

Abstract
Human exposure to arsenic in drinking water is known to be associated with the development of bladder, lung, kidney, and skin cancers. The molecular mechanisms underlying the carcinogenic effects of arsenic species remain incompletely understood. DNA interstrand cross-links (ICLs) are among the most cytotoxic type of DNA lesions that block DNA replication and transcription, and these lesions can be induced by endogenous metabolism and by exposure to exogenous agents. Fanconi anemia (FA) is a congenital disorder manifested with elevated sensitivity toward DNA interstrand cross-linking agents, and monoubiquitination of FANCD2 by FANCL is a crucial step in FA-mediated DNA repair. Here, we demonstrated that As3+ could bind to the PHD/RING finger domain of FANCL in vitro and in cells. This binding led to compromised ubiquitination of FANCD2 in cells and diminished recruitment of FANCD2 to chromatin and DNA damage sites induced by 4,5′,8-trimethylpsoralen plus UVA irradiation. Furthermore, clonogenic survival assay results showed that arsenite coexposure rendered cells more sensitive toward DNA interstrand cross-linking agents. Together, our study suggested that arsenite may compromise genomic stability via perturbation of the Fanconi anemia pathway, thereby conferring its carcinogenic effect.
Graphical abstract
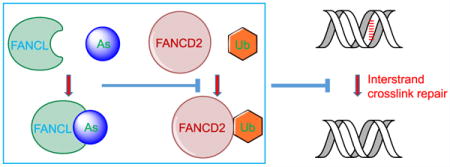
Arsenic is a naturally occurring metalloid that is ubiquitously present in the environment throughout the world. Prolonged exposure to arsenic through drinking and cooking using contaminated groundwater poses significant threats to public health. In this vein, human exposure to arsenic compounds is known to be associated with a variety of cancers including those of the skin, bladder, kidney, liver, and lung, as well as other human diseases encompassing neurological disorders, cardiovascular diseases, and diabetes.1
Several mechanisms have been proposed to account for the carcinogenic effects of arsenic species, including perturbation of growth factor signaling, induction of oxidative stress, inhibition of DNA repair, alterations of DNA and histone methylation, and binding to vicinal thiols in proteins.2,3 In this respect, in vitro studies with model synthetic peptides revealed that As3+ binds much more strongly to Cys3His (C3H)- or Cys4 (C4)-type zinc fingers than those of the Cys2His2 (C2H2)-type.4
Emerging recent studies have demonstrated that As3+ may compromise DNA repair by binding directly to DNA repair proteins, or through modulating histone epigenetic marks that are crucial in DNA repair. As3+ could bind to the zinc finger motifs of xeroderma pigmentosum complementation group A (XPA)5,6 and poly(ADP-ribose) polymerase 1 (PARP1),7 thereby inhibiting nucleotide excision repair (NER) and base excision repair (BER),5–7 respectively. Our recent study showed that As3+ could bind to the RING finger motifs of RNF20/RNF40 histone E3 ubiquitin ligase, which leads to diminished ubiquitination of histone H2B at lysine 120.8 Consistent with the notion that this ubiquitination is important for decompacting the 30 nm chromatin fiber and for establishing a biochemically accessible chromatin environment required for DNA double strand break (DSB) repair,9 we observed that As3+ exposure could result in compromised repair of DNA DSBs via the homologous recombination (HR) and nonhomologous end-joining pathways.8 Along this line, As3+ was also observed to bind to the C3H-type zinc fingers in ten-eleven translocation (Tet) family of enzymes, which perturbs DNA epigenetic marks through inhibition of Tet-mediated oxidation of 5-methylcytosine to 5-hydroxymethylcytosine.10 On the other hand, arsenic trioxide has been successfully employed in the clinical remission of acute promyelocytic leukemia (APL), especially for those patients carrying the PML-RARα fusion oncogene.11 In this vein, the fusion with PML led to the constitutive activation of the RARα transcription factor, and As3+ was found to bind to the RING-finger domain of PML, resulting in the oligomerization and proteasomal degradation of the PML-RARα fusion protein.11,12 Thus, As3+ induces the clinical remission of APL and perturbs DNA DSB repair on the basis of the same chemical rationale, i.e., through targeting zinc finger proteins, though the cellular protein targets differ under these two scenarios.
Not much is known about whether As3+ also interferes with the repair of DNA interstrand cross-link lesions (ICLs). In this respect, DNA ICLs may arise from endogenous metabolism or from exposure to chemotherapeutic agents such as mitomycin C (MMC).13 Owing to the covalent linkage of two strands of DNA and the ensuing blockage of crucial cellular processes including DNA replication and transcription, the ICLs are extremely cytotoxic.13 Thus, multiple repair pathways have been evolved to repair ICLs, which encompass the NER, homologous recombination (HR), and Fanconi anemia (FA) pathways.13 A number of genes involved in the FA pathway have been identified, including 15 bona fide FA genes and 14 FA-like genes,14–16 where individuals deficient in the FA-like genes are characterized by the lack of a clear genotype–phenotype correlation in Fanconi anemia and most of the FA-like genes are only represented by a unique family case.16 In FA-mediated repair of ICL lesions, the anchor complex, comprised of FANCM, FAAP24, MHF1, and MHF2, is activated by recognition of the ICL sites, which leads to the recruitment of the FA core complex that consists of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FAAP20, and FAAP100.14,15 Within the core complex, FANCL harbors a PHD/RING finger domain and is an E3 ubiquitin ligase, which monoubiquitinates FANCI and FANCD2. These monoubiquitination events are thought to be crucial for the recruitment of nucleases and downstream repair factors in the NER and HR pathways to remove DNA ICL lesions.14,15
On the grounds of the previous findings that As3+ was capable of binding to the zinc finger domain of proteins, we reasoned that As3+ may also interact with the RING finger domain of FANCL, thereby blocking its E3 ubiquitin ligase activity and perturbing the repair of DNA ICLs. In the present study, we demonstrated the interaction between As3+ and FANCL in vitro and in cultured human cells by using streptavidin agarose affinity assay, mass spectrometry (MS), and fluorescent microscopy measurements. We also showed that arsenite exposure could compromise MMC-induced monoubiquitination of FANCD2 and reduce the recruitment of FANCD2 to chromatin and DNA damage sites in cultured human cells. Additionally, we found that arsenite exposure rendered cells sensitive toward ICL-inducing agents. Together, our study established a novel mechanism underlying the carcinogenic effects of arsenite.
