

Abstract
Background
Fetal growth impacts cardiovascular health throughout postnatal life in humans. Various animal models of intrauterine growth restriction exhibit reduced heart size at birth, which negatively influences cardiac function in adulthood. The mechanistic target of rapamycin complex 1 (mTORC1) integrates nutrient and growth factor availability with cell growth, thereby regulating organ size. This study aimed at elucidating a possible involvement of mTORC1 in intrauterine growth restriction and prenatal heart growth.
Methods and Results
We inhibited mTORC1 in fetal mice by rapamycin treatment of pregnant dams in late gestation. Prenatal rapamycin treatment reduces mTORC1 activity in various organs at birth, which is fully restored by postnatal day 3. Rapamycin‐treated neonates exhibit a 16% reduction in body weight compared with vehicle‐treated controls. Heart weight decreases by 35%, resulting in a significantly reduced heart weight/body weight ratio, smaller left ventricular dimensions, and reduced cardiac output in rapamycin‐ versus vehicle‐treated mice at birth. Although proliferation rates in neonatal rapamycin‐treated hearts are unaffected, cardiomyocyte size is reduced, and apoptosis increased compared with vehicle‐treated neonates. Rapamycin‐treated mice exhibit postnatal catch‐up growth, but body weight and left ventricular mass remain reduced in adulthood. Prenatal mTORC1 inhibition causes a reduction in cardiomyocyte number in adult hearts compared with controls, which is partially compensated for by an increased cardiomyocyte volume, resulting in normal cardiac function without maladaptive left ventricular remodeling.
Conclusions
Prenatal rapamycin treatment of pregnant dams represents a new mouse model of intrauterine growth restriction and identifies an important role of mTORC1 in perinatal cardiac growth.
Keywords: cardiac function, cardiac growth, cardiac mass, fetal programming, heart development, intrauterine growth restriction, mechanistic target of rapamycin
Subject Categories: Developmental biology, Myocardial Biology, Physiology
Clinical Perspective
What Is New?
Pharmacological inhibition of the mechanistic target of rapamycin complex 1 (mTORC1) pathway in late gestation by rapamycin treatment of pregnant mice causes intrauterine growth restriction, thereby reducing body and organ size in the offspring at birth.
Rapamycin‐sensitive mTORC1 functions are required for perinatal cardiac growth, primarily impacting cardiomyocyte size and survival but not proliferation in the neonatal heart.
Prenatal mTORC1 inhibition reduces cardiac output at birth due to diminished left ventricular dimensions, but contractility is not affected.
After prenatal mTORC1 inhibition body and heart weight partially normalize until early adulthood, and cardiac output fully recovers despite a reduced number of cardiomyocytes in the adult heart.
What Are the Clinical Implications?
Prenatal mTORC1 inhibition could play a role in intrauterine growth restriction caused by various maternal or environmental conditions, such as maternal mal‐ or undernutrition, placental insufficiency, or fetal hypoxia.
mTORC1 function is required for fetal cardiac growth, and its inhibition might be involved in developmental programming of heart disease in adulthood.
Introduction
The intrauterine environment is a major determinant of embryonic and fetal development. Various maternal or environmental conditions can impair fetal growth, resulting in intrauterine growth restriction (IUGR), which often presents with low birth weight and reduced organ size.1 Maternal under‐ or malnutrition, placental insufficiency, fetal hypoxia, or drugs (eg, glucocorticoids) are among the factors causing IUGR in humans and animal models.2 IUGR is considered a major risk factor for various chronic diseases later in life and thereby contributes to developmental (or fetal) programming.3 Hence, it appears imperative to uncover cellular and molecular mechanisms that regulate intrauterine growth. Relatively little is known about the underlying mechanisms, however, but signaling pathways regulating cell growth and proliferation are likely to be involved.
IUGR can be induced in animal models by caloric or protein restriction in the maternal diet during pregnancy,2 which suggests that a sufficient nutritional supply to the embryo and fetus is required for normal intrauterine growth. The mechanistic (or previously named “mammalian”) target of rapamycin (mTOR) pathway is an important sensor of the metabolic and nutritional state of a cell and thereby integrates energy homeostasis and amino acid availability with cell size and proliferation.4 mTOR is a serine/threonine kinase present in 2 multiprotein complexes, mTORC1 and mTORC2, which differ in the composition of regulatory and adaptor protein binding partners. Whereas mTORC2 is best characterized for its involvement in cell survival, growth, cytoskeletal organization, and cell polarity, mTORC1 is a master regulator of cell growth by activating protein, lipid, and nucleic acid biosynthesis while inhibiting catabolic mechanisms such as autophagy.4 One of the best characterized mTORC1 functions is the activation of cap‐dependent mRNA translation by phosphorylating its downstream targets eukaryotic translation initiation factor 4E binding protein 1 (4E‐BP1) and ribosomal S6 kinase (S6K1). 4E‐BP1 phosphorylation results in its dissociation from eukaryotic translation initiation factor 4E, thereby releasing its inhibitory effect and allowing translation initiation. In addition, phosphorylation and activation of S6K1 and its downstream target S6 ribosomal protein favor mRNA biogenesis as well as translation initiation and elongation. Upstream inputs that regulate mTORC1 activity include cellular energy status (ie, glucose and ATP levels), oxygen, growth factors, and amino acid availability.4 Importantly, some (but not all) downstream effects of mTORC1 (in contrast to mTORC2) can be efficiently inhibited by the immunosuppressive drug rapamycin.
In various model organisms the mTOR pathway has been shown to control organ size,5 which also includes the mammalian heart. Although its role during postnatal physiological as well as pathological cardiac hypertrophic growth has been studied extensively,6 much less is known about the involvement of mTOR signaling in prenatal cardiac development. The heart conditional knockout of the gene encoding mTOR in mice using Cre/loxP recombination mediated by different cardiac Cre drivers has been shown to be lethal during late gestation or at early postnatal stages.7, 8, 9 Efficient inhibition of mTORC1 signaling in the heart, however, appears to be either transiently restricted to a period shortly after midgestation9 or is only achieved after birth,7, 8 which hampers final conclusions about its role in the fetal heart. In addition, inactivating the mTOR gene affects both mTORC1 and mTORC2, such that precisely defining the role of one versus the other is not feasible. Similarly, the heart conditional knockout of the upstream mTORC1 activator Ras homolog enriched in brain (Rheb) causes mTORC1 inhibition only after birth, subsequently leading to postnatal lethality.10 Thus, the role of mTORC1 during embryonic and fetal cardiac growth is incompletely understood.
Restricting amino acid availability in the maternal diet or lowering ambient oxygen concentration during pregnancy as well as reducing blood supply to the placenta cause IUGR in animal models.1, 2 Given that mTORC1 is inhibited by amino acid starvation, hypoxia, and low cellular energy levels, thereby impairing cell growth and proliferation,4 we hypothesized that it might be involved in the IUGR phenotype. In addition, most IUGR conditions reduce heart size at birth and cause neonatal cardiac hypoplasia, which is considered a major cardiovascular risk factor in adulthood.3 In this regard we have recently described a mouse model of embryonic heart regeneration based on inactivation of the X‐linked gene encoding holocytochrome c synthase (Hccs) specifically in the developing heart.11 The HCCS enzyme is required for electron transport along the mitochondrial respiratory chain. Heterozygous heart conditional Hccs knock‐out (KO) females (hereafter referred to as cHccs +/−) develop a tissue mosaic in the ventricular myocardium composed of 50% cardiomyocytes harboring mitochondrial dysfunction and 50% healthy cells at midgestation. Compensatory proliferation of the healthy cardiomyocyte population allows embryonic heart regeneration, however, such that the neonatal cHccs +/− heart is composed of 90% healthy cells and contains only 10% diseased cells.11 Nevertheless, embryonic heart regeneration is not sufficient to completely build up the myocardium, resulting in cHccs +/− hearts being hypoplastic at birth due to a reduced number of cardiomyocytes.12 This reduction in cell number is postnatally compensated for by accelerated and augmented cardiomyocyte hypertrophic growth, such that heart size normalizes by early adulthood.12 Given the established role of mTORC1 in heart and organ size control,5, 6 we speculated that mTORC1 activity might be important for fetal cardiac growth and regulation of neonatal heart size in general as well as for compensatory growth of cHccs +/− hearts in particular.
Here we show that inhibiting mTORC1 in the final quarter of gestation by rapamycin treatment of pregnant mice causes IUGR and reduces heart size and cardiac output at birth. Body and heart size partially normalize during postnatal life, and despite a reduction in cardiomyocyte number, cardiac function is not compromised in adult mice after prenatal mTORC1 inhibition. cHccs +/− mice after prenatal rapamycin treatment exhibit cellular differences in the myocardium at birth compared with controls but no major alterations of postnatal heart size or function.
Methods
Mice
The generation and characterization of heart conditional Hccs KO mice have been described previously.11 Briefly, floxed (fl) Hccs mice were bred to mice expressing Cre recombinase under the control of the Nkx2.5 promoter. All mice were maintained on a mixed 129Sv/C57Bl6 genetic background, and all experiments throughout the study were performed on heterozygous Hccs KO females (Hccs fl/+ /Nkx2.5Cre, referred to as cHccs +/−) and their respective Cre positive female littermate controls (Hccs +/+ /Nkx2.5Cre, referred to as Hccs +/+). The latter were furthermore used to compare the effects of rapamycin versus vehicle treatment in control animals, implying that all mice used in the study were carrying the Nkx2.5Cre knock‐in.13 Unless specifically annotated, all results refer to control mice, whereas data of cHccs +/− mice are explicitly labeled in figures or figure legends. The total number of mice included in the different experiments of this study is as follows: vehicle‐treated neonates, Hccs +/+ n=31, cHccs +/− n=30; rapamycin‐treated neonates, Hccs +/+ n=39, cHccs +/− n=41; vehicle‐treated adults, Hccs +/+ n=9, cHccs +/− n=15; rapamycin‐treated adults, Hccs +/+ n=9, cHccs +/− n=11. All animal procedures were performed following institutional guidelines and had previously been approved by the responsible authorities (Landesamt für Gesundheit und Soziales Berlin, approval number G 0027/10).
Rapamycin Injection
Female mice were mated to the respective males for 1 night and separated the next morning (ie, at 0.5 days postconception [dpc]). Dams with visible pregnancy at 14.5 dpc were randomly assigned to vehicle or rapamycin treatment starting in the morning at 15.5 dpc. Rapamycin (Cayman Chemical Co, Ann Arbor, MI) was dissolved as stock solution (20 mg/mL) in dimethylacetamide (DMA, Sigma‐Aldrich, St. Louis, MO). Pregnant dams were treated with rapamycin or vehicle by subcutaneous injections every 12 hours from 15.5 dpc until delivery. Each injection contained 5 mg rapamycin/kg body weight diluted in 200 μL vehicle (10% PEG 300 [Sigma], 17% Tween 80 [Sigma] in 0.9% NaCl solution) or DMA‐containing vehicle only. Injections were administered alternating into the loose skin of the interscapular or the right and left femoral area.
Echocardiography
Adult mice (11 weeks old) were anesthetized by inhalation of a 2.5% isoflurane/oxygen mixture using the Vevo compact dual anesthesia system (VisualSonics, Toronto, ON, Canada). Body temperature was kept constant at 37°C using a heat lamp and a rectal temperature probe. Measurements on neonates were performed on awake pups. Echocardiography was recorded using the Vevo 2100 high‐frequency ultrasound system (VisualSonics, Toronto, ON, Canada) with a MicroScanTM transducer MS400 set to 30 MHz for adult mice or MS700 set to 50 MHz for neonatal mice. Operators were blinded for mouse genotypes and treatment groups during echocardiographic recordings as well as data analyses.
Organ Preparation and Histology
Hearts, kidneys, and lungs from neonatal mice were prepared on postnatal day 1, and hearts, livers, kidneys, and spleens from adult mice were prepared at the age of 11 weeks. Adult mice were euthanized by cervical dislocation, and neonates were decapitated. Organs were excised, rinsed in cold PBS, weighed, snap‐frozen in liquid nitrogen, and stored at −80°C. If organs were used for histological analyses, they were fixed in 4% paraformaldehyde/PBS (Sigma) for 24 to 48 hours. The tissue was subsequently dehydrated through an increasing ethanol series, cleared in toluol, and embedded in paraffin. Five‐micrometer paraffin sections were stained with hematoxylin and eosin (Carl Roth, Karlsruhe, Germany) to assess overall cardiac morphology and tissue composition or with Sirius Red (Direct Red 80, Sigma) to visualize myocardial fibrosis.
Quantification of Fibrosis in Adult Hearts
Light microscopy images of Sirius Red–stained heart sections from 11‐week‐old adult mice were taken using the Biozero BZ‐8100 microscope (Keyence, Osaka, Japan). Under ×5 optical magnification, 6 random fields of the left ventricular (LV) myocardium, including free wall and interventricular septum, were imaged, thereby covering the entire LV tissue. Two nonadjacent cross sections per heart were used. The percentage of interstitial fibrotic tissue was quantified using the color‐threshold plugin of ImageJ software (https://imagej.nih.gov/ij/), which measures the red‐stained area in relation to the total LV myocardial area. Values of all 12 images were averaged to give the mean fibrotic tissue content for each heart. Perivascular fibrosis was excluded and deleted from the images before analysis.
