

Abstract
Background
The mechanisms and relevance of impaired glucose homeostasis in advanced heart failure (HF) are poorly understood. The study goals were to examine glucose regulation, pancreatic endocrine function, and metabolic factors related to prognosis in patients with nondiabetic advanced HF.
Methods and Results
In total, 140 advanced HF patients without known diabetes mellitus and 21 sex‐, age‐, and body mass index–matched controls underwent body composition assessment, oral glucose tolerance testing, and measurement of glucose‐regulating hormones to model pancreatic β‐cell secretory response. Compared with controls, HF patients had similar fasting glucose and insulin levels but higher levels after oral glucose tolerance testing. Insulin secretion was not impaired, but with increasing HF severity, there was a reduction in glucose, insulin, and insulin/glucagon ratio—a signature of starvation. The insulin/C‐peptide ratio was decreased in HF, indicating enhanced insulin clearance, and this was correlated with lower cardiac output, hepatic insufficiency, right ventricular dysfunction, and body wasting. After a median of 449 days, 41% of patients experienced an adverse event (death, urgent transplant, or assist device). Increased glucagon and, paradoxically, low fasting plasma glucose displayed the strongest relations to outcome (P=0.01). Patients in the lowest quartile of fasting plasma glucose (3.8–5.1 mmol·L−1, 68–101 mg·dL−1) had 3‐times higher event risk than in the top quartile (6.0–7.9 mmol·L−1, 108–142 mg·dL−1; relative risk: 3.05 [95% confidence interval, 1.46–6.77]; P=0.002).
Conclusions
Low fasting plasma glucose and increased glucagon are robust metabolic predictors of adverse events in advanced HF. Pancreatic insulin secretion is preserved in advanced HF, but levels decrease with increasing HF severity due to enhanced insulin clearance that is coupled with right heart failure and cardiac cachexia.
Keywords: cachexia, glucagon/glucagon‐like peptide, glucose, heart failure, insulin, metabolism, obesity paradox, right ventricular dysfunction, starvation
Subject Categories: Heart Failure; Diabetes, Type 2; Metabolism; Mortality/Survival
Clinical Perspective
What Is New?
Insufficient data exist regarding pancreatic function and glucose regulation in patients with advanced heart failure (HF).
Pancreatic insulin secretion is preserved in nondiabetic advanced HF, but with increasing HF severity, there was a decline in systemic insulin levels and reduced insulin/C‐peptide ratio that correlated with the degree of right ventricular dysfunction.
Low fasting plasma glucose and elevated glucagon (ie, markers of starvation) were the strongest metabolic predictors of adverse outcome in nondiabetic patients with advanced HF.
What Are the Clinical Implications?
Targeting HF‐related metabolic abnormalities may require a staged approach: Although insulin resistance is an established target in early HF, its relevance in advanced HF may be overridden by cachexia and starvation.
Enhanced insulin clearance (low insulin/C‐peptide ratio) in nondiabetic patients with advanced HF could contribute to body catabolism and wasting.
More research is required to determine whether interventions to improve glucose homeostasis, to prevent low fasting glucose, or to reduce insulin degradation might improve clinical outcomes in advanced HF.
Introduction
Impaired glucose homeostasis is common in patients with heart failure (HF), but little is understood about its mechanisms or how it should be managed.1 Impaired glucose regulation and hyperinsulinemia are present even in the absence of diabetes mellitus (DM) in HF and likely contribute to disease progression.1, 2, 3, 4, 5, 6, 7 Insufficient data exist regarding glucose regulation in severe HF. In a recent trial in advanced HF patients that aimed to decrease insulin resistance, the GLP‐1 (glucagon‐like peptide 1) analogue liraglutide failed to improve long‐term clinical stability, regardless of DM status.8 These results raise questions about whether insulin resistance is a relevant contributor to pathophysiology in the advanced phases of HF. It is possible that other factors such as splanchnic congestion, liver dysfunction due to low cardiac output, cachexia, or metabolic effects of natriuretic peptides drive the disease progression at advanced stages,9, 10, 11, 12 and targeting HF‐related metabolic abnormalities may require different strategies in early and advanced stages of disease.
With increasing HF severity and with the development of cachexia, circulating fasting insulin levels tend to decrease for reasons that as yet remain unknown.13, 14 Diminished signaling of insulin, which is the main human anabolic hormone, may promote catabolism and glucose deregulation, which are commonly observed in advanced HF; however, pancreatic β‐cell secretory responses and systemic insulin clearance in HF patients have never been examined.
We sought to examine the impact of advanced HF on glucose homeostasis in patients without known DM compared with control participants of similar age, sex, and body mass index. We further sought to identify the determinants of glucose regulation and pancreatic insulin secretion and the metabolic parameters related to outcome in order to characterize potential novel therapeutic targets.
Methods
The study enrolled patients with chronic (>6 months) stable advanced systolic HF who were hospitalized electively for consideration of advanced therapies. From ≈450 screened patients, 140 were enrolled. Patients with known DM (or on glucose‐lowering therapy), acute HF decompensation, hemodynamic instability (needing inotropes), reversible cardiac dysfunction, active malignancy, endocrine disease, chronic infection, and history of intentional weight loss were excluded. A group of healthy control participants who were also free of DM and matched by age, sex, and body mass was also enrolled. The study was approved by the local ethical committee, and written informed consent was obtained from all participants.
After an overnight fast, all participants underwent standard oral glucose tolerance testing (OGT) with 75 g of glucose in 200 mL of water in the morning (8 am). Venous blood samples were drawn at baseline and at 30, 60, and 120 minutes to measure plasma glucose, insulin, C‐peptide, and glucagon. Medical history, quality of life, anthropometrics, and echocardiography assessments were performed. Left ventricular function and dimensions were measured according to contemporary recommendations. Right ventricular dysfunction was quantified (0–3) in an apical 4‐chamber view by using tricuspid annular plane systolic excursion and tissue systolic velocity, as described previously.12 A subset of patients (n=81) underwent right heart catheterization with thermodilution cardiac output measurement.
Body weight was measured by an electronic scale (HBF‐510W; Omron), and body composition was measured by dual‐energy x‐ray absorptiometry (Lunar Prodigy; GE Healthcare). Antecedent weight 6 months before the evaluation was carefully ascertained by historical recollection and by review of available medical records, as in previous studies.12, 15 Cardiac cachexia was defined by the presence of significant nonintentional weight loss (>5% within 6 months)15 and by simultaneous presence of abnormal biochemistry (C‐reactive protein >5 mg·L−1, hemoglobin <120 g·L−1, or albumin <32 g·L−1), as described previously.12
Laboratory Analyses
Plasma glucose was measured on Glucose Analyzer 2 (Beckman Coulter; coefficient of variation [CV] 1.9%). Routine biochemical parameters were measured using an automated Abbott Architect ci1600 analyzer. The BNP (B‐type natriuretic peptide) concentrations were measured using a microparticle immunoassay (Architect BNP; Abbott Laboratories). Glomerular filtration rate was estimated by the Chronic Kidney Disease Epidemiology Collaboration equation. Insulin and C‐peptide were measured using RIA (Beckman‐Coulter; CV 3.4% and CV 4.0%, respectively) from stored aliquots (−80°C). Glucagon was measured using RIA (CV 4.6%), total GLP‐1 was measured using ELISA (CV 2%).