EXPERIMENTAL SECTION
Cell Culture
All cell culture experiments were conducted at 37 °C in a humidified atmosphere containing 5% CO2. HEK293T human embryonic kidney epithelial cells (ATCC, Manassas, VA) and HeLa cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, ATCC) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Waltham, MA) containing 100 U/mL penicillin and streptomycin.
For stable isotope labeling by amino acids in cell culture (SILAC) experiments, the complete light or heavy media were prepared by adding light and heavy lysine ([13C6,15N2]-L-lysine) and arginine ([13C6]-L-arginine) into RPMI 1640 medium without L-lysine or L-arginine. The light and heavy SILAC medium also contained 10% dialyzed FBS and 100 U/mL penicillin and streptomycin. The GM00637 cells were cultured in a heavy medium for at least 10 days to ensure complete stable isotope labeling prior to further experiments.
Plasmid Construction
The expression plasmids of GFP-FANCL harboring the C312,315A or C364,367A mutations were obtained from the plasmid for expressing the wild-type GFP-FANCL provided by Dr. Grover C. Bagby from Oregon Health and Science University17 by using a GeneArt Site-directed Mutagenesis Kit (Thermo Fisher Scientific, Waltham, MA), following the vendor’s recommended procedures.
In Vitro Arsenite Binding Assay
The RING finger peptide derived from the PHD domain of FANCL (with amino acid sequences of DCGICYAYQL DGTIPDQVCD NSQCGQPFHQ ICLYEWLRGL LTSRQSFNII FGECPYCSK) was obtained from New England Peptide, Inc. (Gardner, MA) and purified by HPLC. The peptide was dissolved in 20 mM Tris-HCl (pH 6.8) containing 1 mM dithiothreitol. Aliquots of 100 μM peptide were incubated with 200 μM NaAsO2 on ice for 1 h and subsequently diluted by 100 fold. The resultant solution was mixed with an equal volume of 2,5-dihydroxybenzoic acid matrix solution followed by spotting onto a sample plate.4 The sample was then analyzed on a Voyager DE STR matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometer (Applied Biosystems, Foster City, CA) in the linear, positive-ion mode.
Streptavidin Agarose Affinity Assay and Western Blot
The streptavidin agarose affinity assay was performed with the use of a biotin-As probe as described previously.18,19 Briefly, HEK293T cells were transfected with GFP-FANCL plasmid for 24 h followed by treatment with 5 μM biotin-As probe18,19 in Opti-MEM medium (Invitrogen, Carlsbad, CA) for 2 h. The cells were subsequently lysed in CelLytic M lysis buffer supplemented with a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) and incubated with streptavidin agarose beads at 4 °C for 3 h. Streptavidin agarose beads were then washed six times with 1 × PBS containing 0.5% NP-40 and resuspended in SDS-PAGE loading buffer.
After SDS-PAGE separation, the proteins on the gel were transferred to a nitrocellulose membrane using a buffer containing 10 mM NaHCO3, 3 mM Na2CO3, and 20% methanol. The membrane was subsequently blocked with 5% nonfat milk in PBS-T at RT for 2 h and incubated with anti-GFP primary antibody (1:1 0000 dilution, Sigma-Aldrich) at 4 °C overnight. After washing with PBS-T six times, the membrane was incubated with anti-rabbit secondary antibody at RT for 1 h, and subsequently washed with PBS-T six times. The secondary antibody was detected using an ECL Advanced Western Blotting Detection Kit (GE Healthcare, Chicago, IL) and visualized with Hyblot CL autoradiography film (Denville Scientific, Inc., Metuchen, NJ). Similar experiments were carried out by transfecting HEK293T cells with FANCL-C312,315A and C364,367A mutants, or by pretreating cells with 10 μM Zn2+, NaAsO2, or (p-aminophenyl)-arsenoxide (PAPAO) for 1 h prior to treatment with the biotin-As probe.
Fluorescence Microscopy
HEK293T cells were seeded on cover glasses in 24-well plates at a density of ~ 1 × 105 cells per well and transfected with 1 μg of plasmid encoding GFP-FANCL, GFP-FANCL-C312,315A, or GFP-FANCL-C364,367A for 24 h. The cells were then incubated with Opti-MEM containing 5 μM ReAsH-EDT2 (Invitrogen) at 37 °C for 2 h, washed three times with BAL buffer, fixed with 4% paraformaldehyde, stained with DAPI, and imaged using a Leica TCS SP2 confocal microscope (Leica Microsystems, Buffalo Grove, IL). Similar experiments were carried out by pretreating cells with 10 μM Zn2+, NaAsO2, or PAPAO for 1 h prior to transfection with GFP-FANCL plasmid and treatment with ReAsH-EDT2.
Isolation of Chromatin-Associated Proteins
Chromatin-associated proteins were isolated following previously described procedures.20 Briefly, cells were first lysed by incubating, for 30 min on ice, in a cytoplasmic lysis buffer, which contained 10 mM Tris-HCl (pH 8.0), 0.34 M sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, and 0.5% NP-40. The intact nuclei were isolated by centrifugation at 5000 rpm for 2 min and lysed with nuclear lysis buffer, which contained 20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 1 mM EDTA, 150 mM KCl, 0.1% NP-40, 1 mM DTT, and 10% glycerol as well as protease and phosphatase inhibitors. The chromatin-enriched fraction was isolated by centrifugation at 14 000 rpm for 30 min, resuspended in a buffer containing 20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 150 mM KCl, 10% glycerol, protease and phosphatase inhibitors, and 0.15 unit/μL benzonase (Sigma-Aldrich, St. Louis, MO). After incubating on ice for 1 h, the chromatin-associated proteins were obtained by collecting supernatant after centrifugation.21
Ubiquitin Remnant Peptide Immunoprecipitation and LC-MS/MS Analysis
GM00637 cells cultured in RPMI 1640 medium containing heavy or light lysine and arginine were treated with or without 5 μM sodium arsenite for 24 h, followed by treatment with 5 μM MG132 for 1 h. The cells were then harvested and lysed in a lysis buffer with 8 M urea. The lysates from light control (or NaAsO2-treated) cells and heavy NaAsO2-treated (or control) cells were mixed at a 1:1 ratio and digested with trypsin overnight at an enzyme/substrate ratio of 1:50 (by mass). The resulting peptides were desalted on a Sep-Pak C18 column (Waters), followed by immunoprecipitation of peptides carrying the K-ε-GG ubiquitin remnant using the PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit (Cell Signaling). The enriched ubiquitinated peptides were eluted with 0.15% trifluoroacetic acid and injected for LC-MS/MS analysis on a Q Exactive Plus quadrupole-Orbitrap Mass Spectrometer (Thermo).
