

Abstract
Background
Cardiac mesenchymal cell (CMC) administration improves cardiac function in animal models of heart failure. Although the precise mechanisms remain unclear, transdifferentiation and paracrine signaling are suggested to underlie their cardiac reparative effects. We have shown that histone deacetylase 1 (HDAC1) inhibition enhances CMC cardiomyogenic lineage commitment. Here, we investigated the impact of HDAC1 on CMC cytokine secretion and associated paracrine‐mediated activities on endothelial cell function.
Methods and Results
CMCs were transduced with shRNA constructs targeting HDAC1 (shHDAC1) or nontarget (shNT) control. Cytokine arrays were used to assess the expression of secreted proteins in conditioned medium (CM) from shHDAC1 or shNT‐transduced CMCs. In vitro functional assays for cell proliferation, protection from oxidative stress, cell migration, and tube formation were performed on human endothelial cells incubated with CM from the various treatment conditions. CM from shHDAC1‐transduced CMCs contained more cytokines involved in cell growth/differentiation and more efficiently promoted endothelial cell proliferation and tube formation compared with CM from shNT. After evaluating key cytokines previously implicated in cell‐therapy–mediated cardiac repair, we found that basic fibroblast growth factor was significantly upregulated in shHDAC1‐transduced CMCs. Furthermore, shRNA‐mediated knockdown of basic fibroblast growth factor in HDAC1‐depleted CMCs inhibited the effects of shHDAC1 CM in promoting endothelial proliferation and tube formation—indicating that HDAC1 depletion activates CMC proangiogenic paracrine signaling in a basic fibroblast growth factor–dependent manner.
Conclusions
These results reveal a hitherto unknown role for HDAC1 in the modulation of CMC cytokine secretion and implicate the targeted inhibition of HDAC1 in CMCs as a means to enhance paracrine‐mediated neovascularization in cardiac cell therapy applications.
Keywords: cardiac mesenchymal stromal cells, histone deacetylase inhibitors, histone deacetylases, paracrine signaling
Subject Categories: Basic Science Research, Growth Factors/Cytokines, Angiogenesis, Cell Therapy, Ischemia
Clinical Perspective
What Is New?
The current study establishes a heretofore unknown role for histone deacetylase 1 in the modulation of cardiac mesenchymal stromal cell trophic factor secretion and paracrine signaling activities.
shRNA‐mediated depletion of histone deacetylase 1 boosts human cardiac mesenchymal stromal cell proangiogenic paracrine signaling in vitro by augmenting the synthesis and secretion of basic fibroblast growth factor.
What Are the Clinical Implications?
These results highlight histone deacetylase 1 as a unique target that could be exploited by pharmacological or genetics means to enhance the therapeutic efficacy of cardiac mesenchymal stromal cells in cell therapy applications.
Introduction
Adoptive transfer of cardiac mesenchymal stromal cells (CMCs) improves cardiac function in animal models of post–myocardial infarction heart failure.1, 2 Although the precise mechanisms underlying their therapeutic benefits remain unclear, both transdifferentiation3 (ie, directly contributing to the generation of vascular endothelium and/or cardiomyocytes) and secretion of paracrine signaling molecules2 (ie, promoting endogenous vasculogenesis and/or myocyte proliferation) have been proposed to underlie the mechanisms of cell therapy. Given that donor cell populations, irrespective of cell type, exhibit poor rates of retention and limited long‐term persistence after delivery in heart failure,4, 5, 6, 7 paracrine signaling is viewed as a major mechanism of cell‐mediated cardiac repair. Donor cell differentiation and paracrine signaling, however, may be related to one another because secretome constituents, including proteinaceous trophic factors, as well as extracellular vesicle contents, may exhibit cell lineage dependency.8, 9 Thus, donor cell lineage specification as well as the magnitude of cardiovascular lineage commitment after delivery could have pronounced consequences on their paracrine signaling activities and, in turn, their cardiac reparative capacity—a notion that has received little attention.
Donor cell cardiovascular lineage commitment may be related to therapeutic efficacy.3, 10, 11 For instance, lineage‐specific depletion of transplanted bone marrow mononuclear cells expressing endothelial nitric oxide synthase inhibits their therapeutic benefits in an acute myocardial infarction model.10 The idea that cardiovascular lineage commitment is an important property influencing donor cell cardiac reparative capacity is also supported by other studies showing that the expression of cardiogenic transcription factors (ie, Nkx2.5, Tbx5, and Mef2c) in donor cell populations (eg, bone‐marrow–derived mesenchymal stem cells11 and CMCs)3 biases cardiac repair; however, the fundamental underlying mechanisms of this phenomenon remain unknown. It is known that different cell types possess distinct trophic factor secretion profiles and paracrine signaling potencies.8, 9 Thus, we posited that donor cell cardiomyogenic lineage commitment may regulate the expression and synthesis of cardiotrophic factors, which could have pronounced effects on their ability to mediate cardiac repair.
Many of the aforementioned studies3, 10, 11 support the development of effective strategies to augment donor cell cardiovascular lineage commitment with the ultimate goal of improving cardiac reparative potential. Various approaches, including ectopic expression of cardiogenic transcription factors (eg, GMT: Gata4, Mef2c, and Tbx5)12 and exposure to chromatin modifying agents (eg, inhibitors of histone deacetylase13, 14 and DNA methyltransferase) activity,15, 16, 17 have been utilized to augment cardiomyogenic lineage commitment of mesenchymal progenitor cells. We recently identified histone deacetylase 1 (HDAC1) as a novel regulator of CMC cell fate decisions.14 Therein, we found that exposure to either the pan HDAC inhibitor, sodium butyrate, or short‐hairpin RNA interference (shRNAi)‐mediated depletion of HDAC1 potentiates the activation of a core cardiogenic transcriptional program in human CMCs—an effect that correlated with heightened competency of these cells to assume a cardiomyogenic‐like phenotype in vitro.14 In the current study, we sought to investigate the effects of augmented cardiomyogenic lineage commitment, through HDAC1 depletion on CMC cytokine secretion patterns and its associated consequences on CMC paracrine signaling in vitro.
Methods
Cells, Cell Culture, and Treatment
Primary patient‐derived CMCs were propagated in Ham's F12 medium (Gibco, Grand Island, NY) supplemented with 10% FBS (Seradigm, Radnor, PA), 20 ng/mL of recombinant human basic fibroblast growth factor (bFGF; PeproTech, Rocky Hill, NJ), 0.2 mmol/L of l‐glutamine (Gibco), 0.005 U/mL of human erythropoietin (Invitrogen, Carlsbad, CA), and 100 U/mL of penicillin/streptomycin (Gibco). CMCs utilized in experiments were not used beyond passage 8. HEK293FT cells (Life Technologies, Carlsbad, CA) used in lentiviral production were grown in DMEM medium (Gibco) containing 10% FBS (Seradigm) and 0.2 mmol/L of l‐glutamine (Gibco). Human aortic endothelial cells (HAECs; ThermoFisher, Waltham, MA) and human umbilical vein endothelial cells (HUVECs; ThermoFisher) were propagated in Phenol Red–Free medium 200 (200PRF; Gibco) supplemented with Low Serum Growth Supplement (LSGS; ThermoFisher) per the manufacturer's instructions. All cell lines were maintained under standard incubation conditions at 37°C with 5% atmospheric CO2 and passaged using TrypLE (ThermoFisher) when approaching ≈70% confluence.
