

Abstract
Background
Percutaneous coronary intervention has been widely used in the treatment of ischemic heart disease, but vascular restenosis is a main limitation of percutaneous coronary intervention. Our previous work reported that caveolin‐1 had a key functional role in intimal hyperplasia, whereas whether Cavin‐1 (another important caveolae‐related protein) was involved is still unknown. Therefore, we will investigate the effect of Cavin‐1 on neointimal formation.
Methods and Results
Balloon injury markedly reduced Cavin‐1 protein and enhanced ubiquitin protein expression accompanied with neointimal hyperplasia in injured carotid arteries, whereas Cavin‐1 mRNA had no change. In cultured vascular smooth muscle cells (VSMCs), Cavin‐1 was downregulated after inhibition of protein synthesis by cycloheximide, which was distinctly prevented by pretreatment with proteasome inhibitor MG132 but not by lysosomal inhibitor chloroquine, suggesting that proteasomal degradation resulted in Cavin‐1 downregulation. Knockdown of Cavin‐1 by local injection of Cavin‐1 short hairpin RNA (shRNA) into balloon‐injured carotid arteries in vivo promoted neointimal formation. In addition, inhibition or overexpression of Cavin‐1 in cultured VSMCs in vitro prompted or suppressed VSMC proliferation and migration via increasing or decreasing extracellular signal‐regulated kinase phosphorylation and matrix‐degrading metalloproteinases‐9 activity, respectively. However, under basic conditions, the effect of Cavin‐1 on VSMC migration was stronger than on proliferation. Moreover, our results indicated that Cavin‐1 regulated caveolin‐1 expression via lysosomal degradation pathway.
Conclusions
Our study revealed the role and the mechanisms of Cavin‐1 downregulation in neointimal formation by promoting VSMC proliferation, migration, and synchronously enhancing caveolin‐1 lysosomal degradation. Cavin‐1 may be a potential therapeutic target for the treatment of postinjury vascular remodeling.
Keywords: caveolin‐1, Cavin‐1, migration, neointimal hyperplasia, polymerase I and transcript release factor, proliferation, vascular smooth muscle
Subject Categories: Atherosclerosis, Coronary Artery Disease, Restenosis, Vascular Disease
Clinical Perspective
What Is New?
In the present study, our results for the first time revealed the role and the mechanisms of Cavin‐1 downregulation in facilitating neointimal formation by promoting VSMC proliferation, migration, and synchronously enhancing caveolae‐related protein caveolin‐1 lysosomal degradation.
What Are the Clinical Implications?
Our findings support the possibility that Cavin‐1 may be a novel therapeutic target for neointimal hyperplasia after arterial injury and for restenosis after angioplasty.
Introduction
Percutaneous coronary intervention, also known as coronary angioplasty, is a main nonsurgical technique for treating ischemic heart disease and an effective strategy to reduce ischemia‐related mortality. Although drug‐eluting stents have reduced the incidence of restenosis considerably,1, 2, 3, 4 restenosis after angioplasty remains a remarkable challenge. Therefore, the precise mechanism of vascular restenosis needs to be further explored. Many studies showed that neointimal hyperplasia is the major cause of restenosis after percutaneous coronary intervention, which is associated with the proliferation and migration of vascular smooth muscle cells (VSMCs). All aspects of VSMCs behavior are under the coordinated control of inflammatory factors,5 cell–matrix and cell–cell interactions. It is reported that matrix‐degrading metalloproteinases (MMPs), such as MMP‐2 and MMP‐9, influence VSMCs behavior by cleaving both matrix and nonmatrix substrates (degrade extracellular collagen).6, 7, 8 However, the exact mechanism underlying VSMCs behavior remains unclear.
Caveolae are specialized cell surface plasma membrane invaginations found in endothelial cells and VSMCs. These structures have been implicated in the development of neointimal hyperplasia and atherosclerosis. Caveolae‐related protein caveolin‐1 (CAV‐1), an important component of caveolae, is known to have proatherogenic or antiatherogenic effects, depending on the cell type during the progression of atherosclerosis. Our previous work indicated that CAV‐1 was involved in neointimal hyperplasia.9 However, caveolae formation requires both CAV‐1 and Cavin‐1 (also known as PTRF: polymerase I and transcript release factor), and the lack of either protein resulted in reduction in the number of caveolae.10, 11 As the key component in structure proteins of caveolae, CAV‐1 has been widely well studied;12, 13, 14 however, the role of Cavin‐1 remains elusive. Recent studies have shown the role of Cavin‐1 in migration of prostate cancer cells.15, 16 On the other hand, the ubiquitin–proteasome pathway is involved in the process of vascular restenosis,17, 18 and Cavin‐1 has been discovered to be the target of ubiquitin.19 These findings suggest that Cavin‐1 may play a pivotal role in vascular remodeling.
Hence, the aim of the present study was to examine the expression of Cavin‐1 in balloon‐injured carotid arteries, investigate the role of Cavin‐1 in neointimal hyperplasia, and explore the underlying mechanism. To achieve this, we used a balloon‐induced carotid arteries injury model to clarify the expression and function of Cavin‐1. We then identified the effect of Cavin‐1 decline in neointimal hyperplasia and explored the underlying mechanism in vivo and in vitro. Finally, we investigated the interaction between Cavin‐1 and CAV‐1 in order to explore their molecular association in neointimal hyperplasia.
Methods
Animals and Balloon Injury Model
Sprague–Dawley rats (200–250 g) were obtained from the Animal Center of Sun Yat‐sen University (Guangzhou, China). Rats were kept in standard cages under a 12‐hour light/dark cycle, and had free access to water and diet. After 1 week of acclimation, animals were anesthetized with an intraperitoneal injection of ketamine (50 mg/kg) and xylazine (6.7 mg/kg), and the left common carotid artery was subjected to a 2F balloon embolectomy catheterization as previously described.9 Briefly, a 2F balloon catheter (Baxter Healthcare Corp, USA) was inserted into the left external carotid artery and advanced into the common carotid artery. The balloon was inflated and then withdrawn to the entry point. The entire procedure was repeated 3 times. Finally, the left external carotid artery was ligated distally, and the incision was closed. A sham group of mice received only exposure of the left external carotid artery. In some local lentivirus short hairpin RNA (shRNA) delivery experiments, concentrated lentiviral solutions containing sh‐Cavin‐1 or control (50 μL) were injected into the arterial segment after balloon injury and incubated for 30 minutes. Then, the external artery was ligated, and common carotid blood flow was restored. At 14 days after surgery, the rats were euthanized and the carotid arteries were perfused with PBS for Western blot or with 4% paraformaldehyde for histological studies. All animal protocols were approved by the Sun Yat‐sen University Animal Care and Use Committee (China).
Histological and Immunofluorescent Staining
The carotid arteries were perfusion fixed with 10% formalin. Carotid artery sections (5 μm) were stained with hematoxylin and eosin and morphometric analysis was performed using 3 individual sections from the middle of each injured arterial segment, by an investigator who was kept blind to the experimental procedure. Cross‐sectional areas and ratios (intima/media) were carried out using the Scion Image Analysis software (Scion Co, Frederick, MA). The sections were incubated with mouse anti‐ubiquitin (sc‐166553, 1:500; Santa Cruz) or goat anti‐Cavin‐1 (sc‐82327; Santa Cruz) for immunohistochemical staining for evaluating the expression of Cavin‐1 and ubiquitin. The immunoreactive signal was visualized using secondary antibodies conjugated with Alexa‐Fluor‐488 or Alexa‐Fluor‐555 (1:500; Thermo Fisher Scientific, Waltham, MA) at 1:200 dilution. The coverslips were mounted with Dapi Fluoromount‐G to label nuclei (SouthernBiotech, Birmingham, AL) and fluorescent images were obtained with a confocal microscope (LSM510; Zeiss). Fluorescent signal intensity was quantified using relative optical density by ImageJ software (National Institutes of Health, Bethesda, MD). The intensity from sham tissues was set as the 100% baseline and the data from other groups were normalized and compared with sham/vehi/Scramble shRNA/pAD Vector group. Three to 5 sections per mouse from 3 to 4 mice were randomly selected for each group. In some experiments, 7 to 8 cells were randomly selected from each image for statistical analysis.
At the cellular level, the efficiency of shRNA transfection was confirmed by Western blot and the immunofluorescence staining was performed as previously described. Briefly, VSMCs were washed with cold PBS 3 times, fixed with cold methanol, and kept at −20°C for 15 minutes. The VSMCs were washed again with PBS for 5 minutes, and then incubated for 1 hour at room temperature with CAV‐1 diluted 1:200 with 3% BSA in PBS. After washing 3 times with PBS on a shaker for 10 minutes, the cells were incubated with Alexa‐Fluor‐488 (1:500) or LysoTracker Red (50 nmol/L) for 1 hour in the dark. Cells were then visualized using a confocal microscope (Zeiss LSM 800).
Drugs
Cycloheximide, angiotensin II (AngII), chloroquine, and MG132 were purchased from Sigma–Aldrich (St. Louis, MO). Cycloheximide, chloroquine, and AngII were dissolved in sterile PBS, while MG132 was dissolved in dimethyl sulfoxide to make the stock solution. All the drugs were stored as a stock solution at −20°C, and diluted in sterile PBS or culture medium to the appropriate concentration (cycloheximide, 25 μmol/L; chloroquine, 50 μmol/L; MG132, 10 μmol/L; and AngII, 100 nmol/L) immediately before administration. The final concentration of dimethyl sulfoxide in culture medium was <0.1%. In some cell culture experiments, we used 0.05% dimethyl sulfoxide in culture medium, which served as vehicle control group (Contr).
VSMCs Proliferation
VSMCs were isolated from rat thoracic aorta (Sprague–Dawley rats, 100–120 g) and prepared by enzymatic dispersion as described previously.20 Briefly, the blood vessel was opened by a longitudinal cut. The thin layer of endothelium was gently scraped with a surgical blade and the media was then stripped from the adventitia with ophthalmological tweezers and minced. The material from arteries was pooled and digested with 1.5 mg/mL of collagenase and 0.5 mg/mL of elastase (Sigma) for 60 minutes at 37°C. The cells were cultured in 10% fetal bovine serum–DMEM (Gibco, US) and incubated in a humidified incubator with 5% CO2 and 95% air. Culture medium was changed every 2 days, and VSMCs at passages 4 to 6 were identified by VSMC markers ɑ‐SM actin and used for experiments. The proliferation of VSMCs was measured using both cell counting and [3H]‐thymidine incorporation (China Institute of Atomic Energy, Beijing, China). For cell counting, the cell number was determined by the daily counting of viable cells (trypan blue exclusion). For [3H]‐thymidine incorporation, VSMCs were starved for 24 hours and then incubated in serum‐free medium supplemented with AngII (100 nmol/L) for another 24 hours with or without pretreatment with proteasome inhibitor MG132 (10 μmol/L) for 1 hour. Incorporated [3H]‐thymidine was precipitated with 10% trichloroacetic acid and quantitated with a liquid scintillation counter.21
Gene Silencing of Cavin‐1 With Small Interfering RNA or Adenovirus‐Mediated Cavin‐1 Overexpression
For in vivo experiments, concentrated Lenti Cavin‐1 shRNA‐GFP or scramble Lenti control shRNA‐GFP (1×107 TU/50 μL, ViGene Biosciences) was instilled into the arterial segment and incubated for 30 minutes after balloon injury. The transfection efficiency was confirmed by immunofluorescence. The inhibition effect of Lenti shRNA‐mediated knockdown on Cavin‐1 expression was tested at the protein levels in vivo.