Recruitment of FANCD2 to DNA Damage Sites Induced by 4,5′,8-Trimethylpsoralen (TMP)
HeLa cells were plated in a 35 mm glass-bottom culture dish. On the next day, the cells were incubated for 15 min in media containing 6 μM TMP with or without NaAsO2 (5 or 20 μM), prior to laser treatment to photoactivate the TMP and generate laser-localized ICLs. Localized irradiation was performed using a Nikon Eclipse TE2000 confocal microscope (Nikon Instruments Inc., Melville, NY) equipped with an SRS NL100 nitrogen laser-pumped dye laser (Photonics Instruments, St. Charles, IL) that fires 5 ns pulses with a repetition rate of 10 Hz at 365 nm, at a power of 0.7 nW, which was measured at the back aperture of the 60× objective. The laser was directed to a specified rectangular region of interest (ROI) within the nucleus of a cell visualized with a Plan Fluor 60×/NA1.25 oil objective. The laser beam was oriented by galvanometer-driven beam displacers and fired randomly throughout the ROI until the entire region was exposed. Throughout the experiment, the cells were maintained at 37 °C, 5% CO2, and 80% humidity using an environmental chamber. For each plate, cells in successive fields along a cross on the glass (made with a diamond pen) were targeted during the course of 20 min. The plate was immediately fixed in freshly prepared 4% formaldehyde in PBS at RT for 10 min, followed by immunostaining for γH2AX (05–636, Millipore) and FANCD2 (NB100–182, Novus). Image processing and subsequent quantification of FANCD2 intensity in the ICL stripe minus the nuclear background were performed in Volocity (PerkinElmer, Waltham, MA).
Clonogenic Survival Assay
HEK293T cells were seeded in six-well plates at densities of 100–600 cells per well. After incubation with 5 μM NaAsO2 for 24 h, the cells were exposed to various doses of MMC (0–72 ng/μL) or TMP (0–75 ng/μL) + 100 J/m2 UVA in DMEM medium and cultured for 10 days. The colonies were then fixed with 6% glutaraldehyde and stained with 0.5% crystal violet. Colonies with more than 50 cells were counted under a microscope.22
RESULTS
As3+ Binds to the RING-Finger Domain of FANCL Protein in Vitro and in Cells
To test our hypothesis that As3+ exposure may compromise DNA interstrand cross-link repair via the FA pathway, we first examined the interaction between As3+ and the PHD-finger-containing protein FANCL. To this end, we performed an in vitro binding assay to examine whether As3+ can interact directly with the RING finger domain of FANCL protein with the use of a synthetic RING finger peptide derived from human FANCL protein (see Experimental section). MALDI-TOF MS data revealed a mass increases of 72 and 144 Da upon incubation of the synthetic peptide with As3+. These mass increases reflect the binding of one and two As3+ to the peptide along with the releases of three and six protons, respectively, from the cysteine residues at the RING finger domain (Figure 1A).4,8 This result supports that As3+ can bind directly to the RING-finger domain of FANCL protein in vitro.
Figure 1.
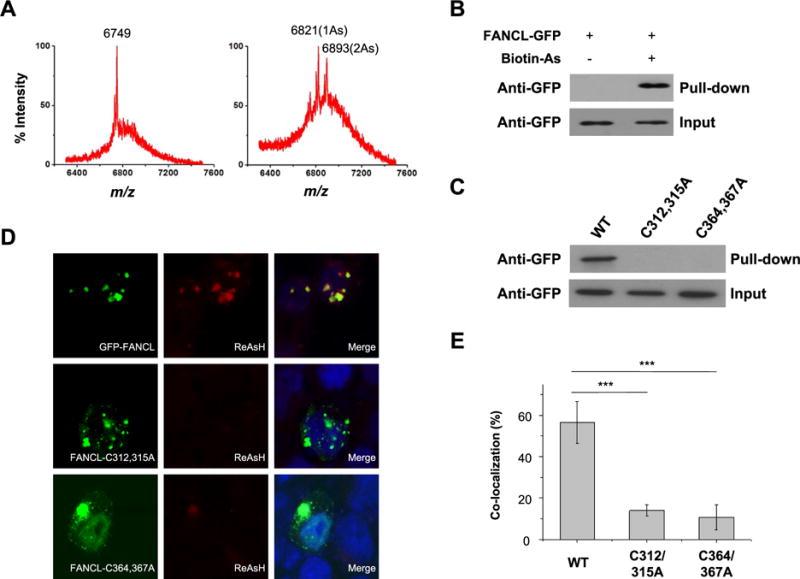
As3+ binding to the RING-finger domain of FANCL in vitro and in cells. (A) MALDI-TOF mass spectrum showing the interaction between As3+ and the RING-finger peptide of FANCL. The peptide was capable of binding up to two As3+. (B) Streptavidin agarose affinity assay revealed the interaction between As3+ and FANCL protein in HEK293T cells. Biotin-As probe was used to assess the binding between As3+ and ectopically expressed GFP-FANCL protein. (C) Streptavidin agarose affinity assay revealed the abolished interaction between As3+ and FANCL protein mutants in HEK293T cells, where the RING-finger cysteine residues 312 and 315 or 364 and 367 were mutated into alanines. (D) Fluorescence microscopy results revealed the colocalization of As3+-bearing ReAsH-EDT2 and GFP-FANCL in HEK293T cells, and mutations of cysteines to alanines in the RING-finger domain diminished the colocalization. (E) Quantitative analysis of the extents of colocalization between ReAsH-EDT2 and FANCL or its mutants. The signal intensities of fluorescence emission in each channel were determined by using ImageJ. The colocalization ratios were calculated as the signal intensity of ReAsH-EDT2 divided by the signal intensity of GFP. The data represent the mean and standard deviation of results obtained from images of 30 different cells. ***P < 0.001. The P values were calculated using two-tailed, unpaired Student’s t test.