Human cardiac mesenchymal stromal cell isolation
Human CMCs were isolated from discarded right atrial appendage specimens collected from patients during routine coronary artery bypass surgery at Jewish Hospital (University of Louisville, Louisville, KY) according to a previously established collagenase digestion protocol.14 De‐identified right atrial appendage specimens were acquired by written consent agreement according to the approved protocol by the Institutional Review Board on human subject research (Institutional Review Board number: 03.052J) at the University of Louisville. Assay experimental units (“n” numbers), where provided, denote the authentic number of biological replicates (each of which consists of CMCs sourced from different patients) used for experimentation.
shRNA‐mediated HDAC knockdown
Mission shRNA (Sigma‐Aldrich, St. Louis, MO) lentiviral particles were derived using the ViraPower Lentiviral Expression System (Life Technologies) per the manufacturer's protocol. Approximately 25 000 CMCs were plated per well of 6‐well tissue culture plates in Ham's F12 complete medium before cell transduction. At 24 hours, viral supernatants containing 8 μg/mL of polybrene were added to cells and incubated for an additional 24 hours, at which time viral medium was subsequently replaced with Ham's F12 complete medium. Seventy‐two hours after initial viral particle exposure, cell transduction efficiency was assessed by fluorescent‐mediated detection of green fluorescent protein (GFP) in cells transduced with MISSION pLKO.1‐puro‐CMV‐TurboGFP Positive Control Vector (SHC003; Sigma‐Aldrich). A complete list of shRNA vectors and shRNA‐scramble controls utilized in experiments is available in Table S1.
Flow Cytometry
Lentiviral transduced CMCs were analyzed using a BD LSR II Flow Cytometer (BD Biosciences, San Jose, CA). Fifty thousand events were collected per sample and analyzed using Flowing Software (version 2.5.0; Perttu Terho, Turku Centre for Biotechnology, Turku, Finland). Live cells were gated according to light scatter characteristics and resultant population FITC fluorescence intensity plotted on dot plots. The percentage of FITC‐positive cells (corresponding to pLKO.1‐puro‐CMV‐TurboGFP lentiviral transduction efficiency) was determined by gating against untransduced (autofluorescence controls) negative control CMCs.
Isolation of Conditioned Medium From shRNA‐Transduced CMCs
Before shRNA transduction, ≈5×105 CMCs were preplated in 75‐cm tissue culture dishes containing Ham's F12 complete medium and incubated at 37°C for 24 hours. Subsequently, CMCs were incubated with viral supernatant (short hairpin RNA nontarget [shNT] or shHDAC1) containing 8 μg/mL of polybrene for 24 hours. Viral medium was then replaced with Ham's F12 complete medium and incubated for an additional 24 hours. Next, transduced cells were washed, counted, and replated (1×106 CMCs total) in 75‐cm tissue culture dishes containing Ham's F12 base medium without supplements. Following 24 hours, the resultant conditioned medium was transferred to conical tubes, centrifuged at 300g for 5 minutes (to pellet any remaining cells and/or debris), and decanted. Conditioned medium was immediately used in experiments or stored at −20°C for later use.
Cytokine Array Assays
Cytokine arrays were generated using the Proteome Profiler Human XL Cytokine Array Kit (R&D Systems, Minneapolis, MN), according to the manufacturer's protocol. For each assay, a total of 500 μL of nonconcentrated cell culture supernatant/condition medium was used. Resultant membranes were imaged with a chemiluminescent image analyzer (MyECL; ThermoFisher Scientific) and analyzed by densitometric quantification (ImageJ; NIH, Bethesda, MD). A total of 4 independent samples (1:3 shNT, 1:3 shHDAC1, 1:10 shNT, and 1:10 shHDAC1) were subject to cytokine array analysis (n=1 for each).
Cell Migration Assays
Approximately 5×104 HAECs were seeded in the upper chamber of 8.0‐μm pore polycarbonate membrane inserts (Transwell; Corning, Corning, NY) containing 100 μL of maintenance medium (15.4 μL of Ham's F12 base media and 84.6 μL of HAEC 200PRF medium containing LSGS). In the bottom chamber of the Transwell, 650 μL of migration medium (100 μL of conditioned medium and 550 μL of 200PRF medium containing LSGS) were added. Cells were then incubated overnight at 37°C. The following day, cells were fixed with 4% formaldehyde for 10 minutes, washed twice with 1× PBS, and stained with 0.1% crystal violet in 20% methanol for 10 minutes. Membrane inserts were then washed and unmigrated cells cleared from the top of the membrane with cotton swabs. Migrated cells (located at the bottom of membranes) were then imaged on an inverted microscope and enumerated using ImageJ software (NIH).
Oxidative Stress Cell Survival Assays
HAECs were seeded at a density of 2×103 cells per well of 96‐well tissue culture plates. Individual wells contained 90 μL of conditioned medium sourced from untransduced, shNT, or shHDAC1 transduced CMCs. HAECs were incubated with conditioned medium for 24 hours at 37°C. After incubation, conditioned medium was replaced with respective conditioned medium containing increasing concentrations of hydrogen peroxide (0, 0.1, 0.2, 0.3, or 0.5 mmol/L). Following 1‐hour incubation at 37°C, the peroxide medium was replaced with respective conditioned medium for an additional 24 hours. Quantity of surviving cells was evaluated by measuring the formation of a fluorescent product following the addition of a metabolically reducible substrate (PrestoBlue reagent; ThermoFisher), according to the manufacturer's instruction. Fluorescence measurements were taken in triplicate for each of 6 independent experiments. The relative number of viable cells remaining at corresponding concentrations of peroxide was calculated as follows:
Growth Assays
Approximately 3×103 HAECs were seeded per well of 96‐well tissue culture plates. Individual wells contained 90 μL of conditioned medium (from untransduced, shNT, or shHDAC1 transduced CMCs) supplemented with 10% FBS. HAECs were incubated at 37°C for 72 hours with daily media changes. After 72 hours, cell growth was assessed by measuring the formation of fluorescent product after the addition of PrestoBlue reagent (ThermoFisher), according to the manufacturer's instruction. Fluorescent measurements were taken in sextuplicate for each of 6 independent experiments. The relative number of viable cells was calculated for each group:
Tube Formation Assays
Geltrex basement membrane (Invitrogen) was added to 24‐well plates (100 μL per well) and allowed to solidify at 37°C for 30 minutes. Next, 7.5×104 HUVECs were suspended in 400 μL of conditioned medium and pipetted atop Geltrex coated wells. After 4 hours of incubation at 37°C, 2 μg/mL of calcein red‐orange (Invitrogen) was added to each well and incubated for 30 minutes at 37°C. Calcein red‐orange stain was then replaced with respective conditioned medium and incubated for 10 minutes at 37°C before fluorescent microscopy imaging. ImageJ software (NIH) was used to enumerate resultant tubes per image field.
qPCR
RNA isolation, reverse transcription, and qPCR was performed as previously described.14 Gene‐specific primers with respective annealing temperatures are listed in Table S2.
Western Blotting
Immunoblotting was performed according to the previously described protocol.14 A detailed list of antibodies with corresponding dilutions is available in Table S3.
Statistical Analyses
Statistical analyses were performed, as previously described18, 19, 20 using the GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla California USA, www.graphpad.com). Flow cytometry data (reporting percentages) were arcsine transformed and subject to 1‐way ANOVA with the post hoc Bonferroni multiple comparison test. Gene expression data were log base 10 (y=log10 y) transformed before performing 1‐way or 2‐way ANOVA with the post hoc Bonferroni multiple comparison test. Immunoblot densitometric data were log base 10 (y=log10 y) transformed before performing 1‐way ANOVA and the post hoc Bonferroni or Holm–Sidak multiple comparison test. Growth assay data with n<10 were analyzed by the Kruskal–Wallis test and P values determined using a post hoc uncorrected Dunn's test. Growth assay data n>10 used a 1‐way ANOVA followed by the post hoc Dunnett multiple comparison test. Enumerated tube formation data were subject to 1‐way ANOVA followed by the post hoc Dunnett multiple comparison test. Data are reported as means±SEM.
