

Abstract
Background
Coagulation factor V (FV) plays a key role in hemostasis, is present in plasma and platelets, and has both pro‐ and anticoagulant properties; however, the contribution of platelet‐derived FV to arterial thrombosis remains undetermined.
Methods and Results
Using transgenic mice with various levels of FV gene expression that was restricted to the plasma or platelets, the roles of platelet FV were evaluated in the regulation of arterial thrombosis and platelet activation. Mice with higher levels of platelet FV exhibited faster thrombotic occlusion of the carotid artery after injury compared with mice with lower platelet FV levels. Infusion of platelets with higher levels of FV into transgenic mice with undetectable levels of platelet FV reduced the time to carotid artery occlusion. In contrast, infusion of purified recombinant plasma FV into mice with undetectable platelet FV levels failed to reduce the carotid occlusion times following injury. Evaluation of isolated platelets revealed that platelet‐derived FV was critical for the regulation of platelet activation. These effects were associated with an increased level of expression of P‐selectin and increased cGMP in platelets.
Conclusions
We established that platelet‐derived FV is a critical mediator of arterial thrombosis that involves platelet activation.
Keywords: arterial, coagulation, factor V, platelet, thrombosis
Subject Categories: Platelets, Thrombosis
Clinical Perspective
What Is New?
Platelet‐derived factor V plays an important role in arterial thrombosis and platelet activation.
What Are the Clinical Implications?
Roles of platelet‐derived factor V in thrombosis could be a potential therapeutic target for prevention of arterial thrombosis.
Introduction
Factor V (FV) is a regulatory coagulation factor that plays dual critical roles in coagulation. It serves as a critical cofactor for factor Xa in the formation of the prothrombinase complex that converts prothrombin to thrombin in the presence of calcium and a phospholipid surface. FV is distributed into distinct plasma and platelet compartments, with ≈80% of the total blood FV in the former and ≈20% in the latter. Clinical and experimental evidence also suggests that FV in human platelets and plasma may be functionally distinct.1, 2 Platelet‐derived FVa is more resistant to activated protein C cleavage and expresses significantly more FXa cofactor activity than plasma‐derived FVa.1 A relationship has been demonstrated between FV and TFPI (tissue factor pathway inhibitor 1), and this interaction may influence the risk of venous thrombosis3, 4; however, there are no reports on the roles of FV in arterial thrombosis.
Patients with undetectable plasma FV but detectable platelet FV demonstrated relatively mild bleeding symptoms, which suggests that platelet FV is critical for coagulation and thrombin generation.3 It is unclear whether the levels of FV are associated with an increased risk for thrombosis, although high procoagulant FV levels caused by enhanced prothrombinase activity and low anticoagulant FV levels caused by reduced activated protein C cofactor activity can both promote thrombosis. Based on the previous study, platelet FV would have shown a correlation with venous thrombosis.5 The importance of platelet‐derived FV in causing a more procoagulant phenotype is supported by several studies.1, 2, 6 Furthermore, although FV is a critical factor in hemostasis, it can also increase thrombotic risk by its impact on thrombin generation. The present study provides new information on the role of platelet FV in the pathogenesis of thrombosis.
In humans, FV is endocytosed from plasma by megakaryocytes,2 whereas murine megakaryocytes synthesize the protein.7 It is unclear if murine platelet FV undergoes the same modifications that are present in the human protein. Murine platelet and plasma FV pools are biosynthetically distinct and sufficient for minimal hemostasis.8 The work of Paula Tracy and Ken Mann9 has shown that human platelet FV released from α‐granules is in a partially activated state, contributing to localized assembly of the prothrombinase complex. Proteolysis takes place in megakaryocytes before packaging within platelet α‐granules.9, 10, 11, 12 In summary, these observations elucidated factors potentially contributing to the prothombotic properties of platelet FV and suggested that platelet FV could significantly influence thrombosis.
Using mice that expressed various levels of the FV gene that were restricted to either plasma or platelets,13 we recently demonstrated that thrombin activity was significantly higher in mice with higher levels of platelet FV.14 In the current study, we demonstrated that platelet FV could play an important role in arterial thrombosis and platelet activation.