Pancreatic β‐Cell Function Modeling
Pre–hepatic insulin secretion rate at baseline and during OGT was estimated by mathematical modeling using a previously validated C‐peptide deconvolution approach.16, 17 The model estimates basal and total insulin secretion and describes β‐cell secretory function by (1) glucose sensitivity, reflecting the ability of the β cell to respond to changes in plasma glucose concentrations; (2) rate sensitivity, reflecting the magnitude of β‐cell response to a given rate of change in plasma glucose concentration; and (3) the potentiation factor, which is related to the release of endogenous incretin hormones, neuronal inputs, and changes in incremental plasma glucose concentrations after OGT.18 Insulin resistance was estimated by HOMA‐IR (homeostatic model assessment of insulin resistance).19
Statistical Methods and Outcome Analysis
Data were analyzed using JMP10 software (SAS Institute). Group differences were tested using the χ2 test, unpaired Student t test, or ANOVA. P˂0.05 was considered statistically significant. Data are presented as mean±SD. The impact on survival was tested by using Kaplan–Meier analysis and a Cox proportional hazards model. Adverse outcomes were defined by the combined end point of death without transplantation, urgent heart transplantation, or implantation of a ventricular assist device, as reported previously.20
Results
Baseline Characteristics
HF patients were predominantly middle‐aged overweight men with nonischemic HF (Table 1). The mean duration of HF was 6.3±8.7 years, and 72% of patients were severely symptomatic (New York Heart Association classes III–IV). Patients were treated with guideline‐recommended pharmacotherapy, and 63% patients had an implantable cardioverter‐defibrillator. By design, the body mass indexes of the control and HF groups were similar. More than one‐third of HF patients (37%) reported ˃5% change of body weight in the previous 6 months, and 18% fulfilled the criteria for cardiac cachexia. Echocardiography demonstrated severe biventricular dysfunction and remodeling, increased biventricular filling pressures, and reduced cardiac output in HF patients.
Table 1.
Baseline Characteristics
| Control (n=21) | HF (n=140) | P Value | |
|---|---|---|---|
| Age, y | 54±6.1 | 56±11 | 0.2 |
| Male sex | 86% | 83% | 0.8 |
| BMI, kg·m−2 | 28±2.0 | 27±4.6 | 0.1 |
| Fat mass, DEXA, kg | 26±6.5 | 25±11 | 0.7 |
| Lean mass, DEXA, kg | 59±8.5 | 54±9.6 | ˂0.04 |
| Body weight, kg | 86±8 | 82±17 | 0.08 |
| Waist circumference, cm | 98±18 | 98±12 | 0.8 |
| Weight change in 6 months, kg | 1.5±2.1 | −3.3±8.4 | <0.001 |
| Cachexia, % | 0 | 18 | 0.006 |
| GFR, mL·min−1·1.73 m2 | 88±12 | 70±19 | ˂0.001 |
| BNP, ng·L−1 | 19±9.7 | 935±948 | <0.001 |
| NYHA class | 1.0±0 | 2.8±0.6 | <0.001 |
| MLHFQ score | ··· | 45±25 | ··· |
| CAD/non‐CAD etiology | ··· | 45%/55% | ··· |
| Furosemide, %, daily dose, mg | ··· | 96, 104±80 | ··· |
| Beta blocker, %, daily dose, mga | ··· | 94, 67±45 | ··· |
| ACEI/ARB, %, daily dose, mgb | ··· | 89, 3.8±3.4 | ··· |
| Cardiovascular assessment | |||
| BP systolic, mm Hg | 121±17 | 113±19 | 0.04 |
| LV diastolic diameter, mm | 50±5.2 | 70±9.7 | <0.001 |
| LVEF, % | 60±1.9 | 24±7.7 | <0.001 |
| RV dysfunction grade (0–3) | 0.0±0.0 | 1.8±1.3 | <0.001 |
| Inferior vena cava diameter, mm | 14±3.8 | 20±6.0 | <0.001 |
| Right atrial pressurec, mm Hg | ··· | 9.7±5.6 | ··· |
| Mean PA pressurec, mm Hg | ··· | 33±12 | ··· |
| PA wedge pressurec, mm Hg | ··· | 23±9.0 | ··· |
| Cardiac outputc, L·m−2 | ··· | 3.6±1.0 | ··· |
| Metabolic assessment | |||
| Hemoglobin A1c, mmol·mol−1 | 38±3.1 | 44±5.8 | <0.001 |
| GLP‐1 total, pM·L−1 | 44±16 | 58±37 | 0.005 |
| FFA, mmol·L−1 | 0.4±0.2 | 0.5±0.3 | 0.002 |
| Total cholesterol, mmol·L−1 | 5.3±0.9 | 4.2±1.1 | 0.0002 |
| Triglycerides, mmol·L−1 | 1.4±0.8 | 1.5±0.8 | 0.6 |
| HOMA‐IR | 2.3±1.4 | 2.6±1.8 | 0.4 |
ACE/ARB indicates angiotensin‐converting enzyme inhibitor or angiotensin receptor blocker; BMI, body mass index; BNP, B‐type natriuretic peptide; BP, blood pressure; CAD, coronary artery disease; DEXA, dual‐energy x‐ray absorptiometry; FFA, free fatty acids; GFR, glomerular filtration rate; GLP‐1, glucagon‐like peptide 1; HF, heart failure; HOMA‐IR, homeostasis model assessment of insulin resistance; LV, left ventricle; LVEF, left ventricular ejection fraction; MLHFQ, Minnesota Living with Heart Failure Questionnaire; NYHA, New York Heart Association; PA, pulmonary artery; RV, right ventricle.
Metoprolol equivalents.
Ramipril equivalents.
Right heart catheterization was available from 80 participants with HF.
