

Abstract
Background
DPP4 (Dipeptidyl peptidase‐4)‐GLP‐1 (glucagon‐like peptide‐1) and its receptor (GLP‐1R) axis has been involved in several intracellular signaling pathways. The Adrβ3 (β3‐adrenergic receptor)/CXCL12 (C‐X‐C motif chemokine 12) signal was required for the hematopoiesis. We investigated the novel molecular requirements between DPP4‐GLP‐1/GLP‐1 and Adrβ3/CXCL12 signals in bone marrow (BM) hematopoietic stem cell (HSC) activation in response to chronic stress.
Methods and Results
Male 8‐week‐old mice were subjected to 4‐week intermittent restrain stress and orally treated with vehicle or the DPP4 inhibitor anagliptin (30 mg/kg per day). Control mice were left undisturbed. The stress increased the blood and brain DPP4 levels, the plasma epinephrine and norepinephrine levels, and the BM niche cell Adrβ3 expression, and it decreased the plasma GLP‐1 levels and the brain GLP‐1R and BM CXCL12 expressions. These changes were reversed by DPP4 inhibition. The stress activated BM sca‐1highc‐Kithigh CD48low CD150high HSC proliferation, giving rise to high levels of blood leukocytes and monocytes. The stress‐activated HSC proliferation was reversed by DPP4 depletion and by GLP‐1R activation. Finally, the selective pharmacological blocking of Adrβ3 mitigated HSC activation, accompanied by an improvement of CXCL12 gene expression in BM niche cells in response to chronic stress.
Conclusions
These findings suggest that DPP4 can regulate chronic stress‐induced BM HSC activation and inflammatory cell production via an Adrβ3/CXCL12‐dependent mechanism that is mediated by the GLP‐1/GLP‐1R axis, suggesting that the DPP4 inhibition or the GLP‐1R stimulation may have applications for treating inflammatory diseases.
Keywords: glucagon‐like peptide‐1, inflammation, stress
Subject Categories: Basic Science Research, Animal Models of Human Disease
Clinical Perspective
What Is New?
Plasma and brain DPP4 activities were increased and plasma GLP‐1 levels were decreased in mice and rats under conditions of chronic immobilized stress.
Genetic and pharmacological interventions toward DPP4 rectified stress‐induced bone marrow hematopoietic stem cell activation and inflammatory via a adrβ3/CXCL12‐dependent mechanism that is mediated by the GLP‐1/GLP‐1R axis in rats and mice.
GLP‐1R activation with its agonist produced a beneficial bone‐marrow response to stress in mice.
What Are the Clinical Implications?
Circulating DPP4 or/and GLP‐1 levels might be serve as a novel biomarker of the presence of stress in animals.
The inhibition of DPP4 or the stimulation of GLP‐1R may have applications in the treatment of chronic stress‐related inflammatory diseases (eg, atherosclerosis‐based cardiovascular disease and metabolic disorders).
Introduction
The organic response to chronic stress implicates 2 major components of the stress system: the hypothalamic–pituitary–adrenal axis and the sympathetic nervous system. The activation of these systems promotes the secretion of glucocorticoid and adrenal catecholamines to the alternate systemic immune and hormonal response.1 Accumulating evidence from the past 2 decades shows that the chronic stress in modern lifestyles is closely linked to the incidence of metabolic syndrome, diabetes mellitus, hypertension, and cardiovascular disease.1
The activation of hematopoietic stem cells (HSCs) that develops as an initial step of disease processes is often targeted in patients with blood or cardiometabolic disorders, in order to control inflammatory and immune responses.1, 2, 3 Laboratory investigations have led to several important observations that helped clarify the molecular mechanisms underlying HSC development and activation. For example, adenosine signaling plays an evolutionary conserved role in HSC formation in vertebrates.4 Tzeng and colleagues demonstrated that the stroma‐secreted CXCL12 (stromal cell‐derived factor‐1) keeps the HSCs in a quiescent state and maintains them in the osteoblastic niche.5 CXCL12 has been shown to be involved in hematopoietic progenitor cell homing and mobilization.6 A recent experimental study documented that under conditions of chronic variable stress in mice, sympathetic nerve fibers released surplus noradrenaline, which signaled bone‐marrow (BM) niche cells to suppress CXCL12 expression through the Adrβ3.2
DPP4, originally known as the lymphocyte cell surface antigen, or as the adenosine deaminase‐binding protein, is distributed in many mammalian tissues, including small intestine, kidney, and heart tissues.7 DPP4 is upregulated in proinflammatory states (eg, obesity, diabetes mellitus, and atherosclerosis‐related coronary artery disease).7, 8, 9 The widespread expression of DPP4 macrophages and immune cells (eg, natural killer cells, monocytes, lymphocytes, and dendritic cells) and the noncatalytic function of DPP4 (also called CD26) as a signaling and binding protein across a wide range of species suggest a teleological role for DPP4 in inflammation and immune response.10, 11, 12 It was also documented that functionally matured macrophages and dendritic cells had increased expressions of DPP4 and CD68 and activation of the nuclear factor‐κB pathway.13, 14 Cordero and colleagues demonstrated that DPP4 regulates T‐cell maturation and migration and the activation of cytotoxic T lymphocytes via an interleukin‐12‐dependent mechanism.11 Additionally, soluble DPP4 binds to insulin‐like growth factor II/mannose‐6‐phosphate receptor and is taken up by CD14+ monocytes, enhancing their antigen‐presenting activity and T‐lymphocyte migration.15 Accumulating evidence shows that DPP4 activity can regulate cellular functions via the degradation of GLP‐1 as well as a variety of other peptides.9 However, DPP4 expression and its role in HSC activation and proliferation have been unknown.
Given that genetic and pharmacological interventions targeted toward DPP4 have produced pleotrophic effects in animals and humans in many pathophysiological conditions,16, 17, 18 we hypothesized that in rodent models, brain GLP‐1/GLP‐1R‐dependent actions provided by a DPP4 inhibitor would have inhibitory effects on increased BM HSC proliferation in response to chronic stress via the improvement of the Adrβ3/CXCL12 axis signal, leading to a decreased output of neutrophils and inflammatory monocytes.
Materials and Methods
Methods
Animals
Seven‐week‐old C57BL/6J male mice and 6‐week‐old F344/Jcl male rats (Chubu Kagaku Shizai, Nagoya, Japan) and 6‐week‐old F344/DuCrj male rats (Charles River Laboratories Japan, Yokohama, Japan) were provided a standard diet and tap water ad libitum. The animal protocols were approved by the Institutional Animal Care and Use Committee of Nagoya University (Protocol No. 27304) and performed according to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health.
Chronic restraint stress procedure and tissue collections
Seven‐week‐old mice (21–24 g) were randomly assigned to the control or stress group (n=10 for each group). First, for the evaluation of chronic stress effect (Study 1), the mice in the stress group were individually subjected to a 2‐hour session of immobilization stress twice daily (morning and afternoon/between 9:00 am and 5:00 pm, 7 days/wk) for 4 weeks, and the control mice were left undisturbed and allowed contact with each other, as described.19 The immobilization stress protocol was as follows (in brief): chronic stress was applied using a 50‐mL conical centrifuge tube with multiple punctures as airholes and a Kimwipe disposable cloth inserted at the head of the tube that immobilized the mouse placed in the tube.
In separate DPP4 inhibitor experiments (Study 2), mice that were to be subjected to immobilization stress were randomly assigned to 2 treatment groups (n=12 in each group): (1) vehicle (distilled water; the Stress group) or (2) the DPP4 inhibitor anagliptin (30 mg/kg per day, the S‐Ana group; the anagliptin was a generous gift from Sawa Kagaku Pharmaceutical Co, Mie, Japan) for 4 weeks. The treatments were given by oral gavage twice daily (8:00 am and 7:00 pm) starting on the day before the stress protocol began.
In the specific GLP‐1R agonist experiments (Study 3), mice that were to be subjected to immobilization stress were randomly assigned to 2 treatment groups (n=8 in each group): (1) vehicle (the saline group) and (2) the S‐Exe group given the GLP‐1R agonist exenatide (5 μg/kg, subcutaneous/twice daily, AstraZeneca, London, UK) for 4 weeks. In the specific Adrβ3 antagonist experiments (Study 4), mice that underwent stress were administered either vehicle or a specific Adrβ3 antagonist, L748337 (0.5 mg/kg per day, intraperitoneal injection; cat. no. L7045, Sigma‐Aldrich, St. Louis, MO), for 4 weeks. In the rat experiments (Study 5), 6‐week‐old male wild‐type and DPP4‐deficient (DPP4−/−) rats16 (90–105 g) were subjected to immobilization stress in a KN‐325‐B Square Rat Holder with 3 sluices (Natsume Seisakusho, Tokyo) that was set for a close fit to the rats for 4 weeks as described above (n=12). Control rats were left undisturbed (n=12).
At the end of the 4‐week stress protocol, all mice and rats (all stressed animals were left undisturbed for 2 hours before analysis) were anesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg; Dainippon Pharmaceutical, Osaka, Japan), and the tissues (brains and femurs) and arterial blood samples were collected for biological analyses (including DPP4 activity, an ELISA, and gene and protein assays) and histological analyses.
Histology and immunohistochemistry
For the histological analysis, the mouse and rat brains and femurs were fixed in ice‐cold 4% paraformaldehyde solution for 24 hours, embedded in paraffin, and processed for histology and immunohistochemistry as described.20 Both brain and femur transverse tissue sections (3 μm) were stained with hematoxylin–eosin solution for routine histological examination.
Brain and femur slices on separate slides were processed for the immunohistochemical analysis of CD26, Adrβ3, and proliferating cell nuclear antigen (PCNA). The primary antibodies for CD26 (1:50; sc‐9153; Santa Cruz Biotechnology, Dallas, TX), Adrβ3 (1:50; ab94506; Abcam, Cambridge, MA), and PCNA (1:100, ab29, Abcam), were applied to the sections, which were then left overnight at 4°C. After being washed with phosphate‐buffered saline 5 times, the sections were sequentially treated with appropriate secondary antibodies (1:200, all from Vector Laboratories, Burlingame, CA), respectively for 2 hours at 4°C, and were then visualized with a corresponding substrate kit (Vector Laboratories).21 Five cross‐sections of the femur were quantified and averaged for each animal. We set a threshold to automatically compute the positively stained area for each antibody or histochemical stain and then computed the ratio (percent) of the positively stained area to the total cross‐sectional BM niche area studied.
Bromodeoxyuridine assays and immunofluorescence analysis
For the bromodeoxyuridine (BrdU) assay, we performed in vivo BrdU labeling to determine the number of proliferating cells in BM niches by the detection of DNA synthesis, using a BrdU immunohistochemistry kit (ab125306, Abcam). An intraperitoneal injection of BrdU (BD Pharmingen) was administered to mice at 100 μg per g body weight. Two hours later, the femurs were isolated and fixed in 4% paraformaldehyde at 4°C overnight, and then were imbedded in paraffin. BM sections were then collected for BrdU staining. BM sections were also used for immunofluorescence staining.