Material and Methods
Animals
TgF5+/+, Tg+F5+/−, and Tg+F5−/− mice were previously generated and characterized.14 The mice exhibited various levels of FV gene expression restricted to either the plasma or platelets (Tg‐F5+/+, 100%/100%; Tg+F5+/−, 65%/50%; Tg+F5−/−, 15%/0%). Male Tg‐F5+/+, Tg+F5+/−, and Tg+F5−/− littermates aged 6 to 8 weeks and weighing between 25 and 30 g were used for all experiments. All mice were euthanized by slow‐fill carbon dioxide asphyxiation. The investigation conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, publication No. 85–23, revised 2011, published by National Research Council). All protocols for animal use were reviewed and approved by the animal care committee of Southwest Medical University in accordance with the institutional animal care and use committee guidelines.
Platelet Depletion, Isolation, and Transfusion
To deplete the endogenous platelets, the mice were injected intraperitoneally with 2.5 μg/g mouse of platelet‐depleting antibody (polyclonal anti–mouse GPIbα [glycoprotein Ib platelet α] rat IgG). Following the injection of the GPIbα antibody or control IgG, the platelets were counted at appropriate time points. Thrombocytopenia was evaluated by blood count. The intraperitoneal injection of the depleting antibody resulted in ≥95% reduction in circulating platelets at 12 hours after injection in all mice.
The mice were anesthetized with ketamine/xylazine followed by 2.5 mg/kg intraperitoneally, and whole blood was collected from the inferior vena cava using a 1‐mL syringe containing 0.1 mL sodium citrate anticoagulant. Platelet‐rich plasma (PRP) was obtained from whole blood by centrifugation at 260g, and the supernatant was collected. The PRP was then centrifuged at 740g for 10 minutes. The pellet, which contained the platelets, was then resuspended in 1 mL PBS and allowed to stand for 30 minutes, and the platelets were counted with a hemocytometer. Platelets from the suspension were then diluted with normal saline to 1.56×109 platelets in 1.2 mL, and 0.2 mL of the suspension was transfused via the tail vein into each recipient 1 day after the injection of the GPIbα antibody.
Carotid Artery Thrombosis Model
Carotid artery thrombosis was produced using ferric chloride (FeCl3), as described previously.15, 16 After the mice were anesthetized with ketamine/xylazine followed by a 2.5 mg/kg intraperitoneally, the common carotid arteries were exposed and a filter paper (3×1.0 mm) soaked in FeCl3 solution (10%) was placed on top of the left carotid artery for 3 minutes. After the filter paper was removed, the carotid artery was washed in PBS, and the flow was monitored continuously from the onset of injury until stable occlusion occurred (defined as no flow for 120 minutes).
Infusion of Plasma FV Protein
To determine whether exogenous FV protein contributes to thrombosis formation, human plasma FV protein was formulated in PBS and administered intravenously at the concentration present in normal healthy humans (12 μg per mouse).17, 18 The plasma FV protein was injected intravenously 30 minutes before the FeCl3‐induced injury, the carotid artery thrombosis model was performed, and the flow was monitored.
Platelet Aggregation
Platelet aggregation was measured using a turbidimetric aggregation‐monitoring device. Aggregation of the isolated platelets was performed at a concentration of 2×108 platelets per microliter in Tyrode's–HEPES buffer (pH 7.4). After adjusting the conditions according to the manufacturer's protocol, platelets were activated with ADP (5 μmol/L) under constant stirring conditions. The extent of aggregation was expressed in light‐transmission units.
Measurement of Platelet cGMP Levels
Washed mouse platelets that had been resuspended in modified Tyrode's buffer were incubated with ADP for 30 minutes in a platelet aggregometer with stirring (1000 rpm) at 37°C. The reaction was stopped by the addition of ice‐cold 12% (wt/vol) trichloroacetic acid. The samples were then centrifuged at 2000g for 15 minutes at 4°C, and the supernatant was extracted 4 times with 5 volumes of water‐saturated diethyl ether. The samples were lyophilized, and the cGMP concentrations were determined using a cGMP enzyme immunoassay kit.