Factors Affecting Glucose Regulation in the HF and Control Groups
Compared with control participants, HF patients had identical fasting plasma glucose (FPG) and insulin but higher C‐peptide, glucagon, and GLP‐1 levels (Table 1). C‐peptide, a split product of proinsulin that reflects pancreatic secretion, was increased and, coupled with the reduced insulin/C‐peptide ratio, revealed that insulin clearance was enhanced in patients with HF (Tables 1 and 2). During OGT, HF patients displayed more pronounced elevation of glucose, insulin, glucagon, and C‐peptide levels than control participants (Figure 1, Table 2). Glucose regulation was not affected by HF etiology but was strongly associated with anthropometric and nutritional variables. FPG and insulin correlated with body mass index (r=0.36 and r=0.48, respectively; both P<0.001), body weight (r=0.29 and r=0.46, respectively; both P<0.001), and total fat mass (r=0.38 and r=0.54, respectively; both P<0.001) but not with lean mass (P=0.7 and P=0.06, respectively; Figure 1B). These relations were nonsignificant in control participants. Compared with noncachectic patients, cachectic HF patients had lower FPG (5.6±0.8 versus 5.3±0.8 mmol·L−1, 101±14 versus 95±14 mg·mL−1, P=0.05), lower area under the curve (AUC) of insulin (174±122 versus 131±80 mIU·L−1·h−1, P=0.03), and lower ratio of insulin AUC to C‐peptide AUC (P=0.006). Renal function, liver transaminases, and albumin concentrations were not related to FPG in HF patients.
Table 2.
Glucose Tolerance Test
| Control (n=21) | HF (n=140) | P Value | HF With Adverse Event (n=57) | HF With No Adverse Event (n=83) | P Value | |
|---|---|---|---|---|---|---|
| Glucose, fasting, mmol·L−1; mg·dL−1 | 5.6±0.7; 101±13 | 5.6±0.8; 101±14 | 0.9 | 5.3±0.8; 95±14 | 5.7±0.8; 103±14 | 0.004 |
| Glucose OGT AUC, mmol·L−1·h−1 | 14±2.5 | 18±4.2 | <0.0001 | 18±3.7 | 19±4.4 | 0.1 |
| Insulin, fasting, mIU·L−1 | 9.0±4.9 | 9.9±6.5 | 0.5 | 9.3±7.1 | 10±6.0 | 0.4 |
| Insulin OGT AUC, mIU·L−1·h−1 | 111±63 | 165±117 | 0.002 | 167±134 | 165±103 | 0.9 |
| C‐peptide, fasting, nmol·L−1 | 0. 9±0.3 | 1.5±0.7 | <0.0001 | 1.6±0.7 | 1.5±0.7 | 0.1 |
| C‐peptide OGT AUC, nmol·L−1·h−1 | 5.9±1.8 | 9.0±2.8 | <0.0001 | 9.4±3.1 | 8.9±2.6 | 0.3 |
| Glucagon, fasting, ng·L−1 | 76±17 | 107±40 | <0.0001 | 116±40 | 101±39 | 0.03 |
| Glucagon OGT AUC, ng·L−1·h−1 | 171±38 | 214±80 | 0.0002 | 221±77 | 211±84 | 0.4 |
| Insulin/glucagon ratio, fasting, mIU·ng−1 | 0.12±0.06 | 0.09±0.06 | 0.15 | 0.08±0.06 | 0.11±0.06 | 0.01 |
| Insulin/C‐peptide ratio, fasting, mIU·nmol−1 | 10±3.8 | 6.7±3.4 | 0.0006 | 6.0±4.3 | 7.1±2.6 | 0.07 |
For full time course of changes during OGT, see Figure 1A. Adverse event during follow‐up: death, urgent transplantation, or implantation of a ventricular assist device. AUC indicates area under curve, trapezoid rule; HR, heart failure; OGT, oral glucose tolerance test.
Figure 1.
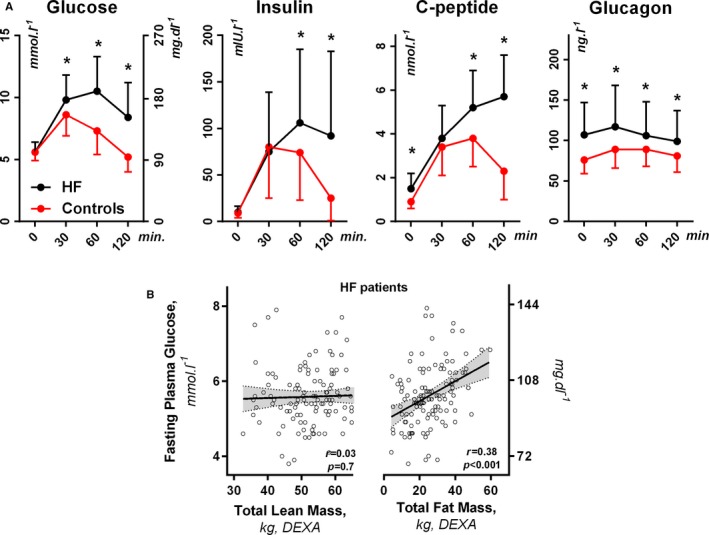
A, Glucose homeostasis parameters during oral glucose tolerance testing in heart failure (HF) patients (n=140) and in age‐, sex‐, and body mass index–matched control participants (n=21). Values are mean±SD. B, Fasting plasma glucose and body composition by dual‐energy x‐ray absorptiometry (DEXA). *P< 0.05 vs controls.
Glucose homeostasis was correlated with HF severity (Figures 2 and 3). FPG, insulin, insulin resistance (HOMA‐IR), and the insulin/glucagon ratio were lower in patients with more advanced HF compared with less symptomatic HF. During OGT, patients in the highest BNP tertile had lower AUC of glucose and insulin.
Figure 2.
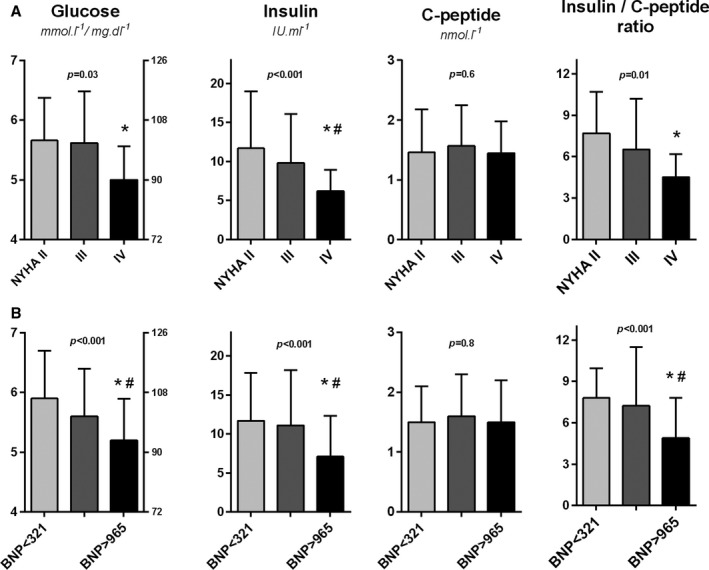
The relation of glucose homeostasis parameters to heart failure severity, quantified by (A) New York Heart Association (NYHA) functional classification or (B) by tertiles of BNP (B‐type natriuretic peptide). ANOVA and post hoc test: *P˂0.05 vs NYHA class II or low BNP tertile, # P˂0.05 vs NYHA class III or mid‐BNP tertile.