Double immunofluorescence was performed using a goat polyclonal antibody to sca‐1 (1:100; Cat. AF1226; R&D Systems, Dallas, TX) and a rabbit polyclonal antibody Ki67 antibody (1:50; #9129; Cell Signaling Technology Inc, Boston, MA) or a mouse monoclonal antibody to PCNA (1:100) and a mouse monoclonal antibody to CD150 (1:200; Biolegend, San Diego, CA). The sections were visualized using Zenon rabbit and mouse immunoglobulin G labeling kits (1:200; Molecular Probes, Eugene, OR) according to the manufacturer's instructions. The slides of the tissues (femurs) and the cells were mounted in glycerol‐based Vectashield medium (Vector Laboratories) containing the nucleus stain 4′,6‐diamidino‐2‐phenylindole. For negative controls, the primary antibodies were replaced with nonimmune immunoglobulin G or xenon‐labeled goat or mouse immunoglobulin G. Staining sections were visualized with a BZ‐X700 microscope (Keyence, Osaka, Japan) using ×20 or ×40 objectives. Images were analyzed with BZ‐X analyzer software (Keyence).22
Gene expression assay
Whole RNA was harvested from cells and tissues with the RNAeasy Mini Kit (Qiagen, Hilden, Germany) according to the recommended protocol. The SuperScript III CellsDirect cDNA Synthesis kit (Invitrogen, Carlsbad, CA) was used to generate cDNA. The resulting cDNA was subjected to a quantitative real‐time polymerase chain reaction analysis with primers specific for matrix metalloproteinase‐9 (MMP‐9), Toll‐like receptor‐2, CXCL12, and Adrβ3 and with the use of an ABI 7300 Real‐Time PCR System (Applied Biosystems, Carlsbad, CA) as described.20 The primer sequences for the targeted genes are provided in Table 1. Targeted gene expression was normalized to the related internal GAPDH gene.
Table 1.
Primer Sequences for Mice and Rats Used for Quantitative Real‐Time PCR
| Product (bp) | Annealing (°C) | ||
|---|---|---|---|
|
CXCL12_Fw (mouse)
CXCL12_Rv |
CGCCAAGGTCGTCGCCG TTGGCTCTGGCGATGTGGC |
118 | 60 |
|
GLP‐1R_Fw (mouse)
GLP‐1R_Rv |
GTACCACGGTGTCCCTCTCA CCTGTGTCCTTCACCTTCCCTA |
1407 | 55 |
|
MMP‐9_Fw (mouse)
MMP‐9_Rv |
CCCTACTGCTGGTCCTTCTGAG AATTGGCTTCCTCCGTGATTCG |
162 | 60 |
|
TLR‐2_Fw (mouse)
TLR‐2_Rv |
AAGAAGCTGGCATTCCGAGGC CGTCTGACTCCGAGGGGTTGA |
151 | 59 |
|
Adrβ3_Fw (mouse)
Adrβ3_Rv |
TGCGCACCTTAGGTCTCATTATGG AAACTCCGCTGGGAACTAGAGAGG |
110 | 60 |
|
GAPDH_Fw (mouse)
GAPDH_Rv |
ATGTGTCCGTCGTGGATCTGA ATGCCTGCTTCACCACCTTCT |
77 | 60 |
|
CXCL12_Fw (rat)
CXCL12_Rv |
GAGCCATGTCGCCAGAGCCAAC CACCTCTCACATCTTGAGCCTCT |
145 | 60 |
|
GAPDH_Fw (rat)
GAPDH_Rv |
GGCACAGTCAAGGCTGAGAATG ATGGTGGTGAAGACGCCAGTA |
143 | 60 |
Adrβ3 indicates adrenergic receptor‐β3; CXCL12 (SDF‐1), C‐X‐C motif chemokine 12 (stromal cell‐derived factor 1); GLP‐1R, glucagon‐like peptide‐1 receptor; MMP‐9, matrix metalloproteinase‐9; PCR, polymerase chain reaction; TLR‐2, Toll‐like receptor‐2.
Western blot analysis
Proteins were lysed from the cells and the tissues using lysis buffer containing 20 mmol/L Tris‐Cl (pH 8.0), 1% Triton X‐100, 150 mmol/L NaCl, 1 mmol/L EDTA, 0.05% sodium dodecyl sulfate (SDS), 1% Na‐deoxycholate, and fresh 1× protease inhibitors. The concentration of each protein was measured by the DC Protein Assay kit (Bio‐Rad Laboratories, Hercules, CA) before the proteins were equally loaded and separated by SDS‐polyacrylamide gel electrophoresis. Proteins were then transferred to FluoroTrans‐W® membranes (Pall, Port Washington, NY) and incubated overnight with primary antibodies against GLP‐1R (ab39072, Abcam; 1:1000), Adrβ3 (ab94508, Abcam; 1:1000), and β‐actin (1:1000; AC‐15, Sigma‐Aldrich). The membranes were then treated with the horseradish peroxidase–conjugated secondary antibody at a 1:10 000 to 15 000 dilution. The Amersham ECL Prime Western Blotting Detection kit (GE Healthcare, Freiburg, Germany) was used for the determination of targeted proteins. Protein levels quantitated from Western blots were normalized by loading β‐actin.
Gelatin zymography
For gelatin zymography, equal amounts of total protein (20 μg) extracted from the BM tissues were mixed with SDS sample buffer without reducing agent and loaded onto a 10% SDS–polyacrylamide gel containing 1 mg/mL gelatin, as described.23 After electrophoresis, the gels were washed twice for 30 minutes with 2.5% Triton X‐100 (vol/vol) to exclude the SDS, and then incubated in the reaction buffer (50 mmol/L Tris‐HCl, 0.15 mol/L NaCl, 10 mmol/L CaCl2, 0.02% NaN3, pH 7.4) at 37°C overnight. Following staining with Coomassie Brilliant Blue for 30 minutes, the gels were destained with destaining buffer for 1 hour. Digestion bands were quantified by an image analyzer software program (NIH image 1.62). One gel was used to examine an equal amount of protein loaded by a directly Coomassie Brilliant Blue staining. The levels of pro‐ and active‐MMP‐9 activities were analyzed, and the total MMP‐9 activity is expressed as MMP‐9 activity.
ELISA and biochemical analyses
Blood samples were obtained directly from the left ventricles of mice and rats after a 10‐hour fast and/or combined stress for 4 hours for the ELISA and biochemical analyses. The levels of plasma GLP‐1 protein (cat. no. EZGLP1T‐36K; EMD Millipore, Billerica, MA) and the cell extract Adrβ3 activity (cat. no. MBS2022034; MyBioSource, San Diego, CA) were determined using the commercially available ELISA kits according to the manufacturer's instructions. The blood cell analysis of the rats and the levels of the mouse and rat plasma glucose, triglyceride, total cholesterol, high‐density lipoprotein cholesterol, aspartate transaminase, blood urea nitrogen, creatinine, nonesterified fatty acid (NEFA), epinephrine, norepinephrine, and catecholamine were examined at a commercial laboratory (SRL, Tokyo, Japan).
Plasma and tissue DPP4 activity analysis
We measured the DPP4 activity by using the DPP4 Glo Protease Assay (Promega, Madison, WI) with an aminoluciferin substrate. The luminogenic substrate contains the Gly‐Pro sequence recognized by DPP4. Following DPP4 cleavage, the substrate for luciferase (aminoluciferin) is released, resulting in the luciferase reaction and the production of light. Targeted tissue samples of the mice and the rats (including brain, subcutaneous fat, visceral fat, lung, aorta, heart, liver, kidney, spleen, small intestine, and muscle; ≈10 mg) were minced using a razor blade and extracted in the lysis buffer (100 mmol/L Tris‐HCl, 20 mmol/L NaCl, 0.5% SDS, pH 8.0) without serine protease inhibitors, by means of a tissue homogenizer. The protein concentration for each result was determined using a protein assay system according to the manufacturer (Bio‐Rad). For the blood DPP4 activity assays, the plasma was isolated using VENOJectII vacuum blood collection tubes containing anticoagulants without serine protease inhibitor (Terumo, Tokyo, Japan) and then diluted in 0.1 mmol/L Tris‐HCl buffer (pH 8.0) by 30‐fold. Equal amounts of protein (200 μg/25 μL) and diluted plasma (25 μL) were subjected to a DPP4 Glo assay (Promega) in the presence or absence of the DPP4 inhibitor anagliptin (20 μmol/L). Human recombinant DPP4 (Sigma‐Aldrich) was used to drive a standard curve. The luminescence intensity was calculated using a POWERSCAN4 (BioTek Instruments Inc, Winooski, VT). The anagliptin‐sensitive value (ie, the absence value minus the presence value as an absolute value of its DPP4 activity) in relative light units per 200 μg of protein (tissue extract) or per mL of plasma was calculated with the standard curve to represent the DPP4 level.
Flow cytometry
Flow cytometry analyses were performed as described.2 On stressed day 28, following removing peripheral blood and BM red blood cells by ammonium chloride, the results were stained with CD45.2 (clone 104; BD Bioscience, 1:300), CD11b (clone M1/70, BD Bioscience, 1:500), Ly6G (clone 1A8, BD Bioscience, 1:500), F4/80 clone BM8, (Biolegend, 1:500) and Ly6c (clone AL‐21, BD Bioscience, 1:500). Based on previous studies,24, 25, 26 we quantified the HSCs in the BM by a flow cytometry–based detection of CD48lowCD150high cells using a c‐Kit (2B8, eBioscience, San Diego, CA; 1:500), Sca‐1 (D7, Biolegend, 1:500), SLAM markers CD48 (clone Hm48‐1, Biolegend, 1:500), and CD150 (clone TC‐12F12.2, Biolegend, 1:500). Monocytes were identified as Ly6GlowCD11bhighF4/80lowLy6Chigh, neutrophils were identified as CD11bhigh/Ly6Ghigh, and HSCs were identified as lin−c‐KithighSca‐1highCD48lowCD150high. The APC mouse lineage antibody Cocktail (448074, BD Pharmingen) used in the fluorescence‐activated cell sorter analysis contains mouse CD3e (clone 145‐2C11), CD11b (clone, M1/70), CD45R.B220 (clone, RA3‐6B2), LY‐76 (clone, TER‐119), Ly6G and Ly6C (clone, RB6‐8C5).
Cell‐cycle assay
We performed a cell‐cycle assay by using a slightly modified propidium iodine–based flow cytometry analysis. In brief, BM was obtained from the experimental groups of mice. BM cells were suspended in 300 μL of PBS and fixed in 70% ethanol at 4°C overnight. Fixed cells were pelleted at 800g for 10 minutes and incubated in KRISHIAN buffer (0.1% sodium citrate, 0.3% NP‐40, 0.02 mg/mL RNAse A [Sigma Aldrich], and 0.05 mg/mL propidium iodide [Invitrogen]) for 1 hour at 4°C in the dark, and then filtered and evaluated for the propidium iodide labeling of DNA by flow cytometry.
Colony‐forming unit assay
Colony‐forming unit assays were performed as described.2 First, 2×104 BM sca‐1+ cells were seeded on a 3‐mm dish in duplicate and incubated for 7 days. Colonies were counted using a low‐magnification inverted microscope.
Statistical Analysis
Data are expressed as mean±SEM. Student t tests (for comparisons of 2 groups) or a one‐way ANOVA (for comparisons of 3 or more groups) followed by Tukey post hoc tests were used for the statistical analyses. The body weight (BW) data were subjected to 2‐way repeated‐measures ANOVA and Bonferroni post hoc tests. SPSS software ver. 17.0 (SPSS, Chicago, IL) was used. A value of P<0.05 was considered significant.