Flow Cytometry
Blood samples, collected in 4% sodium citrate and diluted 1:10 with PBS, were stimulated with ADP (5 μmol/L) for 30 minutes under continuous stirring. Aliquots of 5 μL were then stained with either FITC‐conjugated anti‐CD62P monoclonal antibody (rat LEW IgG1λ) or activated protein C‐conjugated anti‐CD62P monoclonal antibody (mouse IgG1κ) for 30 minutes in the dark. The reaction was stopped with 1 mL of 1% paraformaldehyde. Flow cytometry was carried out with a FACScan to flow cytometer using DIVA software (BD Biosciences). CD62P expression is reported as a percentage of positive cells.
Statistical Analysis
The data are expressed as mean±SEM. In most assays, we used the 2‐sample t test to evaluate statistical significance. In an aggregation assay, we used the nonparametric method.
Results
FV Mediates Arterial Thrombosis After Vascular Injury
In previous studies, we generated transgenic mice with various levels of plasma and platelet pools of FV. FV in either pool is sufficient for hemostasis.6 Of these mouse strains, the Tg+F5−/− mice expressed ≈15% of the wild‐type plasma FV level and had no detectable platelet FV. Using Western blotting, we previously found that the Tg+Fv−/− mice had no detectable FV in the platelets and the lowest levels of FV in platelet‐poor plasma (PPP).14
We used FeCl3 injury of the common carotid artery model to compare thrombus formation in vivo among Tg‐Fv+/+, Tg+Fv+/−, and Tg+Fv−/− mice. FeCl3 injury is a well‐established technique to rapidly and accurately induce the formation of thrombi in an exposed artery. FeCl3 is diffused through the vessel wall, resulting in endothelial cell denudation without exposure of the inner layers. The average time required to form a completely occluded thrombus in the Tg+Fv−/− mice after the initiation of the arterial injury (420±92.7 seconds, n=9) was significantly greater than that for the Tg+Fv+/− mice (182±42.8 seconds, n=9, P<0.04 versus Tg+Fv−/−) or the Tg‐Fv+/+ mice (101±20.4 seconds, n=9, P<0.05 versus Tg+Fv+/−; Figure 1A and 1B), which suggested that FV mediates thrombosis in vivo.
Figure 1.
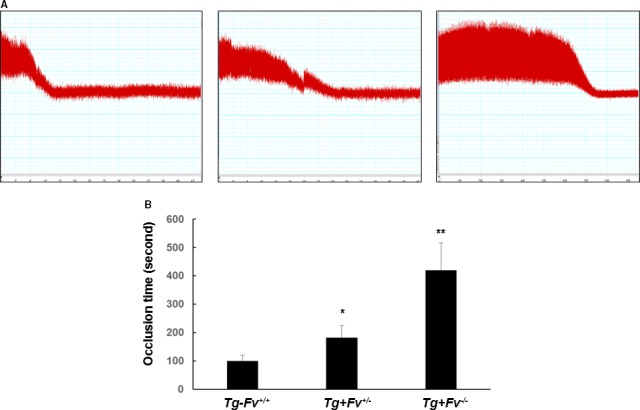
Carotid arterial occlusion after injury induced by ferric chloride (FeCl3). The carotid artery was injured by FeCl3 in the Tg‐Fv+/+, Tg+Fv+/−, and Tg+Fv−/− mice. The carotid artery blood flow tracings (A) and mean times to occlusion of the carotid arteries (B) are shown (n=9 per group). *P<0.05 vs the Tg‐Fv+/+ mice; **P<0.05 vs the Tg+Fv+/− mice.
Platelet‐Derived FV Promotes Arterial Thrombosis
To study the contribution of platelet FV in arterial thrombosis, we used a platelet depletion/transfusion model to examine the occlusion times after injury in the various groups. Consistent with our previous report,14 platelet counts were successfully reduced using rat anti–mouse GPIbα IgG. We transfused Tg‐Fv+/+ and Tg+Fv−/− donor platelets into Tg+Fv−/− recipient mice before photochemical injury. The Tg+Fv−/− recipient mice that received Tg+Fv−/− donor platelets formed occlusive thrombi within 456.7±49.2 seconds (Figure 2A and 2B). Of note, the time required to form the occlusive thrombus was significantly shortened to 210.0±11.8 seconds (P<0.02, n=6 in the Tg+Fv−/− recipient mice that received Tg‐Fv+/+ donor platelets), which indicated a partial restoration of the occlusion time toward that of the Tg‐Fv+/+ mice.