In vitro, VSMCs were grown to 50% to 70% confluency in antibiotic‐free normal growth medium supplemented with 10% fetal bovine serum, and transfected with lentiviral particles of the Cavin‐1 shRNA (sc‐76294‐V; Santa Cruz) and scramble Lenti control shRNA (sc‐108060‐V; Santa Cruz) or adenoviral plasmid‐mediated Cavin‐1 cDNA (pAD Cavin‐1; ViGene Biosciences) and adenoviral plasmid‐mediated Vector (pAD Vector; ViGene Biosciences) according to the manufacturer's instruction. After 3 to 4 days of transient and stable transfections, the VSMCs were cultured under serum‐free condition for 24 hours and prepared for experiments treated with different drugs.
Western Blot
The injured carotid arteries or total cells were lysed in RIPA buffer (R0278; Sigma) and protease inhibitors (P8340; Sigma) and phosphatase Inhibitor Cocktail 1 (P2850; Sigma). For some experiments, plasma membrane was isolated by using ProteoExtract® Native Membrane Protein Extraction Kit (444810; Calbiochem, San Diego, CA) following the manufacturer's instructions as previously described.22 Briefly, after careful removal, the samples were washed with wash buffer followed by a mixture of protease inhibitor cocktail and cold extraction buffer I (1:200). After incubation for 10 minutes at 4°C under gentle agitation, the supernatant was discarded. A mixture of protease inhibitor cocktail and cold extraction buffer II (1:200) again was added to the pellet and incubated for 30 minutes at 4°C under gentle agitation. The supernatant containing the membrane fractions enriched in integral membrane and membrane‐associated proteins was used for further experiments. Insoluble material was removed by centrifugation at 10 000g at 4°C for 10 minutes, and protein concentration was determined using BCA assay (Thermo Fisher Scientific) following the manufacturer's instructions. Equal amounts of protein were separated on 10% SDS–PAGE and transferred to 0.2‐μm polyvinylidene difluoride membrane (Amersham, USA) in the Tris–glycine buffer containing 20% methanol. Membranes were washed with TBST, blocked with 10% skim milk for 1 hour, and incubated with the primary antibodies, such as goat anti‐Cavin‐1 (sc‐82327; Santa Cruz), mouse anti‐Cav‐1 (sc‐53564, 1:500; Santa Cruz), mouse anti‐ extracellular signal‐regulated kinase (ERK) (sc‐514302, 1:800; Santa Cruz), rabbit anti‐ERK phosphorylation (p‐ERK) (#4370, 1:1000 CST), mouse anti‐ubiquitin (sc‐166553, 1:500; Santa Cruz), or mouse anti‐β‐actin (sc‐130065, 1:1000; Santa Cruz). Membranes were washed in TBST buffer, probed with secondary antibodies, and visualized using the enhanced chemiluminescence kit (Thermo Pierce, USA). Densitometric analysis was performed using the densitometer (Gel Doc; Bio‐Rad) to quantify protein expression levels. The protein relative optical density indicated the relative optical density value of protein/β‐actin or phosphorylation protein/total protein and then normalized with the sham group or control group.
Immunoprecipitation
For immunoprecipitation, 200 μg of carotid arteries/VSMCs lysate protein was incubated with 1 μg of mouse anti‐ubiquitin, goat anti‐Cavin‐1 or mouse anti‐CAV‐1 antibody overnight at 4°C on a shaker. Twenty microliter protein A/G plus‐agarose (sc‐2003; Santa Cruz) was added to each sample and incubated for 4 hours. Immunocomplexes were collected by centrifugation at 1500 × g for 5 minutes at 4°C and washed 3 times with lysis buffer with 1.0 mL RIPA. After final washing, the supernatant was discarded and the pellet was resuspended in 40 μL of 1× electrophoresis sample buffer. The samples were boiled for 3 minutes and the beads were removed by centrifugation. Proteins were separated by 7.5% SDS‐PAGE and transferred to a nitrocellulose membrane. After blocking with 10% milk solution in Tris‐buffered saline with 0.1% Tween 20 (TBST) buffer, the nitrocellulose filters were respectively incubated with goat anti‐Cavin‐1 or mouse anti‐CAV‐1 antibody in TBST for 1 hour at room temperature. After washing for 3×5 minutes, the blots were incubated with secondary antibodies conjugated to horseradish peroxidase for an hour. The immunoreactive bands were visualized by enhanced chemiluminescence.22 When cell lysates were immunoprecipitated with anti‐CAV‐1 antibody and then immunoblotted with CAV‐1 and immunoglobulin G antibodies (sc‐51643; Santa Cruz), Cavin‐1 or CAV‐1 was used as internal control in the input and immunoglobulin G was used as negative control.
Real‐Time PCR
Total RNAs were isolated using Trizol (Invitrogen) according to the manufacturer's instruction. Polymerase chain reaction (PCR) was performed on the ABI Prism 7000 using One Step SYBR® PrimeScript™ Reverse Transcriptase (RT)‐PCR Kit (TaKaRa, Shanghai). The following primers were used: Caveolin‐1 (CAV‐1), forward primer CTACAAGCCCAACAACAAGGC and reverse primer AGGAAGCTCTTGATGCACGGT; Cavin‐1/polymerase I and transcript release factor, forward primer CGGCCAGATAAAGAAACTGG and reverse primer CCGGCAGCTTGACTTCAT; MMP9, forward primer GGAGACCTGAGAACCAATCTC and reverse primer TCCAATAGGTGATGTTGTCGT; GAPDH forward primer GCAAGTTCAACGGCACAG and reverse primer TACTCAGCACCAGCATCACC. Data were collected in the extension step. The specificity and identity of PCR products were verified using melting curve analysis following the PCR reaction, which distinguishes specific PCR products from the primer dimmer‐caused nonspecific PCR. Results were analyzed using the software provided by the manufacturer.
Scratch Assays and Transwell Experiments
VSMCs were seeded at 4.0×105 cells/mL. Two days after transfection with lentiviral particles of the Cavin‐1 shRNA or scramble shRNA, confluent cells were linearly scratched using a 20‐μL pipette chip. The scratched region was photographed immediately and 24 hours after scratching using a microscope equipped with a camera.23, 24 The photograph was traced to tracing paper, followed by the coloring of cells with image‐editing software (Fire Alpaca). Subsequently, the area with cells as a percentage of the total area was determined using the area measurement function of VHX‐5000 (Keyence, Osaka, Japan). The cell area in the scramble shRNA control 24 hours after scratching served as 100%, and the area in Cavin‐1 shRNA was recorded by the ratio to scramble shRNA group.
Transwell experiments were performed as previously described.25 In brief, cells were seeded into the upper chamber of the transwell chamber (Corning Life Sciences, Lowell, MA). After incubation with MG132 (10 μmol/L), Ab‐MMP‐9 (sc‐6841, 1:200; Santa Cruz), or Ab‐MMP‐2 (sc‐13594, 1:200; Santa Cruz) for 24 hours, the cells that had invaded the lower surface of the membranes were fixed with methanol for 10 minutes and stained with hematoxylin. The cells on the lower side of the membrane were counted and averaged in 6 high‐power fields with a light microscope.
Gelatin Zymography
VSMCs were transfected as described above. An equal amount of protein from VSMCs‐conditioned medium was loaded onto a 10% polyacrylamide gel with 1 mg/mL gelatin for zymography. The gel was then washed at room temperature for 30 minutes with 2.5% Triton X‐100 and subsequently incubated at 37°C for 24 hours in a buffer containing 10 mmol/L CaCl2, 0.01% NaN3, and 50 mmol/L Tris‐HCl (pH 7.5). The gel was stained with 0.2% Coomassie brilliant blue and photographed on a light box. Proteolysis was detected as a white zone in a dark blue field.16
Statistical Analysis
Group data are presented as mean±SEM. All statistical analysis of the data was performed using SPSS version 8.0 software. For experiments including only 2 groups, we used unpaired Student t test (2 tailed) after confirming that the groups were normally distributed with equal variances. Comparison among 3 or more groups was made using 1‐way ANOVA, Student‐Newman‐Keuls test was used to evaluate significant differences between groups when the 2 groups being compared were normally distributed with equal variances and the nonparametric tests (Mann–Whitney U test) when these conditions were not met. Comparison among different groups on outcomes measured serially in time was made using 1‐way or 2‐way repeated‐measures ANOVA. P<0.05 was considered statistically significant.
Results
Cavin‐1 Protein Expression Is Decreased in Balloon‐Injured Carotid Arteries
To investigate the role of Cavin‐1 in neointimal hyperplasia and the development of restenosis after angioplasty, we first evaluated the change of Cavin‐1 expression during neointimal formation in a rat carotid artery balloon injury model using Western blot. Compared with sham carotids, Cavin‐1 expression in carotid arteries was markedly downregulated at 2, 7, and 14 days after balloon injury (Figure 1A). To confirm that the vascular injury model was successful, we performed hematoxylin and eosin staining and found that balloon injury significantly stimulated arterial neointimal formation at 14 days after injury (Figure 1B), in agreement with previous studies.26, 27, 28 Only monolayer endothelial cells were seen along the inner lumina of sham carotid artery without neointima, while in the balloon‐injured group, the intima was thickening and became a multilayer compartment, and the elastic membrane was disrupted, indicating arterial intimal hyperplasia. Subsequently, we used immunofluorescent staining to examine the expression of Cavin‐1 protein at 14 days after surgery. As compared with the sham group, balloon injury markedly reduced Cavin‐1 protein expression in the injured carotid arterial wall or in a single VSMC after balloon injury group (Figure 1C), consistent with our abovementioned Western blot results (Figure 1A). However, we found that there was no statistical difference in Cavin‐1 mRNA between the 2 groups by using real‐time RT‐PCR (Figure 1D). Taken together, our results demonstrated that the decline of Cavin‐1 protein but not mRNA in the injured carotid arteries may be involved in neointimal hyperplasia.
Figure 1.