Figure 3.
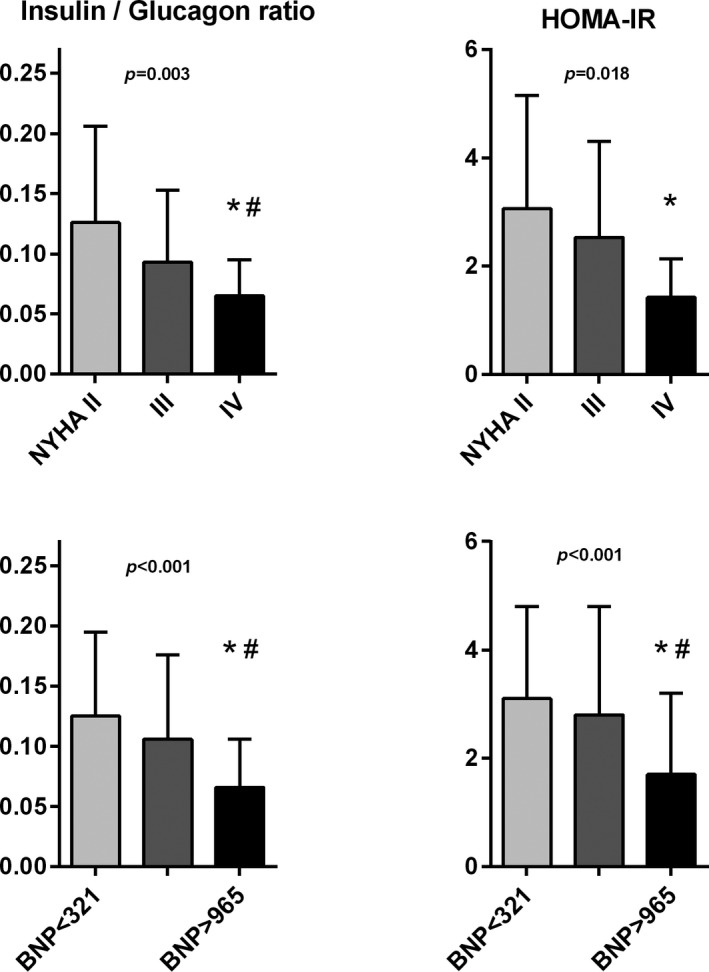
The relation of insulin/glucagon ratio and insulin resistance by HOMA‐IR (homeostatic model assessment of insulin resistance) according to heart failure severity, quantified by New York Heart Association (NYHA) functional classification (top) or tertiles of BNP (B‐type natriuretic peptide; bottom). ANOVA and post hoc test: *P˂0.05 vs NYHA class II or low BNP tertile, # P˂0.05 vs NYHA class III or mid‐BNP tertile.
HF severity correlated inversely with the fasting insulin/C‐peptide ratio (or insulin/C‐peptide AUC ratio), suggesting enhanced insulin clearance in advanced HF (Figure 2, right). The fasting insulin/C‐peptide ratio correlated with cardiac output (r=0.32, P=0.004) and inversely with right (but not left) ventricular dysfunction (Figure 4), tricuspid regurgitation grade (r=−0.18, P=0.04), and liver γ‐glutamyl transferase activity (r=−0.42, P=0.002), indicating a possible link among insulin clearance, right ventricular dysfunction, and HF‐related liver impairment. The fasting insulin/C‐peptide ratio was directly correlated with body mass index (r=0.36, P˂0.001; Figure 4), and decreased with unintentional weight loss (r=−0.19, P=0.02), suggesting a relation to cachexia. The fasting insulin/C‐peptide ratio was unrelated to kidney glomerular filtration.
Figure 4.
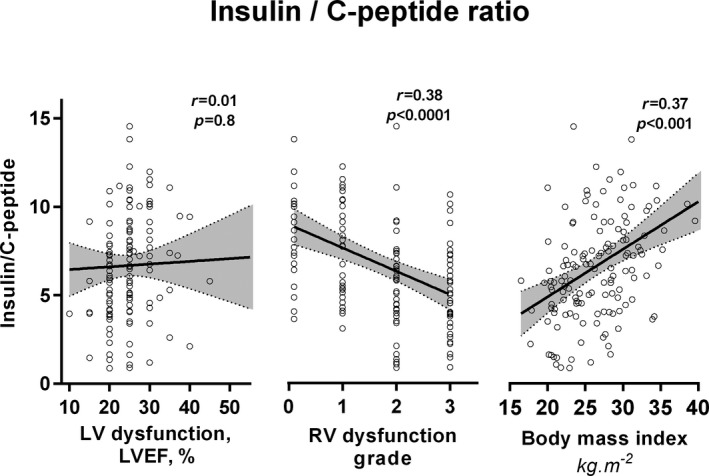
The association of insulin/C‐peptide ratio with left ventricular (LV) or right ventricular (RV) function and body mass in heart failure patients, indicating an association among enhanced insulin systemic clearance, right heart failure, and body wasting. r indicates Pearson correlation index and P‐ value. LVEF indicates left ventricular ejection fraction.
Pancreatic β‐Cell Function in the HF and Control Groups
Patients with HF had increased basal and total insulin secretion rates, and the β‐cell response to glucose (glucose sensitivity and rate sensitivity) was preserved (Table 3). Basal insulin secretion correlated with insulin resistance (HOMA‐IR) and was systematically increased in HF patients compared with control participants (Figure 5). The insulin secretion rate in HF patients (baseline and total) correlated with glucagon (r=0.38, P<0.001, and r=0.26, P=0.002) and GLP‐1 (r=0.35, P<0.001, and r=0.25, P=0.003), which were increased in HF patients, likely stimulating insulin secretion (Figure 5). The insulin secretion rate and β‐cell response to glucose were not related to HF etiology, medication use, or symptom severity. Reduced fasting insulin levels in the most symptomatic HF subgroup were caused by enhanced insulin clearance rather than diminished insulin secretion.
Table 3.
