

Abstract
Background
Increased platelet aggregation during antiplatelet therapy may predict cardiovascular events in patients with coronary artery disease. The majority of these patients receive aspirin monotherapy. We aimed to investigate whether high platelet‐aggregation levels predict cardiovascular events in stable coronary artery disease patients treated with aspirin.
Methods and Results
We included 900 stable coronary artery disease patients with either previous myocardial infarction, type 2 diabetes mellitus, or both. All patients received single antithrombotic therapy with 75 mg aspirin daily. Platelet aggregation was evaluated 1 hour after aspirin intake using the VerifyNow Aspirin Assay (Accriva Diagnostics) and Multiplate Analyzer (Roche; agonists: arachidonic acid and collagen). Adherence to aspirin was confirmed by serum thromboxane B2. The primary end point was the composite of nonfatal myocardial infarction, ischemic stroke, and cardiovascular death. At 3‐year follow‐up, 78 primary end points were registered. The primary end point did not occur more frequently in patients with high platelet‐aggregation levels (first versus fourth quartile) assessed by VerifyNow (hazard ratio: 0.5 [95% CI, 0.3–1.1], P=0.08) or Multiplate using arachidonic acid (hazard ratio: 1.0 [95% CI, 0.5–2.1], P=0.92) or collagen (hazard ratio: 1.4 [95% CI, 0.7–2.8], P=0.38). Similar results were found for the composite secondary end point (nonfatal myocardial infarction, ischemic stroke, stent thrombosis, and all‐cause death) and the single end points. Thromboxane B2 levels did not predict any end points. Renal insufficiency was the only clinical risk factor predicting the primary and secondary end points.
Conclusions
This study is the largest to investigate platelet aggregation in stable coronary artery disease patients receiving aspirin as single antithrombotic therapy. We found that high platelet‐aggregation levels did not predict cardiovascular events.
Keywords: antiplatelet drug resistance, aspirin, coronary artery disease, prognosis
Subject Categories: Platelets, Clinical Studies
Clinical Perspective
What Is New?
In this large study, we investigated platelet aggregation in 900 stable coronary artery disease patients receiving aspirin 75 mg as single antithrombotic therapy.
Using 2 different platelet‐function tests, high platelet‐aggregation levels did not predict cardiovascular events.
High serum thromboxane B2 levels did not predict cardiovascular events either.
What Are the Clinical Implications?
Our results do not support routine risk stratification based on platelet aggregation in stable coronary artery disease patients receiving aspirin as single antithrombotic therapy.
Introduction
Aspirin is recommended for cardiovascular prevention in patients with stable coronary artery disease (CAD).1, 2 The antiplatelet effect of aspirin is exerted by reducing platelet aggregation through irreversible acetylation of cyclooxygenase‐1 (COX‐1) thereby inhibiting the conversion of arachidonic acid (AA) to thromboxane A2. COX‐1 activity is assessed most specifically by measurement of thromboxane metabolites or by AA‐induced platelet aggregation.3 Within recent years, whole‐blood platelet‐aggregation assays such as the VerifyNow Aspirin Assay (Accriva Diagnostics) and Multiplate Analyzer (Roche) have become widely applied. These assays have been shown to predict clinical outcome in patients receiving dual antiplatelet therapy with aspirin and clopidogrel.4, 5 Furthermore, inadequate inhibition of COX‐1 has been associated with adverse clinical outcomes in aspirin‐treated CAD patients.6
Two meta‐analyses showed that aspirin‐treated CAD patients with high platelet aggregation carried a nearly 4‐fold risk of developing major cardiovascular events.7, 8 However, most of the included studies were small and hampered by the use of COX‐1–nonspecific tests and inclusion of patients receiving dual antiplatelet therapy. We aimed to investigate whether high platelet‐aggregation levels predicted cardiovascular events in patients treated with aspirin only.
Methods
Study Population
In this observational study, we included 900 patients with angiographically documented stable CAD receiving single antithrombotic therapy with 75 mg aspirin daily. The study cohort represents a high‐risk population because all patients had documented CAD and either prior myocardial infarction, type 2 diabetes mellitus, or both. The study population and blood collection procedure were described previously in detail.9 Platelet‐aggregation data and their relation to prior myocardial infarction and stent thrombosis, type 2 diabetes mellitus, and renal insufficiency have been published previously.10, 11, 12, 13 Patients were recruited from November 2007 to January 2011 from the Western Denmark Heart Registry, which collects data on all interventional procedures performed in the western part of Denmark.14
Patients with cardiovascular events within the past 12 months were not included because they were likely to receive dual antiplatelet therapy. All diabetic patients were diagnosed with type 2 diabetes mellitus and treated with oral antidiabetic drugs and/or insulin. All nondiabetic patients had fasting plasma glucose levels <7.0 mmol/L at the time of inclusion.
The study was conducted in agreement with the Helsinki‐II declaration and approved by the Central Denmark Region Committees on Health Research (project numbers 2007‐0180, 2008‐0188, 2008‐0189, M‐2009‐0110) and by the Danish Data Protection Agency. The study is registered at ClinicalTrials.gov (identifier NCT01383304). All patients gave written informed consent before inclusion.