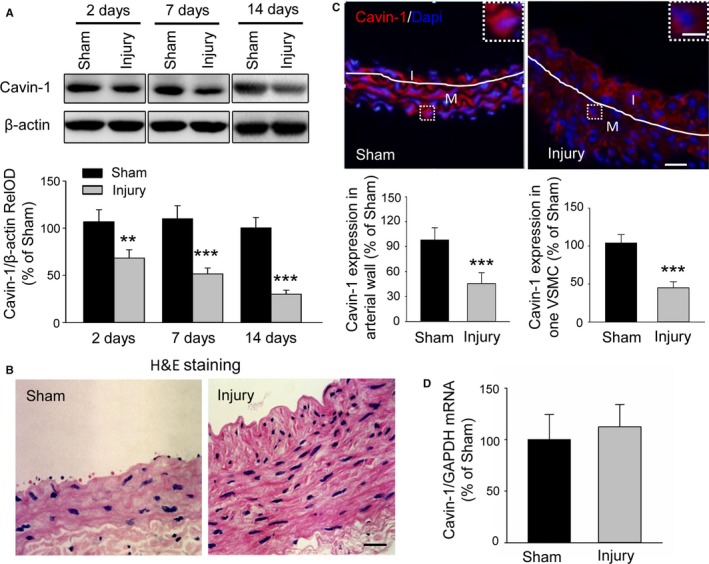
Balloon injury results in a decline of Cavin‐1 protein expression but not mRNA in injured carotid arteries. A, Western blot showing the effect of balloon injury on Cavin‐1 protein expression at 2, 7, and 14 days after balloon injury or sham operation to injured carotid artery. Cavin‐1 relative optical density (RelOD) indicates the percentage of the relative optical density value of Cavin‐1/β‐actin to sham group. n=3 to 4 mice/per group, **P<0.01, ***P<0.001, vs sham group. B, Typical photographs of hematoxylin and eosin (H&E) staining showing the vascular structure of carotid at 14 days in sham and injured groups. Scale bar: 25 μm. C, Representative immunofluorescent staining and the statistical histogram showing the expression of Cavin‐1 in the injured carotid arterial wall or in 1 single vascular smooth muscle cell (VSMC) (inset) at 14 days after sham operation or left balloon injury. Red: Cavin‐1, blue: 4',6‐ diamidino‐2‐phenylindole (Dapi). The white lines show the luminal border between the intima (I) or media (M) layer. The insets at the top right corner show higher‐magnification from the white boxes in the large images. Scale bars: 25 μm, inset: 5 μm. ***P<0.001, vs sham group, n=3 to 4 mice/per group and 3 to 5 images/per mice, and n=7 to 8 cells/per image. D, Cavin‐1 mRNA in carotid artery from sham or balloon‐injured group was determined by real‐time reverse transcriptase polymerase chain reaction (n=6–8).
Increased Proteasomal Degradation Accounting for Inhibition of Cavin‐1 Protein Expression in Injured Arteries
As our above RT‐PCR result showing no change of Cavin‐1 mRNA after balloon injury may not explain why Cavin‐1 protein expression was reduced in balloon‐injured carotids, we turned to some post‐transcription pathways to determine the possible mechanism, such as a protein degradation pathway. First, we performed in vitro cell culture experiments to test which protein degradation pathway was mainly involved in the degradation of Cavin‐1 protein (Figure 2A). Cycloheximide is a most common protein synthesis inhibitor, and is also used to test protein degradation.29, 30, 31 We used it to inhibit the new Cavin‐1 protein synthesis and then clarified which protein degradation pathway was involved in Cavin‐1 protein degradation at the cellular level. Cycloheximide (25 μmol/L) was pretreated in rat VSMCs for 1 hour to block the new Cavin‐1 synthesis, and then the proteasome inhibitor MG132 (10 μmol/L) or the lysosomal inhibitor chloroquine (50 μmol/L) was added for 24 hours. Cycloheximide alone produced a significant decline in Cavin‐1 expression as detected by Western blot assays, suggesting that the post‐transcription pathway played a critical role in the regulation of Cavin‐1 protein expression. The cycloheximide‐associated decline of Cavin‐1 was markedly blocked by the proteasomal inhibitor, but not by lysosomal inhibitor, indicating that the proteasomal degradation may result in the decreased expression of Cavin‐1. In order to further verify our results, we sequentially applied immunocytochemical staining and Western blot experiments to examine the expression of ubiquitinated protein and the correlation of protein ubiquitination and Cavin‐1 in vivo. The immunochemical staining demonstrated that the ubiquitinated protein levels were higher in balloon‐injured proliferated intima of arteries than in the sham group (Figure 2B). The following Western blot data also confirmed that the ubiquitinated protein was upregulated after balloon injury, while immunoprecipitation results indicated that Cavin‐1 protein was downregulated in the same tissues (Figure 2C). Taken together, our results indicated that the decreased expression of Cavin‐1 protein in balloon‐injured rat carotid arteries was attributed to the increased ubiquitin‐mediated protein degradation pathway.
Figure 2.
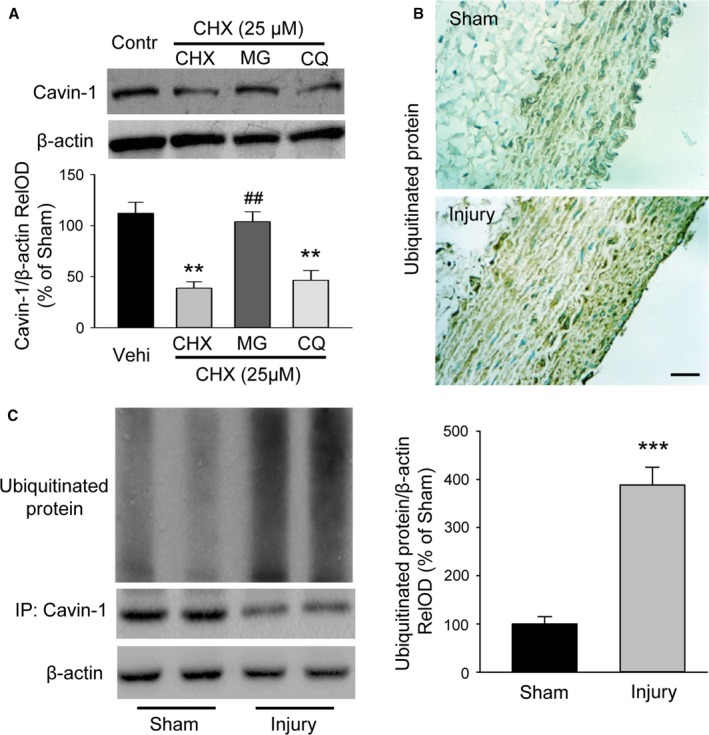
Increased proteasomal degradation results in the decrease of Cavin‐1 protein expression in injured arteries. A, Western blot data showing the degradation of Cavin‐1 protein was mediated by the ubiquitin proteasome pathway, but not by the lysosomal pathway. Rat aortic VSMCs were pretreated with or without cycloheximide (CHX, 25 μmol/L) for 1 hour, followed by treatment with lysosomal inhibitor chloroquine (CQ, 50 μmol/L) or proteasome inhibitor MG132 (10 μmol/L) for an additional 24 hours (n=5, **P<0.01, vs Vehi; ## P<0.01, vs CHX). B, Immunochemical staining showing the ubiquitinated protein in carotid artery at 2 weeks' postangioplasty from sham and injury groups. Scale bar: 25 μm. C, Immunoprecipitation (IP) and Western blot analysis revealed that the ubiquitinated protein levels were higher while the expression of Cavin‐1 were lower in the injured carotid than in sham group (n=6–8, ***P<0.001, vs sham). RelOD indicates relative optical density; Vehi, vehicle; VSMCs, vascular smooth muscle cells.
Inhibition of Cavin‐1 Protein Expression by Cavin‐1 shRNA Promotes Neointimal Formation in Injured Arteries In Vivo
To explore the role of the decline of Cavin‐1 protein in vascular neointimal hyperplasia, we locally injected Cavin‐1 shRNA lentiviral particles into the right external carotid artery after balloon‐injury to knock down Cavin‐1 protein expression and scramble shRNA as a control for shRNA transfection. The transfection efficiency was confirmed by immunofluorescence (Figure 3A, GFP). Hematoxylin and eosin staining with the same samples showed that the balloon‐injured vascular wall was thicker and the neointimal hyperplasia was more severe in the Cavin‐1 shRNA group than in the scramble shRNA group (Figure 3B). The histogram of intima‐to‐media ratio indicated that knockdown of Cavin‐1 by shRNA further enhanced neointima formation in Cavin shRNA‐treated rats at 14 days after local delivery (Figure 3C). Furthermore, the inhibition of Cavin‐1 shRNA on Cavin‐1 expression in balloon‐injured carotid artery was also confirmed by Western blot (Figure 3D). These results suggested that knockdown of Cavin‐1 protein by Cavin‐1 shRNA further promotes neointimal formation in injured arteries in vivo.
Figure 3.
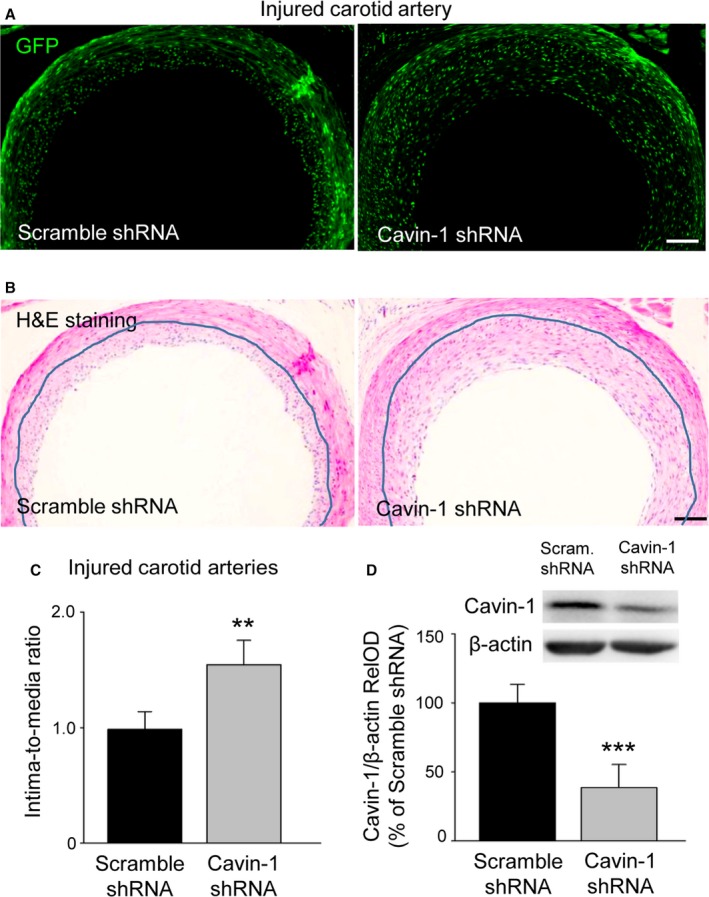
Inhibition of Cavin‐1 protein expression by lentivirus‐mediated Cavin‐1 shRNA promotes neointima formation in balloon‐injured arteries. A, Representative images of GFP immunofluorescence (green) of vessel cross‐sections indicating transfection efficiency of local delivery of lentivirus mediated Cavin‐1 shRNA plasmid at 14 days after injection into balloon‐injured arteries. B, H&E staining from the same samples showing the change of carotid vascular structure after injection of scramble shRNA or Cavin‐1 shRNA. The blue lines show the media or neointima layer. Scale bars (in A and B): 200 μm. C, The thickness histogram of intima‐to‐media ratio showing the effect of scramble shRNA or Cavin‐1 shRNA on neointima formation at 14 days after injection (1.544 vs 0.986; **P<0.01, vs scramble groups). D, Western blot of the injured carotid arteries showing a significant inhibition of Cavin‐1 protein expression in Cavin‐1 shRNA groups (n=6–8, ***P<0.001, vs scramble groups). GFP indicates green fluorescent protein; H&E, hematoxylin and eosin; RelOD, relative optical density; shRNA, short hairpin RNA.