Figure 2.
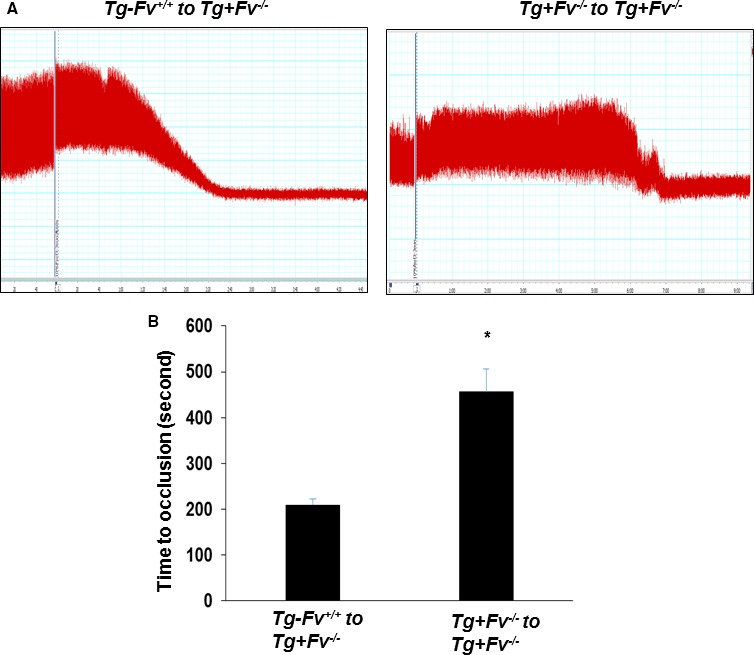
Platelet factor V promoted arterial thrombosis after carotid artery injury. Platelet depletion/transfusion was performed in Tg+Fv−/− recipient mice before photochemical injury. Representative carotid artery blood flow tracings (A) and mean times to occlusion of the carotid arteries (B) are shown (n=8 per group). * P<0.05 vs Tg‐Fv+/+ to Tg+Fv−/− mice.
Infusion of plasma FV had little effect on the occlusion time in the Tg+Fv −/− mice.
To further evaluate whether plasma‐derived FV plays a role in the regulation of the time required to occlude the carotid artery after injury, we infused a normal human physiological dose of purified human FV (12 μg per mouse) into the Tg+Fv −/− mice.14 Intravenous infusion of the purified FV had no significant effect on the thrombotic occlusion time (390±18.2 versus 372±57.6 seconds for saline control infusion; P>0.05; n=6 per group; Figure 3A and 3B), which suggested that the platelet‐derived FV was responsible for thrombotic action.
Figure 3.
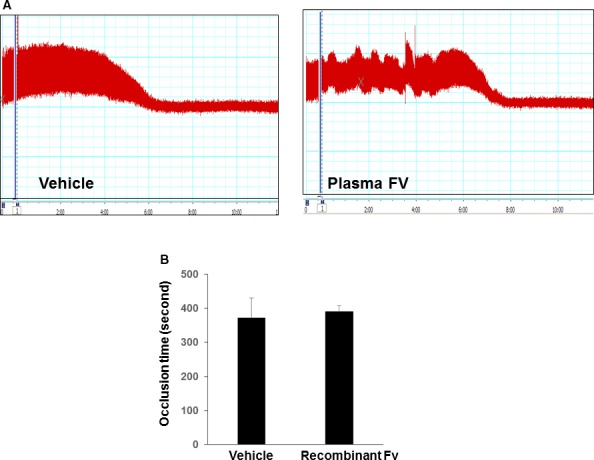
Effect of plasma factor V (FV) on carotid arterial occlusion in Tg+Fv −/− mice. Plasma FV protein was injected intravenously 30 minutes before the FeCl3‐induced injury. The carotid artery thrombosis model was performed. Representative carotid artery blood flow tracings in Tg+Fv −/− mice (A) and mean times to occlusion of the carotid arteries (B) are shown (n=8 per group).