We next asked whether this interaction could also occur in cells. Toward this end, we incubated HEK293T cells with a synthetic biotin-As probe followed by streptavidin agarose affinity pull-down assay to assess the interaction between As3+ and FANCL. Western blot analysis showed that incubation of HEK293T cells with the biotin-As probe facilitates the pull-down of ectopically expressed FANCL protein, indicating that As3+ is indeed able to bind to FANCL in cells (Figure 1B). Mutations of two critical cysteine residues in the RING-finger domain to alanines led to marked diminution in this interaction, suggesting that the interaction between FANCL and As3+ necessitates the intact RING-finger domain of FANCL (Figure 1C). To further substantiate this finding, we performed fluorescence microscopy experiments using an As3+-containing dye, ReAsH-EDT2, which displays red fluorescence after its arsenic moieties bind to four cysteine residues in proteins.23 Indeed, fluorescence microscopy results revealed that the GFP-tagged FANCL, but not the FANCL mutants, colocalizes with ReAsH-EDT2 (Figure 1D,E), again supporting the binding between As3+ and the RING-finger domain of FANCL in cells. However, pretreatment of cells with NaAsO2 or PAPAO, but not Zn2+, led to significant reductions in the colocalization between ReAsH-EDT2 and FANCL (Figure 2A–C), supporting the competitive binding of As3+ to cysteine residues in the RING finger motif of FANCL.
Figure 2.
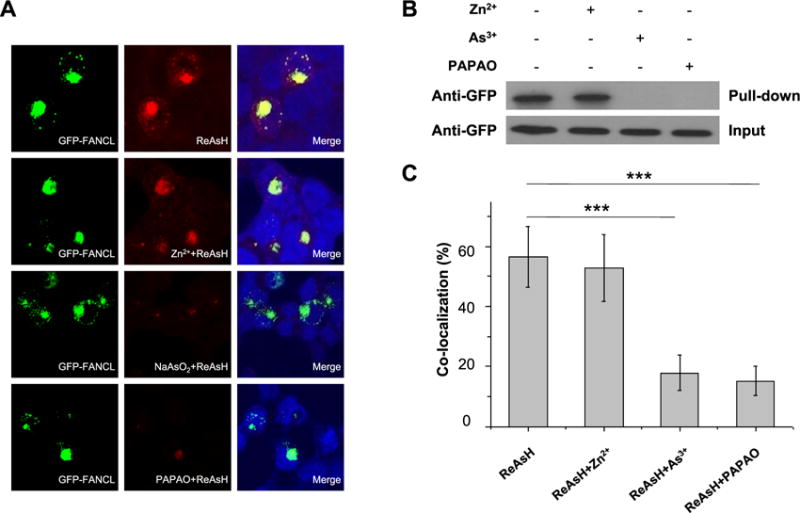
Competitive binding of As3+ to the RING-finger domain of FANCL. (A) Fluorescence microscopy showing the colocalization between As3+-bearing ReAsH-EDT2 and GFP-FANCL in HEK293T cells. The colocalization was abolished in cells pretreated with 10 μM NaAsO2 or PAPAO, but not Zn2+, prior to staining with ReAsH-EDT2. (B) Streptavidin agarose affinity assay revealed the interaction between As3+ and FANCL protein in HEK293T cells. The interaction was diminished after pretreatment with 10 μM NaAsO2 or PAPAO, but not Zn2+. (C) Quantitative analysis of the frequencies of colocalization between ReAsH-EDT2 and FANCL. The data represent the mean and standard deviation of results obtained from images of 30 different cells. ***P < 0.001. The P values were calculated using two-tailed, unpaired Student’s t test.
Arsenite Binding to FANCL Perturbs the Ubiquitination and Chromatin Localization of FANCD2
Having demonstrated the interaction between As3+ and FANCL, we next investigated the effect of this binding on the function of FANCL. To this end, we treated HEK293T and HeLa cells with MMC to induce ICLs24–26 in cells and subsequently incubated the cells with As3+. Western blot results illustrated that the level of monoubiquitination of FANCD2 was significantly elevated after MMC treatment; however, the MMC-stimulated ubiquitination of FANCD2 was dramatically reduced after incubation with arsenite (Figure 3A,B). Considering that FANCL is the major E3 ligase involved in the monoubiquitination of FANCD2,27 the above results supported that the binding of arsenite to the RING finger motif of FANCL diminishes its E3 ubiquitin ligase activity. In addition, chromatin fractionation assay results showed that the ubiquitinated FANCD2 was primarily localized in the chromatin fraction (Figure 3C,E), which agrees with the notion that the recruitment of ubiquitinated FANCD2 to chromatin is an important step in DNA ICL repair.28 In this respect, it is of note that, similar to what were previously observed,29–31 we also detected nonubiquitinated form of FANCD2 in the chromatin fraction, which might be attributed to the presence of other mechanisms for the chromatin recruitment of FANCD2 that is independent of its ubiquitination. Moreover, treatment of cells with increasing concentrations of arsenite led to a dose-dependent decrease in the localization of FANCD2 to chromatin (Figure 3D,F). These results support that arsenite binding to the RING finger motif of FANCL perturbs its E3 ligase activity, thereby resulting in diminished ubiquitination and chromatin localization of FANCD2.
Figure 3.