Inhibition of Cavin‐1 Protein Expression Results in a Little Promotion of VSMC Proliferation
To validate the effects of Cavin‐1 decline on neointima formation and explore the mechanisms, we used Lenti‐mediated shRNA in VSMCs in vitro. The efficiency of Cavin‐1 shRNA on the inhibition of Cavin‐1 expression and the effect on neointima formation were investigated with rat in vivo experiments. We further examined the effect of Cavin‐1 shRNA in VSMCs. Western blot results indicated that transfection of Cavin‐1 shRNA in VSMCs not only inhibited Cavin‐1 expression but also reduced caveolin‐1 (CAV‐1) protein expression (Figure 4A). Then, we evaluated the effect of Cavin‐1 knockdown on the proliferation of VSMCs. Surprisingly, there was no significant difference in VSMC proliferation when VSMCs were cultivated under normal DMEM growth medium condition containing 10% fetal bovine serum (Figure 4B). On the other hand, we investigated the effect of Cavin‐1 decline on VSMCs phenotype and found that treatment of VSMCs with Cavin‐1 shRNA did not affect the expression of the contractile phenotype markers smooth muscle α‐actin and calponin compared with scramble shRNA group (Figure 4C). That is to say, Cavin‐1 did not play a critical role in normal basal cell proliferation and phenotype. We next sought to analyze the effect of scramble shRNA and Cavin‐1 shRNA on AngII‐mediated cell proliferation, which is determined by [3H]‐thymidine incorporation (Figure 4D). After transfected with shRNA for 3 to 4 days, VSMCs were first cultured under serum‐free condition for 24 hours and then treated with or without AngII (100 nmol/L) or MG132 (10 μmol/L) for another 24 hours. Consistent with previous studies, treatment with AngII in VSMCs also induced higher proliferation than in the only scramble shRNA control group. In addition, VSMCs treated with AngII and Cavin‐1 shRNA had significantly increased proliferation than VSMCs treated with AngII and scramble shRNA. Both of the increased proliferation of VSMCs could be partly inhibited by pretreatment of proteasome inhibitor MG132 (10 μmol/L) for 1 hour (Figure 4D). It is widely reported that ERK is involved in different cell proliferation.21, 32, 33 Subsequently, we examined the Cavin‐1 shRNA effects on p‐ERK during VSMC proliferation. We observed that under normal culture conditions, there was no significant difference of p‐ERK between the scramble shRNA and Cavin‐1 shRNA group (Figure 4E), whereas p‐ERK was dramatically phosphorylated in response to AngII treatment. To then assess whether Cavin‐1 shRNA could promote AngII signaling on MAP kinase, VSMCs were transfected with scramble shRNA or Cavin‐1 shRNA for 3 to 4 days before being cultured with serum‐free medium 24 hours and then stimulated with AngII for another 24 hours. As shown in Figure 4E, AngII‐mediated p‐ERK was further aggravated by the Cavin‐1 shRNA treatment; MG132 treatment of the cells partly inhibited AngII‐mediated p‐ERK with or without Cavin‐1 shRNA. These results indicated that inhibition of Cavin‐1 expression may have no effect in proliferation of VSMCs under normal physiological conditions, but could prompt the proliferation of VSMCs under pathophysiological conditions, such as balloon‐induced vascular injury. These effects could be defined as CAV‐1 dependent, which related to reducing CAV‐1 protein expression (Figure 4A).
Figure 4.
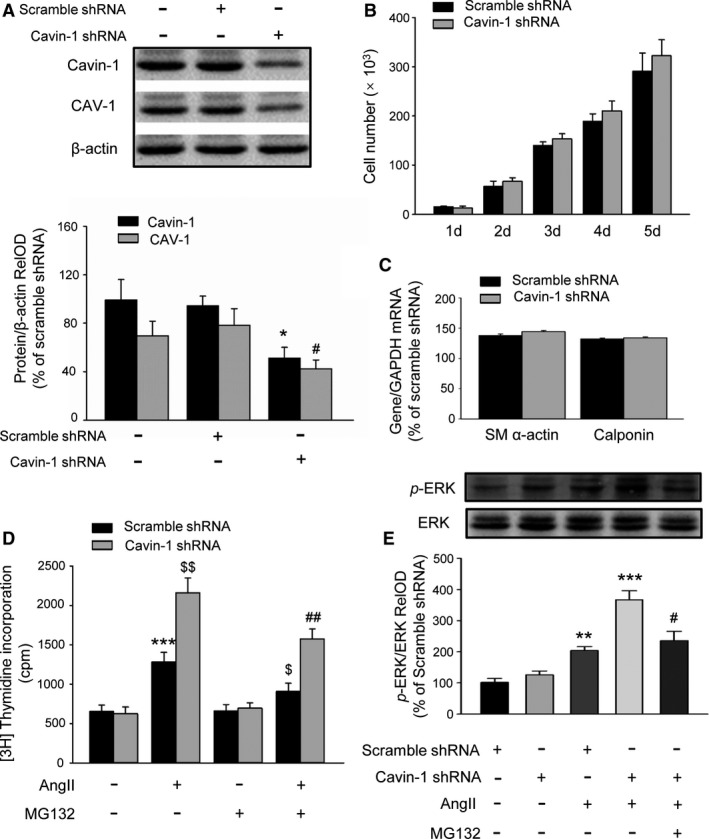
Inhibition of Cavin‐1 expression by shRNA promotes AngII‐induced VSMC proliferation via further activation of extracellular signal‐regulated kinase (ERK). A, Representative Western blot and statistical analysis showing the efficiency of Cavin‐1 shRNA on the inhibition of Cavin‐1 expression and even on CAV‐1 expression in VSMC culture (n=3–4/per group, Cavin‐1 expression: *P<0.05 vs Scramble shRNA alone; CAV‐1 expression: # P<0.05 vs Scramble shRNA alone). B, The histogram of VSMC number showing the effect of Cavin‐1 shRNA on VSMCs proliferation in normal growth culture medium (10% FBS DMEM). C, Knockdown of Cavin‐1 protein expression by Cavin‐1 shRNA had no effect on VSMC phenotypic marker α‐SM actin and calponin (n=6–8/per group). D, [3H]‐thymidine incorporation measured as count per minute (cpm) showing the effect of Cavin‐1 knockdown on VSMC proliferation in stimulation with AngII (100 nmol/L) for 24 hours with or without pretreatment with proteasome inhibitor MG132 (10 μmol/L) for 1 hour. n=9, ***P<0.001 vs Scramble shRNA alone, $ P<0.05, $$ P<0.01 vs Scramble shRNA treated with AngII and ## P<0.01 vs Cavin‐1 shRNA treat with AngII. E, Western blot showing the effect of Cavin‐1 knockdown on AngII‐induced ERK phosphorylation (p‐ERK) with or without pretreatment with MG132. p‐ERK/ERK RelOD indicated the percentage of the relative optical density value of p‐ERK/ERK to Scramble shRNA group. n=3, **P<0.01, ***P<0.001 vs Scramble shRNA treated with AngII, # P<0.05 vs Cavin‐1 shRNA treated with AngII. Ang II indicates angiotensin II; FBS, fetal bovine serum; RelOD, relative optical density; shRNA, short hairpin RNA; VSMCs, vascular smooth muscle cells.
Inhibition of Cavin‐1 Protein Expression Leads to More Promotion of VSMC Migration
To assess whether Cavin‐1 also influences neointima formation by regulating VSMC migration, we carried out the cell scratch wound‐healing assay. After creating a wound in normal cultured VSMCs with 10% fetal bovine serum DMEM, the percentage of closure was measured 24 hours later. The higher migration percentage in the Cavin‐1 shRNA group indicated that knockdown of Cavin‐1 facilitated VSMC migration and promoted gap closure (Figure 5A). Next, we performed transwell experiments to further explore the role of Cavin‐1 in VSMCs migration by measuring the cell numbers in the bottom surface. At 24 hours after seeding, the number of Dapi in Cavin‐1 shRNA group was much larger than that of scramble shRNA VSMCs (Figure 5B), suggesting that knockdown of Cavin‐1 promoted the VSMC migration under normal culture condition. As it is reported that matrix metallopeptidases (such as MMP‐2 or MMP‐9), a type of gelatinase, play an important role in VSMC migration,6, 7, 8 we defined the molecular mechanisms involved in the promotion of Cavin‐1 decline on VSMC migration. The facilitation of Cavin‐1 decline on VSMC migration was almost abolished by pretreatment with proteasome inhibitor MG132 (10 μmol/L) or gelatinase MMP‐9 antibody (1:200 dilution), but was not affected by pretreatment with gelatinase MMP‐2 antibody (1:200 dilution). To further investigate the mechanism of the influence of Cavin‐1 on the activity of gelatinase, we used gelatin zymography to test the effect of Cavin‐1 knockdown by Cavin‐1 shRNA in VSMCs. As shown in Figure 5C, MMP‐9 activity was significantly elevated while another type of gelatinase, MMP‐2, remained unchanged. Therefore, we concluded that Cavin‐1 affected VSMC migration by changing the activity of MMP‐9. Taken together, under the normal condition, inhibition of Cavin‐1 expression caused neither VSMC proliferation nor phenotypic change (Figure 4B and 4C), but resulted in the promotion of VSMC migration (Figure 5A and 5B). These data demonstrated that the effect of Cavin‐1 decline on VSMC migration may be stronger than that on VSMC proliferation.
Figure 5.