Modeling Analysis of Pancreatic β‐Cell Function From OGT Data
| Control (n=21) | HF (n=140) | P Value | HF With Adverse Events | HF With No Adverse Events | P Value | |
|---|---|---|---|---|---|---|
| Basal insulin secretion, pmol·min−1·m−2 | 113±34 | 194±86 | <0.0001 | 208±90 | 184±81 | 0.1 |
| Total insulin secretion, nmol·m−2 | 55±18 | 91±28 | <0.0001 | 94±31 | 91±27 | 0.5 |
| Glucose sensitivity, pmol·min−1·m−2·mM−1 | 189±98 | 151±81 | 0.1 | 154±80 | 149±81 | 0.8 |
| Rate sensitivity, pmol·m−2·mM−1 | 2217±3047 | 1046±1376 | 0.1 | 1287±1434 | 896±1320 | 0.1 |
| Potentiation factor ratio | 1.8±1.6 | 1.3±0.5 | 0.1 | 1.3±0.4 | 1.3±0.5 | 0.8 |
For explanation of parameters, see Methods. Adverse event during follow‐up: death, urgent transplantation, or implantation of a ventricular assist device. HR indicates heart failure; OGT, oral glucose tolerance test.
Figure 5.
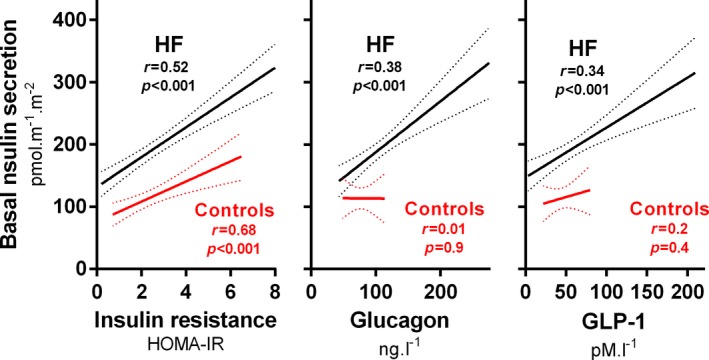
Basal insulin secretion rate in heart failure (HF) patients (n=140) or age‐, sex‐, and body mass index–matched control participants (n=21) derived by a β‐cell function model (see Methods) in relation to insulin resistance (HOMA‐IR), glucagon, and GLP‐1 (glucagon‐like peptide 1) levels. Lines: regression and 95% confidence intervals. HOMA‐IR indicates homeostatic model assessment of insulin resistance.
Association of Glucose Homeostasis With Outcome
Outcomes were ascertained in all participants. After 449 days (interquartile range: 144–1500 days) of follow‐up, 57 patients (41%) experienced an end point (death without transplant, urgent transplant, or implantation of a ventricular assist device). Patients experiencing an event were leaner, with less fat mass (21±11 versus 28±11 kg, P=0.001) and were more likely to be cachectic (28% versus 11%, P=0.01), with higher BNP, lower blood pressure, and more severe right ventricular dysfunction.
Fasting and 30‐minute plasma glucose levels were lower, whereas glucagon levels were higher in patients with an event (Table 2). Among glucose homeostasis parameters, low FPG demonstrated the strongest association with adverse outcomes (Table 4, Figure 6A). Patients in the lowest quartile of FPG (3.8–5.1 mmol·L−1, 68–101 mg·dL−1) had an event risk >3 times higher than the highest quartile (6.0–7.9 mmol·L−1, 108–142 mg·dL−1; relative risk: 3.05 [95% confidence interval, 1.46–6.77]; P=0.002). Increased fasting glucagon was also associated with poor outcomes, independent of and partly additive to FPG (Figure 6B).
Table 4.
Risk of Adverse Outcomes by Glucose Homeostasis Parameters
| RR and 95% CI per 1‐SD Increase (z score) | χ2 | P Value | |
|---|---|---|---|
| Glucose, fasting | 0.69 (0.53–0.86) | 8.0 | 0.005 |
| OGT at 30 min | 0.70 (0.53–0.91) | 7.0 | 0.008 |
| OGT at 60 min | 0.79 (0.62–1.01) | 3.6 | 0.06 |
| OGT at 120 min | 0.99 (0.78–1.26) | 0.1 | 0.9 |
| Insulin, fasting | 0.85 (0.62–1.12) | 1.2 | 0.3 |
| OGT at 30 min | 1.01 (0.76–1.26) | 0.1 | 0.9 |
| OGT at 60 min | 0.99 (0.76–1.25) | 0.1 | 0.9 |
| OGT at 120 min | 1.05 (0.78–1.32) | 0.1 | 0.7 |
| C‐peptide, fasting | 1.30 (1.0–1.67) | 3.9 | 0.05 |
| OGT at 30 min | 1.13 (0.87–1.43) | 0.9 | 0.4 |
| OGT at 60 min | 1.09 (0.84–1.42) | 0.5 | 0.5 |
| OGT at 120 min | 1.14 (0.88–1.48) | 1.0 | 0.3 |
| Glucagon, fasting | 1.32 (1.06–1.60) | 5.8 | 0.01 |
| OGT at 30 min | 1.09 (0.83–1.37) | 0.4 | 0.5 |
| OGT at 60 min | 1.00 (0.99–1.01) | 0.1 | 0.8 |
| OGT at 120 min | 1.17 (0.91–1.44) | 1.7 | 0.2 |
CI indicates confidence interval; OGT, oral glucose tolerance test; RR, relative risk.
Figure 6.
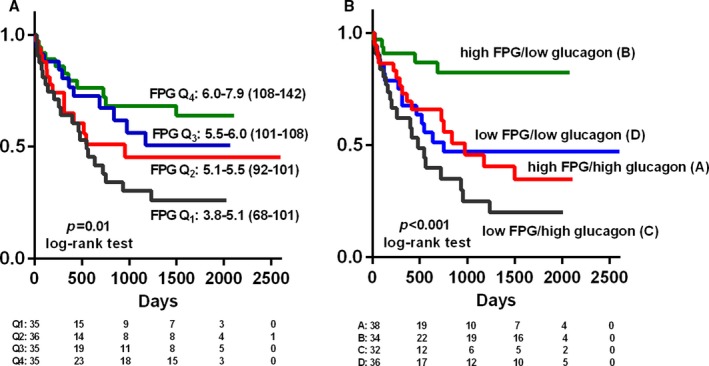
Kaplan–Meier analysis of event‐free survival (combined end point of death, urgent transplantation, or implantation of a ventricular assist device) according to (A) quartiles of fasting plasma glucose (FPG; in mmol·L−1 and mg·dL−1) and (B) combination of FPG above or below median and fasting glucagon above or below median. Number of participants at risk in each group is shown below the graphs.