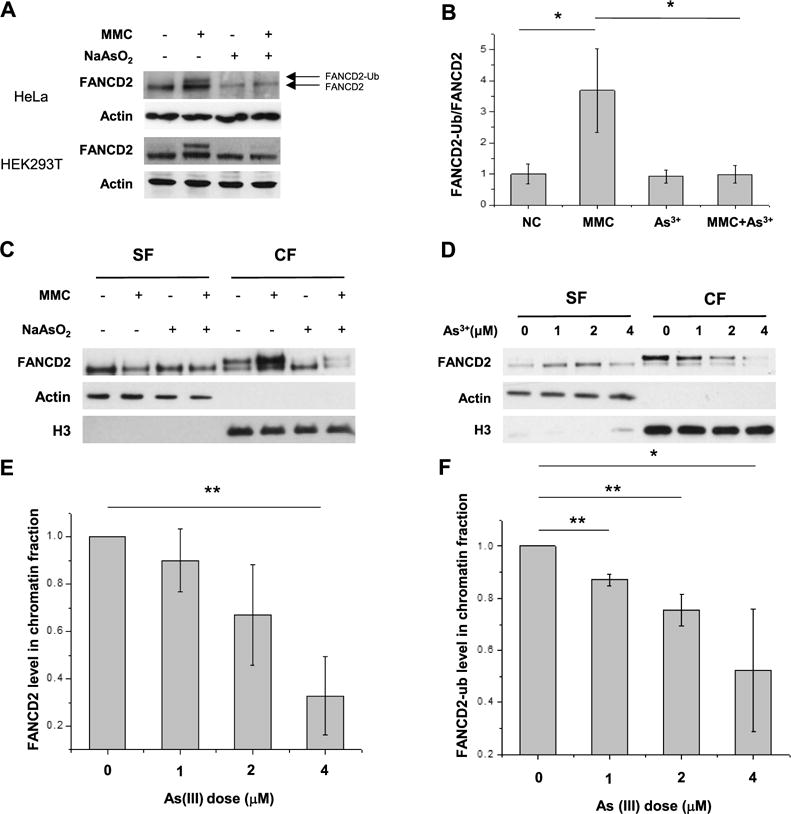
As3+ exposure leading to decreased ubiquitination and chromatin localization of FANCD2. (A) The Western blot result revealed that As3+ exposure elicited reduced ubiquitination of FANCD2 in the lysate of HeLa cells after As3+ exposure upon ICL induction by MMC. (B) Quantitative data of relative levels of FANCD2 ubiquitination after MMC treatment. The data represent the mean and standard deviation of results obtained from three biological replicates. (C) As3+ exposure diminished the ubiquitination level of FANCD2 and perturbed its chromatin localization. “CF” and “SF” designate the chromatin and soluble fractions, respectively. (D) As3+ exposure resulted in a dose-dependent reduction in the chromatin localization of FANCD2. (E) Quantitative data of total FANCD2 level in chromatin fractions for panel D. The levels of FANCD2 were represented as the ratio of FANCD2/histone H3 in chromatin fractions, and the relative level of FANCD2 in the control group was normalized to unity. (F) Quantitative data of FANCD2 ubiquitination for panel (D). The relative levels of FANCD2 ubiquitination were calculated as the ratios of FANCD2-ub over unmodified FANCD2 in chromatin fractions, and the ratio for the control group was normalized into unity. *P < 0.05; **P < 0.01; ***P <0.001. The P values were calculated using two-tailed, unpaired Student’s t test.
To further confirm the diminished ubiquitination of FANCD2 after arsenite treatment, we conducted a quantitative proteomic experiment to assess the level of this ubiquitination with the use of SILAC labeling, immunoprecipitation of tryptic peptides containing the ubiquitin remnant (i.e., diglycine-modified lysine), and LC-MS/MS analysis (Figure 4 and see Experimental Section). We were able to detect the [M + 2H]2+ ions for both the light (m/z 576.3217 for the monoisotopic peak) and heavy (m/z 584.3246 for the monoisotopic peak) forms of the FANCD2 peptide harboring the ubiquitin remnant at Lys561 (Figure 4A,C). The LC-MS data revealed that a 24-h treatment of GM00637 cells with 5 μM NaAsO2 leads to a reduction in the level of this ubiquitination to 46% relative to the control untreated cells (Figure 4B). This result parallels the finding made from Western blot analysis, which showed a ~ 2-fold decrease in the level of Lys561 ubiquitination in FANCD2 after HeLa cells were treated with 4 μM NaAsO2 for 24 h (Figure 3F).
Figure 4.
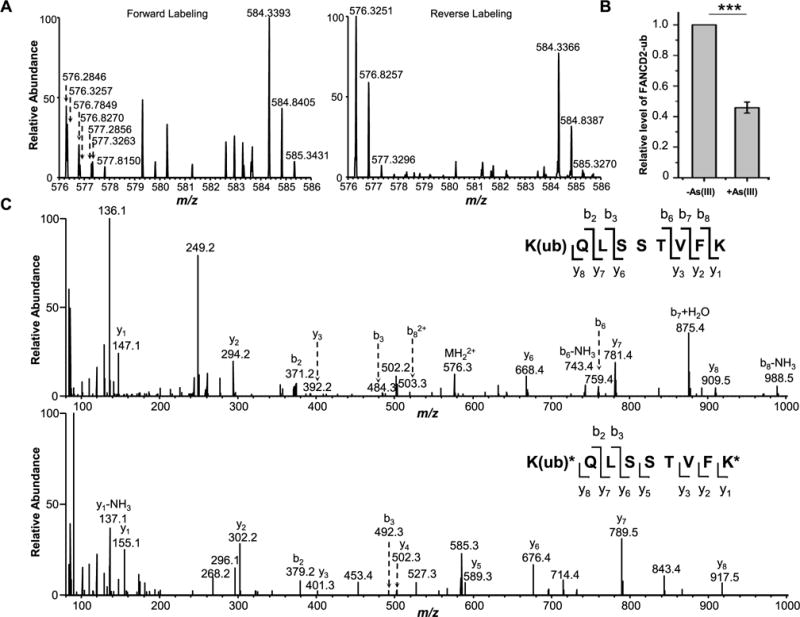
Diminished monoubiquitination of FANCD2 after arsenite treatment in GM00637 cells as revealed by LC-MS/MS data. (A) MS results for FANCD2 K561 ubiquitination from the forward and reverse SILAC labeling samples. In the forward sample, cells cultured in light SILAC medium were treated with 5 μM NaAsO2 for 24 h and mixed with an equal amount of lysate from untreated cells cultured in heavy SILAC medium. In the reverse sample, lysate of cells cultured in heavy medium and treated with 5 μM NaAsO2 was mixed with an equal amount of lysate of untreated cells cultured in light medium. The spectrum shows the [M + 2H]2+ ions for FANCD2 K(ub)QLSSTVFK (light, m/z 576.3217) and K(ub)*QLSSTVFK* (heavy, m/z 584.3246) peptides. (B) Relative levels of ubiquitination of K561 in FANCD2 with or without arsenite treatment. The data represent the mean and standard deviation of results obtained from three independent experiments. The level of FANCD2 ubiquitination in the control group was normalized into 1.0. ***P < 0.001, and the P values were calculated using a two-tailed, unpaired Student’s t test. (C) MS/MS results for K(ub)QLSSTVFK (light) and K(ub)*QLSSTVFK* (heavy) peptides.