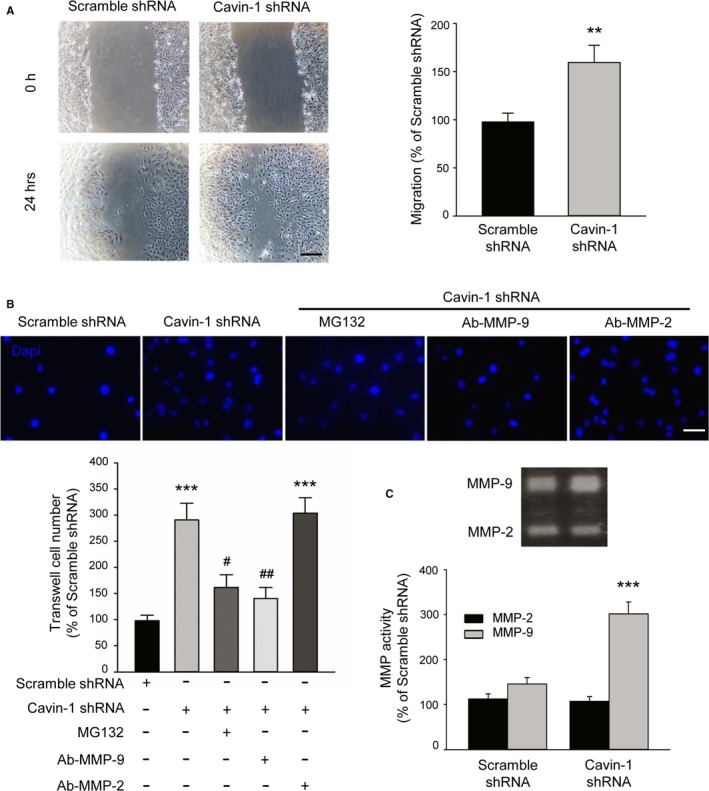
Inhibition of Cavin‐1 expression promotes the VSMC migration under normal culture condition via upregulation of MMP‐9. A, The scratch wound healing assay showing knockdown of Cavin‐1 expression by Cavin‐1 shRNA facilitates VSMC migration. Scale bar: 100 μm. The percentage of closure was measured at 24 hours after wounding (n=6, **P<0.01 vs scramble shRNA group). B, Representative micrographs of Dapi staining and statistical analysis showing the promotion of Cavin‐1 knockdown on VSMC migration at 24 hours after seeding cells to the upper chambers to the bottom surface of the transwell. Scale bar: 25 μm. Scramble shRNA and Cavin‐1 shRNA VSMCs were seeded into the upper chamber, treated with and without MG132 (10 μmol/L), Ab‐MMP‐9 (1:200 dilution), or Ab‐MMP‐2 (1:200 dilution) for 24 hours. (n=6, ***P<0.001 vs shRNA alone, # P<0.05, ## P<0.01 vs Cavin‐1 shRNA). C, The 48‐hour conditioned medium of VSMCs transiently transfected with Scramble shRNA and Cavin‐1 shRNA was collected and equal amounts of protein were subjected to gelatin zymography. Gelatin zymography results showing the effect of Cavin‐1 knockdown on the activity of MMP‐2 or MMP‐9 in VSMCs (n=6, ***P<0.001 vs scramble shRNA). Dapi indicates 4′,6‐diamidino‐2‐phenylindole; MMP‐9, matrix‐degrading metalloproteinase‐9; shRNA, short hairpin RNA; VSMCS, vascular smooth muscle cells.
Overexpression of Cavin‐1 Impairs the Proliferation and Migration of VSMCs
Our above data showed that knockdown of Cavin‐1 further promoted VSMC proliferation and migration and then significantly aggravated the neointimal hyperplasia induced by balloon angioplasty. We next investigated the effect of Cavin‐1 overexpression on VSMC proliferation and migration. Compared with the adenovirus‐mediated vector, using adenovirus‐mediated Cavin‐1 cDNA efficiency enriched the expression of Cavin‐1 in VSMCs, and the expression of CAV‐1 was also increased (Figure 6A). In agreement with the effect of Cavin‐1 knockdown, Cavin‐1 overexpression also had no effect on phenotypic change of VSMCs, and no influence on VSMCs proliferation in normal growth culture medium (10% fetal bovine serum). However, Cavin‐1 overexpression inhibited AngII‐induced [3H]‐thymidine incorporation (data not shown). p‐ERK plays a key role in AngII‐induced VSMCs proliferation. We found that inhibition of Cavin‐1 expression aggravated phosphorylation of ERK. On the other hand, the increase of p‐ERK induced by AngII was overwhelmingly reduced by Cavin‐1 overexpression (Figure 6B), while Cavin‐1 overexpression alone did not change the p‐ERK in rat VSMCs. Subsequently, the data from scratch assay and transwell experiment demonstrated that Cavin‐1 overexpression significantly inhibits VSMC migration (Figure 6C). By using gelatin zymography and RT‐PCR, we found that MMP‐9 protein and mRNA were reduced by Cavin‐1 overexpression (Figure 6D), suggesting that overexpression of Cavin‐1 blocked VSMC migration via attenuating the activity of MMP‐9.
Figure 6.
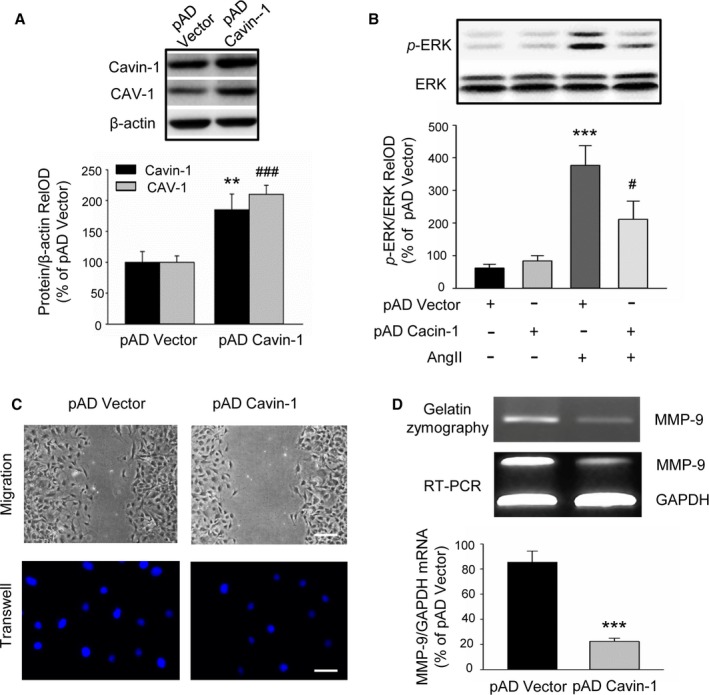
Effect of Cavin‐1 overexpression on VSMC proliferation and migration. A, Representative Western blot and statistical analysis showing the efficiency of adenovirus‐mediated Cavin‐1 cDNA on Cavin‐1 expression and even on CAV‐1 expression (n=3, **P<0.01 vs pAD‐Vector, Cavin‐1; ### P<0.001 vs pAD‐Vector,CAV‐1). B, p‐ERK were markedly increased after treatment VSMC with AngII (100 nmol/L) for 30 minutes, but significantly reduced by Cavin‐1 overexpression. Only overexpression of Cavin‐1 had no effect on VSMC ERK activity (n=3, ***P<0.001 vs pAD alone, # P<0.05 vs pAD Vector plus AngII). C, The scratch assay and transwell experiment showing the effect of overexpression Cavin‐1 on VSMC migration. Scale bars: upper, 50 μm, lower, 25 μm. D, The results from gelatin zymography and RT‐PCR showing the change of MMP‐9 protein and mRNA after Cavin‐1 overexpression. The 48‐hour conditioned medium from VSMCs transiently transfected with pAD Vector or pAD Cavin‐1 was collected and equal amounts of protein were subjected to gelatin zymography. Equal amounts of mRNA from VSMCs transfected with pAD Vector or pAD Cavin‐1 were subjected to RT‐PCR using primers to identify MMP‐9 or GAPDH. PCR products of the expected size were identified by agarose gel electrophoresis (n=6, ***P<0.001 vs pAD Vector). Ang II indicates angiotensin II; MMP‐9, matrix‐degrading metalloproteinase‐9; pAD, adenoviral plasmid; p‐ERK, extracellular signal‐related kinase phosphorylation; RelOD, relative optical density; RT‐PCR, reverse transcriptase polymerase chain reaction; VSMCs, vascular smooth muscle cells.
Inhibition of the Interaction of Cavin‐1 and CAV‐1 by Cavin‐1 shRNA Promotes the Lysosomal Degradation of CAV‐1
A large amount of research shows that CAV‐1, a key component in structure proteins of caveolae, plays an important role in the proliferation and migration of VSMCs.9, 34, 35 Recent studies have demonstrated that Cavin‐1 is also essential to the formation and function of caveolae.10, 11, 36 Since Cavin‐1 and CAV‐1 are always expressed together, we wondered whether there is a relationship between them. According to our abovementioned results (Figures 4A and 6A), we found Cavin‐1 shRNA or cDNA not only decreased or increased Cavin‐1 expression, but also reduced or elevated CAV‐1 expression. Then we continued to examine the effect of Cavin‐1 knockdown on CAV‐1 mRNA at the transcriptional level. The RT‐PCR result indicated that inhibition of Cavin‐1 expression by Cavin‐1 shRNA did not cause any change in CAV‐1 mRNA level of VSMCs (Figure 7A), but promoted the shift of CAV‐1 expression from the cell membrane to the cytoplasm (Figure 7B), thus confirming the effect of Cavin‐1 on CAV‐1 expression. Subsequently, our result of co‐immunoprecipitation of Cavin‐1 and CAV‐1 in VSMCs exhibited that Cavin‐1 knockdown decreased the interrelationship of Cavin‐1 and CAV‐1, and this effect was blocked by inhibition of protein degradation via MG132. It not only confirmed that inhibition of Cavin‐1 expression by Cavin‐1 shRNA resulted in the decline of CAV‐1 expression (Figure 7C), but also suggested the natural and structural interaction between the 2 molecules. This association was blocked by proteasome inhibitor MG132, indicating the regulation of Cavin‐1 on CAV‐1 expression is in the aspect of lysosomal degradation. In addition, we further investigated the relationship between CAV‐1 and Cavin‐1 by immunofluorescent staining. Compared with the scramble shRNA control group, Cavin‐1 shRNA not only inhibited the expression of CAV‐1 protein (Figure 7D, green), but also upregulated lysosomal degradation (LysoTracker Red, red). The increased colocalized signals (yellow) in the Cavin‐1 shRNA group revealed that more CAV‐1 protein was degraded by the lysosomal pathway. Surprisingly, this effect could be blocked by pretreatment of MG132 (Figure 7D, bottom images). Therefore, we proposed that the proteasomal degradation of Cavin‐1 also interacted with the lysosomal degradation of CAV‐1. Furthermore, our work indicated that the interaction between Cavin‐1 and CAV‐1 can maintain the stability of each other, and inhibition of Cavin‐1 expression can also promote CAV‐1 degradation via the lysosomal degradation pathway.