Platelet FV Modulates Platelet Reactivity
To determine whether FV affects platelet reactivity, the aggregation of platelets in response to a suboptimal dose of ADP was examined in the various groups of mice. We observed a significant increase in the extent and rate of aggregation of the platelets from the Tg‐Fv+/+ mice compared with the Tg+Fv+/− and Tg+Fv−/− mice (Figure 4A and 4B), which indicated that FV plays an important modulatory role in platelet function.
Figure 4.
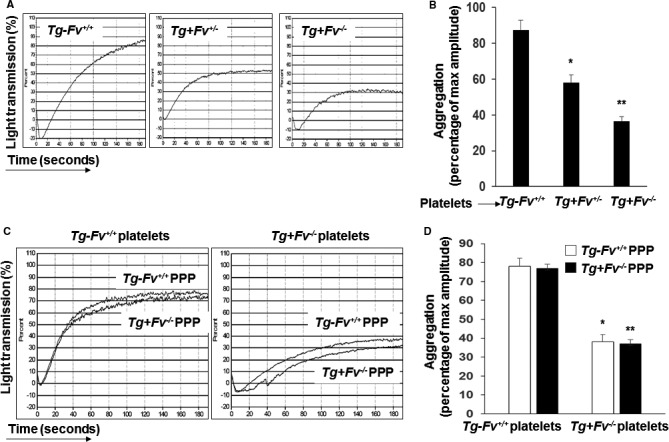
Platelet factor V modulated platelet reactivity. Platelet aggregation in platelet‐rich plasma from mice of the indicated genotypes was induced by ADP and optically monitored. A, Representative aggregation curves are shown. B, The bar graph shows the aggregation results expressed as maximal amplitude of aggregation (n=6 per group; *P<0.05 vs the Tg‐Fv+/+ mice; **P<0.05 vs the Tg+Fv+/− or Tg‐Fv+/+ mice). C, Isolated platelets from either Tg‐Fv+/+ or Tg+Fv −/− mice, as described in Methods. Citrated, pooled, platelet‐poor plasma (PPP) from either Tg‐Fv+/+ or Tg+Fv −/− mice was isolated and combined with platelets of the indicated genotype. Aggregation was induced by ADP and optically monitored. Representative aggregation curves are shown. D, The bar graph shows the aggregation results expressed as maximal amplitude of aggregation (n=8 per group; *, **P<0.05 vs Tg‐Fv+/+ platelets).
Next, platelets from Tg‐Fv+/+ and Tg+Fv−/− mice were separated from PRP, combined with citrated PPP from either the Tg‐Fv+/+ or Tg+Fv−/− mice, and examined in an aggregation assay.
The aggregation response of the platelets from the Tg‐Fv+/+ mice in the PPP from the Tg‐Fv+/+ mice was significantly higher than that observed with platelets and plasma from Tg+Fv−/− mice. However, no significant differences between the 2 groups were observed between the PPP from the Tg‐Fv+/+ and Tg+Fv−/− mice following the addition of ADP (Figure 4C and 4D). Thus, plasma FV was insufficient to enhance the aggregation response state: Instead, this effect required platelet‐derived FV.
We then investigated whether the observed platelet activation in the mice was caused by a direct effect of the platelet‐derived FV. To test this idea, whole blood was collected from the Tg‐Fv+/+, Tg+Fv+/−, and Tg+Fv−/− mice and treated with 5 μmol/L ADP, a known platelet activator, at 37°C for 30 minutes. Platelet activation and dense granule release were then analyzed by measuring surface expression of CD62P (P‐selectin) using flow cytometry. The reduced levels of FV in the platelets from the Tg+Fv−/− mice resulted in a significant decrease in the ADP‐induced CD62P expression compared with that in the platelets from the Tg‐Fv+/+ and Tg+Fv+/− mice (Figure 5A), which suggested a critical role for the platelet FV.
Figure 5.