Results
HDAC1 Depletion Results in Pronounced Alterations in Human CMC Cytokine Secretion Patterns
We hypothesized that methods promoting donor cell cardiovascular lineage commitment will influence CMC cardiotrophic factor secretion and paracrine signaling potency. To begin to address this hypothesis, we sought to first evaluate the consequences of altered CMC cardiovascular lineage commitment, through shRNA‐mediated depletion of HDAC1,14 on CMC cytokine secretion patterns in vitro. Human CMCs were isolated from patient‐derived right atrial auricles using mechanical and enzymatic digestion procedures (Figure 1) and, subsequently, characterized by flow cytometric–mediated detection of prototypical mesenchymal markers, as previously shown (data not shown).14 Resultant CMCs were transduced with either an expression vector encoding an shRNA targeting HDAC1 or shNT using 2 different viral titer dilutions (1:3 or 1:10). At the same time, the efficiency of lentiviral transduction at corresponding viral dilutions was determined by transducing CMCs with a pLKO.1‐puro‐CMV‐TurboGFP control vector (Figure 2A). Approximately 72 hours after viral incubation, the percentage of GFP+ CMCs were enumerated by flow cytometry and showed an approximate transduction efficiency of 69±5.8% and 44±6.1% using 1:3 and 1:10 viral titer dilutions, respectively (untransduced versus 1:3 dilution, P=0.0004; untransduced versus 1:10 dilution, P=0.0071; 1:10 dilution versus 1:3 dilution, P=0.0383; Figure 2B and 2C). Next, the efficacy of HDAC1 knockdown was evaluated by qPCR (Figure 2D) and immunoblot (Figure 2E and 2F). Both viral dilutions yielded significant, comparable reductions in HDAC1 mRNA (shNT 1:3 versus shHDAC1 1:3, P<0.0001; shNT 1:10 versus shHDAC1 1:10, P=0.0425) and protein expression levels (shNT 1:3 versus shHDAC1 1:3, P=0.0002; shNT 1:10 versus shHDAC1 1:10, P<0.0001) relative to their respective shNT‐transduced controls (Figure 2D through 2F). After validating the extent of HDAC1 knockdown, we performed a cytokine proteome array (R&D Systems) to comprehensively evaluate trophic factor secretion (including cytokines, chemokines, and growth factors) in conditioned medium (CM) derived from cardiomyogenic committed (shHDAC1‐transduced) or naïve (shNT‐transduced) CMCs (Figure 3). HDAC1‐depleted CMCs exhibited markedly distinct cytokine secretion patterns relative to shNT‐transduced controls (Figure 3B and 3C), with pronounced differences in soluble factors involved in various biological processes including angiogenesis, apoptosis, cell survival, differentiation, growth, metabolism, and immune modulation. The most pronounced changes were those of paracrine factors involved in cell growth, differentiation, migration, and angiogenesis (summarized in Figure 3D). These observations suggest that HDAC1 depletion may afford CMCs enhanced paracrine signaling potency.
Figure 1.
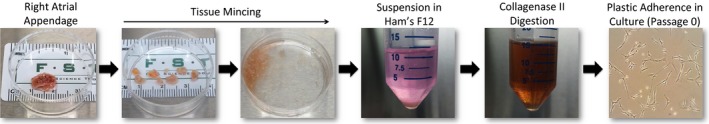
Human cardiac mesenchymal stromal cell isolation schematic.
Figure 2.
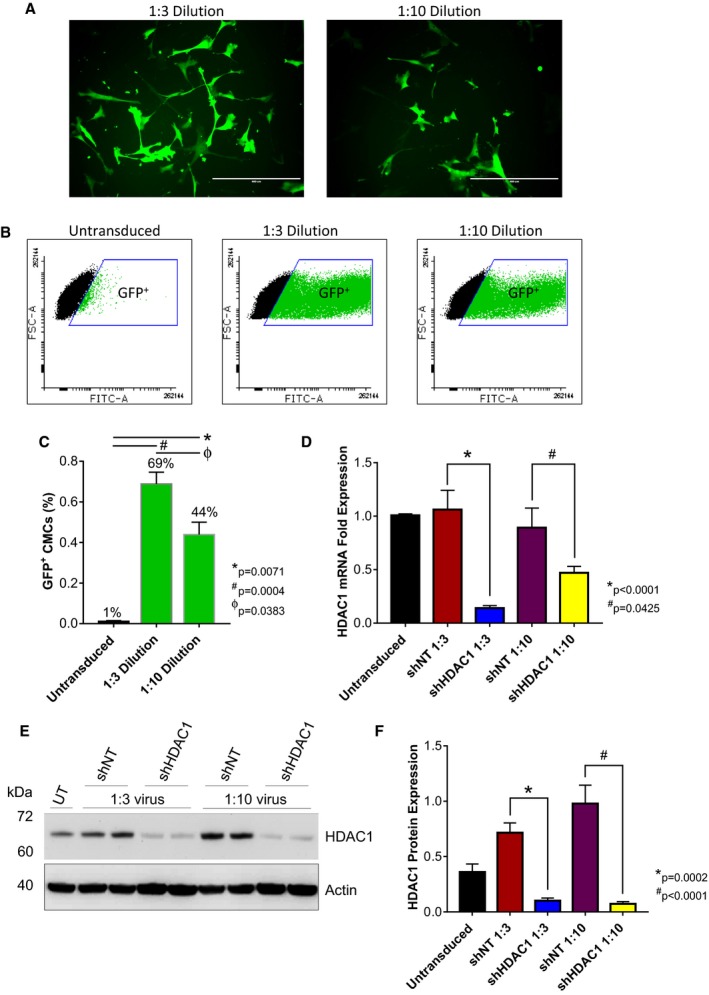
shRNA‐mediated depletion of HDAC1 in human CMCs. Representative (A) epifluorescence microscopy images (n=3; scale=400 μm) and (B and C) flow cytometric analysis (values are mean±SEM; n=3) of CMCs 72 hours after transduction with the MISSION pLKO.1‐puro‐CMV‐TurboGFP Positive Control Vector (1:3 or 1:10 viral titer dilutions). Flow cytometry data were arcsine‐transformed and analyzed by unpaired, 1‐way ANOVA. P values were calculated using the post hoc Bonferroni multiple comparison test. The efficacy of HDAC1 knockdown in CMCs assessed by (D) qPCR (values are mean±SEM; n=4) and (E) Western blot (representative image; n=4). F, Bar graph denoting densitometric quantification of resulting HDAC1 immunoblots. Values represent mean protein expression (relative to β‐actin)±SEM (n=4). qPCR and Western blot data were log base 10 (y=log10 y) transformed and analyzed by unpaired, 1‐way ANOVA. P values were calculated using the post hoc Bonferroni multiple comparison test. CMCs indicates cardiac mesenchymal stromal cells; FSC‐A, forward scatter pulse area; GFP, green fluorescent protein; HDAC1, histone deacetylase 1; shHDAC1, short hairpin RNA‐histone deacetylase 1; shNT, short hairpin RNA nontarget; UT, untransduced.
Figure 3.