Evaluation of Cell Proliferation
To assess proliferation rates in neonatal hearts, immunofluorescence staining for Ki67 was performed. Paraffin sections were deparaffinized and rehydrated, and heat‐mediated antigen retrieval was performed in sodium citrate buffer (10 mmol/L, pH 6.0) for 20 minutes. After blocking in antibody solution containing 5% normal goat serum (Jackson ImmunoResearch, West Grove, PA) for 1 hour, sections were incubated overnight with a rabbit monoclonal anti‐Ki67 antibody (RM‐9106, Thermo Scientific, Waltham, MA) at 4°C. Secondary antibody detection was performed at room temperature for 1 hour using a goat anti‐rabbit Alexa Fluor 555‐conjugated secondary antibody (Invitrogen, Carlsbad, CA). Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) (Invitrogen). Longitudinal sections were imaged with ×40 optical magnification using the Biozero BZ‐8100 fluorescence microscope (Keyence, Osaka, Japan), and 10 random fields per section were taken within the LV myocardium, including the free wall and interventricular septum. Cells that exhibited colocalization of Ki67 and DAPI were considered to be cycling. Ki67‐positive nuclei and the total number of DAPI‐stained nuclei were manually quantified using the cell counter plugin of the ImageJ software. A total number of ≈5000 nuclei per heart were evaluated, and data from all 10 images were averaged to give the mean proliferation rate for each heart.
Evaluation of Cell Death
Apoptotic cells were detected in cardiac paraffin sections using a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (ApopTag® Fluorescein Apoptosis Detection Kit, Merck Millipore, Billerica, MA) according to the manufacturer's instructions. Nuclei were stained with DAPI (Invitrogen). Two longitudinal nonadjacent TUNEL‐stained sections per heart and 6 random fields per section were imaged using the Biozero BZ‐8100 fluorescence microscope (Keyence) with ×15 optical magnification. Cells that exhibited colocalization of TUNEL and DAPI staining were considered apoptotic. TUNEL staining as well as the total number of DAPI‐stained nuclei were manually counted using ImageJ. A total number of ≈15 000 nuclei per neonatal heart were analyzed, and data from all 12 images were averaged to give the mean apoptosis rate for each heart.
Cardiomyocyte Cross‐Sectional Area, Length, and Volume
To evaluate cardiomyocyte cross‐sectional area (CSA), cardiac paraffin sections were stained with fluorochrome‐conjugated wheat germ agglutinin (WGA, Alexa Fluor 555; Invitrogen) to visualize cell membranes. Nuclei were stained with DAPI or TO‐PRO‐3 (Invitrogen). For neonates, images were taken with the confocal laser scanning microscope TSC SPE (Leica Microsystems, Wetzlar, Germany) applying ×120 optical magnification, whereas adult hearts were imaged with the Biozero BZ‐8100 fluorescence microscope (Keyence) applying ×20 optical magnification. Fifteen random fields of the LV myocardium including free wall and interventricular septum were imaged per section, and 2 nonadjacent WGA‐stained sections per heart were used. At least 200 cardiomyocytes per heart were measured using the area measurement tool of the Biozero BZ image analysis application software (Keyence). Only cardiomyocytes that were cut along their transverse axis and matched the following selection criteria were included: visible central nucleus, cell shape close to circular, clear cell borders discernible, and visible cytoplasm. Values of all cardiomyocytes were averaged to give a mean estimate of cardiomyocyte CSA for each heart.
Cardiomyocyte length was measured in adult hearts following immunofluorescence staining of paraffin sections with WGA and an antibody against N‐cadherin (sc‐7939, Santa Cruz Biotechnology, Dallas, TX) to identify intercalated disks (general staining procedure same as for evaluation of proliferation described above). Two nonadjacent sections per heart were imaged at ×40 magnification using a Zeiss Axio Scope.A1 fluorescence microscope. Ten to 15 images per section were taken from areas of the LV myocardium (free wall and interventricular septum) that showed cardiomyocytes in their longitudinal orientation. Rod‐shaped cardiomyocytes with clearly visible cell borders, nuclei and intercalated disks were included. The distance between intercalated disks of ≈70 to 100 cardiomyocytes per heart was measured using the ZEN blue software (Zeiss), and the average cardiomyocyte length was calculated.
Calculation of cardiomyocyte volume was based on the simplified assumption that cardiomyocytes are of cylindrical shape (as previously described14), such that mean CSA was multiplied by mean cardiomyocyte length for each adult heart.
Calculation of Cardiomyocyte Number Per Heart
Cardiomyocyte number per adult heart was estimated based on previously published protocols15 with minor modifications. Heart volume was calculated by multiplying cardiac wet weight or LV mass (determined by echocardiography) with the tissue density of rat myocardium, ie, 1.048 g/mm3.15 The relative contribution of cardiomyocytes to the cardiac tissue was estimated by determining cardiomyocyte area fraction on WGA‐stained paraffin sections (staining as described above for CSA). Briefly, LV myocardium of 2 nonadjacent sections was imaged at ×40 magnification, resulting in 20 to 30 images per heart. For each image the WGA‐positive area (including nonmyocytes, extracellular matrix, and cell membranes) was measured using the color threshold plug‐in in ImageJ and related to the overall tissue area. The remaining WGA‐negative tissue area was considered to represent cardiomyocytes. Cardiomyocyte area fraction was averaged from all images per heart. Cardiomyocyte (CM) number was calculated according to the following equation:
Western Blot Analyses
For isolation of total protein extracts fresh or snap‐frozen tissue samples (heart, lung, kidney) were homogenized in RIPA buffer supplemented with protease (Complete Protease Inhibitor Cocktail Tablets, Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitors (PhosSTOP Phosphatase Inhibitor Cocktail Tablets, Roche Diagnostics) and incubated at 4°C for 2 hours with gentle agitation. For samples on the same gel, equal protein amounts were loaded (20 to 50 μg), separated using denaturing SDS‐PAGE and blotted onto nitrocellulose (GE Healthcare, Chicago, IL) or polyvinylidene fluoride (Merck Millipore) membranes. Membranes were blocked for 1 hour in 5% nonfat dry milk (Carl Roth) in Tris‐buffered saline with Tween 20 and incubated with the following primary antibodies at 4°C overnight: phospho‐S6K1 Thr389 (#9234), total S6K1 (#2708), phospho‐S6 Ser235/236 (#4858), phospho‐S6 Ser240/244 (#4838), total S6 (#2217), phospho‐4E‐BP1 Thr37/46 (#2855), phospho‐4E‐BP1 Ser65 (#9451), total 4E‐BP1 (#9644), phospho‐mTOR Ser2448 (#2971), phospho‐mTOR Ser2481 (#2974), total mTOR (#2983), RICTOR (#2114), RAPTOR (#2280), phospho‐AMPKα Thr172 (#4188), total AMPKα (#2532), phospho‐ULK1 Ser555 (#5869), phospho‐ULK1 Ser757 (#6888), total ULK1 (#8054), LC3B (#3868), p62 (#5114), phospho‐Akt Thr308 (#13038), phospho‐Akt Ser473 (#4060), total Akt (#4691) (all from Cell Signaling Technology, Danvers, MA). Antibodies against GAPDH (MA1‐22670, Thermo Scientific, Waltham, MA), α‐tubulin (T9026, Sigma‐Aldrich, St. Louis, MO), and vinculin (V9131, Sigma‐Aldrich) were used for loading control. Secondary detection was performed using horseradish peroxidase–linked secondary antibodies (Cell Signaling Technology). Enhanced chemiluminescence reaction was performed and detected with the imaging system Odyssey Fc (LI‐COR Biosciences, Lincoln, NE) or by exposure of the membrane to chemiluminescence‐sensitive CL‐XPosure films (Thermo Scientific). Intensity of detected protein bands was quantified by densitometry using ImageJ.
Quantitative Real‐Time Polymerase Chain Reaction
Fresh or snap frozen cardiac tissue samples were homogenized in TRIzol reagent (Invitrogen), and total RNA was isolated according to the manufacturer's instructions. RNA was subsequently purified using RNeasy spin columns (Qiagen, Hilden, Germany), including digestion of genomic DNA on the column (RNase‐free DNase set, Qiagen). Isolated cardiac RNA was reversely transcribed into cDNA using M‐MuLV reverse transcriptase (New England BioLabs, Ipswich, MA) and random hexamer primers. Quantitative real‐time polymerase chain reaction (PCR) was performed using the Power SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA) on the ViiA™ 7 real‐time PCR system (Applied Biosystems). Primers were obtained from BioTeZ (Berlin, Germany), and sequences are as follows: Nppa forward 5′‐CAGCATGGGCTCCTTCTCCAT‐3′, Nppa reverse 5′‐ TGTACACAGGATTTGCTCCAATATG‐3′, Myh7 forward 5′‐CTAGAGTCAAAGTGGGCAACG‐3′, Myh7 reverse 5′‐GTGTCACCATCCAGTTGAACA‐3′, Nppb forward 5′‐AGGACCAAGGCCTCACAAAA‐3′, Nppb reverse 5′‐TTGAGATATGTGTCACCTTGGAATTT‐3′. Target gene expression was normalized against Gapdh: Gapdh forward 5′‐AGGTTGTCTCCTGCGACTTCA‐3′, Gapdh reverse 5′‐CCAGGAAATGAGCTTGACAAAGTT‐3′. All primers and PCR conditions were optimized to PCR efficiencies between 90% and 110% and a correlation coefficient ≥0.990 using cDNA dilution series. All samples were analyzed in triplicate. Relative expression differences between groups were determined using the ΔΔCT method.
Statistical Analyses
All data are presented as mean±standard error of the mean. Data sets were tested for normal distribution by the Kolmogorov‐Smirnov test, and homogeneity of variances between groups was assessed by the Levene test using SPSS (IBM, Armonk, NY). If these criteria were met, differences between 2 groups were evaluated with an unpaired, 2‐sided Student t test using Excel 2010 (Microsoft, Redmond, WA) and those among multiple groups with 1‐way ANOVA followed by Bonferroni post hoc test using SPSS (IBM). Differences among multiple groups with unequal variances were evaluated with nonparametric Kruskal‐Wallis 1‐way analysis of variance followed by Mann‐Whitney post hoc test using SPSS (IBM). Genotype distributions were compared to expected Mendelian distribution by a chi‐squared test using Microsoft Excel. Kaplan‐Meier survival curves were plotted using SPSS, and differences between groups were determined by a Log Rank (Mantel‐Cox) test. A probability (P) value less than 0.05 was considered to indicate statistical significance.
Results
Efficient Inhibition of mTORC1 in Neonatal Mice by Rapamycin Treatment of Pregnant Dams
In order to inhibit mTORC1 in fetal mice, we treated pregnant dams during the final quarter of gestation (starting at 15.5 dpc until delivery) by subcutaneous injections of rapamycin (5 mg/kg body weight) or vehicle every 12 hours (Figure S1). mTORC1 activity was tested by Western blot analyses on different tissues harvested from newborn mice on postnatal day 1 (P1). Prenatal rapamycin treatment efficiently reduced phosphorylation of the mTORC1 downstream targets S6K1 and S6 ribosomal protein in the offspring heart, lung, and kidney (Figure 1A). 4E‐BP1 was unaffected, consistent with previous reports showing different responsiveness of mTORC1 targets to rapamycin.16, 17
Figure 1.