Results
Compared with the control mice, the mice that underwent the 4‐week stress protocol lost significant amounts of subcutaneous fat and inguinal fat (Figure 1A and 1B). Consistent with previous research,19, 27 the stressed mice maintained decreased BW throughout the 4‐week follow‐up period (Figure 1C). The plasma triglyceride level was decreased and the plasma NEFA level was increased in the stressed mice (Table 2). However, there were no significant changes in the levels of plasma total cholesterol, high‐density lipoprotein cholesterol, glucose, aspartate transaminase, blood urea nitrogen, or creatine between the control and stressed mice.
Figure 1.
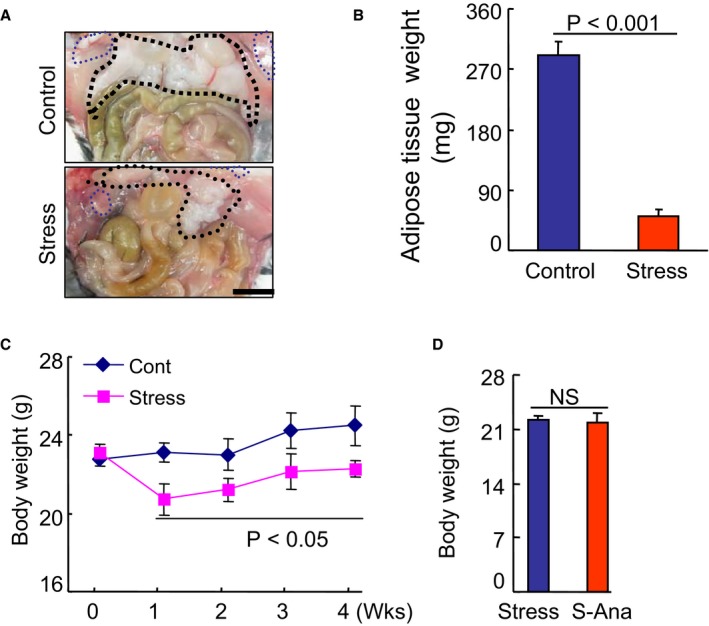
The effects of stress on body weight (BW) in the control and stressed mice. A and B, Representative pictures and quantitative data showing the loss of subcutaneous fat and inguinal fat in the stressed mice (Student t test). C, The changes in BW during the 4‐week follow‐up period in both groups (2‐way repeated‐measures ANOVA and Bonferroni post hoc test). D, There were no significant differences in BW in the stress group mice (Student t test). Scar bar, 50 μm. Values are mean±SE (n=8–10). P<0.05 vs corresponding controls; NS indicates not significant; S‐Ana, stressed anagliptin treatment.
Table 2.
Levels of Lipids and Other Parameters in the Control Stress Groups at 4 Weeks
| Parameter | Control | Stress |
|---|---|---|
| TG, mg/dL | 43.3±8.7 | 25.3±3.0a |
| T‐cho, mg/dL | 57.0±3.4 | 65.6±4.1 |
| HDL‐C, mg/dL | 30.0±1.7 | 25.9±1.4 |
| NEFA, μEq/L | 497.0±33.9 | 672.8±70.4a |
| Glucose, mg/dL | 98.9±7.2 | 106.5±8.9 |
| BUN, mg/dL | 30.7±2.2 | 32.1±1.8 |
| Creatinine, mg/dL | 0.1±0.01 | 0.12±0.02 |
| AST, pg/mL | 78.7±7.3 | 91.7±15.2 |
| Adrenaline, pg/mL | 1545±56 | 4067±302a |
| Noradrenaline, pg/mL | 2012±138 | 5054±451a |
| GLP‐1, pmol/L | 19.9±1.1 | 5.3±1.6a |
| MMP‐9 gene | 39.1±3.2 | 89.2±5.6a |
| TLR‐2 gene | 18.1±2.3 | 46.7±4.1a |
Values are mean±SE (n=6–10). AST indicates aspartate transaminase; BUN, blood urea nitrogen; GLP‐1, glucagon‐like protein‐1; HDL‐C, high density lipoprotein cholesterol; MMP‐9, matrix metalloproteinase‐9; NEFA, nonesterified fatty acid; T‐cho, total cholesterol; TG, triglyceride; TLR‐2, Toll‐like receptor‐2.
P<0.01, vs control group by Student t test.
Stress Increased the Plasma and Tissue DPP4 Levels
As a first step to examine the relationship between chronic stress and DPP4 levels in the blood and organs, we exposed mice to chronic immobilization stress (Figure 2A), and we examined the changes in DPP4 levels in blood and several types of tissue (brain, heart, lung, spleen, small intestine, subcutaneous fat, inguinal fat, kidney, and liver) (Figure 2B through 2D). We observed only low DPP4 levels in the blood and the targeted tissues of the unstressed (control) mice. In the stressed mice, with the exception of the liver tissue, the blood and other targeted tissues showed dramatically increased DPP4 levels on day 28 of the 4‐week stress protocol. The change in DPP4 level was the highest in the brain (by >10‐fold) compared with that of the unstressed mice brains. Compared with the unstressed rat brains, the DPP4 level was increased by over 20‐fold in the brain of the stressed DPP4+/+ rats (Figure 2E). Hematoxylin–eosin staining showed the structure of the brains in both experimental groups (Figure 3A). Immunostaining using CD26 antibody revealed that exposure to chronic stress caused an enhancement of the positive‐stained signaling in the brain cells (Figure 3B). Figure 2F illustrates the time‐dependent increases in blood DPP4 level, suggesting that increased plasma DPP4 is linked to the presence of stress in mice and rats. However, we observed that there was no DPP4 in the extracts of BM cells from not only nonstressed but also stressed mice and rats. Likewise, CD26 staining exhibited no positive staining signaling in BM niche cells of either experimental group (Figure 3C).
Figure 2.
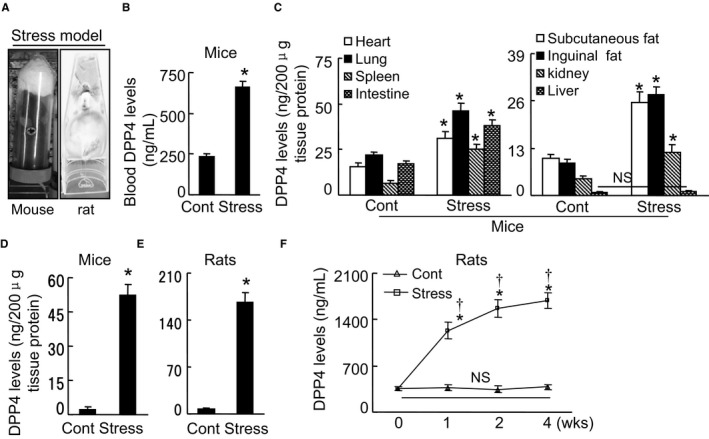
Chronic stress increased the blood and tissue DPP4 levels. A, The mouse/rat immobilized stress model. B through D, In the mice, the levels of DPP4 protein in the blood (B, Student t test), eight tissues (C, heart, lung, spleen, intestine, subcutaneous fat, inguinal fat, kidney, liver; ANOVA and Tukey's post hoc test), and brains (D, Student t test). E, The levels of DPP4 protein in the rat brains (Student t test). F, The changes in blood DPP4 levels during the follow‐up period (2‐way repeated‐measures ANOVA and Bonferroni post hoc test). Data are means±SEM (n=6–8). *P<0.01 vs controls; † P<0.01 vs corresponding controls; DPP4 indicates dipeptidyl peptidase‐4; H&E, hematoxylin–eosin; NS, not significant.
Figure 3.
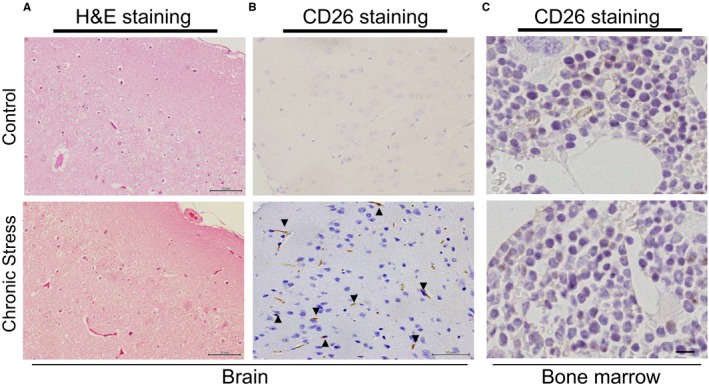
Stress increased the CD26 protein expression in the brain. A, Routine H&E staining of the brain tissue samples of nonstressed and stress mice. B and C, Immunostaining with CD26 antibody showed the expression of CD26 in the brains and the BM of both experimental groups. Scale bar, 50 μm. BM indicates bone marrow; H&E, hematoxylin–eosin.
Stress Activated the HSC Proliferative Ability and Increased the Output of Neutrophils and Monocytes
To explore the impact of stress on leukocytosis resulting from increased leukocyte production, we analyzed the blood of the control mice and stressed mice. Compared with the nonstressed controls, the stressed mice had significantly increased leukocytes (4.4±0.9 versus 2.2±0.2×106/mL, n=8; P<0.01), neutrophils, and monocytes in the blood (Figure 4A and 4B), which is consistent with a previous clinical study.2 These inflammatory cells (neutrophils and monocytes) were more numerous in the BM (Figure 4C).
Figure 4.
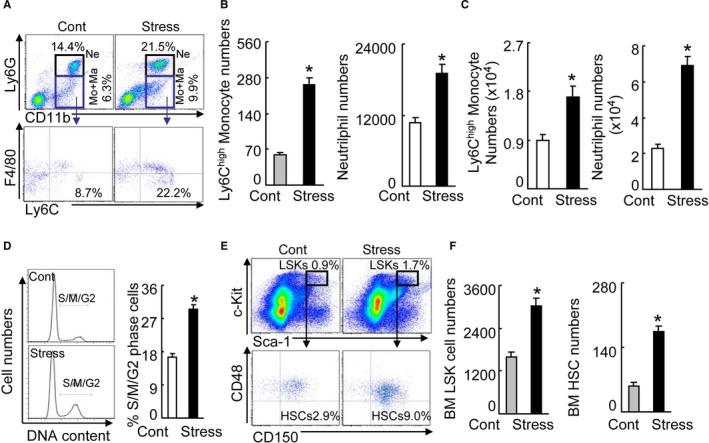
Chronic stress‐activated HSC proliferation in the bone marrow (BM). A, Gating for the enclosed 2 populations isolated from peripheral blood (PB) cells of 2 experimental groups (upper panels) and identifying for neutrophils as LyG6high CD11bhigh (top‐right quadrants) and subanalyzing LyG6lowLy6Chigh (bottom‐right quadrants) for monocytes as LyG6low CD11bhighF4/80lowLyChigh (lower panels). B, PB monocytes and neutrophil numbers in nonstressed (Cont) and stressed mice (Stress) mice after 4 weeks of stress (n=6–8, Student t test). C, BM monocytes and neutrophil numbers after 4 weeks of stress (n=6–8, Student t test). D, Representative histogram of DNA content during the cell cycle (left) and the distribution of S/M/G2 cells expressed as a percentage of total BM cells (right) (n=6–8, Mann–Whitney U test). E, Gating for the enclosed lin−sca‐1+c‐Kit+ cell (LSK) population isolated from BM cells of 2 experimental groups (upper panels) and subanalyzed lin−c‐KithighSca‐1high CD48low CD150high hematopoietic stem cell (HSC; lower panels). F, BM LSK and HSC numbers next to gates represent population frequencies (%) of nonstressed and stressed mice after 4 weeks of stress (n=6–8, Student t test). Data are mean±SEM. *P<0.01 vs nonstressed control mice; NS indicates not significant.