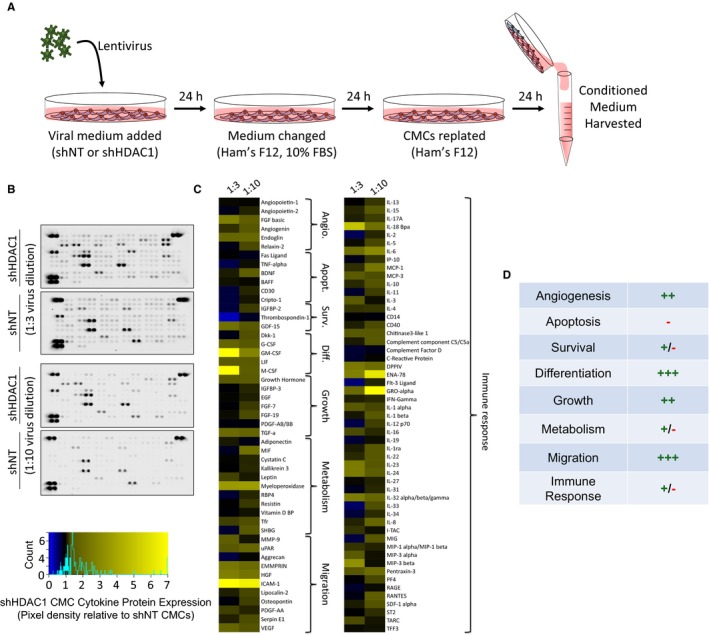
HDAC1 depletion alters human CMC cytokine secretion patterns. A, Schematic detailing the isolation of conditioned medium (CM) from virally transduced CMCs. CM was harvested from shRNA nontarget (shNT) or shRNA HDAC1 (shHDAC1) transduced CMCs 72 hours after lentiviral exposure. Lentivirus dilutions (1:3 or 1:10) were used. B, CM was incubated with cytokine array membranes (Proteome Profiler Human XL Cytokine Array Kit; R&D Systems, Minneapolis, MN) and detected using enhanced chemiluminescence. C, Proteome cytokine array heatmap illustrating post hoc densitometric quantification. Relative pixel density is shown (shHDAC1 relative to shNT controls). D, Cytokine expression changes stratified according to function. (+) denotes upregulation; (−) denotes downregulation. Angio. indicates angiogenesis; Apopt., apoptosis; CMCs, cardiac mesenchymal stromal cells; Diff., differentiation; HDAC1, histone deacetylase 1; shHDAC1, short hairpin RNA‐histone deacetylase 1; shNT, short hairpin RNA‐non target; Surv., survival; UT, untransduced.
Conditioned Medium From HDAC1‐Depleted CMCs Exhibit Elevated Paracrine Signaling Activity in In Vitro Assays of Endothelial Cell Function
The substantial and widespread alterations in CMC cytokine secretion patterns observed with HDAC1‐depletion (Figure 3) suggest the potential for significant modification of their paracrine signaling activities. Given that paracrine‐mediated induction of neovascularization has been implicated as a contributing component to the reparative effects of cell therapy in preclinical heart failure models21, 22, 23 and, further, that we observed a pronounced increase in the secretion of proangiogenic factors (eg, bFGF, angiogenin, and endoglin) from CMCs depleted of HDAC1 (Figure 3), we sought to investigate the consequences of HDAC1 knockdown‐mediated alterations in CMC trophic factor secretion on endothelial cell function. Accordingly, CM isolated from shHDAC, shNT, and untransduced CMCs were utilized in in vitro assays of endothelial cell migration, protection from peroxide‐induced oxidative stress, proliferation, and tube assembly (Figure 4). In cell migration assays, shHDAC1 CM did not more efficiently promote HAEC migration compared to CM from shNT (number of migrated cells, 1110±477 versus 452±151; shHDAC1 CM versus shNT, P=0.4221) or CM from untransduced cells (number of migrated cells, 1110±477 versus 141±32; shHDAC1 CM versus untransduced CM, P=0.1322; Figure 4A and 4B). Incubation with CM derived from shHDAC1‐transduced CMCs did not afford HAECs greater protection from peroxide‐induced oxidative stress than that of CM from shNT or untransduced controls (Figure 4C), at any investigated concentration (0.1–0.5 mmol/L H2O2). Contrary to the respective limited or nonexistence effects of CM on HAEC migration and oxidative stress, HAECs incubated with shHDAC1 CM exhibited an 82% increase in cell proliferative capacity compared with those treated with CM from shNT‐treated (relative number of viable cells, 2.0±0.26 versus 1.1±0.16; shHDAC1 versus shNT, P=0.0454) or untransduced cells (relative number of viable cells, 2.0±0.26 versus 0.95±0.15; shHDAC1 versus untransduced, P=0.0042; Figure 4D and 4E). In addition to enhancing endothelial cell proliferation, shHDAC1 CM efficiently promoted human umbilical vein endothelial cell (HUVEC) tube assembly in vitro, compared with CM from shNT or untransduced CMCs, which showed limited induction (Figure 4F and 4G). Specifically, shHDAC1 CM promoted a 225% to 260% increase in the number of enumerated tubes compared with that of shNT (number of tubes per field, 65±12 versus 18±5; shHDAC1 versus shNT, P=0.0028) or untransduced (number of tubes per field, 65±12 versus 20±6; shHDAC1 versus untransduced, P=0.0026) CMCs (Figure 4G). These data suggest that HDAC1 depletion boosts CMC paracrine signaling‐mediated activation of endothelial cell growth and differentiation.
Figure 4.
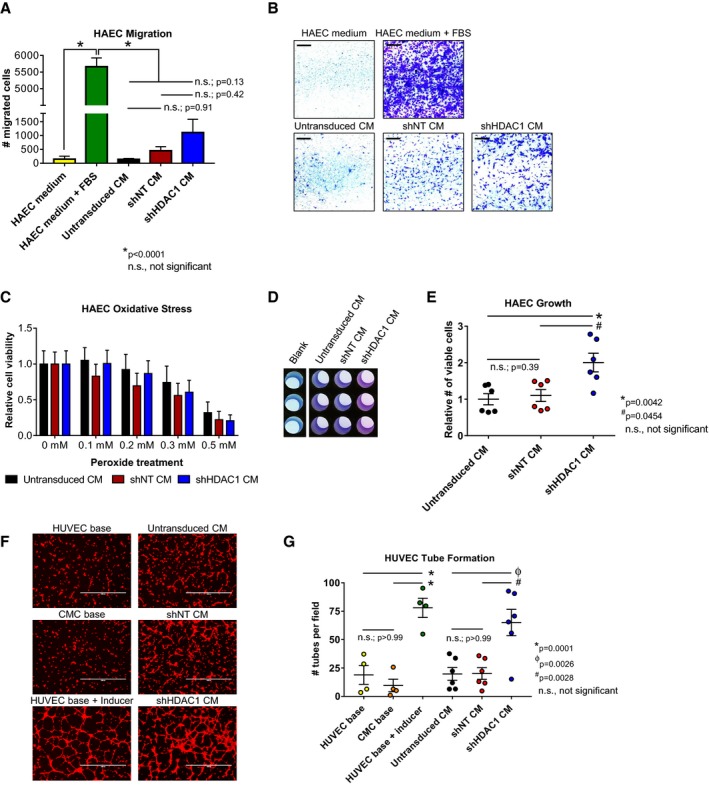
Conditioned medium from HDAC1‐depleted CMCs exhibit altered paracrine signaling potency in vitro. A and B, HAEC transwell migration assays in response to CM from shNT, shHDAC1, or untransduced CMCs. HAEC medium (−Ctrl) and HAEC medium+FBS (+Ctrl) are shown. Values are mean±SEM (n=3). Representative images of HAEC transwell migration assays (scale=1 mm) are shown in (B). Data were analyzed by unpaired, 1‐way ANOVA and P values determined using the post hoc Tukey multiple comparison test. C, Oxidative stress assays were performed on HAECs pretreated with CM (shNT, shHDAC1, or untransduced CMCs) by their incubation with increasing concentrations of H2O2. Relative cell viability is reported. Values are mean±SEM (n=6). Data were analyzed by 2‐way ANOVA and subject to post hoc analysis using the Tukey multiple comparison test. D and E, HAEC growth was assessed following their propagation in CM from shHDAC1, shNT, or untransduced CMCs. Representative images of CM‐treated HAECs incubated with PrestoBlue metabolizable fluorometric reagent are depicted in (D). Values are mean±SEM (n=6). Growth data were analyzed by the Kruskal–Wallis test and P values determined using a post hoc uncorrected Dunn's test. F, Representative fluorescent microscopy images (scale=1000 μm) and (G) graph enumerating HUVEC tube formation in response to incubation with CM. HUVEC base (−Ctrl), CMC base (−Ctrl), and HUVEC base+inducer (low serum growth supplement [LSGS]; +Ctrl) controls are shown (n=4 each). Values are mean±SEM (n=6 for experimental groups). Resultant tube formation data were analyzed by unpaired, 1‐way ANOVA and P values determined using the post hoc Tukey multiple comparison test. All experiments utilized 1:3 shRNA viral titer dilutions. CM indicates conditioned medium; CMC, cardiac mesenchymal stromal cell; HAEC, human aortic endothelial cell; HDAC1, histone deacetylase 1; HUVEC, human umbilical vein endothelial cell; shHDAC1, short hairpin RNA‐histone deacetylase 1; shNT, short hairpin RNA‐non target; UT, untransduced.