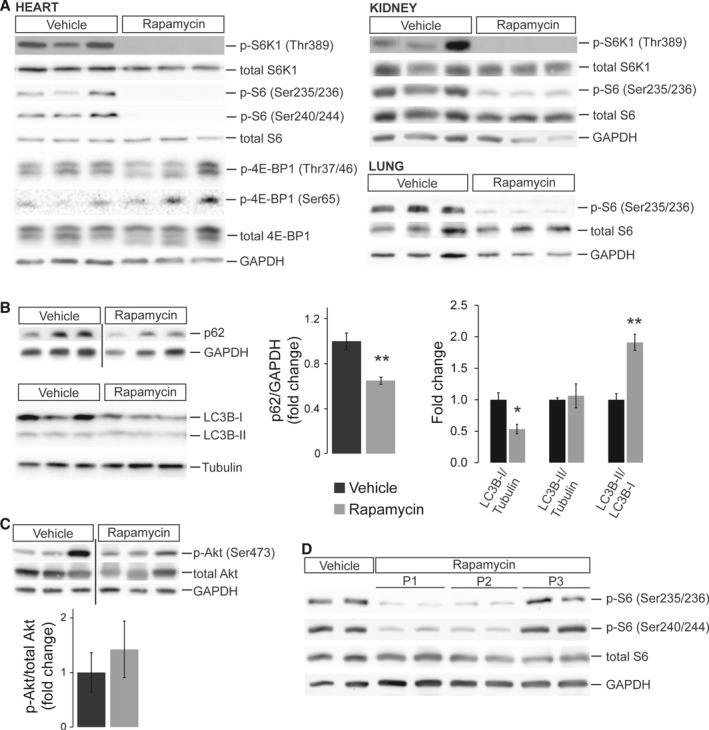
Efficient inhibition of mTORC1 in neonatal mice by rapamycin treatment of pregnant dams. A, Western blots of protein extracts from neonatal (P1) heart, kidney, and lung tissue after prenatal vehicle or rapamycin treatment. Reduced phosphorylation of the mTORC1 downstream targets S6K1 and S6 ribosomal protein confirmed successful mTORC1 inhibition by rapamycin in all 3 organs. In contrast, phosphorylation of 4E‐BP1 was unaffected in the neonatal heart. B, Western blots revealed reduced p62 (SQSTM1) and LC3B‐I protein levels, resulting in an increased LC3B‐II/LC3B‐I ratio in hearts of rapamycin‐ compared to vehicle‐treated neonates (densitometric quantification n=6 per group for p62, n=4 for LC3B, *P<0.05, **P<0.01). C, Western blots of Akt Ser473 phosphorylation normalized to total Akt revealed no significant differences between hearts of rapamycin‐ and vehicle‐treated neonates (densitometric quantification n=5 per group). D, Western blots of heart protein extracts from vehicle‐treated neonates at P1 and rapamycin‐treated littermates at P1, P2, and P3 illustrating phosphorylation status of S6 ribosomal protein. mTORC1 activity is still inhibited at P2 but restored by P3. B and C, Samples were detected on the same membrane but were noncontiguous, indicated by a black line.
mTORC1 regulates autophagy in concert with AMP‐activated protein kinase (AMPK) by phosphorylating ULK1, a kinase important for the initiation of autophagy.18 Phosphorylation of AMPKα was increased in rapamycin‐ compared with vehicle‐treated neonatal hearts (Figure S2A), as previously described in the mouse liver.19 Importantly, total ULK1 protein amounts are elevated in the P1 heart by prenatal rapamycin treatment (Figure S2B). In agreement with AMPK activation, ULK1 phosphorylation at the AMPK‐dependent residue Ser555 (which promotes autophagy) was increased in rapamycin‐ compared with vehicle‐treated hearts, even after normalization to total ULK1 (Figure S2B). In contrast, phosphorylation of the mTOR‐sensitive Ser757, which inhibits autophagy, was unaltered when normalized to total ULK1 (Figure S2B). In conclusion, besides opposing phosphorylation by AMPK and mTOR, regulation of ULK1 activity in rapamycin‐treated neonatal hearts seems furthermore to involve altered ULK1 protein stability and/or synthesis (as previously proposed).20, 21
To investigate the net effect of these interactions, we tested protein levels of p62 (SQSTM1), which is rapidly degraded if autophagy is activated,22 and found reduced p62 in hearts of rapamycin‐ compared with vehicle‐treated neonates (Figure 1B). In addition, we detected reduced LC3B‐I but unaltered LC3B‐II protein amounts in rapamycin‐ compared to vehicle‐treated hearts (Figure 1B), a pattern also observed in heart conditional Rheb KO mice.10 Consequently, the LC3B‐II/LC3B‐I ratio is increased in rapamycin‐treated hearts, a parameter widely used to indicate activation of autophagy. The fact that LC3B‐II levels are not elevated, as often reported, could indicate accelerated autophagic flux such that, after conversion of LC3B‐I to LC3B‐II, the latter is rapidly degraded via lysosomal turnover.22 Based on these data we conclude that autophagy is activated in neonatal hearts after prenatal rapamycin treatment.
It has been proposed that rapamycin can inhibit mTORC2 if applied in high doses or for prolonged treatment periods.23, 24 We did not detect differences in Akt phosphorylation at Ser473 (Figure 1C), a well‐established mTORC2 phosphorylation site, thereby excluding major inhibitory effects of rapamycin on mTORC2 in the neonatal heart. Finally, given that newborn pups did not receive further treatment after birth, we wanted to know at what postnatal stage mTORC1 activity is fully restored. Western blot analyses revealed that S6 phosphorylation in the heart is still reduced on postnatal day 2 (P2) but returns to normal levels at P3 (Figure 1D). In summary, rapamycin treatment of pregnant mice during late gestation results in efficient inhibition of rapamycin‐sensitive mTORC1 functions in the offspring at birth, which in the heart is restored by P3.
Prenatal Rapamycin Treatment Causes IUGR and Reduces Heart Size at Birth
Prenatal rapamycin treatment in late gestation (starting at 15.5 dpc) does not cause fetal lethality, evident as unaltered litter size in rapamycin‐ compared to vehicle‐treated dams on postnatal day 1 (Figure 2A). In contrast, initiation of rapamycin treatment at 11.5 dpc resulted in spontaneous abortions around 16.5 dpc and fetal lethality with severe growth restriction and malformations. Consequently, all further analyses were performed with treatment commencing at 15.5 dpc (as depicted in Figure S1). Pups born from rapamycin‐treated females were smaller in size at birth compared to vehicle‐treated pups, resulting in a 16.4% reduction in body weight (BW) (Figure 2B). Similarly, a reduction in heart size was already evident upon dissection and was furthermore confirmed by histological examination (Figure 2C). The latter does not reveal any major structural or morphological cardiac defects after prenatal mTORC1 inhibition, however. Strikingly, heart weight (HW) was reduced by 34.5%, resulting in a significantly reduced HW/BW ratio in rapamycin‐ versus vehicle‐treated pups (Figure 2D). In contrast, kidney weight (KW) was reduced in accordance with BW by 19.7%, resulting in unaltered KW/BW ratios between groups (Figure 2E). In conclusion, rapamycin treatment during late gestation causes IUGR evident as reduced body and organ size. Heart size is disproportionately affected, suggesting that fetal cardiac growth is specifically sensitive to mTORC1 inhibition.
Figure 2.
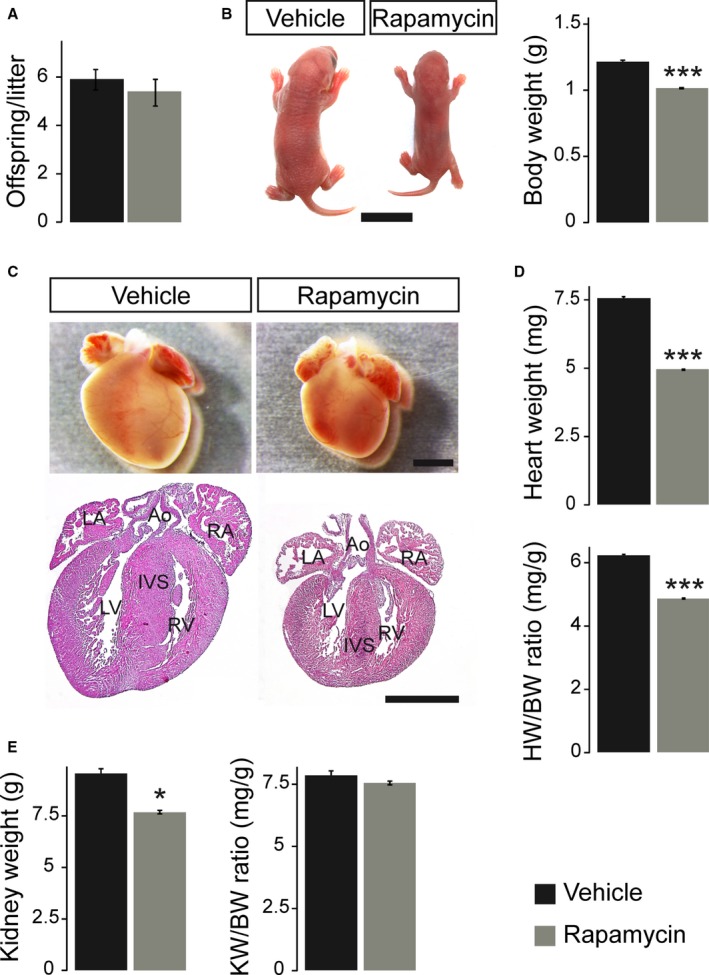
Prenatal rapamycin treatment causes IUGR and reduces heart size at birth. A, Average litter size of vehicle‐ and rapamycin‐treated dams did not differ on postnatal day 1 (n=9 litters per treatment group). B, Newborn offspring after prenatal rapamycin treatment were smaller compared to vehicle‐treated controls (scale bar=1 cm), resulting in significantly reduced body weight (BW). C, Neonatal mice after prenatal rapamycin treatment had smaller hearts compared to their vehicle controls but did not exhibit major structural or morphological cardiac defects (hematoxylin and eosin staining in lower panel, scale bars=1 mm; Ao indicates aorta; IVS, interventricular septum; IUGR, intrauterine growth restriction; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle). D, Rapamycin‐treated neonates demonstrated significantly lower heart weight (HW) and HW/BW ratio compared to vehicle‐treated animals. E, Even though kidney weight (KW) of rapamycin‐treated neonates was significantly reduced compared to vehicle‐treated animals, the KW/BW ratio was not altered. B, D, and E, Vehicle n=9, rapamycin n=10. (***P<0.001, *P<0.05).
Prenatal Rapamycin Treatment Reduces Cardiomyocyte Size and Induces Apoptosis but Does Not Affect Proliferation in the Neonatal Heart
The rapamycin‐induced reduction in neonatal heart size can be the result of different cellular mechanisms: a reduction in cardiomyocyte size or number, and the latter can be caused by induction of cell death or by inhibition of proliferation. We determined cell cycle activity in neonatal (P1) hearts of rapamycin‐ compared to vehicle‐treated pups by Ki67 immunostaining but did not find differences between the groups (Figure 3A). In contrast, when we assayed programmed cell death (apoptosis) by TUNEL staining, a significant increase in the number of TUNEL‐positive cells was observed in rapamycin‐treated P1 hearts (Figure 3B). Most strikingly, cardiomyocyte CSA was significantly reduced in the latter compared to vehicle‐treated pups, indicating a reduction in cell size (Figure 3C). Taken together, these data suggest that prenatal rapamycin treatment reduces cardiac organ size primarily by reducing cardiomyocyte size in combination with induction of apoptosis but not by interfering with proliferation.
Figure 3.
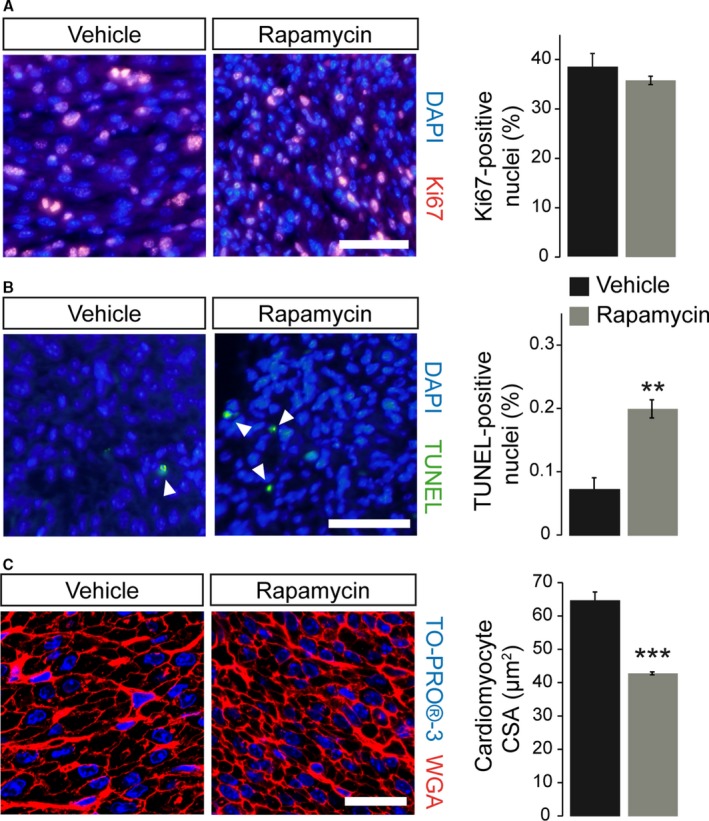
Prenatal rapamycin treatment reduces cardiomyocyte size and induces apoptosis but does not affect proliferation in the postnatal day‐1 heart. A, Quantification of immunofluorescence images of Ki67‐labeled nuclei (red) revealed unchanged proliferation rates within the left ventricular (LV) myocardium of vehicle‐ and rapamycin‐treated neonatal hearts. Nuclei were stained in blue with DAPI (scale bar=50 μm, vehicle n=6, rapamycin n=12). B, Quantification of TUNEL‐positive nuclei (green, see arrowheads) revealed significantly increased apoptosis within the LV myocardium of neonatal hearts after prenatal mTORC1 inhibition compared to vehicle‐treated controls. Nuclei were stained in blue with DAPI (scale bar=50 μm, vehicle n=4, rapamycin n=6). C, Fluorescence staining of cardiomyocyte membranes with wheat germ agglutinin (WGA, red) within the LV myocardium revealed a significantly reduced cardiomyocyte cross‐sectional area (CSA) in rapamycin‐ compared to vehicle‐treated neonates. Nuclei were stained in blue with TO‐PRO‐3 (confocal microscopy, scale bar=25 μm, vehicle n=5, rapamycin n=3). (***P<0.001, **P<0.01). TUNEL indicates terminal deoxynucleotidyl transferase dUTP nick end labeling.