Figure 7.
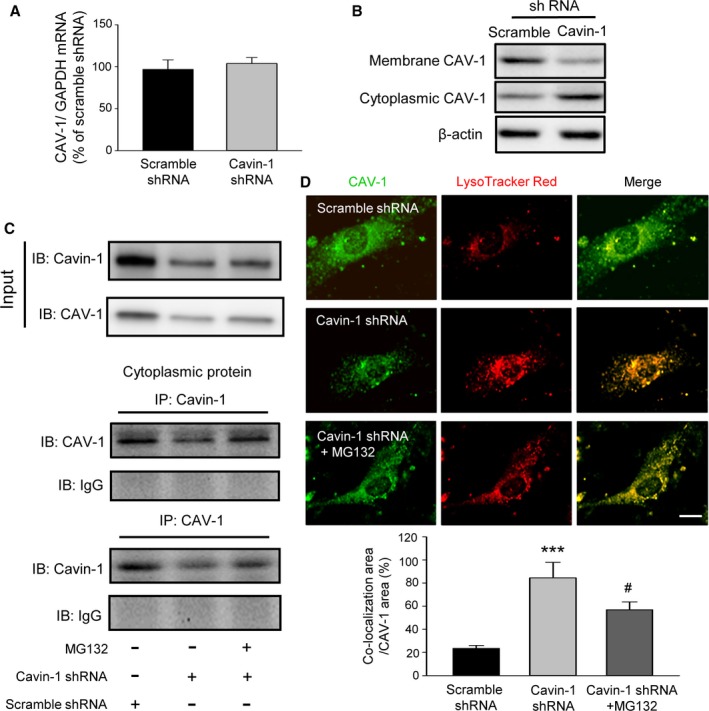
Inhibition of the interaction of Cavin‐1 and CAV‐1 by Cavin‐1 shRNA promotes the lysosomal degradation of CAV‐1. A, RT‐PCR results showed that Cavin‐1 knockdown by Cavin‐1 shRNA had no effect on CAV‐1 mRNA expression in VSMCs (n=5). B, Western blot results from VSMC plasma membrane and cytoplasmic fractions samples showing the promotion of Cavin‐1 knockdown on the moving of CAV‐1 from plasma membrane to cytoplasm. C, Immunoprecipitation was used to assess the effect of Cavin‐1 knockdown with or without MG132 on the interactions of Cavin‐1 and CAV‐1 with or without Cavin‐1 knockdown. Cavin‐1 or CAV‐1 was used as internal control in the input and IgG was used as negative control. Inhibition of Cavin‐1 decreased the interaction, and this effect was blocked by inhibition of protein degradation via MG132. D, Immunofluorescence staining showing the expression of CAV‐1 (green), which mainly distributed in plasma membrane of VSMCs and LysoTracker (red) represented lysosomal degradation of CAV‐1. Scale bar: 5 μm. The statistical analysis histogram illustrates that the percentage of the co‐localization area (yellow, CAV‐1 and LysoTracker) to CAV‐1 (green) was increased after knockdown of Cavin‐1, but was reversed by proteasome inhibitor MG132. n=3, ***P<0.001 vs Scramble shRNA, # P<0.05 vs Cavin‐1 shRNA. IgG indicates immunoglobulin G; IP, immunoprecipitation; RT‐PCR, reverse transcriptase polymerase chain reaction; VSMCs, vascular smooth muscle cells.
Discussion
In the present study, we investigated the role of Cavin‐1 in neointimal hyperplasia after balloon injury. First, we revealed that the decrease in Cavin‐1 protein after vascular injury was attributed to the increased ubiquitin‐mediated proteosomal degradation pathway. Second, we showed that inhibition of Cavin‐1 expression by Cavin‐1 shRNA exacerbated neointimal formation in injured carotid artery. By inhibition or overexpression of Cavin‐1 in cultured VSMCs, we found that Cavin‐1 can regulate AngII induced‐VSMC proliferation or migration via the ERK or MMP‐9 signal pathway, respectively. Furthermore, the influence of Cavin‐1 was stronger on migration than on proliferation. Finally, our results suggested that inhibition of Cavin‐1 expression promoted CAV‐1 degradation via a lysosomal degradation pathway, clarifying that interaction between Cavin‐1 and CAV‐1 can maintain the stability of each other.
Neointimal formation is the main cause of percutaneous coronary intervention treatment failure in ischemic heart disease, and the resulting restenosis has made a significant impact on the patency of percutaneous coronary intervention.37, 38 The proliferation and migration of VSMCs play crucial roles in neointimal formation and restenosis after angioplasty.39 The ubiquitin–proteasome system (UPS) is the major pathway for intracellular proteins degradation by which cells get rid of misfolded proteins.40 A growing body of evidence has implicated UPS in neointimal formation.18 Recent studies have identified increased activity of ubiquitination–proteasome in neointimal areas and furthermore, inhibition of UPS by MG132 effectively reduced neointima formation in vivo, which corresponds to strong antiproliferative effects in vitro and in vivo, suggesting the UPS as a new target in the prevention of vascular restenosis.41, 42 However, the specific protein targets of UPS remain largely unknown. In our present study, the results from immunochemical staining and Western blot demonstrated that the ubiquitinated protein levels were higher in balloon‐injured proliferated intima of arteries than in the sham group (Figure 2B and 2C) and the immunoprecipitation data indicated that Cavin‐1 protein was downregulated in the same tissues. These data suggest that Cavin‐1 may be one of proteins targeted for ubiquitination in neointima formation. This result was consistent with another study showing that the phosphoinositide‐binding site of Cavin‐1 acts as the major ubiquitination site responsible for Cavin‐1 turnover.19 We further explored whether the change in expression of UPS affected the expression and function of Cavin‐1 in the cultured VSMCs. Indeed, we found that the promotion of VSMC proliferation and migration by inhibition of Cavin‐1 expression was reversed by pretreatment with proteasome inhibitor MG132 (Figures 4D, 4E, and 5B). Our data further confirmed that inhibition of Cavin‐1 by shRNA in vivo aggravated the neointimal hyperplasia after vascular injury, with obvious thickening of carotid intima according to the neointima‐to‐media ratio. It was also in agreement with the results from Cavin‐1 knockout mice showing that the media thickness was increased in KO arteries compared with WT arteries.43 Therefore, our results indicated that Cavin‐1 could be a target protein for ubiquitination, the inhibition of which may prevent vascular intimal hyperplasia.
Neointimal formation is characterized by medial VSMC proliferation and migration from the media to the neointima, which leads to blood vessel occlusion.27, 44 Our studies showed that the inhibition of Cavin‐1 protein expression affected both proliferation and migration of VSMCs. Reduction of Cavin‐1 expression induced no change in cell numbers or phenotypic markers in normally cultured VSMCs (Figure 4B and 4C). However, inhibition of Cavin‐1 displayed the promotion of VSMC migration under the same conditions (Figure 5A and 5B). We speculated that the influence of the role of Cavin‐1 in VSMC migration may be stronger than that in proliferation. Interestingly, it was reported that the decline of Cavin‐1 expression is a very common phenomenon in some tumors. Some studies have reported recently that Cavin‐1 protein was downregulated in breast cancer,45 prostate cancer,16 tumorigenic human bronchial epithelial (HBE),46 and non–small cell lung cancer samples.47 Taken together, loss of Cavin‐1 expression has been reported to be relevant to cell proliferation and migration in the pathogenesis of various cancers and neointimal formation.
Subsequently, we applied AngII in vitro cultures to simulate the significant increase of AngII in blood after vascular injury.48 Our results showed that AngII‐induced VSMC proliferation was further enhanced by knockdown of Cavin‐1 via the increase of p‐ERK, while inhibited by overexpression of Cavin‐1 via decrease of ERK phosphorylation. These effects could be inhibited by proteasome inhibitor MG132 (Figures 4E and 6B). The above data indicated that the higher proliferation of injured vascular VSMCs could be accelerated by downregulation of Cavin‐1 expression and inhibited by inhibition of UPS. We proposed that the increment of proteasome pathway after vascular injury downregulates Cavin‐1 expression, and promotes cell proliferation. Cavin‐1 was found to be co‐localized with CAV‐1, and the change of Cavin‐1 was always accompanied by CAV‐1. In addition, CAV‐1 has been recognized as a proliferative inhibitor of VSMCs,49 and the underlying mechanism of CAV‐1 in VSMC proliferation has already been well demonstrated.50, 51 Therefore, VSMC proliferation induced by inhibition of Cavin‐1 could be the results from the decrease in CAV‐1.
It is well known that caveolae play key roles in the biology of the cell. Loss of Cavin‐1/polymerase I and transcript release factor resulted in decreased caveolae formation and the altered associations of signaling pathways within the membrane. Within caveolae, Cavin‐1 is co‐localized with CAV‐1.52 CAV‐1 has been reported to interact directly with ERK through its residues 32 to 95. Peptides derived from this region inhibit the kinase activity of ERK.53 Interestingly, although Cavin‐1 and CAV‐1 are present close to each other, there are no binding domains to allow the direct interaction with each other.54 However, Cavin‐1 also appears to interact with CAV‐1 during cellular senescence, and phosphorylation of Cavin‐1 at Ser365 and Ser366 is required for this interaction.55 This means that there is indirect interaction between Cavin‐1 and CAV‐1. Furthermore, studies from the same group reported that the interaction of Cavin‐1/polymerase I and transcript release factor with platelet‐derived growth factor receptors leads to inhibition of p‐ERK and the interaction is increased in senescent cells.56 Our data demonstrated that knockdown of Cavin‐1 expression induced the downregulation of CAV‐1, and thus we speculated that the mechanism of Cavin‐1 on inhibition of VSMC proliferation may be CAV‐1 dependent. However, the exact mechanism still needs further investigation.