HDAC1 depletion augments bFGF expression in human CMCs
In an effort to identify those secreted factor(s) that may be responsible for the elevated paracrine signaling‐mediated activities of HDAC1‐depleted CMCs on endothelial cell function, we sought first to evaluate the expression of known secreted cytokines, which have been previously implicated in cell‐mediated cardiac repair and angiogenesis (angiopoietin 1, platelet‐derived growth factor, vascular endothelial growth factor, bFGF, hepatocyte growth factor, stem cell factor, and stromal cell‐derived factor‐1).24, 25, 26, 27 qPCR was used to assess the expression of these factors in total RNA isolates derived from shHDAC1, shNT, or untransduced CMCs (Figure 5A). Of the paracrine factors investigated, only bFGF exhibited a marked and statistically significant increase in shHDAC1‐depleted CMCs compared with shNT‐transduced CMCs (bFGF fold expression, 3.98±0.97 versus 1.13±0.09; shHDAC1 versus shNT, P=0.0484) and untransduced controls (bFGF fold expression, 3.98±0.97 versus 1.01±0.09; shHDAC1 versus untransduced, P=0.0291; Figure 5A). Upregulation of bFGF mRNA in shHDAC1‐transduced CMCs was accompanied by a significant increase in bFGF protein levels, as detected by immunoblotting (Figure 5B). Relative to shNT or untransduced controls, densitometric quantification revealed shHDAC1‐transduced CMCs to possess an ≈5‐fold increase in bFGF protein levels (relative bFGF expression, shHDAC1 versus NT, 0.86±0.08 versus 0.14±0.09, P=0.0147; shHDAC1 versus untransduced, 0.86±0.08 versus 0.15±0.07, P=0.0330; Figure 5C).
Figure 5.
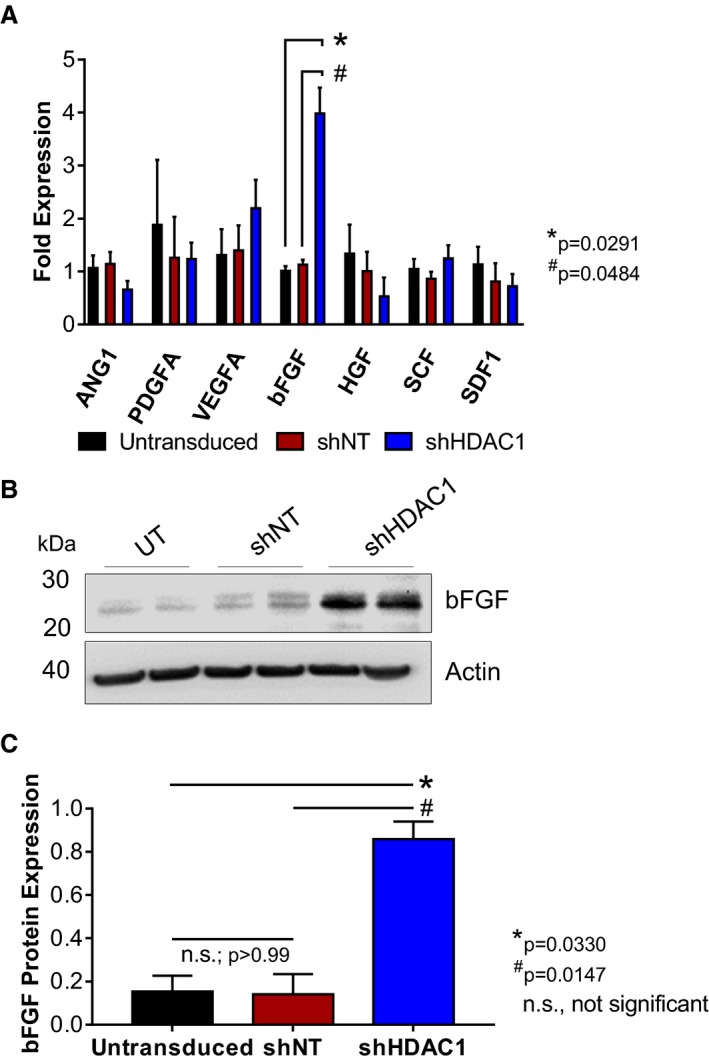
HDAC1‐depletion stimulates basic fibroblast growth factor (bFGF) expression in human CMCs. A, qPCR assays evaluating the expression of known trophic factors involved in cell‐mediated cardiac repair in shHDAC1, shNT, or untransduced CMCs. Values are mean±SEM (n=4). qPCR data were log base 10 (y=log10 y) transformed and analyzed by 2‐way ANOVA. P values were calculated using the post hoc Bonferroni multiple comparison test. B, Representative immunoblot evaluating bFGF expression in total protein isolates derived from shHDAC1, shNT, or untransduced CMCs (n=4). C, Densitometric quantification of bFGF immunoblots (expression relative to β‐actin). Values are mean±SEM (n=4). Western blot data were log base 10 (y=log10 y) transformed and analyzed by unpaired, 1‐way ANOVA. P values were calculated using the post hoc Bonferroni multiple comparison test. All experiments utilized 1:3 shRNA viral titer dilutions. HDAC1 indicates histone deacetylase 1; shHDAC1, short hairpin RNA‐histone deacetylase 1; shNT, short hairpin RNA‐non target; UT, untransduced.
bFGF knockdown diminishes paracrine signaling‐mediated activation of endothelial cell proliferation and tube assembly in HDAC1‐depleted CMCs
Based on the robust upregulation (Figure 5) and enhanced secretion (Figure 3) of bFGF with the depletion of HDAC1 in human CMCs, we posited that HDAC1 inhibition‐mediated upregulation of this factor may be required for their elevated proangiogenic activity. To test this hypothesis, CMCs were first co‐transduced with shRNA constructs targeting both HDAC1 and bFGF to confirm that we could efficiently block the induction of bFGF in HDAC1‐depleted CMCs (Figure 6A). In the accompanying immunoblot (Figure 6A), co‐transduced CMCs (shHDAC1+shbFGF) exhibited levels of bFGF that were similar to that of the control samples, for example, shNT, shbFGF alone, and untransduced CMCs (Figure 6A and 6B). Next, CM was harvested from all CMC cell lines and subjected to in vitro assays of endothelial cell function. HAECs were incubated with corresponding CM and their growth monitored for 72 hours (Figure 6C). shHDAC1 CM resulted in a 75% increase in the relative number of viable HAECs compared with those incubated with CM from shNT‐transduced CMCs (1.80±0.15 versus 1.03±0.13; shHDAC1 CM versus shNT CM, P=0.0001; Figure 6C). CM derived from CMCs co‐transduced with shHDAC1 and shbFGF showed a −24% decrease in the relative number of viable HAECs compared with that of CM from shHDAC1 CMCs (1.37±0.13 versus 1.80±0.15; shHDAC1+shbFGF CM versus shHDAC1 CM, P=0.0475; Figure 6C). Furthermore, CM from shHDAC1+shbFGF co‐transduced CMCs exhibited no statistical difference in their effects on HAEC proliferation compared to that of control samples (1.37±0.13, 1.07±0.10, 1.03±0.03, and 1.00±0.07; shHDAC1+shbFGF CM, shbFGF CM, shNT CM, and untransduced CM, respectively; Figure 6C).