Discussion
We report new observations on how substrate metabolism is affected by the presence of advanced HF. Impaired glucose homeostasis and hyperinsulinemia were common in HF patients compared with control participants, even in the absence of DM. Pancreatic insulin secretion was not impaired in HF; however, with increasing HF severity, there was a decline in systemic insulin levels and insulin/C‐peptide ratio, indicating enhanced insulin clearance as HF progresses. The insulin/C‐peptide ratio correlated with lower cardiac output, worsening liver impairment, and right ventricular dysfunction, supporting this pathophysiologic model. Low FPG and high glucagon were the strongest metabolic predictors of adverse events, with a steep gradient of the risk that even falls within the range of “normal” fasting glucose levels. These findings suggest that with advancing HF severity, the impact of cachexia overrides the impact of insulin resistance, and patients with advanced HF display hormonal features of starvation, particularly when perfusion and right heart function become most compromised. Insulin is an important anabolic hormone, and our data suggest that enhanced insulin degradation, probably occurring in the liver, may be a key, previously unrecognized mechanism promoting cardiac cachexia in patients with advanced HF.
Insulin in Advanced Nondiabetic HF
Consistent with previous reports, our study confirmed that non‐DM HF patients have hyperglycemic and hyperinsulinemic responses to OGT, reflecting HF‐related insulin resistance4, 6, 7, 13, 21, 22; however, the severity of insulin resistance in our cohort was less pronounced than in previous studies. This may reflect the fact that insulin resistance in HF depends on age, body composition, and norepinephrine levels,5 and our HF patients were younger, more often cachectic, and more extensively medicated with neurohumoral antagonists that lower norepinephrine.23
Under normal conditions, insulin is secreted by pancreatic β cells in response to increases in plasma glucose and hormonal influences. Almost 50% of insulin is degraded during its first passage through the liver.24 C‐peptide, a split product of proinsulin that is produced in pancreatic β cells in equimolar ratio with insulin, escapes liver degradation and thus reflects portal insulin secretion. Consequently, the ratio of insulin to C‐peptide is an indicator of hepatic insulin clearance.25
Circulating insulin concentrations, particularly in the fasted state, were reduced in the most symptomatic HF patients, consistent with previous observations in humans,13, 26, 27 an in an animal model.28 We showed in this study that low insulin concentrations in the most symptomatic HF patients were not caused by diminished secretion and pancreatic hypoperfusion, as proposed previously.27 Rather, HF patients had preserved β‐cell function and increased insulin secretion rates, reflecting compensation for insulin resistance and the stimulation from elevated glucagon and GLP‐1 levels. Based on the pancreatic β‐cell function model and insulin/C‐peptide ratios, the likely explanation for lower insulin concentrations in the most advanced HF patients is enhanced insulin breakdown.
The decreased insulin/C‐peptide ratio was specifically related to right heart dysfunction, low cardiac output, increased γ‐glutamyl transferase, and body wasting (Figure 4). Low hepatic vein insulin concentration, suggestive of enhanced insulin liver clearance, was reported in children with HF caused by congenital heart disease.29 Insulin is degraded by insulinase (IDE [insulin‐degrading enzyme]) that is mostly expressed in the liver. IDE processes other proteins, including the natriuretic peptides,30 so upregulation of IDE in patients with an advanced HF state might be expected. We speculate that right HF and liver congestion might be responsible for enhanced insulin clearance.29 Low insulin levels may be of clinical relevance. Because insulin inhibits lipolysis and proteolysis, relative insulin deficiency associated with congestion may promote body wasting through loss of its positive anabolic effects. This positive feedback loop mechanism may explain why patients with right ventricular dysfunction have accelerated cachexia and fat tissue loss.12
Signature of Starvation in Nondiabetic Patients With Advanced HF
The hormonal and metabolic profiles of advanced HF patients in this study with low glucose, low insulin, and elevated glucagon (Figure 3) correspond to changes typically observed in the early phases of starvation,31, 32, 33 a situation characterized by low liver glycogen stores, enhanced liver gluconeogenesis, and increased ketone production.26, 34
Low FPG was the strongest metabolic predictor of adverse events in this advanced HF population, in striking contrast to prior studies in non‐HF populations, in which hyperglycemia and insulin resistance identified patients at higher risk. The results accord with studies suggesting that FPG does not have a linear but rather U‐shaped relation to prognosis.35, 36 An association between low FPG and increased risk in patients with preexisting cardiovascular disease was previously explained by the adverse effect of glucose‐lowering medication.37 In this study, we showed that even spontaneous reduction in FPG is tightly linked to adverse outcomes (“fasting glucose paradox”), with a steep gradient of risk already within the range of what are considered “normal” plasma glucose levels. The tight regulation that maintains FPG homeostasis may be disrupted in advanced HF by the combined effects of low body weight, liver glycogen depletion,38 and low cardiac output with diminished liver perfusion.39 This situation may culminate in spontaneous fasting hypoglycemia sometimes observed in patients with advanced HF.40, 41
Fasting plasma glucose correlated with low fat mass (Figure 1B). Adipose tissue might help to preserve normoglycemia in HF patients by providing glycerol for liver gluconeogenesis.31 Glycerol and fatty acid release from adipose tissue is stimulated in HF by catecholamines and natriuretic peptides.11, 31 Diminished glycerol availability in cachectic HF patients may be the rate‐limiting step for gluconeogenesis and could explain the correlation of FPG with body mass.
Besides low FPG, adverse outcomes were also predicted by elevated baseline glucagon. An elevation of glucagon in HF patients may reflect sympathetic activation,42 but it also may represent a response to the “starved state,” as an adaptation to restore normal glycemia via gluconeogenesis and replenish liver glycogen stores at the expense of catabolism.43 Low insulin and increased glucagon levels in advanced HF patients facilitates utilization of fatty acids and ketogenesis.26, 34 Patients with advanced HF display myocardial upregulation of ketone‐utilizing enzymes44; however, further stimulation of ketone production in the setting of ketogenesis by using SGLT‐2 (sodium glucose cotransporter 2) inhibitors might lead to ketoacidosis,45 and this risk should be monitored in trials testing the effects of SGLT‐2 inhibition in populations with non‐DM advanced HF. The observation that low FPG is a strong metabolic predictor of adverse events indicates that advanced HF patients with low‐normal FPG may benefit from judicious glucose infusion before a major procedure (eg, implantation of a ventricular assist device) or during fasting to refill liver glycogen stores, particularly if patients have low body fat and are nondiabetic. This hypothesis warrants testing in prospective studies. In contrast, hyperinsulinemia does not seem to contribute to the risk to death in non‐DM advanced HF.