Impact of Fetal mTORC1 Inhibition on a Mouse Model of Prenatal Compensatory Cardiac Growth
Given the importance of mTOR for cardiac growth and organ size control,5, 6 it might be specifically required for embryonic heart regeneration in cHccs +/− mice.11 Interestingly, neonatal cHccs +/− hearts show increased phosphorylation of S6K1 and S6 ribosomal protein (Figure 4A) but not of mTORC1 (ie, 4E‐BP1, ULK1) or mTORC2 (ie, Akt) (Figure S3) downstream targets compared to controls. This suggests that perinatal compensatory cardiac growth, maturation, or cell survival in cHccs +/− mice might depend on mTORC1 signaling to S6K1, which has been shown to fulfill various functions in the heart.6 To address this question, we subjected cHccs +/− fetuses to the same regime of prenatal rapamycin treatment as described above (ie, starting at 15.5 dpc). Similarly to control hearts, rapamycin efficiently inhibited mTORC1 activity in cHccs +/− hearts on postnatal day P1 (Figure S4A). In litters from rapamycin‐treated dams, genotype distribution at P1 was as expected for X‐chromosomal inheritance of Hccs and not different from vehicle‐treated litters (Figure S4B), excluding prenatal lethality of cHccs +/− females upon mTORC1 inhibition. Importantly, the rapamycin‐induced reduction in BW, HW, and HW/BW ratio was similar in cHccs +/− females compared to rapamycin‐treated controls (Hccs +/+) (Figure 4B). The reduction in heart weight in neonatal cHccs +/− compared to Hccs +/+ females due to the reduced number of cardiomyocytes described previously12 was evident in both treatment groups but was not aggravated by rapamycin. At the cellular level, rapamycin does not specifically impair proliferation in cHccs +/− hearts at P1 (Figure 4C) but further induces apoptosis (Figure 4D) and reduces cardiomyocyte CSA (Figure 4E) when compared to rapamycin‐treated Hccs +/+ females. In summary, whereas rapamycin does not seem to affect late‐gestational regulation of overall heart size specifically in cHccs +/− females, it does have more pronounced effects on cardiomyocyte size and cell survival. The latter implies that rapamycin‐sensitive mTORC1 functions control certain cellular aspects of perinatal compensatory growth and tissue homeostasis in cHccs +/− hearts, which could impact on their morphology and function in adulthood (see Discussion in Data S1).
Figure 4.
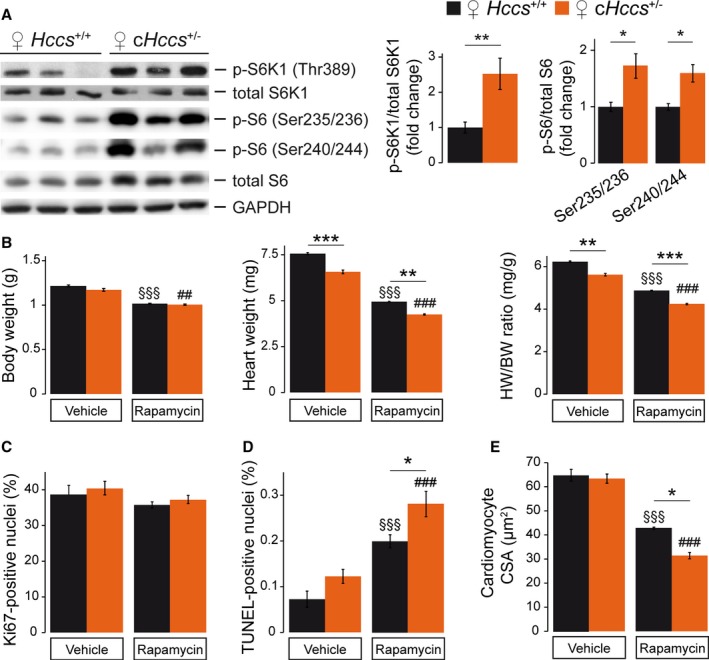
Impact of fetal mTORC1 inhibition on a mouse model of prenatal compensatory cardiac growth. A, Western blots illustrating the phosphorylation status of S6K1 and S6 ribosomal protein revealed enhanced mTORC1 activity in neonatal (P1) cHccs +/− hearts compared to littermate controls (densitometric quantification for S6K1: Hccs +/+ n=15, cHccs +/− n=14, for S6: n=6 per group). B, Body weight (BW) of rapamycin‐treated neonates was significantly reduced compared to vehicle‐treated animals, but no difference was observed between genotypes. Rapamycin‐treated Hccs +/+ and cHccs +/− neonates demonstrated similar reductions in heart weight (HW) and HW/BW ratio compared to vehicle‐treated animals. Note that the reduced HW and HW/BW ratio in cHccs +/− compared to Hccs +/+ newborns reported previously12 persisted after prenatal mTORC1 inhibition (vehicle groups n=9, rapamycin groups n=10). C, Quantification of Ki67‐positive nuclei revealed unchanged proliferation rates within the LV myocardium of vehicle‐ and rapamycin‐treated hearts and between genotypes (vehicle groups n=6, rapamycin Hccs +/+ n=12, rapamycin cHccs +/− n=13). D, Quantification of TUNEL‐positive nuclei revealed significantly increased apoptosis in hearts after prenatal mTORC1 inhibition, with apoptosis in rapamycin‐treated cHccs +/− hearts being significantly higher than in rapamycin‐treated Hccs +/+ controls (vehicle Hccs +/+ n=4, all other groups n=6). E, Prenatal rapamycin treatment significantly reduced cardiomyocyte cross‐sectional area (CSA) in both genotypes, and CSA in rapamycin‐treated cHccs +/− hearts was significantly smaller compared to rapamycin‐treated Hccs +/+ controls (vehicle groups n=5, rapamycin groups n=3). B through E, Note that data used for Hccs +/+ animals are the same as depicted in Figures 2 and 3, respectively. (*P<0.05, **P<0.01, ***P<0.001; §§§ P<0.001 vs vehicle Hccs +/+; ## P<0.01, ### P<0.001 vs vehicle cHccs +/−). mTORC1 indicates mechanistic target of rapamycin complex 1; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Normal Contractility but Reduced Stroke Volume and Cardiac Output in Neonatal Hearts After Rapamycin Treatment
Considering the significant effects of prenatal mTORC1 inhibition on neonatal heart size, we performed echocardiography on rapamycin‐ and vehicle‐treated pups at P1 in order to evaluate cardiac function. In agreement with the heart weight and morphology data described above, rapamycin‐treated neonates had a significantly reduced systolic LV wall thickness (Figure 5A), a reduced LV internal diameter (Figure 5B), and a reduced calculated LV mass (Figure 5C). LV contractility, however, was not affected by prenatal rapamycin treatment (Figure 5D). The smaller LV dimensions result in a significantly reduced LV stroke volume and cardiac output in rapamycin‐ versus vehicle‐treated pups, given that heart rate was not different between groups (Figure 5E). In addition, prenatal mTORC1 inhibition does not have more severe effects on cardiac dimensions or function in cHccs +/− compared to Hccs +/+ females (Table S1). In summary, prenatal rapamycin treatment reduces LV size and cardiac output at birth.
Figure 5.
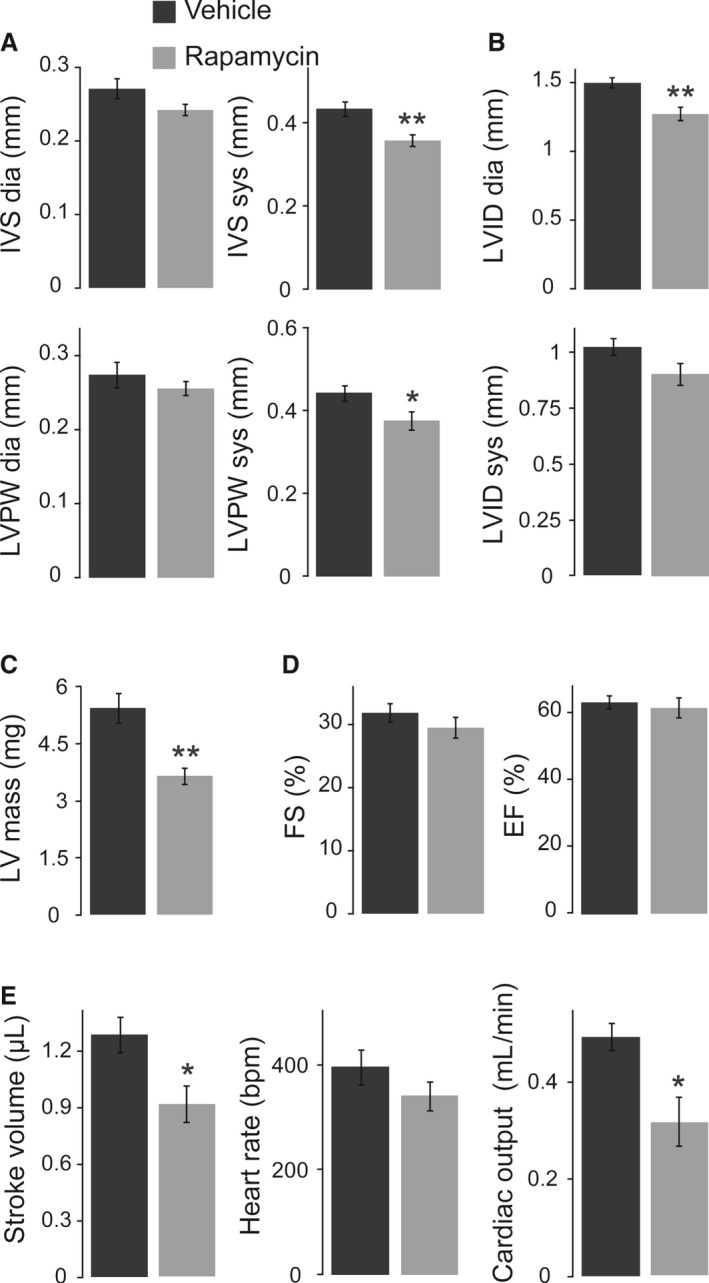
Echocardiography revealed normal contractility but reduced cardiac output in neonatal hearts after rapamycin treatment. A, Interventricular septum (IVS) and left ventricular posterior wall (LVPW) thickness in end‐diastole (dia) and end‐systole (sys) were reduced after prenatal rapamycin treatment, even though only systolic wall thickness reached statistical significance. B, End‐diastolic and end‐systolic left ventricular internal diameter (LVID) was reduced in neonatal mice after prenatal mTORC1 inhibition, whereat only diastolic LVID was significantly different. C, Neonatal LV mass calculated from echocardiography data was significantly reduced after prenatal rapamycin treatment. D, LV contractility at birth, measured as fractional shortening (FS) and ejection fraction (EF), was not affected by prenatal rapamycin treatment. E, The smaller LV dimensions result in significantly reduced LV stroke volume and consequently cardiac output in rapamycin‐ vs vehicle‐treated pups, given that heart rate was not different between groups. A through E, Vehicle n=7, rapamycin n=6. *P<0.05, **P<0.01.
Body and Organ Weights Partially Normalize After Prenatal Rapamycin Treatment Until Adulthood
To monitor the postnatal fate of pups born after prenatal mTORC1 inhibition, we analyzed rapamycin‐ and vehicle‐treated animals at the age of 11 weeks. As shown above, mTORC1 activity is restored in the rapamycin‐treated heart 3 days after birth (Figure 1D), suggesting that body and organ growth should be uncompromised thereafter. Nevertheless, we noticed postnatal lethality in a subset of rapamycin‐treated pups within the first 12 days after birth, resulting in 67.3% survival compared with 95.8% in vehicle‐treated mice (Figure 6A). After day 12 no further deaths were recorded, and all animals survived to early adulthood. Comparison of genotype distribution at weaning (ie, at 21 days of age) revealed no evidence for preferential lethality of rapamycin‐treated cHccs +/− mice after birth (Figure S5). At the age of 11 weeks, rapamycin‐treated animals still had a significantly reduced BW and tibia length (TL) compared with vehicle‐treated mice (Figure 6B). BW was reduced by 10% compared with 16% at birth, indicating a certain degree of postnatal catch‐up growth, however. Importantly, HW completely normalizes in rapamycin‐ compared with vehicle‐treated mice, resulting in normal HW/BW and HW/TL ratios after 11 weeks (Figure 6C). Histological examination confirmed mainly normal cardiac morphology, although hearts appeared slightly smaller, and LV walls slightly thinner, compared with the vehicle group (Figure 6D). Liver and spleen weights were not different in adult rapamycin‐ versus vehicle‐treated mice, whereas KW was significantly reduced (Figure 6E). Nevertheless, organ weights normalized to BW or TL were not different between treatment groups (Figure 6F and Table S2). Moreover, no specific effect of rapamycin treatment on body or organ size was observed in cHccs +/− compared to Hccs +/+ females (Table S2). Taken together, after prenatal rapamycin treatment, pups that survived the early postnatal period showed incomplete catch‐up growth of body and kidney size, whereas heart, liver, and spleen weights all normalized until adulthood.
Figure 6.