Figure 6.
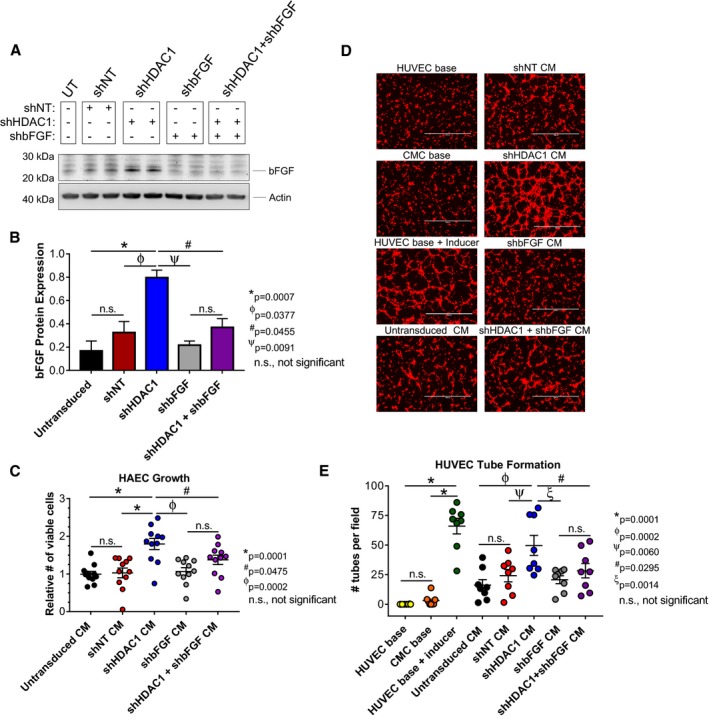
Basic fibroblast growth factor (bFGF) knockdown inhibits paracrine signaling‐mediated activation of endothelial cell proliferation and tube assembly in HDAC1‐depleted CMCs. A, Immunoblots evaluating the expression of bFGF in total protein extracts derived from shRNA‐transduced CMCs (n=4). B, Densitometric analysis of bFGF immunoblots (expression relative to β‐actin). Values are mean±SEM (n=4). Western blot data were log base 10 (y=log10 y) transformed and analyzed by unpaired, 1‐way ANOVA. P values were calculated using the post hoc Holm–Sidak multiple comparison test. C, HAEC growth was assessed following their propagation in CM from all shRNA‐transduced CMC groups (untransduced, shNT, shHDAC1, shbFGF, and shHDAC1+shbFGF). Values are mean±SEM (n=11). Growth data were analyzed by 1‐way ANOVA and P values determined using the post hoc Dunnett multiple comparison test. D, Representative fluorescent microscopy images (scale=1000 μm) and (E) graph enumerating HUVEC tube formation in response to incubation with CM from untransduced, shNT, shHDAC1, shbFGF, or shHDAC1+shbFGF transduced CMCs. HUVEC base (−Ctrl), CMC base (−Ctrl), and HUVEC base+inducer (low serum growth supplement [LSGS]; +Ctrl) controls are included. Values are mean±SEM (n=8 for control and experimental groups). Tube formation data were analyzed using 1‐way ANOVA and P values determined using the post hoc Dunnett multiple comparison test. All experiments utilized 1:3 shRNA viral titer dilutions. CM indicates conditioned medium; CMC, cardiac mesenchymal stromal cell; HAEC, human aortic endothelial cell; HDAC1, histone deacetylase 1; HUVEC, human umbilical vein endothelial cell; shbFGF, short hairpin RNA basic fibroblast growth factor; shHDAC1, short hairpin RNA‐histone deacetylase 1; shNT, short hairpin RNA nontarget; UT, untransduced.
The ability of the aforementioned harvested CM to promote HUVEC tube assembly/formation was then tested in vitro. HUVECS were incubated with CM from shHDAC1+shbFGF, shHDAC1, shbFGF, shNT, or untransduced CMCs for 4 hours, at which time they were stained and imaged (Figure 6D). Consistent with our previous results, shHDAC1 CM more efficiently promoted HUVEC tube organization than CM derived from shNT (number of tubes per field, 50±8 versus 24±5; shHDAC1 CM versus shNT CM, P=0.0060) and untransduced (number of tubes per field, 50±8 versus 16±5; shHDAC1 CM versus untransduced CM, P=0.0002) CMCs (Figure 6D and 6E). Intriguingly, CM from shHDAC1+shbFGF co‐transduced CMCs demonstrated a 44% reduction in tube formation capacity relative to CM derived from shHDAC1 only transduced CMCs (number of tubes per field, 28±6 versus 50±8; shHDAC1+shbFGF CM versus shHDAC1 CM, P=0.0295; Figure 6D and 6E). Furthermore, the tube formation capacity of CM sourced from shHDAC1+shbFGF co‐transduced CMCs was found to not significantly differ from that of the controls (shbFGF CM, shNT CM, and untransduced CM; Figure 6E). Taken together, these data suggest that the heightened proangiogenic paracrine activities of CMCs following HDAC1 depletion are, in part, attributed to a mechanism requiring enhanced synthesis and secretion of the proangiogenic factor, bFGF.
Discussion
The perception that donor cell lineage commitment may be an important determinant of the outcomes of cell therapy3, 10, 11 has driven the development of various strategies to augment donor cell cardiovascular lineage commitment—with the intention of improving cardiac repair.12, 13, 14, 15, 16 We recently demonstrated that HDAC1 activity regulates CMC cardiomyogenic lineage commitment, wherein its inhibition or genetic depletion promoted CMC cardiogenic program activation and cardiomyogenic‐like differentiation.14 Building upon these previous observations, the goal of the current study was to interrogate the effects of HDAC1 inhibition‐mediated alterations in cardiomyogenic lineage commitment on CMC cytokine secretion patterns and its consequences on CMC paracrine signaling dynamics in vitro.14 We found that the depletion of HDAC1 results in pronounced alterations in CMC cytokine secretion patterns. Such changes resulted in significant modification of CMC paracrine signaling activities in vitro, whereas CM derived from HDAC1‐depleted CMCs more efficiently promoted endothelial cell proliferation and tube formation compared with control. Gene expression assays of paracrine signaling factors implicated as important mediators of cell therapy‐mediated cardiac repair (eg, angiopoietin 1, platelet‐derived growth factor, vascular endothelial growth factor, bFGF, hepatocyte growth factor, stem cell factor, and stromal cell‐derived factor‐1)24, 25, 26, 27 revealed a marked increase in the proangiogenic factor, bFGF, following HDAC1 knockdown. The importance of this factor in regulating the paracrine signaling activities of HDAC1‐depleted CMCs was stressed by preventing its induction. Specifically, shRNA‐mediated knockdown of bFGF in HDAC1‐depleted CMCs efficiently blocked the enhanced paracrine signaling‐mediated activation of endothelial cell migration and tube assembly. These data suggest that HDAC1 inhibition affords CMCs heightened proangiogenic paracrine signaling activities, and, furthermore, that this phenomenon is, in part, attributed to a mechanism requiring enhanced synthesis and secretion of the proangiogenic factor, bFGF.