Limitations
Because of matching for age and body mass index, control participants were not completely healthy but rather mildly insulin resistant, and as such, the variance in some metabolic indicators was larger than would be expected in a completely healthy population. The control group was smaller than the HF group, which can increase the possibility of type I error, compromise the power analysis, and have other statistical undesirable effects. Nevertheless, for both logistical and ethical reasons, we were not able to recruit more otherwise healthy volunteers for our protocol. The justification for the control group (even of suboptimal size) is to provide internal reference for measurements that are not routinely performed and that would be difficult to interpret without having such controls. Insulin resistance was not directly measured using the euglycemic clamp technique but rather was estimated from fasting insulin and glucose. The study population represented severely symptomatic but relatively young HF patients free of known DM. Consequently, the findings may not be relevant to older, less morbid HF populations or to patients with DM. HF patients with treated DM were excluded, but some HF patients displayed glucose abnormalities consistent with a mild form of DM. Even after more strict exclusion of HF patients (n=49) who had increased hemoglobin A1c (˃48 mmol·mol−1), FPG (˃7 mmol·L−1, 126 mg·dL−1), or 2‐hour postload plasma glucose (˃11.1 mmol·L−1, 200 mg·dL−1), FPG was still inversely associated with outcome (relative risk: 0.58 [95% confidence interval, 0.35–0.96]; P=0.032). Our findings also may be applicable to advanced HF with preserved ejection fraction,46 although these patients were not included in this study.
In conclusion, we demonstrated that glucose homeostasis is impaired in non‐DM patients with advanced HF. Pancreatic insulin secretion was preserved in HF, and the decline of insulin levels with increasing HF severity arose from enhanced insulin clearance associated with right HF—a link potentially contributing to the development of cardiac cachexia. Low FPG and increased glucagon, markers of starvation, were the strongest metabolic predictors of adverse events in this advanced HF population, rather than hyperglycemia or hyperinsulinemia. These findings emphasize the prognostic value of insulin resistance, present in the early and moderate phases of HF, that becomes overridden in the advanced stages of HF. More research is required to determine whether interventions to improve glucose homeostasis, to prevent low FPG, or to reduce insulin degradation might improve clinical outcomes in advanced HF.
Sources of Funding
This study was supported by Ministry of Health of the Czech Republic by grant AZV 16‐27496A. All rights reserved.
Disclosures
None.
Acknowledgments
We would like to thank Blazena Vodickova, Jitka Purrova and Petr Stavek for excellent help with this project.
(J Am Heart Assoc. 2017;6:e005290 DOI: 10.1161/JAHA.116.005290.)28784650
References
- 1. Bozkurt B, Aguilar D, Deswal A, Dunbar SB, Francis GS, Horwich T, Jessup M, Kosiborod M, Pritchett AM, Ramasubbu K, Rosendorff C, Yancy C. Contributory risk and management of comorbidities of hypertension, obesity, diabetes mellitus, hyperlipidemia, and metabolic syndrome in chronic heart failure: a scientific statement from the American Heart Association. Circulation. 2016;134:e535–e578. [DOI] [PubMed] [Google Scholar]
- 2. Ingelsson E, Sundstrom J, Arnlov J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA. 2005;294:334–341. [DOI] [PubMed] [Google Scholar]
- 3. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29–34. [DOI] [PubMed] [Google Scholar]
- 4. Witteles RM, Tang WH, Jamali AH, Chu JW, Reaven GM, Fowler MB. Insulin resistance in idiopathic dilated cardiomyopathy: a possible etiologic link. J Am Coll Cardiol. 2004;44:78–81. [DOI] [PubMed] [Google Scholar]
- 5. Paolisso G, De Riu S, Marrazzo G, Verza M, Varricchio M, D'Onofrio F. Insulin resistance and hyperinsulinemia in patients with chronic congestive heart failure. Metabolism. 1991;40:972–977. [DOI] [PubMed] [Google Scholar]
- 6. Doehner W, Rauchhaus M, Ponikowski P, Godsland IF, von Haehling S, Okonko DO, Leyva F, Proudler AJ, Coats AJ, Anker SD. Impaired insulin sensitivity as an independent risk factor for mortality in patients with stable chronic heart failure. J Am Coll Cardiol. 2005;46:1019–1026. [DOI] [PubMed] [Google Scholar]
- 7. Swan JW, Anker SD, Walton C, Godsland IF, Clark AL, Leyva F, Stevenson JC, Coats AJ. Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol. 1997;30:527–532. [DOI] [PubMed] [Google Scholar]
- 8. Margulies KB, Hernandez AF, Redfield MM, Givertz MM, Oliveira GH, Cole R, Mann DL, Whellan DJ, Kiernan MS, Felker GM, McNulty SE, Anstrom KJ, Shah MR, Braunwald E, Cappola TP. Effects of liraglutide on clinical stability among patients with advanced heart failure and reduced ejection fraction: a randomized clinical trial. JAMA. 2016;316:500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Margulies KB. Evolving challenges for targeting metabolic abnormalities in heart failure. JACC Heart Fail. 2016;4:567–569. [DOI] [PubMed] [Google Scholar]
- 10. Collins S. A heart‐adipose tissue connection in the regulation of energy metabolism. Nat Rev Endocrinol. 2014;10:157–163. [DOI] [PubMed] [Google Scholar]
- 11. Polak J, Kotrc M, Wedellova Z, Jabor A, Malek I, Kautzner J, Kazdova L, Melenovsky V. Lipolytic effects of B‐type natriuretic peptide1‐32 in adipose tissue of heart failure patients compared with healthy controls. J Am Coll Cardiol. 2011;58:1119–1125. [DOI] [PubMed] [Google Scholar]
- 12. Melenovsky V, Kotrc M, Borlaug BA, Marek T, Kovar J, Malek I, Kautzner J. Relationships between right ventricular function, body composition, and prognosis in advanced heart failure. J Am Coll Cardiol. 2013;62:1660–1670. [DOI] [PubMed] [Google Scholar]
- 13. Anker SD, Chua TP, Ponikowski P, Harrington D, Swan JW, Kox WJ, Poole‐Wilson PA, Coats AJ. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation. 1997;96:526–534. [DOI] [PubMed] [Google Scholar]
- 14. Doehner W, Pflaum CD, Rauchhaus M, Godsland IF, Egerer K, Cicoira M, Florea VG, Sharma R, Bolger AP, Coats AJ, Anker SD, Strasburger CJ. Leptin, insulin sensitivity and growth hormone binding protein in chronic heart failure with and without cardiac cachexia. Eur J Endocrinol. 2001;145:727–735. [DOI] [PubMed] [Google Scholar]