We next investigated the effect of chronic stress on blood cell production and cell cycling by flow cytometry. The BM harvest from the stressed mice had increased percentages of cells in the S/M/G2 phase, which is indicative of increased proliferative ability in BM niche cells in response to stress (Figure 4D). This enhanced proliferation resulted in higher numbers of lin−sca‐1+c‐Kit+ cells and lin−c‐KithighSca‐1highCD48lowCD150high HSCs (Figure 4E and 4F). We also observed that stress increased the numbers of Ki67+/CD150+ cells as compared with control mice (Figure 5). The sca‐1+ harvested from stressed mice had augmented colony‐forming capacity, indicative of increased hematopoietic progenitor cell proliferation (Figure 6A). Routine hematoxylin–eosin staining showed the structure of BM niches of both experimental groups (Figure 7A). Consistent with the data obtained in the BM cell‐cycle assay, the stressed mice had increased BrdU‐incorporated cells in BM niches (Figure 7B). Immunofluorescence revealed that the stress increased the numbers of PCNA+CD150+ cells in BM niches and sca‐1+/Ki67+ cells in cultured BM cells (Figure 7C and 7D), indicating that stress‐induced leukocytosis and monocytosis resulted from increased bone marrow HSC proliferation in mice. It was reported that GLP‐1R was expressed exclusively in neurons, with the strongest expression levels in cortical pyramidal neurons.28 Clinical and experimental evidence suggest that a DPP4‐GLP‐1/GLP‐1R axis plays an important role in brain damage in response to ischemic injury.28 To explore the cellular mechanism of the stress‐induced activation of HSC proliferation, we examined the levels of plasma GLP‐1 and brain GLP‐1R in the mouse (Figure 7E). We observed that stress resulted in reductions in the level of plasma GLP‐1 and the brain GLP‐1R protein expression (Table 2), suggesting that impaired brain GLP‐1/GLP‐1R signaling may be involved in the BM HSC activation in stressed mice.
Figure 5.
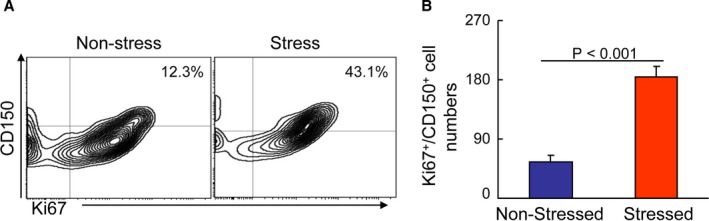
Chronic stress‐activated BM CD150+ cell proliferation. A and B, Representative images of the histograms and combined quantitative for the Ki67+/CD150+ cells in the bone marrow (BM) of the nonstressed and stressed mice. Values are mean±SEM (n=6, Student t test).
Figure 6.
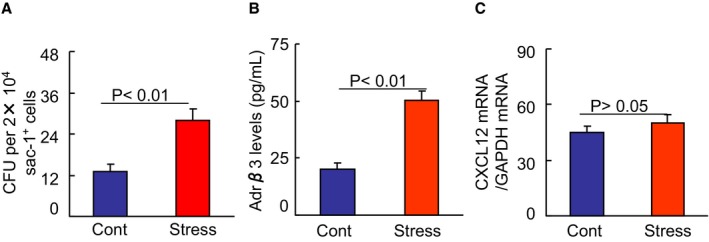
Chronic stress augmented colony‐forming capacity and Adrβ3 activity in BM sca‐1+ cells. A, First, 2×104 BM sca‐1+ cells were seeded on a 3‐mm dish in duplicate and incubated for 7 days. Colonies were counted using a low‐magnification inverted microscope. Colony‐forming unit (CFU) assay showed an increased colony‐forming capacity of the sac‐1+ cells from stressed BM. B, The extracts of the sac‐1+ cells (100 μg/well) from the BM of nonstressed and stressed mice were subjected to an Adrβ3 ELISA. Stress had increased levels of Adrβ3 activity in the BM‐derived sac‐1+ cells. C, Following culturing in serum‐free DMEM overnight, the sac‐1+ cells were cultured in the presence and absence of the GLP‐1R agonist exenatide (5 nmol/L) for 24 h, and the levels of CXCL12 mRNA were analyzed by quantitative real‐time PCR. GLP‐1 had no effect on CXCL12 mRNA. Values are mean±SEM (n=5–8, Student t test). BM indicates bone marrow; CXCL12, C‐X‐C motif chemokine 12; GLP‐1, glucagon‐like peptide‐1; PCR, polymerase chain reaction.
Figure 7.
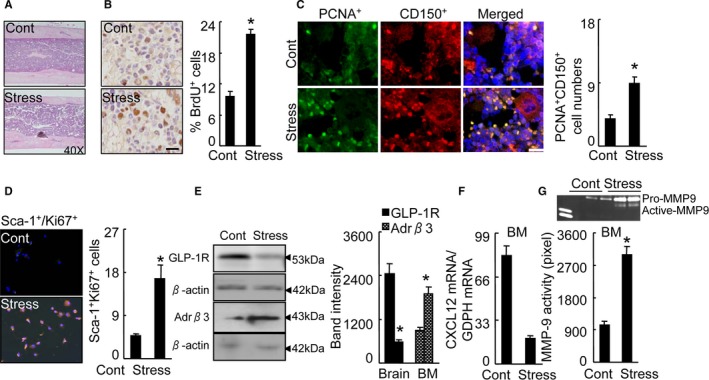
Stress‐activated BM CD150+ HSC proliferation. A, Routine H&E staining on BM niche structures of control and stressed mice. B, Immunoreactive staining of BM niches for BrdU. Bar graphs: The percentage of positive cells (per 2×102 cells) (n=5, Student t test). C, Double immunofluorescence of the BM niches for PCNA (green) and CD150 (red). Bar graphs: The numbers of double‐positive PCNA + CD150+ cells (orange, n=6, Student t test). D, Double immunofluorescence of cultured BM cells for sca‐1 and Ki67. Bar graphs: The numbers of double‐positive sca‐1+Ki67+ cells (orange, n=6, Student t test). E, Representative Western images and quantitative data for brain GLP‐1R protein and Adrβ3 protein of BM sca‐1+ cells (n=3, Student t test). F, Quantitative real‐time PCR for CXCL12 mRNA in BM sca‐1+ cells of 2 experimental group mice (n=6, Student t test). G, Representative gelatin zymography images and quantitative data for MMP9 activity in BM sca‐1+ cells (n=3, Student t test). Data are mean±SEM. *P<0.01 vs control mice. Adrβ3 indicates adrenergic receptor β3; BM, bone marrow; BrdU, bromodeoxyuridine; CXCL12, C‐X‐C motif chemokine 12; GLP‐1R, glucagon‐like peptide‐1 receptor; H&E, hematoxylin and eosin; HSC, hematopoietic stem cells; MMP9, matrix metalloproteinase 9; PCR, polymerase chain reaction; PCNA, proliferating cell nuclear antigen.
A recent study reported that an association between the Adrβ3 signal and CXCL12 expression may be a mechanism linking stress and BM immune action.2 In the present study, the plasma biological analysis revealed that the stressed mice had increased levels of plasma adrenaline and noradrenaline (Table 2). The stress decreased the CXCL12 gene expression and increased the Adrβ3 protein expression in the sca‐1+ cells of BM cells (Figure 7E and 7F). The ELISA data revealed that the level of Adrβ3 activity was higher in BM‐derived sca‐1+ cells from stressed mice compared with that of the control mice (Figure 6B). The quantitative analysis revealed that stress caused an increase in the MMP‐9 activity of the activated BM sca‐1+ cells (Figure 7G). The real‐time polymerase chain reaction analysis also revealed that the levels of MMP‐9 gene as well as Toll‐like receptor‐2 genes were higher in the BM sca‐1+ cells of the stressed mice compared with that of the control mice (Table 2). It was also reported that psychological stress stimulated inflammatory gene expression in the leukocyte transcriptome via a β‐adrenergic induction of myelopoiesis,29 and the loss of CXCL12/stromal cell‐derived factor‐1 was shown to decrease the quiescent state of hematopoietic stem cells in mice.5 Taking all of these findings together, it appears that in the stressed mice studied herein, the DPP4‐GLP‐1/GLP‐1R axis may have controlled the bone marrow HSC proliferative behavior and the elevated output of neutrophils and inflammatory monocytes through the modulation of β‐adrenergic induction and CXCL12 expression in the BM niche cells. However, a GLP‐1R agonist had no effect on CXCL12 gene expression in the BM sca‐1+ cells (Figure 6C), suggesting that GLP‐1 lacks an effect on CXCL12 expression in the BM stem cells of the mice.
DPP4 Is Required for BM HSC Activation
As a second step to test our hypothesis, we exposed mice to DPP4 inhibitor treatment with either vehicle or anagliptin for 4 weeks. The oral administration of anagliptin had no significant effect on BW on day 28 of the 4‐week stress protocol (Figure 1D). Except for the plasma NEFA, the DPP4 inhibition also had no effect on plasma glucose, lipids, or other parameters of renal and liver function (Table 3). The anagliptin treatment almost completely decreased the systematic and local brain tissue DPP4 levels. We assessed the effect of this DPP4 inhibitor on the sympathetic nervous system by measuring the plasma adrenaline and noradrenaline levels. To our surprise, the DPP4 inhibition significantly suppressed the stress‐induced increases of both of these neurohormones (Table 3).
Table 3.
Levels of Lipids and Other Parameters in the 2 Experimental Groups at 4 Weeks
| Parameter | Stress | S‐Ana |
|---|---|---|
| BW, g | 22.3±0.4 | 21.9±1.3 |
| TG, mg/dL | 25.9±2.9 | 21.7±1.9 |
| T‐cho, mg/dL | 64.5±5.0 | 66.3±4.1 |
| HDL‐C, mg/dL | 33.9±1.7 | 32.3±0.8 |
| NEFA, μEq/L | 689.1±77.1 | 418±34a |
| Glucose, mg/dL | 105.9±8.1 | 90.9±9.9a |
| BUN, mg/dL | 29.5±2.1 | 26.9±1.9 |
| Creatinine, mg/dL | 0.1±0.02 | 0.1±0.01 |
| AST, pg/mL | 90.7±7.9 | 88.7±9.2 |
| Adrenaline, pg/mL | 4323±378 | 1568±190a |
| Noradrenaline, pg/mL | 4892±382 | 2412±172a |
| GLP‐1, pmol/L | 5.9±1.0 | 14.9±1.5a |
| Blood DPP4 level, ng/mL | 688.4±34.0 | 274.9±24.8a |
| Brain DPP4 level, ng/200 μg tissue | 49.4±3.2 | 25.6±3.0a |
Values are mean±SE (n=6–10). AST indicates aspartate transaminase; BUN, blood urea nitrogen; BW, body weight; DPP4, dipeptidyl peptidase‐4; GLP‐1, glucagon‐like protein‐1; HDL‐C, high‐density lipoprotein cholesterol; NEFA, non‐esterified fatty acid; S‐Ana, stressed‐anagliptin treatment; T‐cho, total cholesterol; TG, triglyceride.