The results of this study establish a heretofore unknown role for HDAC1 in the regulation of CMC trophic factor secretion and paracrine signaling activities and, furthermore, identify HDAC1 as a unique target that could be exploited by pharmacological or genetic means to enhance the therapeutic efficacy of CMCs. These results also suggest an alternative perspective concerning the mechanism of action of donor cells in cell therapy—given that donor cell cardiovascular differentiation and their paracrine signaling activities are generally regarded as distinct mechanisms contributing to their cardiac reparative capacity.28 Our data could be extended to suggest that cardiovascular lineage commitment‐dependent variations in paracrine signaling could significantly modify donor cell reparative capacity. Nevertheless, there are a number of limitations—most notably the method utilized to augment CMC lineage commitment. Indeed, chromatin modifying agents such as inhibitors of histone deacetylase14 and DNA methyltransferase15, 16, 17 activity can promote cardiomyogenic lineage commitment in mesenchymal progenitor cells, but such methods could alter other genetic programs beyond those dictating cardiovascular cell lineage commitment/specification. Thus, this study alone cannot definitively determine whether alterations in CMC cytokine secretion patterns were attributed only to enhancement of CMC cardiovascular lineage commitment; such changes may also be associated with HDAC1 inhibition‐mediated derepression of cytokine‐specific promoters (resulting from hyperacetylation of chromosomal histone proteins). Although this is beyond the scope of the current study, future investigations could interrogate the effects of lineage‐commitment alterations in CMC cytokine secretion and signaling by forced expression of cardiogenic transcription factors, namely, GATA4, MEF2C, and TBX5.12 This will limit the potential of off‐target effects commonly associated with the inhibition of epigenetic modifiers.
The precise mechanism underlying the significant upregulation and secretion of bFGF in HDAC1‐depleted CMCs is unclear, but could be the result of alterations in cardiovascular cell lineage commitment, as well as modification in acetylation status of known HDAC1 substrates (both histone and nonhistone proteins). In regard to the former, GATA4, the early cardiac lineage commitment transcription factor whose expression is subject to HDAC1 regulation,14 has been previously identified as a modulator of mesenchymal stem cell paracrine activity.29 In that study, Li et al demonstrated mesenchymal stem cells ectopically expressing GATA4 more efficiently express proangiogenic factors, promote paracrine‐mediated activation of HUVEC tube formation in vitro, and stimulate postinfarction angiogenesis and functional recovery in vivo.29 Thus, the upregulation of bFGF in response to CMC HDAC1 depletion may be, in part, directed by the enhanced expression of the early cardiogenic transcription factor, GATA4; however, given the sundry array of known, as well as unknown, targets of HDAC1 activity, additional modes of regulation contributing to enhanced bFGF expression cannot be excluded.
Given the heightened in vitro paracrine signaling activities of HDAC1‐depleted CMCs, we predict that such cells will more efficiently promote cardiac repair by paracrine‐mediated activation of endogenous vasculogenesis. Moreover, based on the widespread alterations in cytokine secretion profiles, we anticipate that HDAC1‐depleted CMCs will target additional endogenous cell types and/or modulate supplementary cellular processes that could also contribute to post–myocardial infarct repair/remodeling. For instance, in addition to the upregulation of proangiogenic factors, we also observed a marked increase in the secretion of cytokines involved in differentiation, growth, migration, and survival. Thus, it is tempting to speculate that HDAC1 inhibition or depletion may also afford CMCs the ability to more efficiently promote myocyte proliferation, inhibit myocardial apoptosis, recruit reparative endogenous progenitors, and/or mitigate fibroblast activation (eg, ventricular fibrosis)—processes that have been suggested to be influenced by the paracrine activities of transplanted progenitor cell types in preclinical heart failure models (reviewed in a previous work28). To this end, future studies will be expanded to investigate the impact of augmented cardiovascular lineage commitment on other CMC paracrine signaling‐mediated activities in vitro and, importantly, the associated effects of lineage commitment‐dependent variations in paracrine signaling on CMC‐mediated cardiac repair in vivo.
Author Contributions
Moore: Conception and design, financial support, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; Zhao: Conception and design, collection and assembly of data, data analysis and interpretation, final approval of manuscript; Fischer: Collection and assembly of data, data analysis and interpretation; Keith: Provision of study materials, collection and assembly of data, data analysis and interpretation, final approval of manuscript; Hagan: Collection and assembly of data, data analysis and interpretation; Wysoczynski: Conception and design, data analysis and interpretation, final approval of manuscript; Bolli: Conception and design, financial support, manuscript writing, and final approval of manuscript.
Sources of Funding
This work was supported by NIH grants P01 HL078825 (Program Project Grant; Wysoczynski and Bolli), P20 GM103492 (Centers of Biomedical Research Excellence; Wysoczynski), and UM1 HL‐113530 (Cardiovascular Cell Therapy Research Network [CCTRN]; Bolli). Additional funding was provided by the American Heart Association Scientist Development Grant 13SDG14560005 (Scientist Development Grant; Wysoczynski) and the University of Louisville School of Medicine Basic Grant Program (Moore).
Disclosures
None.
Supporting information
Table S1. shRNA Vectors and shRNA‐Scramble Controls Utilized in Experiments (Plasmids)
Table S2. Gene‐Specific Primers With Respective Annealing Temperatures (Primers)
Table S3. Antibodies With Corresponding Dilutions
Acknowledgments
Special thanks to Drs Kaushal Solanki, Vishnu Priya Mallipeddi, and Swati Shokeen for their time and attention in the collection and assembly of data.
(J Am Heart Assoc. 2017;6:e006183 DOI: 10.1161/JAHA.117.006183.)28679560
Portions of the data in this manuscript were presented at the Experimental Biology Conference, April 22 to 26, 2017, in Chicago, IL.