Arsenite Exposure Led to Compromised Recruitment of FANCD2 to DNA Damage Sites and Sensitized Cells toward DNA ICL Agents
We next utilized immunofluorescence microscopy to examine the effect of arsenite treatment on the recruitment of FANCD2 to DNA damage sites upon ICL induction. In this vein, HeLa cells were exposed to TMP plus UVA light (TMP+UVA)32 to induce ICLs and subsequently stained with γH2AX33 and FANCD2 antibodies to reveal the colocalization of damage sites and FANCD2 protein in the presence or absence of arsenite treatment. Compared to the control group, the recruitment of FANCD2 to ICL sites was substantially reduced upon arsenite treatment (Figure 5A). Moreover, recruitment of FANCD2 to DNA damage sites decreases with the dose of arsenite used (Figure 5B). This finding, in conjunction with the results obtained from the chromatin fractionation assay, led us to conclude that, upon ICL induction by MMC or TMP+UVA, arsenite inhibits the recruitment of FANCD2 to ICL sites, which may compromise the repair of ICLs via the FA pathway.
Figure 5.
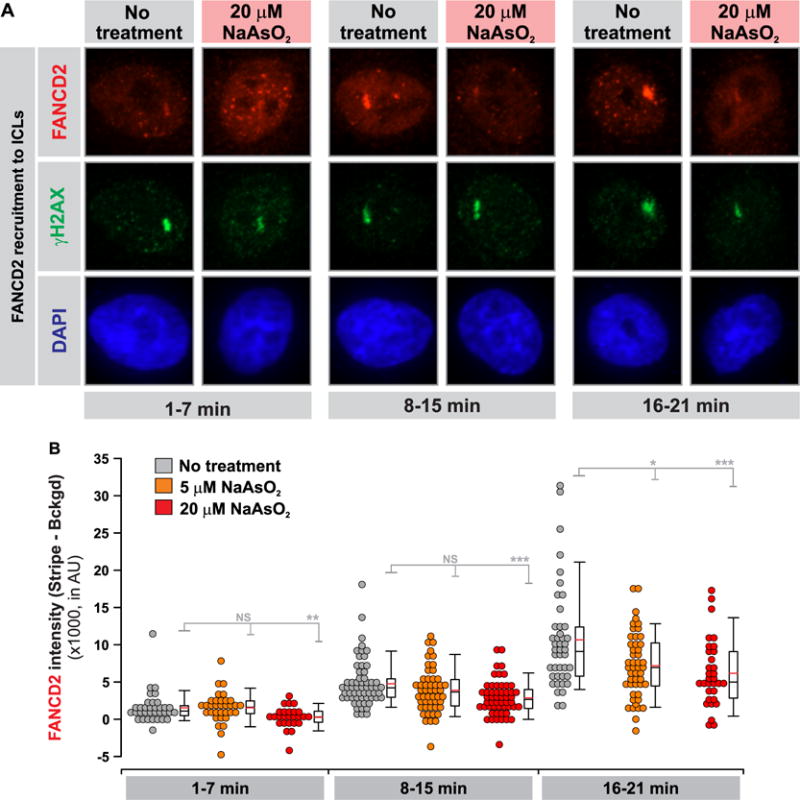
NaAsO2 exposure perturbs the recruitment of FANCD2 to laser-localized ICLs. (A) Cells were treated with TMP ± NaAsO2 (5 or 20 μM) and targeted with a 365 nm laser in a defined ROI in the cell nuclei to photoactivate the 4,5′,8-trimethylpsoralen and introduce ICLs. The cells were subsequently fixed and stained for γH2AX and FANCD2 to quantify the recruitment of FANCD2 to laser-localized ICLs as a function of time. Representative images showing γH2AX (damage marker) and the colocalizing FANCD2 stripe for each condition at the different time intervals following ICL induction. (B) Quantification results for the data shown in A. The intensity of FANCD2 in the stripe and the nuclear background was quantified in at least 30 cells per time point per condition analyzed, where the images were acquired under identical exposure conditions. Three independent experiments were performed and showed equivalent results. Means were compared using a Rank-sums test (*P < 0.05; **P < 0.01;***P < 0.001).
To further substantiate the above findings, we employed a clonogenic survival assay to assess whether arsenite exposure sensitizes cultured human cells toward DNA interstrand cross-linking agents. The cells were pretreated with 5 μM NaAsO2 for 24 h prior to their exposure to different doses of MMC or TMP +UVA. After culturing the cells for 10 days, we quantified the survival rates of cell colonies. Our results showed that the number of surviving colonies decreased with the concentrations of MMC or TMP, and importantly the clonogenic survival was exacerbated in cells cotreated with arsenite (Figure 6A,B). In this vein, it is worth noting that exposure to 5 μM NaAsO2 for 24 h alone also led to reduced clonogenic survival relative to control cells (data not shown). Nevertheless, the dose-dependent decrease in cell survival is much more pronounced in cells cotreated with NaAsO2 than in cells exposed only to interstrand cross-linking agents (Figure 6). This result suggests that arsenite conferred elevated sensitivities of cells toward the cytotoxic effects of DNA interstrand cross-linking agents.
Figure 6.