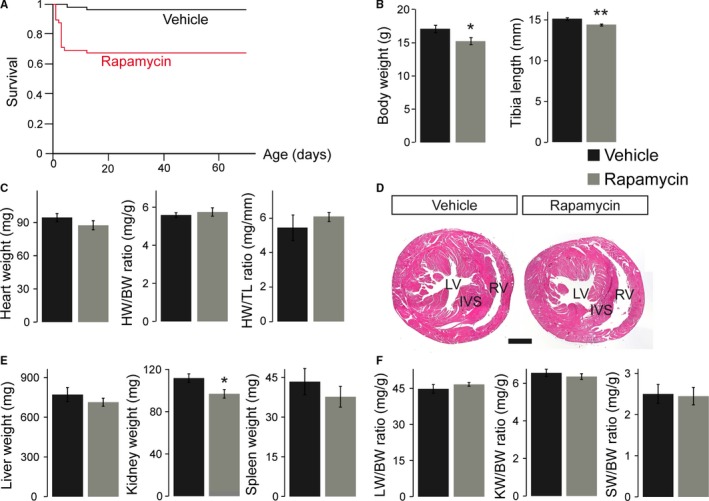
Body and organ weights partially normalize after prenatal rapamycin treatment until adulthood. A, Survival curves show death of some rapamycin‐treated pups within the first 12 days after birth but not thereafter. Consequently, postnatal survival after 70 days is significantly reduced in rapamycin‐ (n=55) vs vehicle‐treated (n=48) animals (P<0.0005). Survival curves include all possible genotypes and both sexes. B, Body weight (BW) and tibia length (TL) in 11‐week‐old adult mice after prenatal rapamycin treatment were reduced compared with vehicle‐treated animals, indicating incomplete postnatal catch‐up growth. C, Heart weight (HW) in adult rapamycin‐treated animals completely normalizes during postnatal life, resulting in normal HW/BW and HW/TL ratios. D, Slightly reduced left ventricular (LV) wall thickness, but no major morphological changes within the LV, interventricular septum (IVS) or right ventricle (RV), were observed after prenatal mTORC1 inhibition compared to vehicle‐treated animals (hematoxylin and eosin staining, scale bar=1 mm). E, Kidney weight (KW), but not liver (LW) or spleen weight (SW), was significantly reduced in rapamycin‐ vs vehicle‐treated 11‐week‐old mice. F, LW/BW, KW/BW and SW/BW ratios in adult mice displayed no significant differences between the treatment groups. B, C, E, and F, Vehicle n=7, rapamycin n=9. (*P<0.05, **P<0.01). mTORC1 indicates mechanistic target of rapamycin complex 1.
Reduced LV Mass but Normal Cardiac Contractility and Output in Adult Mice After Prenatal Rapamycin Treatment
Long‐term consequences of prenatal rapamycin treatment on postnatal heart morphology and function were evaluated by echocardiography at the age of 11 weeks. As suggested by histological examinations (Figure 6D), rapamycin‐treated animals had a significantly reduced LV wall thickness compared with vehicle‐treated controls (Figure 7A). The fact that LV diameter was not different between the groups (Figure 7B) resulted in a significantly reduced LV mass in rapamycin‐treated mice calculated from echo data, whereas the LV mass/BW ratio was unaffected (Figure 7C). Similar to neonatal stages, rapamycin‐treated adults did not show impairment of LV contractility (Figure 7D). Interestingly and in contrast to newborns, LV stroke volume, heart rate, and cardiac output were normal in rapamycin‐ compared with vehicle‐treated adults (Figure 7E). The latter suggests functional recovery during postnatal life, considering reduced cardiac output in rapamycin‐treated animals at birth (Figure 5E). No major impairment of cardiac morphology and function was observed in rapamycin‐treated cHccs +/− compared to Hccs +/+ females (Table S3), indicating that prenatal mTORC1 inhibition does not negatively influence the long‐term outcome specifically in cHccs +/− females despite their reliance on compensatory cardiac growth. In summary, prenatal inhibition of rapamycin‐sensitive mTORC1 functions results in a sustained reduction of LV mass until early adulthood. Despite unaltered overall cardiac wet weight (Figure 6C), we conclude that LV dimensions are not completely restored in rapamycin‐treated mice within the first 11 weeks of life, although heart size (ie, wet weight and LV mass) appears largely appropriate when normalized to BW.
Figure 7.
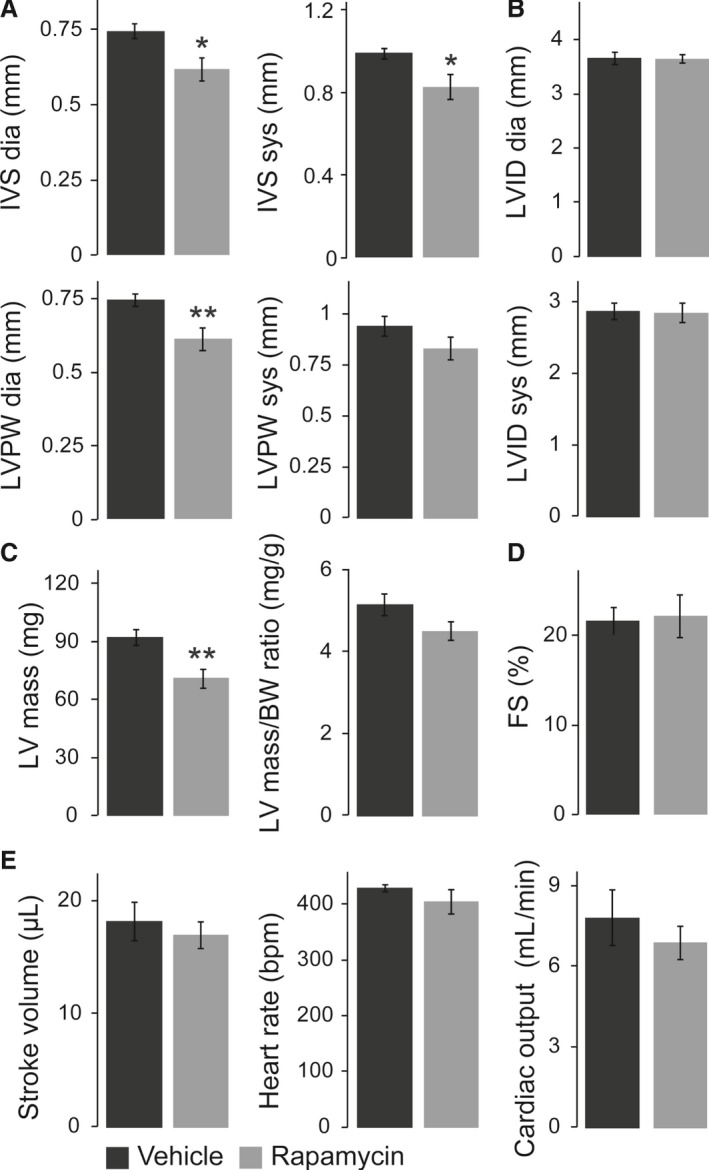
Echocardiography revealed reduced left ventricular (LV) mass but normal cardiac function in adult mice after prenatal rapamycin treatment. A, Interventricular septum (IVS) and left ventricular posterior wall (LVPW) thickness in end‐diastole (dia) and end‐systole (sys) were reduced in adult hearts after prenatal rapamycin compared with vehicle treatment, even though systolic LVPW missed statistical significance. B, End‐diastolic and end‐systolic left ventricular internal diameter (LVID) was not different between the groups. C, LV mass calculated from echocardiography data was significantly reduced in adult mice after prenatal mTORC1 inhibition compared with vehicle‐treated animals, whereas the LV mass/BW ratio was unaffected. D, Rapamycin‐treated adults did not show impairment of LV contractility, as indicated by normal fractional shortening (FS). E, LV stroke volume, heart rate, and cardiac output were normal in rapamycin‐ compared with vehicle‐treated adults. A through E, Vehicle n=7, rapamycin n=9. (*P<0.05, **P<0.01). mTORC1 indicates mechanistic target of rapamycin complex 1.
Reduced Cardiomyocyte Number but No Maladaptive Remodeling in Adult Hearts After Prenatal Rapamycin Treatment
The reduced LV wall thickness and mass in adult hearts after prenatal rapamycin treatment could be caused by a reduction in cardiomyocyte number or size. Surprisingly, cardiomyocyte CSA was increased in rapamycin‐ compared with vehicle‐treated mice (Figure 8A), suggesting an increase in cell size. In addition, we measured cardiomyocyte length in tissue sections by N‐cadherin immunofluorescence staining of the intercalated disks between longitudinally oriented cells (Figure 8B). Cardiomyocyte length was not different between rapamycin‐ and vehicle‐treated adult hearts (Figure 8B). Assuming a simplistic model of cardiomyocytes being of cylindrical shape, multiplying CSA by cell length allowed us to calculate the average cardiomyocyte volume per heart. This revealed a significant increase in cardiomyocyte volume in rapamycin‐ compared with vehicle‐treated adult hearts (Figure 8C). Considering reduced LV mass in echocardiography and unchanged cardiac wet weight in rapamycin‐treated animals, the latter would argue for compensatory cardiomyocyte hypertrophy to account for a reduction in cell number. To estimate cardiomyocyte number per heart, we first determined the relative contribution of cardiomyocytes to the myocardium in WGA‐stained LV tissue sections. These data revealed a slightly but significantly reduced cardiomyocyte area fraction in rapamycin‐ compared with vehicle‐treated adult hearts (Figure 8D). Based on LV mass or cardiac wet weight, cardiomyocyte area fraction, and cardiomyocyte volume, we were able to calculate the number of cardiomyocytes per heart (see Methods). Although this allows only a rough assessment and likely underestimates the absolute cardiomyocyte number, it should be suitable for relative comparisons between treatment groups. The results indeed revealed a significantly reduced number of cardiomyocytes per heart in rapamycin‐ compared with vehicle‐treated mice in adulthood (Figure 8E). The reduction in cell number could be compensated for by excessive deposition of extracellular matrix and therefore result in maladaptive myocardial remodeling. We did not detect differences in interstitial fibrosis within the LV myocardium between rapamycin‐ and vehicle‐treated mice, however (Figure 8F). Furthermore, RNA expression of fetal genes to indicate a molecular signature of pathological conditions was not different between groups (Figure 8G). Finally, prenatal rapamycin treatment does not have more severe consequences on CSA, fibrosis, or fetal gene expression in adult cHccs +/− compared to Hccs +/+ females (Figure S6), indicating that prenatal mTORC1 inhibition can be compensated in cHccs +/− hearts. Taken together, prenatal rapamycin treatment reduces the number of cardiomyocytes in the postnatal heart, which can be partially compensated by increased cell volume to achieve a near normal organ size.
Figure 8.
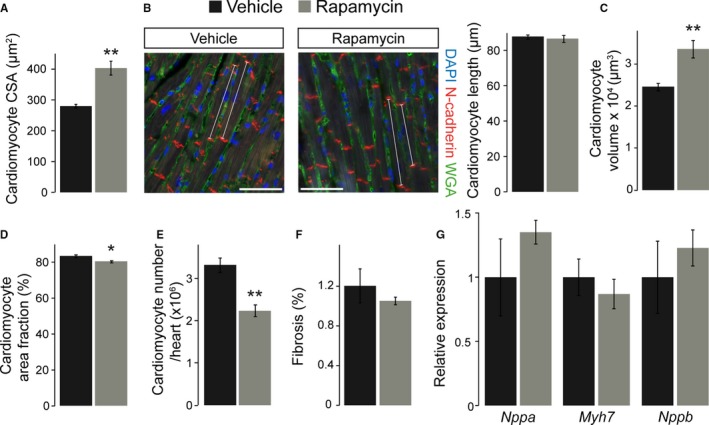
Cardiomyocyte number is reduced, but cell volume is increased in adult hearts after prenatal rapamycin treatment. A, Cardiomyocyte cross‐sectional area (CSA) was significantly larger in adult hearts exposed to prenatal rapamycin treatment compared with vehicle (vehicle n=6, rapamycin n=7). B, Immunofluorescence staining for N‐cadherin to visualize intercalated disks (red), WGA to detect cell membranes (green), and DAPI to stain nuclei (blue) in adult hearts. White lines indicate representative longitudinally oriented cardiomyocytes used to measure the distance between 2 intercalated disks (scale bar=50 μm). Cardiomyocyte length was not different between vehicle‐ and rapamycin‐treated adult hearts. C, Calculated cardiomyocyte volume is significantly increased in adult hearts after prenatal rapamycin vs vehicle treatment. D, Cardiomyocyte area fraction is slightly but significantly lower in rapamycin‐ vs vehicle‐treated adult hearts. E, Calculation of cardiomyocyte number revealed a significant reduction in adult hearts after prenatal rapamycin treatment compared with vehicle controls (n=5 per group in B through E). F, Quantification of interstitial fibrosis within the left ventricle myocardium of adult mice did not reveal differences between the treatment groups (vehicle n=6, rapamycin n=8). G, RNA expression of Nppa (natriuretic peptide type A), Myh7 (β‐myosin heavy chain 7) and Nppb (natriuretic peptide type B) was determined in adult hearts by quantitative real‐time polymerase chain reaction. For all 3 genes no significant difference was observed after prenatal rapamycin compared to vehicle treatment (n=8 per group). (*P<0.05, **P<0.01).