P<0.01 vs control group by ANOVA and Tukey's post hoc test.
To further examine the consequences of DPP4 inhibition, we used flow cytometry to analyze the bone marrow for HSC activation and inflammatory cell production. The DPP4 inhibition by anagliptin treatment resulted in decreased numbers of lin−sca‐1+c‐Kit+ LSK cells and lin−c‐KithighSca‐1highCD48lowCD150high HSCs (Figure 8A and 8B) in the BM. DPP4 inhibition also suppressed the numbers of BM Ly6chigh monocytes and neutrophils (Figure 8C); both Ly6chigh monocytes and neutrophils were also less numerous in the blood of the S‐Ana groups (Figure 8D and 8E). The cell‐cycle and BrdU assays revealed that the BM harvested from the DPP4‐treated mice had decreased percentages of S/M/G2 and BrdU+ cells (Figures 8F and 9A). We also observed that the S‐Ana mice had decreased numbers of Ki67+/CD150+ cells in the BM (Figure 8G). Figure 9B shows that the stress‐induced activation of CD150+ cell proliferation was sensitive to DPP4 inhibitor loading, which indicates the restoration of the stress‐induced HSC hyperproliferative action by DPP4 inhibition. As sympathetic nervous activation and signaling seems to be tightly associated with CXCL12 change in response to stress,2 we next addressed the question of whether DPP4 inhibitor treatment would rectify the changes in the expression of Adrβ3 and CXCL12 in the BM. We observed that DPP4 inhibition resulted in decreased levels of Adrβ3 protein and increased levels of CXCL12 mRNA by the BM sca‐1+ cells (Figure 9C and 9D). In addition, the levels of brain GLP‐1R protein were restored in the S‐Ana mice (Figure 9D). As anticipated, DPP4 inhibition produced a reduction of MMP‐9 activity in the activated BM sca‐1+ cells (Figure 9E). The ELISA data demonstrated that DPP4 inhibition prevented the degradation of plasma GLP‐1 (Table 3). It has been reported that increased plasma noradrenaline signals BM niche cells to reduce CXCL12 expression via the Adrβ3 signaling pathway.2 DPP4 inhibition has been shown to protect brain against injury.28 Hisadome and colleagues reported that a peripheral GLP‐1 signal could modulate preproglucagon cells of the lower brainstem via the vagal afferents.30 We therefore concluded that DPP4 inhibition could produce an inhibitory effect on both increased HSC proliferation and the outputting of neutrophils and monocytes through an Adrβ3/CXCL12‐dependent mechanism that is mediated by the GLP‐1/GLP‐1R axis, suggesting the potential clinical usefulness of DPP4 inhibition as a therapeutic strategy.
Figure 8.
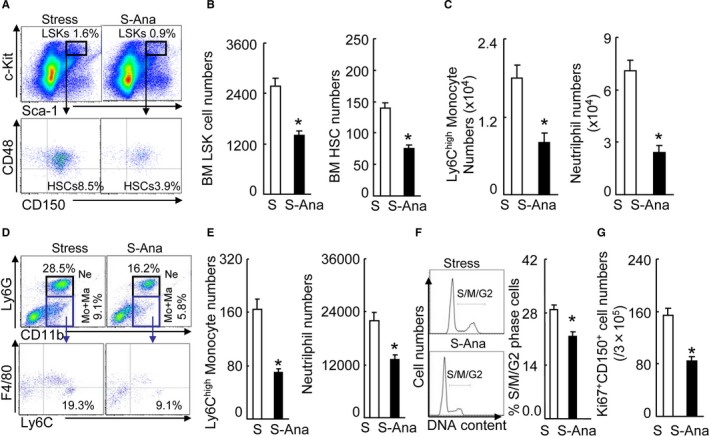
DPP4 inhibition suppressed the activated HSC proliferation in the BM. A, Gating for the LSK cell population isolated from BM cells of stress‐alone (S) and stressed‐anagliptin treatment (S‐Ana) group mice (upper panels) and subanalyzed lin−c‐KithighSca‐1high CD48low CD150high HSC (lower panels). B, BM LSK and HSC numbers next to gates represent population frequencies (%) of 2 groups after 4 weeks of stress (n=6–8, Student t test). C, Ly6Chigh monocytes and neutrophils in the BM (n=6–8, Student t test). D, Gating for the enclosed 2 populations isolated from PB cells of 2 groups (upper panels) and identifying for neutrophils as LyG6high CD11bhigh (top‐right quadrants) and subanalyzing LyG6lowLy6Chigh (bottom‐right quadrants) for monocytes as LyG6low CD11bhighF4/80lowLyChigh (lower panels). E, PB monocytes and neutrophil numbers in 2 groups of mice after 4 weeks of stress (n=6–8, Student t test). F, Representative histogram of DNA content during the cell cycle (left) and the distribution of S/M/G2 cells expressed as a percentage of total BM cells (right) (n=5, Mann–Whitney U test). G, FACS analysis show the numbers of Ki67/+ CD150+ cells in the BM. Data are mean±SEM. *P<0.01 vs stressed‐alone control. BM indicates bone marrow; DPP4, dipeptidyl peptidase‐4; FACS, fluorescence activated cell sorter; HSC, hematopoietic stem cells; LSK, lin−sca‐1+c‐Kit+; PB, peripheral blood; S‐Ana, stressed‐exenatide treatment.
Figure 9.
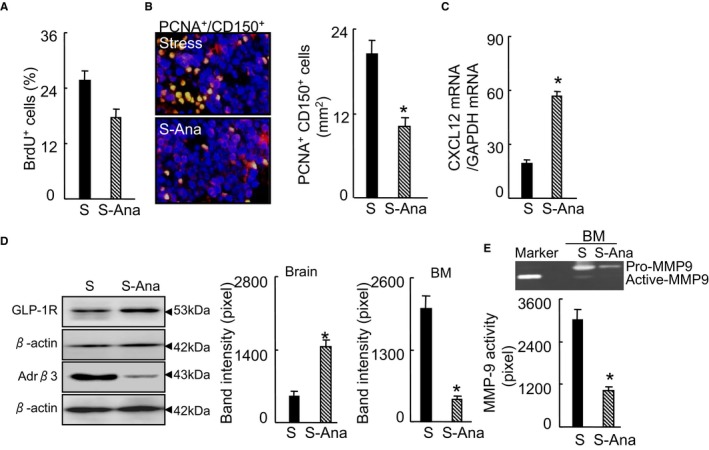
DPP4 inhibition rectified the stress‐induced BM CD150+ cell hyperproliferation. A, Quantitative data for the percentage of BrdU+ proliferating cells (per 2×102 BM cells) in the BM niches of 2 experimental group mice (n=5, Student t test). B, Double immunofluorescence of BM niches for proliferating CD150+ cells (PCNA + CD150+). Bar graphs: The numbers of double‐positive PCNA + CD150+ cells (n=6, Student t test). C, CXCL12 mRNA in BM sca‐1+ cells of the experimental group mice (n=6, Student t test). D, Representative Western blotting images and quantitative data for brain GLP‐1R protein and Adrβ3 protein of BM sca‐1+ cells (n=3, Student t test). E, Representative gelatin zymography images and quantitative data for MMP9 activity in BM sca‐1+ cells (n=3, Student t test). Data are mean±SEM. *P<0.01 vs stress‐alone control mice. Adrβ3 indicates adrenergic receptor β3; BM, bone marrow; BrdU, bromodeoxyuridine; CXCL12, C‐X‐C motif chemokine 12; DPP4, dipeptidyl peptidase‐4; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GLP‐1R, glucagon‐like peptide‐1 receptor; MMP9, matrix metalloproteinase 9; PCNA, proliferating cell nuclear antigen; S‐Ana, stressed‐anagliptin treatment.
GLP‐1/GLP‐R Signaling Negatively Regulates Stress‐Induced HSC Activation
As GLP‐1R activation and signaling seemed to be tightly associated with their pleiotrophic effects on the brain and cardiovascular injuries,30, 31, 32 we extended our examination to the effect of GLP‐1R stimulation on HSC activation. The stressed mice were subjected to treatment with either saline or the GLP‐1R agonist exenatide by subcutaneous injection twice daily for 4 weeks. At the end of the 4‐week stress protocol, the harvested BM was subjected to flow cytometry and immunostaining assays. The S‐Exe mice showed significant decreases in the numbers of the lin−sca‐1+c‐Kit+ LSK cells and the lin−c‐KithighSca‐1highCD48lowCD150high HSCs (Figure 10A and 10B). Exenatide attenuated the numbers of PCNA+CD150+ cells and the percentages of S/M/G2 cells compared with the corresponding numbers in the control mice (Figure 10C and 10E). Fluorescence‐activated cell sorter analysis showed that exenatide treatment reduced the numbers of Ki67+/CD150+ in the BM (Figure 10D). The BM niches of the S‐Exe mice showed decreased Adrβ3 staining signal (Figure 10F). The Western blotting analysis revealed that the brain GLP‐1R protein expression was increased in the S‐Exe mice (Figure 10G). Moreover, the BM sca‐1+ cells of the S‐Exe mice showed significantly reduced levels of MMP9 activity (799±63 versus 1809±203, P<0.05) (Figure 10H). GLP‐1R activation was reported to have a protective effect against brain damage in animal models.33 Thus, in the present experiment, the GLP‐1R stimulation by its agonist appears to have restored the stress‐induced bone marrow HSC action via the Adrβ3 signaling inactivation‐mediated restoration of CXCL12 expression in BM niche cells. In addition, except for plasma NEFA, there were no significant differences in plasma lipid or the parameters of renal and liver functions between the stress and S‐Exe mice (Table 4).
Figure 10.