Several studies have investigated the role of Cav‐1 in migration. However, the results were inconclusive. For example, in endothelial cells and fibroblasts, loss of Cav‐1 is known to affect cell polarization and impede directional migration via Rho family small GTPases,57, 58 whereas other studies have demonstrated that compared with wild‐type cells, VSMCs from CAV‐1‐deficient mice showed an increase in migration.59 Therefore, the role of CAV‐1 in migration may be dependent on the type of expressing cells. In the present study, our results showed that reduced CAV‐1 expression induced by Cavin‐1 shRNA promoted VSMC migration. Cavin‐1 expression decreased the migration of PC3 prostate cancer cells. Although Cavin‐1 is always associated with CAV‐1, some studies using PC3 cell line, which expresses CAV‐1 but lacks Cavin‐1, reveal the different role of Cavin‐1 and CAV‐1 in cell migration.15, 16 Those results showed that Cavin‐1 inhibited cell migration by reduction of MMP‐9. This was shown to be independent of CAV‐1‐related caveola formation. Our results confirmed the effect of Cavin‐1 on the activity of MMP‐9 and proposed that the mechanism of promoting VSMC migration by Cavin‐1 inhibition may be by a CAV‐1‐dependent (via influence of CAV‐1 protein expression) and ‐independent (via increasing MMP‐9 activity) pathway, respectively. Furthermore, using RT‐PCR, we found that Cavin‐1 can inhibit the expression of MMP‐9 mRNA at the transcriptional level, thereby attenuating the activity of MMP‐9 (Figure 6D). However, recent studies implied that CAV‐1 is also linked to MMP‐9 activity. One of them reported that the MMP‐9 activity in serum from the ligation model was higher in CAV‐1 knockout mice.34 This discrepancy may be because they did not measure the Cavin‐1 expression, and could not reveal the different role of Cavin‐1 and CAV‐1 in VSMC migration in this model. It did not exclude the possibility that inhibition of Cavin‐1 in Cav‐1 knockout might induce the increase of MMP‐9 activity. However, other studies also showed that CAV‐1 promoted Ewing sarcoma metastasis via regulating MMP‐9 expression.60 In VSMCs Cavin‐1 and CAV‐1 are always associated with each other, so we had no direct evidence to show whether this effect of Cavin‐1 in MMP‐9 activity is CAV‐1 dependent or not. Hence, we are very cautious about drawing a conclusion about the role of CAV‐1 in VMSC proliferation and migration.
The most important limitation of the present study is that we cannot distinguish the effects of Cavin‐1 and CAV‐1 on the function of VSMCs. Since Cavin‐1 and CAV‐1 always interrelated together and influenced each other, knocking down either Cavin‐1 or CAV‐1 is responsible for the decrease in expression of the other protein. Moreover, a recent study has shown that Cavin‐1 and CAV‐1 are both required for cell proliferation and migration in rhabdomyosarcoma and indicated that the cooperation between Cavin‐1 and CAV‐1 underlies the cell growth in myogenic tumors.61 In the present study, we found that decreased Cavin‐1 expression did not inhibit the CAV‐1 mRNA at the transcript level, which was confirmed using the Cavin‐1 knockout mice.36 Also, our data demonstrated that the lysosomal degradation pathway was in response to the decrease in CAV‐1. Our and others' results confirmed that the degradation pathway differs in Cavin‐1 and CAV‐1, via the proteasomal pathway and lysosomal pathway, respectively.19, 21 Moreover, Cavin‐1 was co‐localized with CAV‐1, and Cavin‐1 expression modulates the subcellular localization of CAV‐1 in migrating cells.15 Furthermore, our studies confirmed the hypothesis that inhibition of Cavin‐1 expression can promote CAV‐1 degradation via the lysosomal degradation pathway (Figure 7C and 7D).
In summary, we have explored the mechanism of the decrease in Cavin‐1 protein via the proteasomal degradation pathway after balloon injury–induced neointimal proliferation and suggested that Cavin‐1 may act as a novel therapeutic target for the treatment of restenosis.
Sources of Funding
This work was supported by National Natural Science Foundation of China (81270377, 81371198), Science and Technology Department of Guangdong (2015A020212020) and Natural Science Foundation of Guangdong (2014A030313062).
Disclosures
None.
Acknowledgments
We thank Dr Madhuvika Murugan (Rutgers University) and Dr Suzhao Li (University of Colorado Denver) for English revision.
(J Am Heart Assoc. 2017;6:e005754 DOI: 10.1161/JAHA.117.005754.)28751541
References
- 1. Nagaraja V, Ooi SY, Nolan J, Large A, De BM, Ludman P, Bagur R, Curzen N, Matsukage T, Yoshimachi F, Kwok CS, Berry C, Mamas MA. Impact of incomplete percutaneous revascularization in patients with multivessel coronary artery disease: a systematic review and meta‐analysis. J Am Heart Assoc. 2016;5:e004598 DOI: 10.1161/JAHA.116.004598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alfonso F, Byrne RA, Rivero F, Kastrati A. Current treatment of in‐stent restenosis. J Am Coll Cardiol. 2014;63:2659–2673. [DOI] [PubMed] [Google Scholar]
- 3. Misumida N, Aoi S, Saeed M, Ota T, Eda T, Umeda H, Kanei Y. The role of angiographic follow‐up after percutaneous coronary intervention. Int J Cardiol. 2016;222:911–920. [DOI] [PubMed] [Google Scholar]
- 4. Siontis GC, Stefanini GG, Mavridis D, Siontis KC, Alfonso F, Perez‐Vizcayno MJ, Byrne RA, Kastrati A, Meier B, Salanti G, Juni P, Windecker S. Percutaneous coronary interventional strategies for treatment of in‐stent restenosis: a network meta‐analysis. Lancet. 2015;386:655–664. [DOI] [PubMed] [Google Scholar]
- 5. Simon DI. Inflammation and vascular injury: basic discovery to drug development. Circ J. 2012;76:1811–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (MMP)‐3 activates MMP‐9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol. 2011;31:e35–e44. [DOI] [PubMed] [Google Scholar]
- 7. Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, Greene WC, Valente AJ. Interleukin‐18‐induced human coronary artery smooth muscle cell migration is dependent on NF‐kappaB‐ and AP‐1‐mediated matrix metalloproteinase‐9 expression and is inhibited by atorvastatin. J Biol Chem. 2006;281:15099–15109. [DOI] [PubMed] [Google Scholar]
- 8. Cho A, Reidy MA. Matrix metalloproteinase‐9 is necessary for the regulation of smooth muscle cell replication and migration after arterial injury. Circ Res. 2002;91:845–851. [DOI] [PubMed] [Google Scholar]
- 9. Liu HM, Zhao XF, Guo LN, Tan Z, Wang TH. Effects of caveolin‐1 on the 17beta‐estradiol‐mediated inhibition of VSMC proliferation induced by vascular injury. Life Sci. 2007;80:800–812. [DOI] [PubMed] [Google Scholar]
- 10. Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, Walser P, Abankwa D, Oorschot VM, Martin S, Hancock JF, Parton RG. PTRF‐Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu L, Pilch PF. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem. 2008;283:4314–4322. [DOI] [PubMed] [Google Scholar]
- 12. Fu Y, Moore XL, Lee MK, Fernandez‐Rojo MA, Parat MO, Parton RG, Meikle PJ, Sviridov D, Chin‐Dusting JP. Caveolin‐1 plays a critical role in the differentiation of monocytes into macrophages. Arterioscler Thromb Vasc Biol. 2012;32:e117–e125. [DOI] [PubMed] [Google Scholar]
- 13. Jia G, Sowers JR. Caveolin‐1 in cardiovascular disease: a double‐edged sword. Diabetes. 2015;64:3645–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nwosu ZC, Ebert MP, Dooley S, Meyer C. Caveolin‐1 in the regulation of cell metabolism: a cancer perspective. Mol Cancer. 2016;15:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hill MM, Daud NH, Aung CS, Loo D, Martin S, Murphy S, Black DM, Barry R, Simpson F, Liu L, Pilch PF, Hancock JF, Parat MO, Parton RG. Co‐regulation of cell polarization and migration by caveolar proteins PTRF/Cavin‐1 and caveolin‐1. PLoS One. 2012;7:e43041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aung CS, Hill MM, Bastiani M, Parton RG, Parat MO. PTRF‐cavin‐1 expression decreases the migration of PC3 prostate cancer cells: role of matrix metalloproteinase 9. Eur J Cell Biol. 2011;90:136–142. [DOI] [PubMed] [Google Scholar]
- 17. Meiners S, Laule M, Rother W, Guenther C, Prauka I, Muschick P, Baumann G, Kloetzel PM, Stangl K. Ubiquitin‐proteasome pathway as a new target for the prevention of restenosis. Circulation. 2002;105:483–489. [DOI] [PubMed] [Google Scholar]
- 18. Qin Z, Cui B, Jin J, Song M, Zhou B, Guo H, Qian D, He Y, Huang L. The ubiquitin‐activating enzyme E1 as a novel therapeutic target for the treatment of restenosis. Atherosclerosis. 2016;247:142–153. [DOI] [PubMed] [Google Scholar]
- 19. Tillu VA, Kovtun O, McMahon KA, Collins BM, Parton RG. A phosphoinositide‐binding cluster in cavin1 acts as a molecular sensor for cavin1 degradation. Mol Biol Cell. 2015;26:3561–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Christen T, Bochaton‐Piallat ML, Neuville P, Rensen S, Redard M, van Eys G, Gabbiani G. Cultured porcine coronary artery smooth muscle cells. A new model with advanced differentiation. Circ Res. 1999;85:99–107. [DOI] [PubMed] [Google Scholar]
- 21. Tan Z, Zhou LJ, Li Y, Cui YH, Xiang QL, Lin GP, Wang TH. E(2)‐BSA activates caveolin‐1 via PI(3)K/ERK1/2 and lysosomal degradation pathway and contributes to EPC proliferation. Int J Cardiol. 2012;158:46–53. [DOI] [PubMed] [Google Scholar]
- 22. Tan Z, Zhou LJ, Mu PW, Liu SP, Chen SJ, Fu XD, Wang TH. Caveolin‐3 is involved in the protection of resveratrol against high‐fat‐diet‐induced insulin resistance by promoting GLUT4 translocation to the plasma membrane in skeletal muscle of ovariectomized rats. J Nutr Biochem. 2012;23:1716–1724. [DOI] [PubMed] [Google Scholar]
- 23. Zheng S, Huang J, Zhou K, Xiang Q, Zhang Y, Tan Z, Simoncini T, Fu X, Wang T. Progesterone enhances vascular endothelial cell migration via activation of focal adhesion kinase. J Cell Mol Med. 2012;16:296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zheng S, Huang J, Zhou K, Zhang C, Xiang Q, Tan Z, Wang T, Fu X. 17beta‐estradiol enhances breast cancer cell motility and invasion via extra‐nuclear activation of actin‐binding protein ezrin. PLoS One. 2011;6:e22439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang P, Xu J, Zheng S, Huang J, Xiang Q, Fu X, Wang T. 17beta‐estradiol down‐regulates lipopolysaccharide‐induced MCP‐1 production and cell migration in vascular smooth muscle cells. J Mol Endocrinol. 2010;45:87–97. [DOI] [PubMed] [Google Scholar]