Discussion
Intrauterine growth restriction can be induced by a variety of maternal or environmental conditions, most of which converge on the (single or combined) restriction of nutrients, energy, or oxygen to the fetus.1 The cellular mechanisms that sense such shortage and slow down intrauterine growth, however, are not well understood. Here we show that rapamycin‐sensitive mTORC1 function regulates fetal growth and determines body and organ size at birth. Given that mTORC1 integrates nutrient, amino acid, and oxygen availability with cell growth and proliferation,4 it seems likely that inhibition of mTORC1 by different IUGR conditions is causally involved in fetal growth restriction and aberrant organ maturation. Indeed, a low‐protein diet (LPD) fed to female mice throughout pregnancy reduces pancreatic β‐cell mass in the newborn offspring, eventually leading to impaired insulin secretion and glucose intolerance.25 LPD causes reduced mTORC1 activity in β‐cells at birth, and transiently restoring mTORC1 function rescues β‐cell mass and prevents the diabetic phenotype in the offspring.25 In addition, food restriction in pregnant baboons or sheep reduces mTORC1 activity in the fetal liver and skeletal muscle, respectively.26, 27 Although corresponding data for the growth‐restricted fetal or newborn heart are missing, it is tempting to speculate that mTORC1 inhibition is a general consequence of IUGR in various organs, thereby contributing to developmental programming of adult disease.
Rapamycin treatment of pregnant dams represents a new IUGR model in rodents, which could prove useful to study developmental programming in various organ systems. We have shown efficient inhibition of rapamycin‐sensitive (ie, S6K1, S6) but not rapamycin‐insensitive (ie, 4E‐BP1) mTORC1 downstream targets in the newborn heart, kidney, and liver and a concomitant reduction in heart and kidney weight. For animal studies of developmental programming, however, it has been recommended to use only 1 representative male and/or female offspring per litter in order to account for differences in the intrauterine or postnatal environment specific for certain pregnancies or dams. A limitation of the current study in this regard is that we included several Hccs +/+ and cHccs +/− females from the same litter, which is common practice when analyzing genetically modified mice. Consequently, we cannot exclude that the close relationship of littermates in contrast to randomly chosen mice might have an effect on certain study results. The rapamycin treatment protocol applied in this study (ie, starting at 15.5 dpc) results in a quite stable ≈16% reduction in neonatal body weight, the degree of which is comparable to other IUGR studies.28, 29, 30 In contrast, the widely used maternal protein restriction during pregnancy has shown quite remarkable variations in neonatal body weight between different studies and investigators, which are likely to be influenced by the mouse or rat strain used, the exact diet composition, the precise onset of LPD, the duration of LPD after birth, and other variables (reviewed by Zohdi et al31). In this regard we believe that rapamycin treatment of pregnant dams is easy to control and can be standardized to achieve consistent degrees of growth restriction and therefore reproducible results when studying fetal programming in adulthood. Furthermore, the rapamycin dose can potentially be adjusted to regulate the degree of IUGR, and an adapted treatment regime might even allow to precisely time the period of mTORC1 inhibition by starting and terminating rapamycin application at the desired gestational stage. Such a protocol could determine time windows during embryonic or fetal development that are most susceptible to growth restriction and therefore most relevant for developmental programming.
An important consideration in assessing the new IUGR model described here is the effect of rapamycin on the maternal organism. Because it is applied systemically to pregnant mice, it is likely to interfere with placental growth and function, even though placental development is already well established at the onset of treatment (ie, 15.5 dpc in this study).32 Nevertheless, mTORC1 signaling has been shown to play an important role in placental nutrient sensing and consequently maternal‐to‐fetal nutrient exchange.33 Interestingly, however, mTORC1 activity is reduced in the placenta by maternal protein or nutrient restriction in rats and baboons, respectively,28, 29 as well as in human placentas upon IUGR.34, 35 Systemic rapamycin administration to pregnant dams could furthermore interfere with mammary gland development, thereby impairing lactation and milk intake of newborn offspring. Indeed, treatment of pregnant mice with the mTORC1 inhibitor RAD001 or rapamycin starting on the day before delivery and continuing for 5 or 12 days, respectively, disturbs mammary gland tissue architecture and reduces organ size, milk protein production, and pup weight.36, 37 Rapamycin furthermore reduces milk protein production in murine and bovine mammary epithelial cells in vitro, which was also observed upon amino acid starvation.38 Importantly, maternal LPD during pregnancy in rats impairs late gestation mammary gland development,39 and uteroplacental insufficiency induced by uterine artery ligation in rats impairs mammary gland development and function as well as milk production and composition, thereby restraining postnatal pup growth.30 These studies indicate that impaired mammary gland function as well as placental mTORC1 inhibition appear to be general features of various IUGR animal models, such that a potential maternal impact by rapamycin treatment would not be fundamentally different. The degree of the latter will have to be compared to other IUGR models in future studies, however. This appears furthermore warranted considering lethality in ≈30% of rapamycin‐treated pups during the first 12 postnatal days (Figure 6A).The latter could well be caused by lactation insufficiency or metabolic alterations in the maternal organism, resulting in undernourishment of the offspring. Alternatively, postnatal lethality could be independent of maternal factors and might be the result of variable severity of rapamycin‐induced IUGR and organ dysfunction or impaired postnatal compensatory processes (eg, insufficient catch‐up growth) in some pups.
Our data show that full mTORC1 activity is essential for fetal heart growth during late gestation, given that neonates after prenatal rapamycin treatment have small hearts and a reduced HW/BW ratio compared with vehicle‐treated controls. The latter is the result of a disproportional (34.5%) reduction in HW compared with BW (16.4%). These findings are in contrast to maternal LPD or placental insufficiency in rats, in which reduction in offspring HW parallels the reduction in BW, such that HW/BW ratio is normal.30, 31 Interestingly, the reduction in KW (19.7%) in rapamycin‐treated pups was proportional to body weight, resulting in normal KW/BW ratios. These data suggest that fetal heart growth more heavily depends on rapamycin‐sensitive mTORC1 activity as compared with other organs and that the prenatal heart might be specifically susceptible to fetal programming involving mTORC1 inhibition.
Decreased neonatal heart size after prenatal rapamycin treatment is primarily caused by a reduction in cardiomyocyte size, whereas proliferation is unaffected at birth. This is in agreement with rapamycin inhibiting phosphorylation of S6K1 but not 4E‐BP1 in the neonatal heart, as it has been shown that 4E‐BP1 primarily mediates the proliferative effects downstream of mTORC1, whereas S6K1 regulates cell size.40, 41 It is furthermore consistent with the heart conditional knockout of mTOR and the upstream mTORC1 activator Rheb, both of which result in full mTORC1 inhibition shortly after birth with reduced cardiomyocyte size by days 8 and 15, respectively,7, 10 whereas proliferation rates were not directly determined. We furthermore detected increased apoptotic cell death within the myocardium of rapamycin‐treated neonates, which is in agreement with previous studies showing cardiomyocyte apoptosis upon mTORC1 inhibition in the embryonic or early postnatal heart.7, 8, 9 It is tempting to speculate that in addition to diminished cell size a reduced cardiomyocyte number due to rapamycin‐induced cell death might also contribute to the reduction in heart size. A limitation of our study, however, is that we did not differentiate between cell types when determining proliferation and apoptosis, so we cannot exclude that primarily a nonmyocyte cell population is undergoing cell death. Similarly, unaltered overall proliferation rates in the myocardium could obscure slight differences between cardiac cell types. Nevertheless, when studying adult hearts after prenatal rapamycin treatment, morphometric calculations revealed a reduced cardiomyocyte number in rapamycin‐ versus vehicle‐treated hearts in adulthood. An intriguing question is whether this cardiomyocyte deficit is solely due to increased cardiomyocyte apoptosis in the perinatal phase or whether impaired proliferation in the prenatal period also contributes. In addition, given that murine cardiomyocytes can proliferate during the first postnatal week,42 and mTORC1 activity is only restored by day 3, cardiomyocyte proliferation could also be impaired shortly after birth. A limitation of our study in this regard is that we did not investigate fetal hearts or placental morphology and function between commencement of rapamycin treatment at 15.5 dpc and birth. Therefore, we cannot exclude that rapamycin induces metabolic alterations in the mother or molecular changes in the placenta that generally impair nutrient supply to the fetus, thereby potentially inhibiting fetal (cardiac) growth beyond the rapamycin‐sensitive functions of mTORC1. For example, impaired placental amino acid exchange28, 29, 34 could potentially result in full mTOR inhibition, which in turn could inhibit cardiomyocyte proliferation in the fetus. Given that such rapamycin‐independent inhibitory effects would be restored quickly after birth, our current study might be unable to detect the molecular and cellular consequences when investigating hearts on postnatal day 1. So whether rapamycin‐treated neonates are born with a reduced cardiomyocyte number or whether this develops after birth will have to be determined in future studies, in parallel with investigations of placental function.
The reduction in body and organ weight in rapamycin‐treated mice at birth is partially normalized until early adulthood. Body weight and tibia length remain slightly reduced at the age of 11 weeks, which is in agreement with persistent weight reduction in other IUGR rodent models.31, 43 Interestingly, the ability for postnatal catch‐up growth appears to vary among organs. Although absolute heart, liver, and spleen weights are mainly normal in 11‐week‐old rapamycin‐ compared with vehicle‐treated animals, kidney weight is still reduced, although not different when normalized to body weight or tibia length. Nevertheless, these data suggest that the kidney has an impaired postnatal growth plasticity in response to IUGR (as previously proposed by human as well as animal studies43, 44), which might contribute to developmental programming of hypertension and kidney disease. For the heart, we observed a discrepancy between overall gravimetric wet weight and LV mass calculated from echocardiography data. Whereas the latter was reduced in rapamycin‐ versus vehicle‐treated mice in adulthood, wet weight was unchanged. These differences are likely caused by the impact of blood or fluids in the cardiac cavities as well as right ventricular and atrial myocardium on wet weight but not LV mass calculations. Given that histological examinations and echocardiography data confirmed slightly reduced LV wall thickness in rapamycin‐ versus vehicle‐treated adult mice, we conclude that heart dimensions are not fully normalized until early adulthood after prenatal mTORC1 inhibition. This is in contrast to other IUGR animal models, which mainly revealed unaltered heart size in adulthood, although it was often determined as wet weight at later stages.31, 43 Therefore, it is quite possible that LV mass completely normalizes in rapamycin‐treated mice with further aging.
Neonates after prenatal rapamycin treatment show reduced cardiac output caused by smaller LV dimensions and stroke volume but not impaired contractility when compared with vehicle‐treated controls. This is in contrast to mice with an inducible, heart conditional knockout of mTOR or Raptor in adulthood, which develop contractile dysfunction and heart failure under baseline conditions,45, 46 although over a period of 4 to 6 weeks. Importantly, it is also different from heart conditional Rheb KO mice, which show efficient mTORC1 inhibition by postnatal day 5 and contractile dysfunction by day 8.10 Similarly, heart conditional mTOR KO mice that exhibit efficient mTOR protein depletion by postnatal day 7 develop contractile dysfunction by day 15.7 The latter 2 genetic models, however, cause a broad inhibition of mTORC1 (and also mTORC2 in case of the mTOR knockout) in the heart beyond rapamycin‐sensitive functions, evident as decreased phosphorylation of 4E‐BP1, which was unaffected in our current study. These data suggest that LV contractility in fetal and neonatal hearts is relatively insensitive toward rapamycin‐dependent mTORC1 inhibition, and cardiac output is primarily determined by LV volume. Within the first few weeks after birth, however, mTORC1 activity becomes essential to establish and maintain normal LV function. The latter coincides with various milestones of postnatal cardiac maturation, such as changes in cardiomyocyte metabolism, growth pattern, tissue composition, and myocardial workload.47 Thus, it is tempting to speculate that mTORC1 plays an important role in the transition from fetal to postnatal cardiac growth and function.
Despite slightly reduced LV mass and wall thickness and a reduced number of cardiomyocytes, adult hearts after prenatal rapamycin treatment exhibit normal contractility and complete recovery of cardiac output as compared with neonatal stages without signs of maladaptive myocardial remodeling. This is consistent with some rodent studies showing unaltered LV contractility after IUGR under baseline conditions in adulthood,31, 48 but others have reported reduced heart function at 10 to 12 weeks of age.49, 50 Similarly, most IUGR animal models eventually show increased myocardial fibrosis with age, whereas the onset of tissue remodeling in early adulthood is less clear.31, 49, 51 In addition, maternal LPD during pregnancy has recently been proposed to alter the biochemical composition of myocardial tissue in adult offspring.51 Whether this represents a general effect of IUGR or whether it is specific to the LPD model needs to be confirmed, such that characterizing the cardiac lipid, proteoglycan, and carbohydrate profile in rapamycin‐treated hearts might add valuable information to this question. So although prenatal rapamycin treatment obviously results in an IUGR phenotype at birth, it will have to be established whether it furthermore represents a suitable model for fetal programming in adulthood. In this regard it will be interesting to determine the long‐term cardiac outcome of adult mice after prenatal rapamycin treatment upon aging as well as their response to various challenges (such as ischemia, pressure overload, or neurohumoral stimulation) in future studies. Such data would have important implications for both fetal programming in general as well as for the specific role of mTORC1 in long‐term cardiovascular health and disease susceptibility.
Sources of Funding
This work was supported by institutional funds of the Max‐Delbrück‐Center for Molecular Medicine Berlin and the University Hospital Münster but received no specific grant from other funding agencies.
Disclosures
None.
Supporting information
Data S1. Supplemental Discussion.