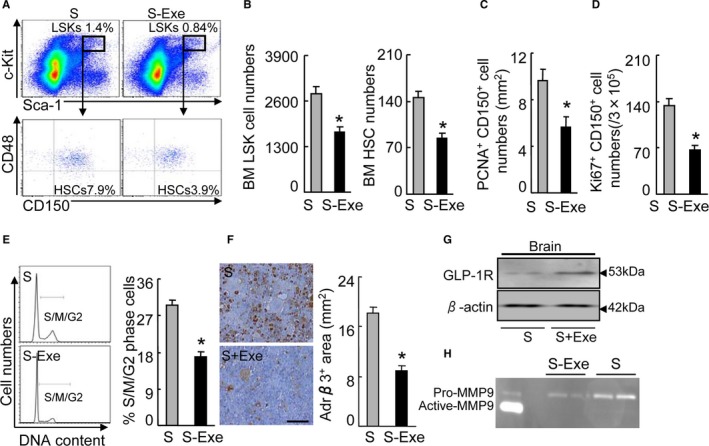
GLP‐1R stimulation redressed the activated CD150+ cell proliferation in response to stress. A, Gating for the LSK cell population isolated from BM cells of stress‐alone (S) and stressed‐exenatide treatment (S‐Ana) group mice (upper panels) and subanalyzed Lin−c‐KithighSca‐1high CD48low CD150high HSC (lower panels). B, BM LSK and HSC numbers next to gates represent population frequencies (%) of 2 experimental groups after 4 weeks of stress (n=6–8, Student t test). C, Quantitative data for the numbers of PCNA +/CD150+ cells in the BM niches. D, FACS analysis shows the numbers of Ki67/+ CD150+ cells in the BM. E, Representative histogram of DNA content during the cell cycle (left) and the distribution of S/M/G2 cells expressed as a percentage of total BM cells (right) (n=6, Mann–Whitney U test). F, Immunoreactive staining of BM niches for Adrβ3. Bar graphs: The percentage of positive area (mm2) (n=6 control, n=5 S‐Exe, Student t test). F, Representative Western bands for brain GLP‐1R protein expression. H, Representative gelatin zymography images for pro‐MMP9 and active‐form MMP9 activities in the BM sca‐1+ cells of the control and S‐Exe mice. Data are mean±SEM. *P<0.01 vs stress‐alone control mice. BM indicates bone marrow; FACS, fluorescence‐activated cell sorter; GLP‐1R, glucagon‐like peptide‐1 receptor; HSC, hematopoietic stem cells; LSK, lin−sca‐1+c‐Kit+; MMP9, matrix metalloproteinase 9; PCNA, proliferating cell nuclear antigen; S‐Ana, stressed anagliptin treatment; S‐Exe, stressed‐exenatide treatment.
Table 4.
Levels of Lipids and Targeted Neural Hormones and Other Parameters and Genes in the Stress and S‐Exe Groups at 4 Weeks
| Parameter | Stress | S‐Exe |
|---|---|---|
| BW, g | 22.0±1.3 | 21.2±1.7 |
| TG, mg/dL | 23.6±3.1 | 26.2±2.7 |
| T‐cho, mg/dL | 66.3±5.1 | 60.9±3.9 |
| HDL‐C, mg/dL | 26.9±3.1 | 32.3±2.2 |
| NEFA, μEq/L | 608.0±53.8 | 490.3±89.1a |
| Glucose, mg/dL | 103.5±8.9 | 94.8±9.8 |
| BUN, mg/dL | 32.5±1.9 | 30.1±3.0 |
| Creatinine, mg/dL | 0.1±0.0 | 0.1±0.0 |
| AST, pg/mL | 91.1±5.3 | 86.1±6.3 |
Values are mean±SE (n=6–8). AST indicates aspartate transaminase; BUN, blood urea nitrogen; BW, body weight; HDL‐C, high‐density lipoprotein cholesterol; NEFA, nonesterified fatty acid; S‐Exe, stressed‐exenatide treatment; T‐cho, total cholesterol; TG, triglyceride.
P<0.05 vs control group by Student unpaired t test.
DPP4 Deletion Mitigates BM HSC Activation
To further obtain direct evidence regarding the role of DPP4 in the BM HSC activation process, we created an immobilization stress model in DPP4+/+ and DPP4−/− rats. Consistent with the DPP4 inhibition, DPP4 deficiency reduced the plasma adrenaline and noradrenaline levels (Table 5). Stressed DPP4−/− rats had higher levels of plasma GLP‐1 compared with the control DPP4+/+ rats (Table 5). Routine hematoxylin–eosin staining showed the structures of BM niches of both rat genotypes (Figure 11A). As anticipated, DPP4 deficiency suppressed the BM niche cell proliferative overreaction in response to stress (Figure 11B and 11C). Moreover, DPP4 deletion reduced the numbers of PCNA+CD150+ cells, accompanied by a reduction of sca‐1+c‐Kit+ cells in the BM (Figure 11D and 11E). DPP4 deletion also lowered the numbers of leukocytes and neutrophils in the blood (Figure 11F). The changes in the levels of brain GLP‐1R protein as well as CXCL12 gene and Adrβ3 protein in the BM were rectified by DPP4 gene deletion (Figure 11G through 11I). These results indicated that DPP4 deficiency improved the expressions of Adrβ3 protein and CXCL12 gene in BM, and that it mitigated stress‐induced BM HSC activation and neutrophilia. In addition, at the baseline, there were no significant differences in BW or the blood lipid profile, glucose, blood urea nitrogen creatinine, aspartate transaminase, or GLP‐1 between the DPP4+/+ and DPP4−/− rats (Table 5). Stress reduced the BW and plasma triglyceride and increased the plasma NEFA in the 2 groups, and the DPP4−/− rats showed decreased plasma NEFA (Table 5).
Table 5.
Levels of Lipids and Targeted Neural Hormones and Other Parameters and Genes in 4 Experimental Groups at 4 Weeks
| Parameter | NS‐WT | NS‐DKO | S‐WT | S‐DKO |
|---|---|---|---|---|
| BW, g | 270.0±2.4 | 277.3±6.9 | 203.4±8.7a | 209.3±5.7a |
| TG, mg/dL | 102.0±19.2 | 96.1±7.5 | 42.3±43.1a | 43.1±1.1a |
| T‐cho, mg/dL | 60.8±2.0 | 60.1±2.4 | 53.8±2.9a | 52.9±4.4a |
| HDL‐C, mg/dL | 27.5±0.8 | 25.9±1.3 | 25.0±0.6 | 23.9±1.9 |
| NEFA, μEq/L | 382.7±21.4 | 443.3±25.6 | 840.0±75.9a | 394.5±26.5b |
| Glucose, mg/dL | 99.4±8.7 | 95.3±4.2 | 105.8±6.8a | 96.1±8.5 |
| BUN, mg/dL | 20.6±0.3 | 22.5±0.7 | 19.2±0.7 | 20.7±0.8 |
| Creatinine, mg/dL | 0.2±0.0 | 0.3±0.0 | 0.2±0.0 | 0.3±0.0 |
| AST, pg/mL | 113.8±9.3 | 123.9±14.5 | 125.7±13.0 | 149.1±34.5 |
| Adrenaline, pg/mL | 1732±189 | 1690±124 | 3309±201a | 1389±117b |
| Noradrenaline, pg/mL | 1581±199 | 1307±221 | 3904±401a | 2021±199b |
| GLP‐1, pmol/L | 61.0±2.6 | 79.0±6.3 | 39.3±3.5a | 52.3±4.7b |
Values are mean±SE (n=6–8). AST indicates aspartate transaminase; BUN, blood urea nitrogen; BW, body weight; GLP‐1, glucagon‐like protein‐1; HDL‐C, high‐density lipoprotein cholesterol; NEFA, nonesterified fatty acid; NS‐DKO, non‐stress‐dipeptidyl peptidase‐4 knockout rats; NS‐WT, non‐stress‐wild type rats; S‐DKO, stresse‐dipeptidyl peptidase‐4 knockout rats; S‐WT stress‐wild type rats; T‐cho, total cholesterol; TG, triglyceride.
P<0.05 vs NS‐WT.
P<0.05 vs S‐WT by ANOVA and Tukey's post hoc tests.
Figure 11.
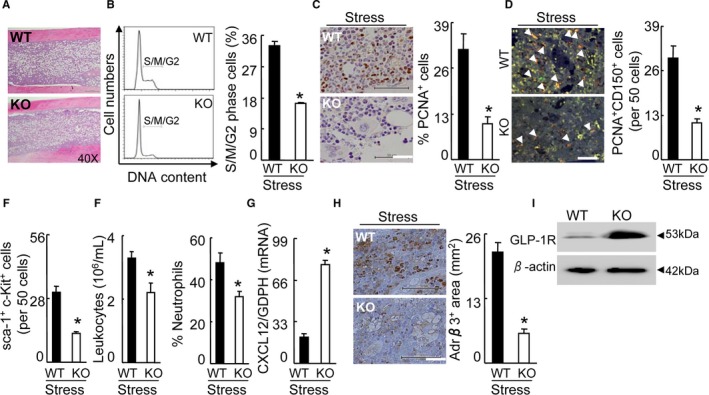
DPP4−/− rectified the activated BM CD150+ cell proliferation in response to stress. A, Routine H&E staining on BM of stressed DPP4+/+ (WT) and DPP4−/− (KO) rats. B, Representative histogram of DNA content during the cell cycle (left) and the distribution of S/M/G2 cells expressed as a percentage of total BM cells (right) (n=5, Student t test). C, Immunoreactive staining of BM niches for PCNA. Bar graphs: Proliferating PCNA + cells (per 2×102 BM cells) (n=6, Student t test). Scale bars, 50 μm. D, Double immunofluorescence of BM niches for PCNA and CD150. Bar graphs: The numbers of double‐positive PCNA + CD150+ cells (orange; n=6, Student t test). Arrowheads indicate double PCNA+ and CD150+ cells. Scale bars, 50 μm. E, Quantitative data for the numbers of sca‐1+c‐Kit+‐positive cells in the BM niches of both groups (n=6, Student t test). Scale bars, 50 μm. F, The numbers of blood leukocytes and the percentage of blood neutrophils in both groups (n=6, stressed DPP4+/+ rats; n=8 stressed DPP4−/− rats; Student t test). G, Quantitative real‐time PCR for CXCL12 genes in the BM sca‐1+ cells (n=6, Student t test). H, Immunoreactive staining of BM niches for Adrβ3. Bar graphs: The percentage of positive area per mm2 (n=5, Student t test). Scale bars, 50 μm. I, Representative Western bands for brain GLP‐1R protein expression in both genotype rats. Data are mean±SE. *P<0.01 vs stress‐alone control rats. Adrβ3 indicates adrenergic receptor β3; BM, bone marrow; CXCL12, C‐X‐C motif chemokine 12; DPP4, dipeptidyl peptidase‐4; GLP‐1R, glucagon‐like peptide‐1 receptor; H&E, hematoxylin and eosin; PCNA, proliferating cell nuclear antigen; PCR, polymerase chain reaction.
Pharmacological Inhibition of Adrβ3 Mitigates BM HSC Activation
As Adrβ3 activation and signaling seemed to be tightly associated with HSC activation in response to chronic stress, we extended our examination to the Adrβ3 blocking on BM HSC activation in the mice that underwent stress. As anticipated, the results indicated that an Adrβ3 selective inhibitor, L748337, suppressed the levels of the lin−sca‐1+c‐Kit+ LSK cells and the lin−c‐KithighSca‐1highCD48lowCD150high HSCs in stressed mice compared with the stress‐alone control mice (Figure 12A and 12B). Compared with the control mice, the S‐L74 mice had decreased levels of BrdU+ cells in BM and CXCL12 gene expression in BM‐derived sca‐1+ cells (Figure 12C and 12D). Stress upregulates inflammatory gene expression in the leukocyte transcriptome via an Adrβ3 induction of myelopoiesis.29 Adrβ3 signaling activation has been shown to reduce CXCL12 expression in myeloid cells.2 CXCL12 is required for neutrophil release from the BM under basal and stress granulopoiesis conditions.34 Thus, Adrβ3 signal appears to regulate HSC proliferation capacity via the reduction of CXCL12 expression in the BM of stressed mice.
Figure 12.