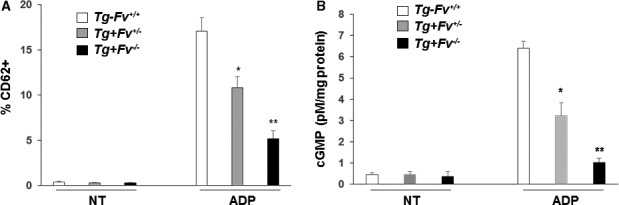
Platelet factor V was associated with platelet activation. A, The expression of CD62P on platelets was strikingly reduced in Tg+Fv−/− mice compared with Tg‐Fv+/+ and Tg+Fv+/− mice (n=3 per group). The percentages of CD62P‐positive platelets in the ADP‐treated and nontreated (NT) blood were comparable. B, Platelets were collected from Tg‐Fv+/+, Tg+Fv+/−, and Tg+Fv−/− mice, and the intracellular levels of cGMP were monitored using an enzyme‐linked immunoassay. Results represent the mean±SEM of 3 independent experiments. *P<0.05 vs the Tg‐Fv+/+ mice; **P<0.05 vs the Tg+Fv+/− mice.
The level of cGMP in platelets is associated with thrombin‐induced platelet aggregation and thrombosis.19 Platelets were collected from Tg‐Fv+/+, Tg+Fv+/−, and Tg+Fv−/− mice, and the intracellular levels of cGMP were monitored using an enzyme‐linked immunoassay (Figure 5B). The platelets of the Tg‐Fv+/+ mice showed a significantly increased cGMP level compared with those of the Tg+Fv+/− and Tg+Fv−/− mice, suggesting a dependence of this parameter on the intracellular FV.
Discussion
In this study, we have generated mice with various levels of FV in the plasma and platelet pools, which led to changes in thrombin generation.14 Three groups of mice were included in a carotid artery thrombosis model. These mice had different levels of FV in their plasma and platelet pools. We found that the mice with higher platelet FV levels exhibited the most rapid thrombotic occlusion of the carotid artery after injury. Infusion of platelets with higher levels of FV into transgenic mice with undetectable platelet FV reduced the prolonged time to carotid artery occlusion. These results indicated that platelet‐derived FV is prothrombotic. This conclusion was supported by the observation that platelet FV was able to enhance platelet aggregation. In addition, FV enhanced the ADP‐induced platelet activation through P‐selectin expression and the cGMP signaling pathway. Our findings suggested that platelet FV may be a key factor in promoting acute arterial thrombosis.
In our previous studies, we demonstrated that the prothrombin time in Tg+Fv−/− mice was significantly increased, which suggested a defect in thrombin generation.14 Furthermore, we found that the thrombin activity in the PPP and PRP of mice with different levels of FV were closely correlated with their FV levels. Specifically, the Tg+Fv−/− mice had the lowest thrombin activity in both the PPP and PRP.14 These mice exhibited a prolonged time to carotid artery occlusion, indicating that FV is involved in the regulation of thrombosis formation, which could be related to thrombin generation. Our further observations showed that transfusion of Tg‐Fv+/+ platelets into Tg+Fv−/− mice shortened the prolonged carotid artery occlusion time, but infusion of purified plasma FV into Tg+Fv−/− mice did not have this effect. Human plasma FV can be endocytosed into platelets, whereas mouse platelet FV is mostly synthesized. In work by Stopa et al,20 preincubation with FVa protein increased platelet‐dependent thrombin generation, suggesting uptake of FVa protein by the platelets; however, uptake of infused plasma FV may not occur in mouse platelets, explaining why infusion of plasma FV had little effect on the occlusion time in the Tg+Fv−/− mice. These results strongly supported the prothrombotic role of platelet FV. Although the difference between human and murine platelet FV may make it difficult to extrapolate the observations in mice to humans, it is worth noting that the separation of murine plasma and platelet FV pools makes it easier to decipher the function of platelet FV without interference from the plasma FV pool, as demonstrated in the plasma FV infusion experiment (Figure 3). Our observation suggested that targeting platelet FV could have significantly more impact on artery thrombosis than plasma FV, although plasma FV could be taken up by platelets in humans, thus also playing a limited role. Targeting platelet FV may be a promising approach for treating cardiovascular diseases. For example, both human embryonic and induced pluripotent stem cells are promising sources of cells for clinical cardiovascular therapies because they are able to undergo differentiation toward polyploid megakaryocytes and, ultimately, the release of platelets into the bloodstream.