Table S1. Echocardiographic Measurements in Neonatal Mice After Prenatal mTORC1 Inhibition
Table S2. Body and Organ Weights in Adult Mice After Prenatal Rapamycin or Vehicle Treatment
Table S3. Echocardiographic Measurements in Adult Mice After Prenatal mTORC1 Inhibition
Figure S1. Inhibition of mTORC1 in fetal and neonatal mice by rapamycin treatment of pregnant dams. Rapamycin was injected subcutaneously at a dose of 5 mg/kg body weight in pregnant dams every 12 hours from 15.5 days postconception until delivery. As controls, pregnant dams were injected with vehicle only.
Figure S2. Evaluation of the autophagy‐regulating kinases AMPK and ULK1 in neonatal hearts after prenatal rapamycin treatment. A, Phosphorylation of the AMPK (AMP‐activated protein kinase) subunit α is increased in neonatal hearts after prenatal rapamycin treatment, indicating AMPK activation. B, The kinase ULK1 (Unc‐51‐like autophagy‐activating kinase 1) is phosphorylated by AMPK at Ser555 to initiate autophagy, whereas mTOR phosphorylates ULK1 at Ser757 to inhibit autophagy. Consistent with AMPK activation, ULK1 Ser555 phosphorylation is increased in neonatal hearts after prenatal rapamycin treatment, whereas Ser757 phosphorylation is unchanged when normalized to total ULK1. Note that the latter is confounded by a significant increase in total ULK1 protein levels in rapamycin‐ compared to vehicle‐treated hearts. (Densitometric quantification n=4 per group in A and B, *P<0.05, **P<0.01, ***P<0.001).
Figure S3. Unaltered phosphorylation of mTOR, ULK1, 4E‐BP1, and Akt in neonatal cHccs +/− hearts. A, Phosphorylation of mTOR at Ser2448 and Ser2481 is unaltered in neonatal cHccs +/− hearts compared to controls. Similarly, no difference in total protein amounts of mTOR or its interacting proteins RAPTOR and RICTOR (specific for mTOR complexes 1 and 2, respectively) is detected between groups (densitometric quantification n=8 per genotype; POI=protein of interest). B, Phosphorylation of the mTORC1 downstream targets 4E‐BP1 and ULK1 (at the mTORC1‐dependent site Ser757) is not different between neonatal cHccs +/− and control hearts. The kinase Akt acts as both an upstream regulator of mTORC1 and as an mTORC2 downstream target. Phosphorylation of Akt at Thr308 (via the PI3K pathway) and Ser473 (by mTORC2) is unaltered in cHccs +/− hearts (densitometric quantification n=5 to 6 per genotype).
Figure S4. Prenatal mTORC1 inhibition in cHccs +/− fetuses does not cause lethality prior to birth. A, Western blots of heart protein extracts from neonatal (P1) cHccs +/− females and their Hccs +/+ female littermate controls after prenatal vehicle or rapamycin treatment illustrating the phosphorylation status of the mTORC1 downstream targets S6K1, S6 and 4E‐BP1. Note the increased mTORC1 activity toward S6K1 and S6 in cHccs +/− females in vehicle‐treated animals (see main text and Figure 4) but similar reduction of S6K1 and S6 phosphorylation in hearts of rapamycin‐ compared to vehicle‐treated offspring, indicating successful mTORC1 inhibition in cHccs +/− hearts. Phosphorylation of 4E‐BP1 was demonstrated to be unaffected by prenatal rapamycin treatment (see main text and Figure 1). B, The genotype distribution within vehicle‐ as well as rapamycin‐treated litters was not significantly different from expected genotype frequencies, indicating that cHccs +/− females do not exhibit prenatal lethality due to mTORC1 inhibition. Statistical significance between treatment groups and the expected genotype distribution was assessed by chi‐squared test. Statistical significance comparing the frequencies of each genotype between vehicle‐ and rapamycin‐treated litters was assessed by unpaired 2‐tailed Student t tests (n=9 litters per group).
Figure S5. There was no preferential death of rapamycin‐treated cHccs +/− females after birth. A, Breeding of males hemizygous for the “floxed” Hccs allele (Hccs floxed/y /Nkx2.5 +/+) to females homozygous for the Nkx2.5Cre allele (Hccs +/+/Nkx2.5 Cre/Cre) generates 2 possible offspring genotypes, ie, control males (Hccs +/y /Nkx2.5 Cre/+) and heterozygous heart‐conditional Hccs knockout females (Hccs floxed/+ /Nkx2.5 Cre/+) at an expected ratio of 50:50. B, Breeding of males homozygous for the Nkx2.5Cre allele (Hccs +/y/Nkx2.5 Cre/Cre) to females heterozygous for the “floxed” Hccs allele (Hccs floxed/+ /Nkx2.5 +/+) generates 3 postnatally viable genotypes, ie, control males (Hccs +/y /Nkx2.5 Cre/+), control females (Hccs +/+ /Nkx2.5 Cre/+), and heterozygous heart‐conditional Hccs knockout females (Hccs floxed/+ /Nkx2.5 Cre/+) at an expected ratio of 33% each. Hemizygous heart‐conditional Hccs knockout males (Hccs floxed/y /Nkx2.5 Cre/+) die in utero at stage 10.5 dpc.11 Genotype distribution within vehicle‐ as well as rapamycin‐treated litters was evaluated at weaning (ie, at 21 days of age). For both breeding schemes no significant difference compared to expected genotype frequencies was observed, indicating that rapamycin‐treated cHccs +/− females do not preferentially die within the first 12 days after birth (see Figure 6A). Statistical significance between treatment groups and the expected genotype distribution was assessed by chi‐squared test (3 litters per group in A; 5 vehicle‐ and 4 rapamycin‐treated litters in B).
Figure S6. Prenatal mTORC1 inhibition does not cause significantly different tissue remodeling in adult cHccs +/− compared to Hccs +/+ hearts. A, Fluorescence images of cross‐sectioned cardiomyocytes within the left ventricular (LV) myocardium of adult hearts. Cardiomyocyte membranes were stained in red with wheat germ agglutinin (WGA) and nuclei in blue with DAPI (scale bar=100 μm). Cardiomyocyte cross‐sectional area (CSA) was significantly larger in Hccs +/+ as well as cHccs +/− hearts exposed to prenatal rapamycin compared to vehicle treatment. Note that compensatory cardiomyocyte hypertrophy in cHccs +/− mice reported previously12 is evident in the vehicle group but lost in rapamycin‐treated hearts (vehicle Hccs +/+ n=6, rapamycin Hccs +/+ n=7, vehicle and rapamycin cHccs +/− n=5). B, Representative images of Sirius Red–stained LV myocardium of adult mice. White arrowheads highlight interstitial fibrosis, whereas black arrowheads indicate perivascular fibrosis, which was excluded from quantification (scale bar=300 μm). The contribution of fibrotic tissue to the LV myocardium of adult mice did not reveal differences between treatment groups or genotypes (n=6 for all groups except vehicle cHccs +/−, n=8). C, Relative Nppa, Myh7, and Nppb RNA expression in adult hearts was determined using quantitative real‐time polymerase chain reaction to evaluate pathological conditions. No major changes in gene expression were observed in rapamycin (R) compared to vehicle (V) groups in either Hccs +/+ or cHccs +/− hearts (n=8 per group). Note that data used for Hccs +/+ animals in A through C are the same as depicted in Figure 8. (*P<0.05, **P<0.01, §§ P<0.01 versus vehicle Hccs +/+, # P<0.05 versus vehicle cHccs +/−).
Acknowledgments
We thank Martin Taube and Stefanie Schelenz for performing echocardiography, Anja Conrad and Michaela Tirre for technical assistance, and the Microscopy Core Facility at the Max‐Delbrück‐Center for technical support.
(J Am Heart Assoc. 2017;6:e005506 DOI: 10.1161/JAHA.117.005506.)28778941
References
- 1. Gaccioli F, Lager S. Placental nutrient transport and intrauterine growth restriction. Front Physiol. 2016;7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Warner MJ, Ozanne SE. Mechanisms involved in the developmental programming of adulthood disease. Biochem J. 2010;427:333–347. [DOI] [PubMed] [Google Scholar]
- 3. Thornburg KL. The programming of cardiovascular disease. J Dev Orig Health Dis. 2015;6:366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15:555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tumaneng K, Russell RC, Guan KL. Organ size control by Hippo and TOR pathways. Curr Biol. 2012;22:R368–R379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mazelin L, Panthu B, Nicot AS, Belotti E, Tintignac L, Teixeira G, Zhang Q, Risson V, Baas D, Delaune E, Derumeaux G, Taillandier D, Ohlmann T, Ovize M, Gangloff YG, Schaeffer L. mTOR inactivation in myocardium from infant mice rapidly leads to dilated cardiomyopathy due to translation defects and p53/JNK‐mediated apoptosis. J Mol Cell Cardiol. 2016;97:213–225. [DOI] [PubMed] [Google Scholar]
- 8. Zhang P, Shan T, Liang X, Deng C, Kuang S. Mammalian target of rapamycin is essential for cardiomyocyte survival and heart development in mice. Biochem Biophys Res Commun. 2014;452:53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu Y, Pires KM, Whitehead KJ, Olsen CD, Wayment B, Zhang YC, Bugger H, Ilkun O, Litwin SE, Thomas G, Kozma SC, Abel ED. Mechanistic target of rapamycin (mTOR) is essential for murine embryonic heart development and growth. PLoS One. 2013;8:e54221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tamai T, Yamaguchi O, Hikoso S, Takeda T, Taneike M, Oka T, Oyabu J, Murakawa T, Nakayama H, Uno Y, Horie K, Nishida K, Sonenberg N, Shah AM, Takeda J, Komuro I, Otsu K. Rheb (Ras homologue enriched in brain)‐dependent mammalian target of rapamycin complex 1 (mTORC1) activation becomes indispensable for cardiac hypertrophic growth after early postnatal period. J Biol Chem. 2013;288:10176–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drenckhahn JD, Schwarz QP, Gray S, Laskowski A, Kiriazis H, Ming Z, Harvey RP, Du XJ, Thorburn DR, Cox TC. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev Cell. 2008;15:521–533. [DOI] [PubMed] [Google Scholar]
- 12. Drenckhahn JD, Strasen J, Heinecke K, Langner P, Yin KV, Skole F, Hennig M, Spallek B, Fischer R, Blaschke F, Heuser A, Cox TC, Black MJ, Thierfelder L. Impaired myocardial development resulting in neonatal cardiac hypoplasia alters postnatal growth and stress response in the heart. Cardiovasc Res. 2015;106:43–54. [DOI] [PubMed] [Google Scholar]
- 13. Stanley EG, Biben C, Elefanty A, Barnett L, Koentgen F, Robb L, Harvey RP. Efficient Cre‐mediated deletion in cardiac progenitor cells conferred by a 3′UTR‐ires‐Cre allele of the homeobox gene Nkx2‐5. Int J Dev Biol. 2002;46:431–439. [PubMed] [Google Scholar]
- 14. Bal MP, de Vries WB, Steendijk P, Homoet‐van der Kraak P, van der Leij FR, Baan J, van Oosterhout MF, van Bel F. Histopathological changes of the heart after neonatal dexamethasone treatment: studies in 4‐, 8‐, and 50‐week‐old rats. Pediatr Res. 2009;66:74–79. [DOI] [PubMed] [Google Scholar]
- 15. Buzello M, Boehm C, Orth S, Fischer B, Ehmke H, Ritz E, Mall G, Amann K. Myocyte loss in early left ventricular hypertrophy of experimental renovascular hypertension. Virchows Arch. 2003;442:364–371. [DOI] [PubMed] [Google Scholar]
- 16. Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E‐BP1 to mediate cell‐type‐specific repression of mRNA translation. Proc Natl Acad Sci USA. 2008;105:17414–17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341:1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chaveroux C, Eichner LJ, Dufour CR, Shatnawi A, Khoutorsky A, Bourque G, Sonenberg N, Giguère V. Molecular and genetic crosstalks between mTOR and ERRα are key determinants of rapamycin‐induced nonalcoholic fatty liver. Cell Metab. 2013;17:586–598. [DOI] [PubMed] [Google Scholar]
- 20. Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self‐association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406–416. [DOI] [PubMed] [Google Scholar]
- 21. Nazio F, Carinci M, Valacca C, Bielli P, Strappazzon F, Antonioli M, Ciccosanti F, Rodolfo C, Campello S, Fimia GM, Sette C, Bonaldo P, Cecconi F. Fine‐tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J Cell Biol. 2016;215:841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. [DOI] [PubMed] [Google Scholar]
- 24. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA. Rapamycin‐induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alejandro EU, Gregg B, Wallen T, Kumusoglu D, Meister D, Chen A, Merrins MJ, Satin LS, Liu M, Arvan P, Bernal‐Mizrachi E. Maternal diet‐induced microRNAs and mTOR underlie β cell dysfunction in offspring. J Clin Invest. 2014;124:4395–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abu Shehab M, Damerill I, Shen T, Rosario FJ, Nijland M, Nathanielsz PW, Kamat A, Jansson T, Gupta MB. Liver mTOR controls IGF‐I bioavailability by regulation of protein kinase CK2 and IGFBP‐1 phosphorylation in fetal growth restriction. Endocrinology. 2014;155:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu MJ, Ford SP, Nathanielsz PW, Du M. Effect of maternal nutrient restriction in sheep on the development of fetal skeletal muscle. Biol Reprod. 2004;71:1968–1973. [DOI] [PubMed] [Google Scholar]