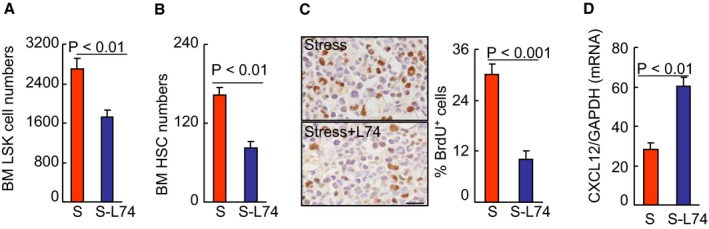
DPP4 inhibition suppressed the activated HSC proliferation in the BM. A and B, BM LSKs and HSC numbers in the BM of stress‐alone (S) and stress+L748337 (S‐L74) mice after 4 weeks of stress (n=6, Student t test). C, Immunoreactive staining of BM niches for BrdU. Bar graphs: The percentage of positive cells (per 2×102 cells) (n=5, Student t test). D, CXCL12 mRNA in BM sca‐1+ cells of the experimental group mice (n=6, Student t test). Data are mean±SE. BM indicates bone marrow; BrdU, bromodeoxyuridine; CXCL12, C‐X‐C motif chemokine 12; DPP4, dipeptidyl peptidase‐4; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HSC, hematopoietic stem cells; LSK, lin−sca‐1+c‐Kit+ cell.
Discussion
Over the past decade, the important roles of the DPP4‐GLP1/GPL‐1R axis in various pathophysiological conditions have been revealed by clinical and experimental studies.9 Although those investigations uncovered GLP‐1‐dependent and ‐independent mechanisms underlying DPP4 inhibition‐ and GLP‐1R activation‐mediated beneficial effects on cerebral and cardiovascular injuries,9 the role of the brain DPP4‐GLP1/GPL‐1R axis in the BM's instinctive response to stress has not been investigated. A close link between the BM niche Adrβ3 signal and CXCL12 expression in a stress‐induced BM immune action was recently reported.2
In the present study, we focused on the novel mechanism and upstream molecular requirements necessary for the regulation of the interaction between Adrβ3 signaling and CXCL12 expression in stress‐induced BM HSC action and the outputting of inflammatory cells into the blood. Our major findings were as follows: (1) Chronic stress reduced body weight and fatty volumes (inguinal and subcutaneous fats). (2) Chronic stress increased DPP4 levels in the blood and several tissues (especially brain) but not in the BM. (3) Genetic or pharmacological disruption of DPP4 activity can rectify stress‐induced BM HSC activation and monocytosis and neutrophilia via an Adrβ3/CXCLl12‐dependent mechanism that is mediated by the GLP‐1/GLP‐1R axis in rats and mice. (4) GLP‐1R activation with its agonist can produce a beneficial BM response to stress in mice. (5) The selective pharmacological blocking of Adrβ3 mitigated HSC activation, accompanied by improvement of CXCL12 gene expression in BM niche cells in response to chronic stress.
Our view of the DPP4‐GLP‐1/GLP‐1R axis action as a potential regulator of the close interaction between the brain and BM has key clinical implications. Under our experimental conditions, the ability of chronic stress to increase DPP4 activity levels is likely to have contributed to the stimulation of the BM HSCs’ response. Our preclinical mouse studies demonstrated that the orally active DPP4 inhibitor anagliptin will rectify the activated BM HSC proliferative ability and outputting of neutrophils and monocytes into the blood in stressed mice. Our present findings demonstrated that stress increased the plasma and brain DPP4 levels; these changes were reversed by pharmacological and genetic disruption of DPP4 gene. DPP4 deletion had no effect on hematopoiesis in the rats under nonstressed conditions. Because DPP4 inhibition can have protective effects against ischemia‐induced animal brain injury,28 we propose that brain DPP4 functions as an important regulator of stress‐induced BM HSC responses. It was reviewed that DPP4 inhibitors often exhibited cardiovascular beneficial effects via an augmentation of GLP‐1/GLP‐1 signaling.9 In the present study, DPP4 inhibition restored impaired plasma GLP‐1 levels and brain GLP‐1R expression in stressed mice. Thus, the upregulation of brain GLP‐1/GLP‐1R signaling by DPP4 inhibition could represent a common mechanism in the prevention of BM responses to stress. This notion is further supported by our finding that GLP‐1R stimulation with exenatide produced a beneficial BM response to stress in mice.
Accumulating evidence indicates that stress activates the sympathetic nervous system and the hypothalamic–pituitary–adrenal axis to alter systemic hormonal and BM immune responses, resulting in harmful effects on targeted organs, leading to the onset of stress‐related brain and cardiovascular diseases. It was reported that psychosocial stress upregulated BM HSC proliferative ability, leukopoiesis, and inflammatory monocyte production in humans and animals.2, 29 Adrβ3 induction of myelopoiesis has been implicated in social stress–induced inflammatory gene expression in the leukocyte transcriptome.29 In addition, a recent study showed that under conditions of chronic variable stress in mice, sympathetic nerve fibers that released surplus noradrenaline can impair CXCL12 expression via the Adrβ3 signaling pathway in BM niche cells.2 In the present study, stress increased the plasma adrenaline and noradrenaline levels and BM niche cell Adrβ3 protein expression and activity, and it decreased the BM niche CXCL12 gene expression; in addition, the ability of stress to enhance the BM HSC action was abrogated in both the DPP4−/− rats and DPP4+/+ mice that were treated with the DPP4 inhibitor. Our observations here show that a selective Adrβ3 blocker (ie, L748337) mitigated HSC activation accompanied by an improvement of CXCL12 gene expression in BM niche cells in response to chronic stress. Thus, increased DPP4 activity appears to promote BM HSCs’ biological behavior in a chronic immobilized stress state through its ability to reduce the BM niche CXCL12 expression that is mediated by Adrβ3 signaling induction, leading to an increased outputting of neutrophils and inflammatory monocytes into the blood. On the other hand, it is well known that activated inflammatory cells (macrophages and neutrophils) are a major source of MMP‐9 in the atherosclerotic lesions of humans and animals.20 Our present findings showed that the levels of MMP‐9 gene and activity were induced by chronic stress; these changes were reversed by DPP4 inhibition and GLP‐1R activation in BM myeloid cells, suggesting that the reduction of MMP‐9 expression and activity by DPP4 inhibition or GLP‐1R activation could present a common mechanism in the protection of cardiovascular tissues from stress.
Another implication of our experiments is the potential use of increased circulating DPP4 and decreased GLP‐1 as biomarkers to predict the presence of stress in animals. Our observations suggest that plasma DPP4 levels were sensitive to the chronic stress, and that the noninvasive measurement of plasma DPP4 levels would be helpful for the assessment of the brain injury in the animals that underwent chronic stress. However, the role of DPP4 in the initiation and progression of various diseases associated with modern stressors (including work‐related stress, natural disasters, environmental stress, and social anxiety) should be further examined in retrospective and prospective cohort studies, in which many of the problems that affect case–control studies are not an issue.
It should also be noted that in the present study there were no DPP4 activities in the BM‐derived sca‐1+ cells and the whole BM cells from not only nonstressed mice but also stressed mice. We also observed no CD26 staining signal in the BM, suggesting that immature BM cells including HSCs have no DPP4 expression. This raises the possibility that a cell‐autonomous role of DPP4 blocking may not have caused the reversal of BM HSC proliferation under our experimental stress conditions. It was reported that DPP4 expression increased with the functional maturation of dendritic cells and macrophages from monocytes.13 Other studies documented that DPP4 enhances T‐cell maturation and migration, cytokine secretions, and the activation of cytotoxic T cells via an interleukin‐12‐dependent mechanism.11 Moreover, a small interfering RNA knockdown of DPP4 on dendritic cells suppressed the proliferation of both mature CD4+ and CD8+ T cells.14 Therefore, a cell‐autonomous role of DPP4 blocking appears to contribute to the regulation of functions of mature blood cells (eg, proliferation and migration) rather than BM‐derived immature stem cells such as HSCs and immune cells.
It has been reported that stressful stimuli applied repetitively enhance activity of the sympathetic system and hypothalamic–pituitary–adrenal axis similarly to increase both plasma catecholamine and cortisol levels.1 As both sympathetic nerve endings and cortisol stimulate lipolysis, adipose tissues are shrunk to release NEFA after 2 weeks of daily stress administration.17 Uchida and colleagues showed atrophic adipose tissue with reduced cell size and elevated NEFA levels after restraint stress without any alteration in food intake.19 We have shown that the mice that underwent the 4‐week stress protocol lost significant amounts of subcutaneous fat and inguinal fat. Our observations here show that the stressed mice had increased levels of plasma NEFA as well as plasma adrenaline and noradrenaline as compared with the unstressed mice. It has been reported that large adipocytes have increased lipolytic capacity and are sensitive to lipolytic stimuli.35 Thus, stress hormone–mediated lipolysis appears to increase NEFA concentration and reduce adipose cell size and adipose volume and BW. However, it should be noted that DPP4 inhibition had no effect on BW and adipose volume in mice that underwent chronic stress conditions. It is known that patients with obesity and diabetes mellitus had increased plasma DPP4 levels.36 Recently, we have reported that the plasma levels were increased in ischemic heart disease patients even without diabetes mellitus.8 These findings suggest that stress‐related DPP4 level changes are not attributable to obesity or the adipose tissue lost but rather to the sympathetic system and hypothalamic–pituitary–adrenal axis.
Study limitations should be considered. First, here we did not detect the levels of tyrosine hydroxylase protein and activity in the BM of the nonstressed and the stressed mice. Secondly, as known, the whole BM contains a small fraction of the lin−sca‐1+/c‐Kit+CD150+/CD48− HSCs. In addition, in the present work, the whole BM cells were subjected for 4 assays (ie, the cell cultures, polymerase chain reaction, Western blotting, and fluorescence‐activated cell sorter assays). Thus, these factors preclude us from purifying lin−c‐KithighSca‐1highCD48lowCD150high HSCs for those cellular experiments. Further studies will be needed to investigate these issues.
Conclusions
Our present findings help explain how chronic stress interferes with hematopoiesis, and they clarified the role of the interaction between the brain DPP4‐GLP‐1/GLP‐1R axis and the BM Adrβ3/CXCL12 signal in HSC activation processes. In animals exposed to chronic stress, the inactivation of GLP‐1/GLP‐1R associated with increased DPP4 signaled BM sca‐1+ cells to decrease CXCL12 levels and increased MMP‐9 activation through the Adrβ3 signaling pathway activation, accelerated HSC colony formation and proliferation capacities, and enhanced neutrophilia and monocytosis. These events resulted in an extensive release of inflammatory leukocytes and monocytes into the circulation and promoted inflammatory disorder. DPP4 inhibition or the alteration of the GLP‐1/GLP‐1R axis limited an imbalance between DPP4 and GLP‐1 in the blood and brain, supporting the notion that GLP‐1/GLP‐1R signaling and targeting of the interaction between an Adrβ3/CXCL12 signaling‐mediated interaction between the brain and BM should be explored as a potential avenue for clinical therapy.
Data Access and Responsibility
The authors had full access to and take full responsibility for the data. All authors have read and agree to the manuscript as written.
Sources of Funding
This work was supported in part by the Scientific Research Fund of the Chinese Ministry of Education (nos. 81560240, 81460082) and by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (nos. 15H04801, 15H04802).
Disclosures
None.