The observation that the platelets from the Tg‐Fv+/+ mice showed increased aggregation responses in vitro provides a direct link between accelerated thrombosis and platelet thrombogenic status. There was no significant difference between the effects of PPP from Tg‐Fv+/+ and Tg+Fv−/− mice on agonist‐stimulated aggregation, indicating that plasma FV was insufficient to enhance platelet activation. Instead, this effect required platelet‐derived FV.
Using flow‐cytometric analysis, we found that the expression of P‐selectin was augmented in the platelets collected from the Tg‐Fv+/+ mice. P‐selectin increases interactions between platelets and endothelial cells. The effect of FV on platelet activation was higher when ADP was used as an agonist, suggesting a potential interaction between FV and ADP that could enhance the prothrombotic properties of FV. In addition, cGMP is implicated in the modulation of platelet aggregation21; therefore, these events may be causally related. Similarly, mice with higher levels of FV demonstrated increased cGMP levels in the platelets in response to agonists. Further work will be required to explore this phenomenon in detail. Platelet FV is an important regulator of platelet activation and thus stimulation of cGMP‐dependent signaling.
Human plasma FV is a single‐chain molecule, and the platelet‐released FV was partially cleaved due to proteolysis11; however, the diagnostic utility of the FV activity test in PPP may be limited. A limitation of experiments using the carotid artery thrombosis model is that the prothrombotic effects of platelet‐derived FV cannot be definitively proven to be caused by thrombin generation. Nevertheless, our current results, which demonstrated that platelet FV promotes arterial thrombosis, support the significance of our proposed molecular mechanisms in a clinically relevant in vivo context. Another limitation is the lack of relevant data on the association between FV and TFPI in the present study. Furthermore, evidence indicates that the platelet pool of FV differs between mice and humans.7 Future studies are needed to investigate the potential interaction between FV and TFPI levels in the context of the risk of thrombosis.
For the first time, to our knowledge, we identified platelet‐derived FV as a critical mediator of arterial thrombosis in vivo. These effects were associated with increased expression of P‐selectin and a higher cGMP level in the platelets. These results support a relationship between platelet‐derived FV and platelet activation.
Sources of Funding
This work was supported by National Natural Science Foundation of China Grant (81570263), and Grant of Sichuan Province Science and Technology Agency Grant (2014FZ0104) to Wu.
Disclosures
Dr Sun owns stocks in Nanova Inc. The other authors do not have any conflicts of interest to disclose.
(J Am Heart Assoc. 2017;6:e006345 DOI: 10.1161/JAHA.117.006345.)28673898
References
- 1. Gould WR, Silveira JR, Tracy PB. Unique in vivo modifications of coagulation factor V produce a physically and functionally distinct platelet‐derived cofactor: characterization of purified platelet‐derived factor V/Va. J Biol Chem. 2004;279:2383–2393. [DOI] [PubMed] [Google Scholar]
- 2. Gould WR, Simioni P, Silveira JR, Tormene D, Kalafatis M, Tracy PB. Megakaryocytes endocytose and subsequently modify human factor V in vivo to form the entire pool of a unique platelet‐derived cofactor. J Thromb Haemost. 2005;3:450–456. [DOI] [PubMed] [Google Scholar]
- 3. Duckers C, Simioni P, Spiezia L, Radu C, Dabrilli P, Gavasso S, Rosing J, Castoldi E. Residual platelet factor V ensures thrombin generation in patients with severe congenital factor V deficiency and mild bleeding symptoms. Blood. 2010;115:879–886. [DOI] [PubMed] [Google Scholar]
