ca2+ handling in transgenic mice of different ages. A, Representative superimposed Ca transients elicited at 1 Hz from myocytes of 3‐month‐old wild‐type (WT) and E163R mice (left), as well as WT and R92Q mice (right). B, Amplitude of Ca transients at 1 Hz (left), 3 Hz (center), and 5 Hz (right) in cells from WT, E163R, and R92Q mice of differente ages (3 months, 6 months, and 12 months). C, Diastolic [Ca] during steady‐state stimulation at 1 Hz (left), 3 Hz (center), and 5 Hz (right) in cells from WT, E163R, and R92Q mice of differente ages (3 months, 6 months, and 12 months). D, Kinetics of Ca transients elicited at 1 Hz in cells from mice of different ages. Time to peak (TTP) on the left, time to 50% decay (center), and time from peak to 90% decay (right). E, Representative Ca fluorescence traces of the protocol used to elicit spontaneous Ca‐release events from 3‐month‐old mice: R92Q above and E163R below. F, Frequency of spontaneous Ca‐release events during pauses in mice of different ages: Ca waves above and Ca transients below. (B through D and F) 3 Months: means±SEM from 38 WT (2 mice), 44 E163R (3 mice), and 39 R92Q (2 mice) cells; 6 months: 65 WT (3 mice), 65 E163R (4 mice), and 55 R92Q (3 mice) cells; 12 months: 55 WT (3 mice), 41 E163R (2 mice), and 53 R92Q (3 mice) cells. Statistics: linear mixed models Tukey correction.