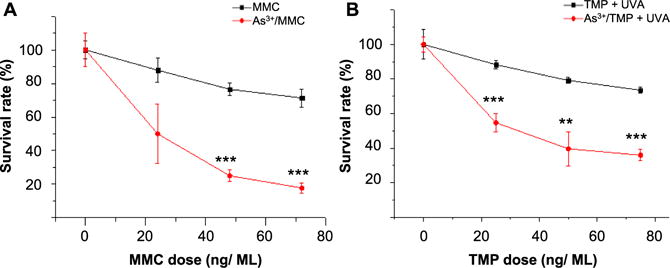
Clonogenic survival assay showing diminished resistance toward ICL agents upon As3+ exposure. (A) HEK293T cells were plated in six-well plates and treated with increasing doses of MMC to induce ICLs and exposed to As3+. After being cultured for 10 days, the cells exposed to As3+ displayed diminished resistance to MMC as reflected by lower survival rates relative to the control group. (B) HEK293T cells were treated with increasing doses of TMP and irradiated with 365 nm light to photoactivate the 4,5′,8-trimethylpsoralen to introduce ICLs. Compared to the control group, the cells exposed to As3+ manifested increased sensitivity to TMP + UVA. **P < 0.01; ***P < 0.001. The P values refer to the comparisons between the groups treated with ICL agents alone and those treated with ICL agents together with As3+, and the values were calculated using a two-tailed, unpaired Student’s t test.
DISCUSSION
Arsenic is a widespread environmental carcinogen,34 and binding to vicinal thiols in proteins is thought to be one of the major mechanisms underlying the cytotoxic and carcinogenic effects of arsenic species.3 Zinc fingers are small structural motifs in proteins characterized by the coordination of one or more zinc ions with their Zn2+-binding regions. They assist to stabilize the folded structures of proteins and maintain their functions.35 It was observed previously that As3+ displayed stronger binding affinity toward C3H and C4 types of zinc fingers than that of the C2H2 type.4 For instance, arsenite was found to bind to the C4-type zinc finger in XPA5,6 and C3H-type zinc finger in PARP-1,7 which impair their functions in DNA repair. RING finger is a specific type of Zn2+-binding domain that contains a C3HC4 amino acid motif and can coordinate two zinc ions.36 RING finger is present in many E3 ubiquitin ligases including, among others, APC/C, MDM2, BRCA1, and the above-mentioned RNF20-RNF40.37 Replacement of Zn2+ ions with As3+ in the RING finger domain of PML was found to lead to the proteasomal degradation of PML-RARα fusion protein.12 In addition, arsenite binding to the RING finger domains of RNF20-RNF40 E3 ubiquitin ligase inhibits the ubiquitination of lysine 120 in histone H2B and produces a chromatin environment that is not biochemically accessible for DNA double-strand break repair.8
The Fanconi anemia syndrome is a genetic disease manifested with progressive bone marrow failure, developmental abnormalities, and increased cancer susceptibility.38 FA arises from germ-line mutation in one of the FA genes, resulting in compromised repair of DNA ICLs as well as stalling of DNA replication and transcription machineries.13–16 Central to the FA-mediated DNA repair pathway is the genotoxic stress-induced monoubiquitination of Lys561 in FANCD2 by FANCL in complex with E2 ligases UBE2T or UBE2W,39 which promote the initiation of downstream nucleolytic incisions by recruiting MTMR15/FAN1 to DNA damage sites.40 Monoubiquitination of FANCD2 promotes its binding to chromatin, and the ubiquitination is required for FANCD2’s interaction with BRCA139 and MTM15/FAN1,41 DNA repair, and normal cell cycle progression. FANCD2 is deubiquitinated by USP1 once DNA repair is complete.42
As a component of the FA core complex that ubiquitinates FANCD2 and FANCI, the E3 ubiquitin ligase FANCL, harboring a C4HC3 RING finger motif,43 plays an important role in the FA pathway for the repair of ICLs. Our present study demonstrated that arsenite binds to the RING finger domain of FANCL both in vitro and in cultured human cells. In keeping with the fact that the coordination of Zn2+ ions with the RING finger motif of FANCL is essential for its structural integrity, protein interaction, and E3 ligase activity,43 we found that the occupancy of As3+ at the Zn2+-binding sites of FANCL led to a diminished ubiquitination and compromised localization of FANCD2 to chromatin and DNA damage sites. The impaired ubiquitination of FANCD2 triggered by arsenite exposure perturbs the FA pathway-mediated repair of DNA ICL lesions. Consistent with this notion, we observed that cells exposed to arsenite exhibited diminished resistance to agents that can induce DNA ICL lesions. Together, we uncovered, for the first time, that arsenite is able to bind to the RING finger domain of FANCL and this binding leads to diminished DNA ICL repair. This novel discovery provides a better understanding of the mechanism underlying the carcinogenic effect of arsenite.
Acknowledgments
This work was supported by the National Institutes of Health (R21 ES025392).
Footnotes
ORCID
Yinsheng Wang: 0000-0001-5565-283X
Notes
The authors declare no competing financial interest.