- 28. Rosario FJ, Jansson N, Kanai Y, Prasad PD, Powell TL, Jansson T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down‐regulates placental amino acid transporters. Endocrinology. 2011;152:1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kavitha JV, Rosario FJ, Nijland MJ, McDonald TJ, Wu G, Kanai Y, Powell TL, Nathanielsz PW, Jansson T. Down‐regulation of placental mTOR, insulin/IGF‐I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J. 2014;28:1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Dowd R, Kent JC, Moseley JM, Wlodek ME. Effects of uteroplacental insufficiency and reducing litter size on maternal mammary function and postnatal offspring growth. Am J Physiol Regul Integr Comp Physiol. 2008;294:R539–R548. [DOI] [PubMed] [Google Scholar]
- 31. Zohdi V, Lim K, Pearson JT, Black MJ. Developmental programming of cardiovascular disease following intrauterine growth restriction: findings utilising a rat model of maternal protein restriction. Nutrients. 2014;7:119–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coan PM, Ferguson‐Smith AC, Burton GJ. Developmental dynamics of the definitive mouse placenta assessed by stereology. Biol Reprod. 2004;70:1806–1813. [DOI] [PubMed] [Google Scholar]
- 33. Jansson T, Aye IL, Goberdhan DC. The emerging role of mTORC1 signaling in placental nutrient‐sensing. Placenta. 2012;33(suppl 2):e23–e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roos S, Jansson N, Palmberg I, Säljö K, Powell TL, Jansson T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down‐regulated in restricted fetal growth. J Physiol. 2007;582(Pt 1):449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yung HW, Calabrese S, Hynx D, Hemmings BA, Cetin I, Charnock‐Jones DS, Burton GJ. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol. 2008;173:451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jankiewicz M, Groner B, Desrivières S. Mammalian target of rapamycin regulates the growth of mammary epithelial cells through the inhibitor of deoxyribonucleic acid binding Id1 and their functional differentiation through Id2. Mol Endocrinol. 2006;20:2369–2381. [DOI] [PubMed] [Google Scholar]
- 37. Fu NY, Rios AC, Pal B, Soetanto R, Lun AT, Liu K, Beck T, Best SA, Vaillant F, Bouillet P, Strasser A, Preiss T, Smyth GK, Lindeman GJ, Visvader JE. EGF‐mediated induction of Mcl‐1 at the switch to lactation is essential for alveolar cell survival. Nat Cell Biol. 2015;17:365–375. [DOI] [PubMed] [Google Scholar]
- 38. Moshel Y, Rhoads RE, Barash I. Role of amino acids in translational mechanisms governing milk protein synthesis in murine and ruminant mammary epithelial cells. J Cell Biochem. 2006;98:685–700. [DOI] [PubMed] [Google Scholar]
- 39. Bautista CJ, Rodríguez‐González GL, Torres N, Hernández‐Pando R, Ramírez V, Rodríguez‐Cruz M, Nathanielsz PW, Zambrano E. Protein restriction in the rat negatively impacts long‐chain polyunsaturated fatty acid composition and mammary gland development at the end of gestation. Arch Med Res. 2013;44:429–436. [DOI] [PubMed] [Google Scholar]
- 40. Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, Sonenberg N, Kelly PA, Sotiropoulos A, Pende M. Atrophy of S6K1(–/–) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol. 2005;7:286–294. [DOI] [PubMed] [Google Scholar]
- 41. Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, Kozma SC, Thomas G, Sonenberg N. mTORC1‐mediated cell proliferation, but not cell growth, controlled by the 4E‐BPs. Science. 2010;328:1172–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Foglia MJ, Poss KD. Building and re‐building the heart by cardiomyocyte proliferation. Development. 2016;143:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wlodek ME, Westcott K, Siebel AL, Owens JA, Moritz KM. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008;74:187–195. [DOI] [PubMed] [Google Scholar]
- 44. Schmidt IM, Chellakooty M, Boisen KA, Damgaard IN, Mau Kai C, Olgaard K, Main KM. Impaired kidney growth in low‐birth‐weight children: distinct effects of maturity and weight for gestational age. Kidney Int. 2005;68:731–740. [DOI] [PubMed] [Google Scholar]
- 45. Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan KL, Brown JH, Chen J, Sonenberg N, Condorelli G. MTORC1 regulates cardiac function and myocyte survival through 4E‐BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, Krishnan J, Lerch R, Hall MN, Rüegg MA, Pedrazzini T, Brink M. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. [DOI] [PubMed] [Google Scholar]
- 47. Jonker SS, Louey S. Endocrine and other physiologic modulators of perinatal cardiomyocyte endowment. J Endocrinol. 2016;228:R1–R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rueda‐Clausen CF, Morton JS, Davidge ST. Effects of hypoxia‐induced intrauterine growth restriction on cardiopulmonary structure and function during adulthood. Cardiovasc Res. 2009;81:713–722. [DOI] [PubMed] [Google Scholar]
- 49. Menendez‐Castro C, Toka O, Fahlbusch F, Cordasic N, Wachtveitl R, Hilgers KF, Rascher W, Hartner A. Impaired myocardial performance in a normotensive rat model of intrauterine growth restriction. Pediatr Res. 2014;75:697–706. [DOI] [PubMed] [Google Scholar]
- 50. Alsaied T, Omar K, James JF, Hinton RB, Crombleholme TM, Habli M. Fetal origins of adult cardiac disease: a novel approach to prevent fetal growth restriction induced cardiac dysfunction using insulin like growth factor. Pediatr Res. 2017;81:919–925. [DOI] [PubMed] [Google Scholar]
- 51. Zohdi V, Wood BR, Pearson JT, Bambery KR, Black MJ. Evidence of altered biochemical composition in the hearts of adult intrauterine growth‐restricted rats. Eur J Nutr. 2013;52:749–758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Discussion.
Table S1. Echocardiographic Measurements in Neonatal Mice After Prenatal mTORC1 Inhibition
Table S2. Body and Organ Weights in Adult Mice After Prenatal Rapamycin or Vehicle Treatment
Table S3. Echocardiographic Measurements in Adult Mice After Prenatal mTORC1 Inhibition
Figure S1. Inhibition of mTORC1 in fetal and neonatal mice by rapamycin treatment of pregnant dams. Rapamycin was injected subcutaneously at a dose of 5 mg/kg body weight in pregnant dams every 12 hours from 15.5 days postconception until delivery. As controls, pregnant dams were injected with vehicle only.
Figure S2. Evaluation of the autophagy‐regulating kinases AMPK and ULK1 in neonatal hearts after prenatal rapamycin treatment. A, Phosphorylation of the AMPK (AMP‐activated protein kinase) subunit α is increased in neonatal hearts after prenatal rapamycin treatment, indicating AMPK activation. B, The kinase ULK1 (Unc‐51‐like autophagy‐activating kinase 1) is phosphorylated by AMPK at Ser555 to initiate autophagy, whereas mTOR phosphorylates ULK1 at Ser757 to inhibit autophagy. Consistent with AMPK activation, ULK1 Ser555 phosphorylation is increased in neonatal hearts after prenatal rapamycin treatment, whereas Ser757 phosphorylation is unchanged when normalized to total ULK1. Note that the latter is confounded by a significant increase in total ULK1 protein levels in rapamycin‐ compared to vehicle‐treated hearts. (Densitometric quantification n=4 per group in A and B, *P<0.05, **P<0.01, ***P<0.001).
Figure S3. Unaltered phosphorylation of mTOR, ULK1, 4E‐BP1, and Akt in neonatal cHccs +/− hearts. A, Phosphorylation of mTOR at Ser2448 and Ser2481 is unaltered in neonatal cHccs +/− hearts compared to controls. Similarly, no difference in total protein amounts of mTOR or its interacting proteins RAPTOR and RICTOR (specific for mTOR complexes 1 and 2, respectively) is detected between groups (densitometric quantification n=8 per genotype; POI=protein of interest). B, Phosphorylation of the mTORC1 downstream targets 4E‐BP1 and ULK1 (at the mTORC1‐dependent site Ser757) is not different between neonatal cHccs +/− and control hearts. The kinase Akt acts as both an upstream regulator of mTORC1 and as an mTORC2 downstream target. Phosphorylation of Akt at Thr308 (via the PI3K pathway) and Ser473 (by mTORC2) is unaltered in cHccs +/− hearts (densitometric quantification n=5 to 6 per genotype).
Figure S4. Prenatal mTORC1 inhibition in cHccs +/− fetuses does not cause lethality prior to birth. A, Western blots of heart protein extracts from neonatal (P1) cHccs +/− females and their Hccs +/+ female littermate controls after prenatal vehicle or rapamycin treatment illustrating the phosphorylation status of the mTORC1 downstream targets S6K1, S6 and 4E‐BP1. Note the increased mTORC1 activity toward S6K1 and S6 in cHccs +/− females in vehicle‐treated animals (see main text and Figure 4) but similar reduction of S6K1 and S6 phosphorylation in hearts of rapamycin‐ compared to vehicle‐treated offspring, indicating successful mTORC1 inhibition in cHccs +/− hearts. Phosphorylation of 4E‐BP1 was demonstrated to be unaffected by prenatal rapamycin treatment (see main text and Figure 1). B, The genotype distribution within vehicle‐ as well as rapamycin‐treated litters was not significantly different from expected genotype frequencies, indicating that cHccs +/− females do not exhibit prenatal lethality due to mTORC1 inhibition. Statistical significance between treatment groups and the expected genotype distribution was assessed by chi‐squared test. Statistical significance comparing the frequencies of each genotype between vehicle‐ and rapamycin‐treated litters was assessed by unpaired 2‐tailed Student t tests (n=9 litters per group).
Figure S5. There was no preferential death of rapamycin‐treated cHccs +/− females after birth. A, Breeding of males hemizygous for the “floxed” Hccs allele (Hccs floxed/y /Nkx2.5 +/+) to females homozygous for the Nkx2.5Cre allele (Hccs +/+/Nkx2.5 Cre/Cre) generates 2 possible offspring genotypes, ie, control males (Hccs +/y /Nkx2.5 Cre/+) and heterozygous heart‐conditional Hccs knockout females (Hccs floxed/+ /Nkx2.5 Cre/+) at an expected ratio of 50:50. B, Breeding of males homozygous for the Nkx2.5Cre allele (Hccs +/y/Nkx2.5 Cre/Cre) to females heterozygous for the “floxed” Hccs allele (Hccs floxed/+ /Nkx2.5 +/+) generates 3 postnatally viable genotypes, ie, control males (Hccs +/y /Nkx2.5 Cre/+), control females (Hccs +/+ /Nkx2.5 Cre/+), and heterozygous heart‐conditional Hccs knockout females (Hccs floxed/+ /Nkx2.5 Cre/+) at an expected ratio of 33% each. Hemizygous heart‐conditional Hccs knockout males (Hccs floxed/y /Nkx2.5 Cre/+) die in utero at stage 10.5 dpc.11 Genotype distribution within vehicle‐ as well as rapamycin‐treated litters was evaluated at weaning (ie, at 21 days of age). For both breeding schemes no significant difference compared to expected genotype frequencies was observed, indicating that rapamycin‐treated cHccs +/− females do not preferentially die within the first 12 days after birth (see Figure 6A). Statistical significance between treatment groups and the expected genotype distribution was assessed by chi‐squared test (3 litters per group in A; 5 vehicle‐ and 4 rapamycin‐treated litters in B).
Figure S6. Prenatal mTORC1 inhibition does not cause significantly different tissue remodeling in adult cHccs +/− compared to Hccs +/+ hearts. A, Fluorescence images of cross‐sectioned cardiomyocytes within the left ventricular (LV) myocardium of adult hearts. Cardiomyocyte membranes were stained in red with wheat germ agglutinin (WGA) and nuclei in blue with DAPI (scale bar=100 μm). Cardiomyocyte cross‐sectional area (CSA) was significantly larger in Hccs +/+ as well as cHccs +/− hearts exposed to prenatal rapamycin compared to vehicle treatment. Note that compensatory cardiomyocyte hypertrophy in cHccs +/− mice reported previously12 is evident in the vehicle group but lost in rapamycin‐treated hearts (vehicle Hccs +/+ n=6, rapamycin Hccs +/+ n=7, vehicle and rapamycin cHccs +/− n=5). B, Representative images of Sirius Red–stained LV myocardium of adult mice. White arrowheads highlight interstitial fibrosis, whereas black arrowheads indicate perivascular fibrosis, which was excluded from quantification (scale bar=300 μm). The contribution of fibrotic tissue to the LV myocardium of adult mice did not reveal differences between treatment groups or genotypes (n=6 for all groups except vehicle cHccs +/−, n=8). C, Relative Nppa, Myh7, and Nppb RNA expression in adult hearts was determined using quantitative real‐time polymerase chain reaction to evaluate pathological conditions. No major changes in gene expression were observed in rapamycin (R) compared to vehicle (V) groups in either Hccs +/+ or cHccs +/− hearts (n=8 per group). Note that data used for Hccs +/+ animals in A through C are the same as depicted in Figure 8. (*P<0.05, **P<0.01, §§ P<0.01 versus vehicle Hccs +/+, # P<0.05 versus vehicle cHccs +/−).