Acknowledgments
We gratefully acknowledge M. Tanaka and I. Mizuguchi of the Division for Medical Research Engineering of Nagoya University Graduate School of Medicine for assistance in technical supports.
(J Am Heart Assoc. 2017;6:e006394 DOI: 10.1161/JAHA.117.006394.)28710180
References
- 1. Gu HF, Tang CK, Yang YZ. Psychological stress, immune response, and atherosclerosis. Atherosclerosis. 2012;223:69–77. [DOI] [PubMed] [Google Scholar]
- 2. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur Muhlen C, Bode C, Fricchione GL, Denninger J, Lin CP, Vinegoni C, Libby P, Swirski FK, Weissleder R, Nahrendorf M. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20:754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Courties G, Herisson F, Sager HB, Heidt T, Ye Y, Wei Y, Sun Y, Severe N, Dutta P, Scharff J, Scadden DT, Weissleder R, Swirski FK, Moskowitz MA, Nahrendorf M. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res. 2015;116:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jing L, Tamplin OJ, Chen MJ, Deng Q, Patterson S, Kim PG, Durand EM, McNeil A, Green JM, Matsuura S, Ablain J, Brandt MK, Schlaeger TM, Huttenlocher A, Daley GQ, Ravid K, Zon LI. Adenosine signaling promotes hematopoietic stem and progenitor cell emergence. J Exp Med. 2008;212:649–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tzeng YS, Li H, Kang YL, Chen WC, Cheng WC, Lai DM. Loss of Cxcl12/Sdf‐1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood. 2011;117:429–439. [DOI] [PubMed] [Google Scholar]
- 6. Khurana S, Melacarne A, Yadak R, Schouteden S, Notelaers T, Pistoni M, Maes C, Verfaillie CM. SMAD signaling regulates CXCL12 expression in the bone marrow niche, affecting homing and mobilization of hematopoietic progenitors. Stem Cells. 2015;32:3012–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhong J, Maiseyeu A, Davis SN, Rajagopalan S. DPP4 in cardiometabolic disease: recent insights from the laboratory and clinical trials of DPP4 inhibition. Circ Res. 2015;116:1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang G, Li Y, Cui L, Jiang H, Li X, Jin C, Jin D, Zhao G, Jin J, Sun R, Piao L, Xu W, Fang C, Lei Y, Yuan K, Xuan C, Ding D, Cheng X. Increased plasma dipeptidyl peptidase‐4 activities in patients with coronary artery disease. PLoS One. 2016;11:e0163027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lei Y, Hu L, Yang G, Piao L, Jin M, Cheng X. Dipeptidyl peptidase‐IV inhibition for the treatment of cardiovascular disease‐ recent insights focusing on angiogenesis and neovascularization. Circ J. 2017;81:770–776. DOI: 10.1253/circj.CJ‐16‐1326. [DOI] [PubMed] [Google Scholar]
- 10. Buhling F, Kunz D, Reinhold D, Ulmer AJ, Ernst M, Flad HD, Ansorge S. Expression and functional role of dipeptidyl peptidase IV (CD26) on human natural killer cells. Nat Immun. 1994;13:270–279. [PubMed] [Google Scholar]
- 11. Cordero OJ, Salgado FJ, Vinuela JE, Nogueira M. Interleukin‐12 enhances CD26 expression and dipeptidyl peptidase IV function on human activated lymphocytes. Immunobiology. 1997;197:522–533. [DOI] [PubMed] [Google Scholar]
- 12. Yu DM, Yao TW, Chowdhury S, Nadvi NA, Osborne B, Church WB, McCaughan GW, Gorrell MD. The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J. 2010;277:1126–1144. [DOI] [PubMed] [Google Scholar]
- 13. Ohnuma K, Yamochi T, Uchiyama M, Nishibashi K, Yoshikawa N, Shimizu N, Iwata S, Tanaka H, Dang NH, Morimoto C. CD26 up‐regulates expression of CD86 on antigen‐presenting cells by means of caveolin‐1. Proc Natl Acad Sci USA. 2004;101:14186–14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhong J, Rao X, Deiuliis J, Braunstein Z, Narula V, Hazey J, Mikami D, Needleman B, Satoskar AR, Rajagopalan S. A potential role for dendritic cell/macrophage‐expressing DPP4 in obesity‐induced visceral inflammation. Diabetes. 2015;62:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ikushima H, Munakata Y, Iwata S, Ohnuma K, Kobayashi S, Dang NH, Morimoto C. Soluble CD26/dipeptidyl peptidase IV enhances transendothelial migration via its interaction with mannose 6‐phosphate/insulin‐like growth factor II receptor. Cell Immunol. 2002;215:106–110. [DOI] [PubMed] [Google Scholar]
- 16. Lei Y, Yang G, Hu L, Piao L, Inoue A, Jiang H, Sasaki T, Zhao G, Yisireyili M, Yu C, Xu W, Takeshita K, Okumura K, Kuzuya M, Cheng XW. Increased dipeptidyl peptidase‐4 accelerates diet‐related vascular aging and atherosclerosis in ApoE‐deficient mice under chronic stress. Int J Cardiol. May16, 2017. DOI: 10.1016/j.ijcard.2017.05.062. Available at: http://www.internationaljournalofcardiology.com/article/S0167-5273(17)30851-3/fulltext. Accessed July 8, 2017. [DOI] [PubMed] [Google Scholar]
- 17. Yisireyili M, Takeshita K, Hayashi M, Wu H, Uchida Y, Yamamoto K, Kikuchi R, Hao CN, Nakayama T, Cheng XW, Matsushita T, Nakamura S, Murohara T. Dipeptidyl peptidase‐ IV inhibitor alogliptin improves stress‐induced insulin resistance and prothrombotic state in a murine model. Psychoneuroendocrinology. 2016;73:186–195. [DOI] [PubMed] [Google Scholar]
- 18. Moon JY, Woo JS, Seo JW, Lee A, Kim DJ, Kim YG, Kim SY, Lee KH, Lim SJ, Cheng XW, Lee SH, Kim W. The dose‐dependent organ‐specific effects of a dipeptidyl peptidase‐4 inhibitor on cardiovascular complications in a model of type 2 diabetes. PLoS One. 2016;11:e0150745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uchida Y, Takeshita K, Yamamoto K, Kikuchi R, Nakayama T, Nomura M, Cheng XW, Egashira K, Matsushita T, Nakamura H, Murohara T. Stress augments insulin resistance and prothrombotic state: role of visceral adipose‐derived monocyte chemoattractant protein‐1. Diabetes. 2012;61:1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng XW, Song H, Sasaki T, Hu L, Inoue A, Bando YK, Shi GP, Kuzuya M, Okumura K, Murohara T. Angiotensin type 1 receptor blocker reduces intimal neovascularization and plaque growth in apolipoprotein E‐deficient mice. Hypertension. 2011;57:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng XW, Kuzuya M, Kim W, Song H, Hu L, Inoue A, Nakamura K, Di Q, Sasaki T, Tsuzuki M, Shi GP, Okumura K, Murohara T. Exercise training stimulates ischemia‐induced neovascularization via phosphatidylinositol 3‐kinase/Akt‐dependent hypoxia‐induced factor‐1 alpha reactivation in mice of advanced age. Circulation. 2010;122:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang H, Cheng XW, Shi GP, Hu L, Inoue A, Yamamura Y, Wu H, Takeshita K, Li X, Huang Z, Song H, Asai M, Hao CN, Unno K, Koike T, Oshida Y, Okumura K, Murohara T, Kuzuya M. Cathepsin K‐mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat Commun. 2014;5:3838. [DOI] [PubMed] [Google Scholar]
- 23. Li X, Cheng XW, Hu L, Wu H, Guo P, Hao CN, Jiang H, Zhu E, Huang Z, Inoue A, Sasaki T, Du Q, Takeshita K, Okumura K, Murohara T, Kuzuya M. Cathepsin S activity controls ischemia‐induced neovascularization in mice. Int J Cardiol. 2015;183:198–208. [DOI] [PubMed] [Google Scholar]
- 24. Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi‐1 is required for maintenance of adult self‐renewing haematopoietic stem cells. Nature. 2003;423:302–305. [DOI] [PubMed] [Google Scholar]
- 25. Morrison SJ, Qian D, Jerabek L, Thiel BA, Park IK, Ford PS, Kiel MJ, Schork NJ, Weissman IL, Clarke MF. A genetic determinant that specifically regulates the frequency of hematopoietic stem cells. J Immunol. 2002;168:635–642. [DOI] [PubMed] [Google Scholar]
- 26. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hayashi M, Takeshita K, Uchida Y, Yamamoto K, Kikuchi R, Nakayama T, Nomura E, Cheng XW, Matsushita T, Nakamura S, Murohara T. Angiotensin II receptor blocker ameliorates stress‐induced adipose tissue inflammation and insulin resistance. PLoS One. 2014;9:e116163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Darsalia V, Ortsater H, Olverling A, Darlof E, Wolbert P, Nystrom T, Klein T, Sjoholm A, Patrone C. The DPP‐4 inhibitor linagliptin counteracts stroke in the normal and diabetic mouse brain: a comparison with glimepiride. Diabetes. 2010;62:1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, Kobor MS, Reader BF, Sheridan JF, Cole SW. Social stress up‐regulates inflammatory gene expression in the leukocyte transcriptome via beta‐adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA. 2013;110:16574–16579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hisadome K, Reimann F, Gribble FM, Trapp S. Leptin directly depolarizes preproglucagon neurons in the nucleus tractus solitarius: electrical properties of glucagon‐like peptide 1 neurons. Diabetes. 2010;59:1890–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ha SJ, Kim W, Woo JS, Kim JB, Kim SJ, Kim WS, Kim MK, Cheng XW, Kim KS. Preventive effects of exenatide on endothelial dysfunction induced by ischemia‐reperfusion injury via KATP channels. Arterioscler Thromb Vasc Biol. 2012;32:474–480. [DOI] [PubMed] [Google Scholar]
- 32. Kim M, Platt MJ, Shibasaki T, Quaggin SE, Backx PH, Seino S, Simpson JA, Drucker DJ. GLP‐1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med. 2013;19:567–575. [DOI] [PubMed] [Google Scholar]
- 33. Darsalia V, Hua S, Larsson M, Mallard C, Nathanson D, Nystrom T, Sjoholm A, Johansson ME, Patrone C. Exendin‐4 reduces ischemic brain injury in normal and aged type 2 diabetic mice and promotes microglial M2 polarization. PLoS One. 2014;9:e103114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eash KJ, Means JM, White DW, Link DC. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood. 2009;113:4711–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laurencikiene J, Skurk T, Kulyte A, Heden P, Astrom G, Sjolin E, Ryden M, Hauner H, Arner P. Regulation of lipolysis in small and large fat cells of the same subject. J Clin Endocrinol Metab. 2011;96:E2045–E2049. [DOI] [PubMed] [Google Scholar]
- 36. Zheng TP, Liu YH, Yang LX, Qin SH, Liu HB. Increased plasma dipeptidyl peptidase‐4 activities are associated with high prevalence of subclinical atherosclerosis in Chinese patients with newly diagnosed type 2 diabetes: a cross‐sectional study. Atherosclerosis. 2015;242:580–588. [DOI] [PubMed] [Google Scholar]