- 4. Dahm AE, Bezemer ID, Sandset PM, Rosendaal FR. Interaction between tissue factor pathway inhibitor and factor V levels on the risk of venous thrombosis. J Thromb Haemost. 2010;8:1130–1132. [DOI] [PubMed] [Google Scholar]
- 5. Kamphuisen PW, Rosendaal FR, Eikenboom JC, Bos R, Bertina RM. Factor V antigen levels and venous thrombosis—risk profile, interaction with factor V Leiden, and relation with factor VIII antigen levels. Arterioscler Thromb Vasc Biol. 2000;20:1382–1386. [DOI] [PubMed] [Google Scholar]
- 6. Bouchard BA, Chapin J, Brummel‐Ziedins KE, Durda P, Key NS, Tracy PB. Platelets and platelet‐derived factor Va confer hemostatic competence in complete factor V deficiency. Blood. 2015;125:3647–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang TL, Pipe SW, Yang A, Ginsburg D. Biosynthetic origin and functional significance of murine platelet factor V. Blood. 2003;102:2851–2855. [DOI] [PubMed] [Google Scholar]
- 8. Sun H, Yang TL, Yang A, Wang X, Ginsburg D. The murine platelet and plasma factor V pools are biosynthetically distinct and sufficient for minimal hemostasis. Blood. 2003;102:2856–2861. [DOI] [PubMed] [Google Scholar]
- 9. Tracy PB, Mann KG. Abnormal formation of the prothrombinase complex: factor V deficiency and related disorders. Hum Pathol. 1987;18:162–169. [DOI] [PubMed] [Google Scholar]
- 10. Viskup RW, Tracy PB, Mann KG. The isolation of human platelet factor V. Blood. 1987;69:1188–1195. [PubMed] [Google Scholar]
- 11. Monković DD, Tracy PB. Functional characterization of human platelet‐released factor V and its activation by factor Xa and thrombin. J Biol Chem. 1990;265:17132–17140. [PubMed] [Google Scholar]
- 12. Ayombil F, Abdalla S, Tracy PB, Bouchard BA. Proteolysis of plasma‐derived factor V following its endocytosis by megakaryocytes forms the platelet‐derived factor V/Va pool. J Thromb Haemost. 2013;11:1532–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun H, Wang X, Degen JL, Ginsburg D. Reduced thrombin generation increases host susceptibility to group a streptococcal infection. Blood. 2009;113:358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang Y, Xiao L, Chen N, Li Y, Deng X, Wang L, Sun H, Wu J. Platelet‐derived factor V promotes angiogenesis in a mouse hind limb ischemia model. J Vasc Surg. 2016;16:30038–30046. [DOI] [PubMed] [Google Scholar]
- 15. Eitzman DT, Westrick RJ, Nabel EG, Ginsburg D. Plasminogen activator inhibitor‐1 and vitronectin promote vascular thrombosis in mice. Blood. 2000;95:577–580. [PubMed] [Google Scholar]
- 16. Eckly A, Hechler B, Freund M, Zerr M, Cazenave JP, Lanza F, Mangin PH, Gachet C. Mechanisms underlying FeCl3‐induced arterial thrombosis. J Thromb Haemost. 2011;9:779–789. [DOI] [PubMed] [Google Scholar]
- 17. Jenny RJ, Pittman DD, Toole JJ, Kriz RW, Aldape RA, Hewick RM, Kaufman RJ, Mann KG. cDNA and derived amino acid sequence of human factor V. Proc Natl Acad Sci USA. 1987;84:4846–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nesheim ME, Katzmann JA, Tracy PB, Mann KG. Factor V. Methods Enzymol. 1981;80:249–274. [DOI] [PubMed] [Google Scholar]
- 19. Isenberg JS, Romeo MJ, Yu C, Yu CK, Nghiem K, Monsale J, Rick ME, Wink DA, Frazier WA, Roberts DD. Thrombospondin‐1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood. 2008;111:613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stopa JD, Neuberg D, Puligandla M, Furie B, Flaumenhaft R, Zwicker JI. Protein disulfide isomerase inhibition blocks thrombin generation in humans by interfering with platelet factor V activation. JCI Insight. 2017;2:e89373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Z, Zhang G, Feil R, Han J, Du X. Sequential activation of p38 and ERK pathways by cGMP‐dependent protein kinase leading to activation of the platelet integrin alphaIIb beta3. Blood. 2006;107:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]