References
- 1. Wysoczynski M, Dassanayaka S, Zafir A, Ghafghazi S, Long BW, Noble C, DeMartino AM, Brittian KR, Bolli R, Jones SP. A new method to stabilize c‐kit expression in reparative cardiac mesenchymal cells. Front Cell Dev Biol. 2016;4:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wysoczynski M, Guo Y, Moore JB IV, Muthusamy S, Li Q, Nasr M, Li H, Nong Y, Wu W, Tomlin AA, Zhu X, Hunt G, Gumpert AM, Book MJ, Khan A, Tang XL, Bolli R. Myocardial reparative properties of cardiac mesenchymal cells isolated on the basis of adherence. J Am Coll Cardiol. 2017;69:1824–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rossini A, Frati C, Lagrasta C, Graiani G, Scopece A, Cavalli S, Musso E, Baccarin M, Di Segni M, Fagnoni F, Germani A, Quaini E, Mayr M, Xu Q, Barbuti A, DiFrancesco D, Pompilio G, Quaini F, Gaetano C, Capogrossi MC. Human cardiac and bone marrow stromal cells exhibit distinctive properties related to their origin. Cardiovasc Res. 2011;89:650–660. [DOI] [PubMed] [Google Scholar]
- 4. Hong KU, Li QH, Guo Y, Patton NS, Moktar A, Bhatnagar A, Bolli R. A highly sensitive and accurate method to quantify absolute numbers of c‐kit+ cardiac stem cells following transplantation in mice. Basic Res Cardiol. 2013;108:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hong KU, Guo Y, Li QH, Cao P, Al‐Maqtari T, Vajravelu BN, Du J, Book MJ, Zhu X, Nong Y, Bhatnagar A, Bolli R. c‐kit+ Cardiac stem cells alleviate post‐myocardial infarction left ventricular dysfunction despite poor engraftment and negligible retention in the recipient heart. PLoS One. 2014;9:e96725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Keith MC, Tokita Y, Tang XL, Ghafghazi S, Moore JB IV, Hong KU, Elmore JB, Amraotkar AR, Guo H, Ganzel BL, Grubb KJ, Flaherty MP, Vajravelu BN, Wysoczynski M, Bolli R. Effect of the stop‐flow technique on cardiac retention of c‐kit positive human cardiac stem cells after intracoronary infusion in a porcine model of chronic ischemic cardiomyopathy. Basic Res Cardiol. 2015;110:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang XL, Li Q, Rokosh G, Sanganalmath SK, Chen N, Ou Q, Stowers H, Hunt G, Bolli R. Long‐term outcome of administration of c‐kit(POS) cardiac progenitor cells after acute myocardial infarction: transplanted cells do not become cardiomyocytes, but structural and functional improvement and proliferation of endogenous cells persist for at least one year. Circ Res. 2016;118:1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahn SY, Chang YS, Sung DK, Yoo HS, Sung SI, Choi SJ, Park WS. Cell type‐dependent variation in paracrine potency determines therapeutic efficacy against neonatal hyperoxic lung injury. Cytotherapy. 2015;17:1025–1035. [DOI] [PubMed] [Google Scholar]
- 9. Charoenviriyakul C, Takahashi Y, Morishita M, Matsumoto A, Nishikawa M, Takakura Y. Cell type‐specific and common characteristics of exosomes derived from mouse cell lines: yield, physicochemical properties, and pharmacokinetics. Eur J Pharm Sci. 2017;96:316–322. [DOI] [PubMed] [Google Scholar]
- 10. Yoon CH, Koyanagi M, Iekushi K, Seeger F, Urbich C, Zeiher AM, Dimmeler S. Mechanism of improved cardiac function after bone marrow mononuclear cell therapy: role of cardiovascular lineage commitment. Circulation. 2010;121:2001–2011. [DOI] [PubMed] [Google Scholar]
- 11. Behfar A, Yamada S, Crespo‐Diaz R, Nesbitt JJ, Rowe LA, Perez‐Terzic C, Gaussin V, Homsy C, Bartunek J, Terzic A. Guided cardiopoiesis enhances therapeutic benefit of bone marrow human mesenchymal stem cells in chronic myocardial infarction. J Am Coll Cardiol. 2010;56:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ieda M, Fu JD, Delgado‐Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang G, Tian J, Feng C, Zhao LL, Liu Z, Zhu J. Trichostatin a promotes cardiomyocyte differentiation of rat mesenchymal stem cells after 5‐azacytidine induction or during coculture with neonatal cardiomyocytes via a mechanism independent of histone deacetylase inhibition. Cell Transplant. 2012;21:985–996. [DOI] [PubMed] [Google Scholar]
- 14. Moore JB IV, Zhao J, Keith MC, Amraotkar AR, Wysoczynski M, Hong KU, Bolli R. The epigenetic regulator HDAC1 modulates transcription of a core cardiogenic program in human cardiac mesenchymal stromal cells through a p53‐dependent mechanism. Stem Cells. 2016;34:2916–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qian Q, Qian H, Zhang X, Zhu W, Yan Y, Ye S, Peng X, Li W, Xu Z, Sun L, Xu W. 5‐Azacytidine induces cardiac differentiation of human umbilical cord‐derived mesenchymal stem cells by activating extracellular regulated kinase. Stem Cells Dev. 2012;21:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moscoso I, Centeno A, Lopez E, Rodriguez‐Barbosa JI, Santamarina I, Filgueira P, Sanchez MJ, Dominguez‐Perles R, Penuelas‐Rivas GP, Domenech N. Differentiation, “in vitro” of primary and immortalized porcine mesenchymal stem cells into cardiomyocytes for cell transplantation. Transpl Proc. 2005;37:481–482. [DOI] [PubMed] [Google Scholar]
- 17. Makino S, Fukuda K, Miyoshi S, Konishi F, Kodama H, Pan J, Sano M, Takahashi T, Hori S, Abe H, Hata J, Umezawa A, Ogawa S. Cardiomyocytes can be generated from marrow stromal cells in vitro. J Clin Invest. 1999;103:697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Q, Bolli R, Qiu Y, Tang XL, Guo Y, French BA. Gene therapy with extracellular superoxide dismutase protects conscious rabbits against myocardial infarction. Circulation. 2001;103:1893–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li XY, McCay PB, Zughaib M, Jeroudi MO, Triana JF, Bolli R. Demonstration of free radical generation in the “stunned” myocardium in the conscious dog and identification of major differences between conscious and open‐chest dogs. J Clin Invest. 1993;92:1025–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang XL, Qiu YM, Park SW, Sun JZ, Kalya A, Bolli R. Time course of late preconditioning against myocardial stunning in conscious pigs. Circulation Research. 1996;79:424–434. [DOI] [PubMed] [Google Scholar]
- 21. Avolio E, Meloni M, Spencer HL, Riu F, Katare R, Mangialardi G, Oikawa A, Rodriguez‐Arabaolaza I, Dang ZX, Mitchell K, Reni C, Alvino VV, Rowlinson J, Livi U, Cesselli D, Angelini G, Emanueli C, Beltrami AP, Madeddu P. Combined intramyocardial delivery of human pericytes and cardiac stem cells additively improves the healing of mouse infarcted hearts through stimulation of vascular and muscular repair. Circ Res. 2015;116:E81–E94. [DOI] [PubMed] [Google Scholar]
- 22. Li TS, Cheng K, Malliaras K, Smith RR, Zhang YQ, Sun BM, Matsushita N, Blusztajn A, Terrovitis J, Kusuoka H, Marban L, Marban E. Direct comparison of different stem cell types and subpopulations reveals superior paracrine potency and myocardial repair efficacy with cardiosphere‐derived cells. J Am Coll Cardiol. 2012;59:942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burchfield JS, Dimmeler S. Role of paracrine factors in stem and progenitor cell mediated cardiac repair and tissue fibrosis. Fibrogenesis Tissue Repair. 2008;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98:1076–1084. [DOI] [PubMed] [Google Scholar]
- 25. Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res. 2008;103:1204–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams AR, Hare JM. Mesenchymal stem cells: biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ Res. 2011;109:923–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duran JM, Makarewich CA, Sharp TE, Starosta T, Zhu F, Hoffman NE, Chiba Y, Madesh M, Berretta RM, Kubo H, Houser SR. Bone‐derived stem cells repair the heart after myocardial infarction through transdifferentiation and paracrine signaling mechanisms. Circ Res. 2013;113:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao J, Ghafghazi S, Khan AR, Farid TA, Moore JB IV. Recent developments in stem and progenitor cell therapy for cardiac repair. Circ Res. 2016;119:E152–E159. [DOI] [PubMed] [Google Scholar]
- 29. Li HX, Zuo S, He ZS, Yang YT, Pasha Z, Wang YG, Xu MF. Paracrine factors released by GATA‐4 overexpressed mesenchymal stem cells increase angiogenesis and cell survival. Am J Physiol Heart Circ Physiol. 2010;299:H1772–H1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. shRNA Vectors and shRNA‐Scramble Controls Utilized in Experiments (Plasmids)
Table S2. Gene‐Specific Primers With Respective Annealing Temperatures (Primers)
Table S3. Antibodies With Corresponding Dilutions