- 15. Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb‐Peploe KM, Harrington D, Kox WJ, Poole‐Wilson PA, Coats AJ. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–1053. [DOI] [PubMed] [Google Scholar]
- 16. Tura A, Pacini G, Kautzky‐Willer A, Gastaldelli A, DeFronzo RA, Ferrannini E, Mari A. Estimation of prehepatic insulin secretion: comparison between standardized c‐peptide and insulin kinetic models. Metabolism. 2012;61:434–443. [DOI] [PubMed] [Google Scholar]
- 17. Mari A, Schmitz O, Gastaldelli A, Oestergaard T, Nyholm B, Ferrannini E. Meal and oral glucose tests for assessment of beta ‐cell function: modeling analysis in normal subjects. Am J Physiol Endocrinol Metab. 2002;283:E1159–E1166. [DOI] [PubMed] [Google Scholar]
- 18. Cersosimo E, Solis‐Herrera C, Trautmann ME, Malloy J, Triplitt CL. Assessment of pancreatic beta‐cell function: review of methods and clinical applications. Curr Diabetes Rev. 2014;10:2–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. [DOI] [PubMed] [Google Scholar]
- 20. Wohlfahrt P, Melenovsky V, Kotrc M, Benes J, Jabor A, Franekova J, Lemaire S, Kautzner J, Jarolim P. Association of fibroblast growth factor‐23 levels and angiotensin‐converting enzyme inhibition in chronic systolic heart failure. JACC Heart Fail. 2015;3:829–839. [DOI] [PubMed] [Google Scholar]
- 21. AlZadjali MA, Godfrey V, Khan F, Choy A, Doney AS, Wong AK, Petrie JR, Struthers AD, Lang CC. Insulin resistance is highly prevalent and is associated with reduced exercise tolerance in nondiabetic patients with heart failure. J Am Coll Cardiol. 2009;53:747–753. [DOI] [PubMed] [Google Scholar]
- 22. Swan JW, Walton C, Godsland IF. Assessment of insulin sensitivity in man: a comparison of minimal model‐ and euglycaemic clamp‐derived measures in health and heart failure. Clin Sci (Lond). 1994;86:317–322. [DOI] [PubMed] [Google Scholar]
- 23. Melenovsky V, Kotrc M, Polak J, Pelikanova T, Bendlova B, Cahova M, Malek I, Jarolim P, Kazdova L, Kautzner J. Availability of energetic substrates and exercise performance in heart failure with or without diabetes. Eur J Heart Fail. 2012;14:754–763. [DOI] [PubMed] [Google Scholar]
- 24. Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev. 1998;19:608–624. [DOI] [PubMed] [Google Scholar]
- 25. Bonora E, Zavaroni I, Coscelli C, Butturini U. Decreased hepatic insulin extraction in subjects with mild glucose intolerance. Metabolism. 1983;32:438–446. [DOI] [PubMed] [Google Scholar]
- 26. Lommi J, Kupari M, Koskinen P, Naveri H, Leinonen H, Pulkki K, Harkonen M. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28:665–672. [DOI] [PubMed] [Google Scholar]
- 27. Sharma B, Majid PA, Pakrashi BC, Dykes JR, Taylor SH. Insulin secretion in heart failure. Br Med J. 1970;2:396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Melenovsky V, Benes J, Skaroupkova P, Sedmera D, Strnad H, Kolar M, Vlcek C, Petrak J, Benes J Jr, Papousek F, Oliyarnyk O, Kazdova L, Cervenka L. Metabolic characterization of volume overload heart failure due to aorto‐caval fistula in rats. Mol Cell Biochem. 2011;354:83–96. [DOI] [PubMed] [Google Scholar]
- 29. Hait G, Corpus M, Lamarre FR, Yuan SH, Kypson J, Cheng G. Alteration of glucose and insulin metabolism in congenital heart disease. Circulation. 1972;46:333–346. [DOI] [PubMed] [Google Scholar]
- 30. Potter LR. Natriuretic peptide metabolism, clearance and degradation. FEBS J. 2011;278:1808–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frayn KN. Metabolic Regulation: A Human Perspective. Oxford: Blackwell Science; 2003. [Google Scholar]
- 32. Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cahill GF Jr, Herrera MG, Morgan AP, Soeldner JS, Steinke J, Levy PL, Reichard GA Jr, Kipnis DM. Hormone‐fuel interrelationships during fasting. J Clin Invest. 1966;45:1751–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lommi J, Koskinen P, Naveri H, Harkonen M, Kupari M. Heart failure ketosis. J Intern Med. 1997;242:231–238. [DOI] [PubMed] [Google Scholar]
- 35. Wandell PE, Theobald H. The association between low fasting blood glucose value and mortality. Curr Diabetes Rev. 2007;3:274–279. [DOI] [PubMed] [Google Scholar]
- 36. Wei M, Gibbons LW, Mitchell TL, Kampert JB, Stern MP, Blair SN. Low fasting plasma glucose level as a predictor of cardiovascular disease and all‐cause mortality. Circulation. 2000;101:2047–2052. [DOI] [PubMed] [Google Scholar]
- 37. Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail‐Beigi F, Grimm RH Jr, Probstfield JL, Simons‐Morton DG, Friedewald WT. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Benzing G III, Schubert W, Sug G, Kaplan S. Simultaneous hypoglycemia and acute congestive heart failure. Circulation. 1969;40:209–216. [DOI] [PubMed] [Google Scholar]
- 39. Samsky MD, Patel CB, DeWald TA, Smith AD, Felker GM, Rogers JG, Hernandez AF. Cardiohepatic interactions in heart failure: an overview and clinical implications. J Am Coll Cardiol. 2013;61:2397–2405. [DOI] [PubMed] [Google Scholar]
- 40. Block MB, Gambetta M, Resnekov L, Rubenstein AH. Spontaneous hypoglycaemia in congestive heart‐failure. Lancet. 1972;2:736–738. [DOI] [PubMed] [Google Scholar]
- 41. Mellinkoff SM, Tumulty PA. Hepatic hypoglycemia; its occurrence in congestive heart failure. N Engl J Med. 1952;247:745–750. [DOI] [PubMed] [Google Scholar]
- 42. Norrelund H, Wiggers H, Halbirk M, Frystyk J, Flyvbjerg A, Botker HE, Schmitz O, Jorgensen JO, Christiansen JS, Moller N. Abnormalities of whole body protein turnover, muscle metabolism and levels of metabolic hormones in patients with chronic heart failure. J Intern Med. 2006;260:11–21. [DOI] [PubMed] [Google Scholar]
- 43. Unger RH. Glucagon and the insulin: glucagon ratio in diabetes and other catabolic illnesses. Diabetes. 1971;20:834–838. [DOI] [PubMed] [Google Scholar]
- 44. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, Rame JE. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rosenstock J, Ferrannini E. Euglycemic diabetic ketoacidosis: a predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care. 2015;38:1638–1642. [DOI] [PubMed] [Google Scholar]
- 46. Melenovsky V, Hwang SJ, Lin G, Redfield MM, Borlaug BA. Right heart dysfunction in heart failure with preserved ejection fraction. Eur Heart J. 2014;35:3452–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]