- 26. Lee CS, Kwon YW, Yang HM, Kim SH, Kim TY, Hur J, Park KW, Cho HJ, Kang HJ, Park YB, Kim HS. New mechanism of rosiglitazone to reduce neointimal hyperplasia: activation of glycogen synthase kinase‐3beta followed by inhibition of MMP‐9. Arterioscler Thromb Vasc Biol. 2009;29:472–479. [DOI] [PubMed] [Google Scholar]
- 27. Afzal TA, Luong LA, Chen D, Zhang C, Yang F, Chen Q, An W, Wilkes E, Yashiro K, Cutillas PR, Zhang L, Xiao Q. NCK associated protein 1 modulated by miRNA‐214 determines vascular smooth muscle cell migration, proliferation, and neointima hyperplasia. J Am Heart Assoc. 2016;5:e004629 DOI: 10.1161/JAHA.116.004629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang W, Zhang X, Gonzalez‐Cobos JC, Stolwijk JA, Matrougui K, Trebak M. Leukotriene‐C4 synthase, a critical enzyme in the activation of store‐independent Orai1/Orai3 channels, is required for neointimal hyperplasia. J Biol Chem. 2015;290:5015–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ichikawa T, Li J, Dong X, Potts JD, Tang DQ, Li DS, Cui T. Ubiquitin carboxyl terminal hydrolase L1 negatively regulates TNFalpha‐mediated vascular smooth muscle cell proliferation via suppressing ERK activation. Biochem Biophys Res Commun. 2010;391:852–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Salcedo A, Mayor F Jr, Penela P. Mdm2 is involved in the ubiquitination and degradation of G‐protein‐coupled receptor kinase 2. EMBO J. 2006;25:4752–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geng J, Xia L, Li W, Zhao C, Dou F. Cycloheximide treatment causes a ZVAD‐sensitive protease‐dependent cleavage of human tau in drosophila cells. J Alzheimers Dis. 2016;49:1161–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang F, Ren X, Zhao M, Zhou B, Han Y. Angiotensin‐(1‐7) abrogates angiotensin II‐induced proliferation, migration and inflammation in VSMCs through inactivation of ROS‐mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci Rep. 2016;6:34621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng H, Liao ZL, Ning LH, Chen HY, Wei SS, Yang XC, Guo H. Alendronate‐anchored PEGylation of ceria nanoparticles promotes human hepatoma cell proliferation via AKT/ERK signaling pathways. Cancer Med. 2017;6:374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma YD, Thiyagarajan V, Tsai MJ, Lue SI, Chia YC, Shyue SK, Weng CF. Pyrogallol abates VSMC migration via modulation of caveolin‐1, matrix metalloproteinase and intima hyperplasia in carotid ligation mouse. Environ Toxicol Pharmacol. 2016;48:63–75. [DOI] [PubMed] [Google Scholar]
- 35. Kiyan J, Smith G, Haller H, Dumler I. Urokinase‐receptor‐mediated phenotypic changes in vascular smooth muscle cells require the involvement of membrane rafts. Biochem J. 2009;423:343–351. [DOI] [PubMed] [Google Scholar]
- 36. Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008;8:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rodriguez AE, Pavlovsky H, Del Pozo JF. Understanding the outcome of randomized trials with drug‐eluting stents and coronary artery bypass graft in patients with multivessel disease: a review of a 25‐year journey. Clin Med Insights Cardiol. 2016;10:195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kwok CS, Hulme W, Olier I, Holroyd E, Mamas MA. Review of early hospitalisation after percutaneous coronary intervention. Int J Cardiol. 2017;227:370–377. [DOI] [PubMed] [Google Scholar]
- 39. Ho‐Tin‐Noe B, Le DJ, Gomez D, Louedec L, Vranckx R, El‐Bouchtaoui M, Legres L, Meilhac O, Michel JB. Early atheroma‐derived agonists of peroxisome proliferator‐activated receptor‐gamma trigger intramedial angiogenesis in a smooth muscle cell‐dependent manner. Circ Res. 2011;109:1003–1014. [DOI] [PubMed] [Google Scholar]
- 40. Herrmann J, Soares SM, Lerman LO, Lerman A. Potential role of the ubiquitin‐proteasome system in atherosclerosis: aspects of a protein quality disease. J Am Coll Cardiol. 2008;51:2003–2010. [DOI] [PubMed] [Google Scholar]
- 41. Herrmann J, Edwards WD, Holmes DR Jr, Shogren KL, Lerman LO, Ciechanover A, Lerman A. Increased ubiquitin immunoreactivity in unstable atherosclerotic plaques associated with acute coronary syndromes. J Am Coll Cardiol. 2002;40:1919–1927. [DOI] [PubMed] [Google Scholar]
- 42. Kovalcsik E, Antunes RF, Baruah P, Kaski JC, Dumitriu IE. Proteasome‐mediated reduction in proapoptotic molecule Bim renders CD4(+)CD28null T cells resistant to apoptosis in acute coronary syndrome. Circulation. 2015;131:709–720. [DOI] [PubMed] [Google Scholar]
- 43. Sward K, Albinsson S, Rippe C. Arterial dysfunction but maintained systemic blood pressure in cavin‐1‐deficient mice. PLoS One. 2014;9:e92428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schlosser A, Pilecki B, Hemstra LE, Kejling K, Kristmannsdottir GB, Wulf‐Johansson H, Moeller JB, Fuchtbauer EM, Nielsen O, Kirketerp‐Moller K, Dubey LK, Hansen PB, Stubbe J, Wrede C, Hegermann J, Ochs M, Rathkolb B, Schrewe A, Bekeredjian R, Wolf E, Gailus‐Durner V, Fuchs H, de Hrabe AM, Lindholt JS, Holmskov U, Sorensen GL. MFAP4 promotes vascular smooth muscle migration, proliferation and accelerates neointima formation. Arterioscler Thromb Vasc Biol. 2016;36:122–133. [DOI] [PubMed] [Google Scholar]
- 45. Bai L, Deng X, Li Q, Wang M, An W, Deli A, Gao Z, Xie Y, Dai Y, Cong YS. Down‐regulation of the cavin family proteins in breast cancer. J Cell Biochem. 2012;113:322–328. [DOI] [PubMed] [Google Scholar]
- 46. Shen J, Behrens C, Wistuba II, Feng L, Lee JJ, Hong WK, Lotan R. Identification and validation of differences in protein levels in normal, premalignant, and malignant lung cells and tissues using high‐throughput Western Array and immunohistochemistry. Cancer Res. 2006;66:11194–11206. [DOI] [PubMed] [Google Scholar]
- 47. Gamez‐Pozo A, Sanchez‐Navarro I, Calvo E, Agullo‐Ortuno MT, Lopez‐Vacas R, Diaz E, Camafeita E, Nistal M, Madero R, Espinosa E, Lopez JA, Fresno Vara JA. PTRF/cavin‐1 and MIF proteins are identified as non‐small cell lung cancer biomarkers by label‐free proteomics. PLoS One. 2012;7:e33752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilson DP, Saward L, Zahradka P, Cheung PK. Angiotensin II receptor antagonists prevent neointimal proliferation in a porcine coronary artery organ culture model. Cardiovasc Res. 1999;42:761–772. [DOI] [PubMed] [Google Scholar]
- 49. Hardin CD, Vallejo J. Caveolins in vascular smooth muscle: form organizing function. Cardiovasc Res. 2006;69:808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwencke C, Schmeisser A, Walter C, Wachter R, Pannach S, Weck B, Braun‐Dullaeus RC, Kasper M, Strasser RH. Decreased caveolin‐1 in atheroma: loss of antiproliferative control of vascular smooth muscle cells in atherosclerosis. Cardiovasc Res. 2005;68:128–135. [DOI] [PubMed] [Google Scholar]
- 51. Grossi M, Rippe C, Sathanoori R, Sward K, Forte A, Erlinge D, Persson L, Hellstrand P, Nilsson BO. Vascular smooth muscle cell proliferation depends on caveolin‐1‐regulated polyamine uptake. Biosci Rep. 2014;34:e00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Low JY, Nicholson HD. Emerging role of polymerase‐1 and transcript release factor (PTRF/Cavin‐1) in health and disease. Cell Tissue Res. 2014;357:505–513. [DOI] [PubMed] [Google Scholar]
- 53. Engelman JA, Chu C, Lin A, Jo H, Ikezu T, Okamoto T, Kohtz DS, Lisanti MP. Caveolin‐mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. A role for the caveolin‐scaffolding domain. FEBS Lett. 1998;428:205–211. [DOI] [PubMed] [Google Scholar]
- 54. Wanaski SP, Ng BK, Glaser M. Caveolin scaffolding region and the membrane binding region of SRC form lateral membrane domains. Biochemistry. 2003;42:42–56. [DOI] [PubMed] [Google Scholar]
- 55. Bai L, Deng X, Li J, Wang M, Li Q, An W, A D, Cong YS. Regulation of cellular senescence by the essential caveolar component PTRF/Cavin‐1. Cell Res. 2011;21:1088–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li Q, Bai L, Liu N, Wang M, Liu JP, Liu P, Cong YS. Increased polymerase I and transcript release factor (Cavin‐1) expression attenuates platelet‐derived growth factor receptor signalling in senescent human fibroblasts. Clin Exp Pharmacol Physiol. 2014;41:169–173. [DOI] [PubMed] [Google Scholar]
- 57. Beardsley A, Fang K, Mertz H, Castranova V, Friend S, Liu J. Loss of caveolin‐1 polarity impedes endothelial cell polarization and directional movement. J Biol Chem. 2005;280:3541–3547. [DOI] [PubMed] [Google Scholar]
- 58. Grande‐Garcia A, Echarri A, de Rooij J, Alderson NB, Waterman‐Storer CM, Valdivielso JM, del Pozo MA. Caveolin‐1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J Cell Biol. 2007;177:683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hassan GS, Williams TM, Frank PG, Lisanti MP. Caveolin‐1‐deficient aortic smooth muscle cells show cell autonomous abnormalities in proliferation, migration, and endothelin‐based signal transduction. Am J Physiol Heart Circ Physiol. 2006;290:H2393–H2401. [DOI] [PubMed] [Google Scholar]
- 60. Lagares‐Tena L, Garcia‐Monclus S, Lopez‐Alemany R, Almacellas‐Rabaiget O, Huertas‐Martinez J, Sainz‐Jaspeado M, Mateo‐Lozano S, Rodriguez‐Galindo C, Rello‐Varona S, Herrero‐Martin D, Tirado OM. Caveolin‐1 promotes Ewing sarcoma metastasis regulating MMP‐9 expression through MAPK/ERK pathway. Oncotarget. 2016;7:56889–56903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Faggi F, Chiarelli N, Colombi M, Mitola S, Ronca R, Madaro L, Bouche M, Poliani PL, Vezzoli M, Longhena F, Monti E, Salani B, Maggi D, Keller C, Fanzani A. Cavin‐1 and Caveolin‐1 are both required to support cell proliferation, migration and anchorage‐independent cell growth in rhabdomyosarcoma. Lab Invest. 2015;95:585–602. [DOI] [PubMed] [Google Scholar]