References
- 1.Kaur S, Kamli MR, Ali A. Role of arsenic and its resistance in nature. Can J Microbiol. 2011;57:769–774. doi: 10.1139/w11-062. [DOI] [PubMed] [Google Scholar]
- 2.Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Pharmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- 3.Shen S, Li XF, Cullen WR, Weinfeld M, Le XC. Arsenic binding to proteins. Chem Rev. 2013;113:7769–7792. doi: 10.1021/cr300015c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou X, Sun X, Cooper KL, Wang F, Liu KJ, Hudson LG. Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J Biol Chem. 2011;286:22855–22863. doi: 10.1074/jbc.M111.232926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwerdtle T, Walter I, Hartwig A. Arsenite and its biomethylated metabolites interfere with the formation and repair of stable BPDE-induced DNA adducts in human cells and impair XPAzf and Fpg. DNA Repair. 2003;2:1449–1463. doi: 10.1016/j.dnarep.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Asmuss M, Mullenders LH, Eker A, Hartwig A. Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis. 2000;21:2097–2104. doi: 10.1093/carcin/21.11.2097. [DOI] [PubMed] [Google Scholar]
- 7.Ding W, Liu W, Cooper KL, Qin XJ, de Souza Bergo PL, Hudson LG, Liu KJ. Inhibition of poly(ADP-ribose) polymerase-1 by arsenite interferes with repair of oxidative DNA damage. J Biol Chem. 2009;284:6809–6817. doi: 10.1074/jbc.M805566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F, Paramasivam M, Cai Q, Dai X, Wang P, Lin K, Song J, Seidman MM, Wang Y. Arsenite binds to the RING finger domains of RNF20-RNF40 histone E3 ubiquitin ligase and inhibits DNA double-strand break repair. J Am Chem Soc. 2014;136:12884–12887. doi: 10.1021/ja507863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol. 2011;7:113–119. doi: 10.1038/nchembio.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Jiang J, Li L, Amato NJ, Wang Z, Wang Y. Arsenite targets the zinc finger domains of Tet proteins and inhibits Tet-mediated oxidation of 5-methylcytosine. Environ Sci Technol. 2015;49:11923–11931. doi: 10.1021/acs.est.5b03386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- 12.Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, Zhang QY, Yang HY, Huang QH, Zhou GB, Tong JH, Zhang Y, Wu JH, Hu HY, de The H, Chen SJ, Chen Z. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010;328:240–243. doi: 10.1126/science.1183424. [DOI] [PubMed] [Google Scholar]
- 13.Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 15.Duxin JP, Walter JC. What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol. 2015;37:49–60. doi: 10.1016/j.ceb.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17:337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 17.Dao KH, Rotelli MD, Brown BR, Yates JE, Rantala J, Tognon C, Tyner JW, Druker BJ, Bagby GC. The PI3K/Akt1 pathway enhances steady-state levels of FANCL. Mol Biol Cell. 2013;24:2582–2592. doi: 10.1091/mbc.E13-03-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, Yang F, Shim JY, Kirk KL, Anderson DE, Chen X. Identification of arsenic-binding proteins in human breast cancer cells. Cancer Lett. 2007;255:95–106. doi: 10.1016/j.canlet.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalef E, Gitler C. Purification of vicinal dithiol-containing proteins by arsenical-based affinity chromatography. Methods Enzymol. 1994;233:395–403. doi: 10.1016/s0076-6879(94)33046-8. [DOI] [PubMed] [Google Scholar]
- 20.Aygun O, Svejstrup J, Liu Y. A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc Natl Acad Sci U S A. 2008;105:8580–8584. doi: 10.1073/pnas.0804424105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai X, Otake K, You C, Cai Q, Wang Z, Masumoto H, Wang Y. Identification of Novel α-N-Methylation of CENP-B That Regulates Its Binding to the Centromeric DNA. J Proteome Res. 2013;12:4167. doi: 10.1021/pr400498y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 23.Martin BR, Giepmans BN, Adams SR, Tsien RY. Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat Biotechnol. 2005;23:1308–1314. doi: 10.1038/nbt1136. [DOI] [PubMed] [Google Scholar]
- 24.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen RB, Ozes A, Kim T, Estep A, Kowalczykowski SC. BRCA2 is epistatic to the RAD51 paralogs in response to DNA damage. DNA Repair. 2013;12:306–311. doi: 10.1016/j.dnarep.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoenfeld AR, Apgar S, Dolios G, Wang R, Aaronson SA. BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Mol Cell Biol. 2004;24:7444–7455. doi: 10.1128/MCB.24.17.7444-7455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muniandy PA, Liu J, Majumdar A, Liu ST, Seidman MM. DNA interstrand crosslink repair in mammalian cells: step by step. Crit Rev Biochem Mol Biol. 2010;45:23–49. doi: 10.3109/10409230903501819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCabe KM, Olson SB, Moses RE. DNA interstrand crosslink repair in mammalian cells. J Cell Physiol. 2009;220:569–573. doi: 10.1002/jcp.21811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhagwat N, Olsen AL, Wang AT, Hanada K, Stuckert P, Kanaar R, D’Andrea A, Niedernhofer LJ, McHugh PJ. XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol Cell Biol. 2009;29:6427–6437. doi: 10.1128/MCB.00086-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaudhury I, Stroik DR, Sobeck A. FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol Cell Biol. 2014;34:3939–3954. doi: 10.1128/MCB.00457-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, Wilson JB, McChesney P, Williams SA, Kwon Y, Longerich S, Marriott AS, Sung P, Jones NJ, Kupfer GM. The Fanconi anemia proteins FANCD2 and FANCJ interact and regulate each other’s chromatin localization. J Biol Chem. 2014;289:25774–25782. doi: 10.1074/jbc.M114.552570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumaresan KR, Ramaswamy M, Yeung AT. Structure of the DNA interstrand cross-link of 4,5′,8-trimethylpsoralen. Biochemistry. 1992;31:6774–6783. doi: 10.1021/bi00144a018. [DOI] [PubMed] [Google Scholar]
- 33.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 34.Agre P, Kozono D. Aquaporin water channels: molecular mechanisms for human diseases. FEBS Lett. 2003;555:72–78. doi: 10.1016/s0014-5793(03)01083-4. [DOI] [PubMed] [Google Scholar]
- 35.Klug A, Rhodes D. Zinc fingers: a novel protein fold for nucleic acid recognition. Cold Spring Harbor Symp Quant Biol. 1987;52:473–482. doi: 10.1101/sqb.1987.052.01.054. [DOI] [PubMed] [Google Scholar]
- 36.Freemont PS, Hanson IM, Trowsdale J. A novel cysteine-rich sequence motif. Cell. 1991;64:483–484. doi: 10.1016/0092-8674(91)90229-r. [DOI] [PubMed] [Google Scholar]
- 37.Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer. 2011;11:629–643. doi: 10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz RS, D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med. 2010;362:1909–1919. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 40.Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacKay C, Declais AC, Lundin C, Agostinho A, Deans AJ, MacArtney TJ, Hofmann K, Gartner A, West SC, Helleday T, Lilley DMJ, Rouse J. Identification of KIAA1018/FAN1, a DNA Repair Nuclease Recruited to DNA Damage by Monoubiquitinated FANCD2. Cell. 2010;142:65–76. doi: 10.1016/j.cell.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nijman SMB, Huang TT, Dirac AMG, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Cole AR, Lewis LP, Walden H. The structure of the catalytic subunit FANCL of the Fanconi anemia core complex. Nat Struct Mol Biol. 2010;17:294–298. doi: 10.1038/nsmb.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]