Laboratory Investigations
Blood sampling
Blood samples were obtained from the antecubital vein with patients in supine position 60 minutes after oral intake of 75 mg of non–enteric‐coated aspirin (Hjerdyl; Sandoz) and after 30 minutes of rest. Vacuum tubes and a large‐bore needle (19‐gauge) were used for blood sampling, and a minimum of stasis was used to minimize platelet activation. The first milliliter of blood was used for hematology analysis.
Platelet aggregation
Blood for platelet‐aggregation analyses was drawn 1 hour after aspirin intake and rested at room temperature for 30 to 120 minutes before analysis. Whole‐blood platelet aggregation was evaluated with 2 different instruments, as described previously.15 For VerifyNow analyses, blood was collected in 2.7‐mL tubes containing 3.2% sodium citrate. Platelet aggregation was reported in aspirin reaction units. For Multiplate analyses, 3.6‐mL tubes containing 3.2% sodium citrate were used. Platelet aggregation was induced with AA 1.0 mmol/L (ASPI test; Triolab AS) or collagen 1.0 μg/mL (Horm; Medinor). Aggregation was reported as the area under the curve (aggregation units×minute).
Adherence
All patients were treated with aspirin before inclusion in the study, but to improve adherence and uniform pharmacokinetics before blood sampling, all patients received a pillbox with 7 tablets of 75‐mg aspirin. Furthermore, serum thromboxane B2 (TXB2) concentrations were measured according to Patrono et al16 with the modification that serum was collected after 1 hour of clotting, and serum TXB2 was measured by ELISA.
Follow‐up and Clinical End Points
Follow‐up was performed using national registries. Unambiguous, individual‐level linkage between registries was made possible by the Danish Civil Registration, which assigns a permanent and unique 10‐digit identification number to Danish inhabitants at birth and to residents on immigration. End point assessment was based on data from the Western Denmark Heart Registry,14 the Danish Stroke Registry,17 and the Danish Register of Causes of Death.18 Coauthors S.D.K. and E.L.G., who were blinded to platelet‐aggregation results, reviewed all end points and source documents regarding myocardial infarction and stent thrombosis to confirm diagnoses. Myocardial infarction was defined using the universal definition as of 2007.19 Stent thrombosis was defined as definite, probable, or possible stent thrombosis, according to the Academic Research Consortium criteria.20 Cardiovascular death was defined by the International Classification of Diseases, 10th Revision (ICD‐10) codes I00–I25, I27, I30–I52, and I60–I72. Ischemic stroke was defined by ICD‐10 codes I63 and I64.
The primary end point was the composite of acute nonfatal myocardial infarction, ischemic stroke, and cardiovascular death. The composite of acute nonfatal myocardial infarction, ischemic stroke, stent thrombosis, and all‐cause death was analyzed as a secondary end point. Finally, acute nonfatal myocardial infarction, ischemic stroke, stent thrombosis, cardiovascular death, and all‐cause death were analyzed as single secondary end points. As new studies appeared during the follow‐up period showing lower ischemic event rates than expected, we extended the study period from 2 to 3 years.
Statistical Analysis
The study was designed with a power of 90%, expecting event rates of the primary end point of 5% and 15% for the first and fourth quartiles of platelet aggregation, as evaluated with the VerifyNow Aspirin Assay. Given these assumptions, a study cohort of 872 patients was needed, and the target recruitment was set at 900 patients.
Continuous data are presented as mean and standard deviation or median and interquartile range, as appropriate. Differences between 2 unpaired groups were tested with a 2‐sided t test or the Mann–Whitney test, as appropriate. Proportions between 2 groups were tested using the χ2 test and presented as absolute counts and percentages. Multivariable Cox proportional hazards survival regression was used to investigate the effect of high platelet aggregation and high TXB2 levels on the primary and secondary outcomes after adjustment for relevant prognostic factors and factors influencing platelet aggregation in the study cohort (covariates: age, sex, prior myocardial infarction, diabetes mellitus, smoking, body mass index, platelet count, and renal function). With the high number of predictor variables relative to the number of events, there was a risk of overfitting the models; therefore, we made sensitivity analyses including each covariate one at a time. These analyses confirmed that the results were very similar to the reported results. This was also supported by the similarity between crude and adjusted analyses. Given the relatively small number of events for the single end points, Cox models investigating these outcomes were adjusted for platelet count only, which was the only covariate significantly affecting the model. The proportional hazards assumption was assessed by a plot of log(−log[survival function]) versus time for combined clinical end points. Survival curves were estimated by the Kaplan–Meier method.
Furthermore, post hoc nested case–control analyses were performed including patients with an end point as cases and matching them at a 1:2 ratio with respect to age, sex, and prior myocardial infarction. Platelet aggregation data were dichotomized according to the median value, and data were analyzed using conditional logistic regression. All analyses were 2‐sided, and a P<0.05 was considered statistically significant. Statistical analyses were performed using Stata version 11.0 (StataCorp).
Results
Baseline characteristics of the entire cohort and patients with a primary end point are presented in Table 1. Platelet aggregation results were divided into quartiles, which were used for further analyses. The ranges of platelet aggregation within the quartiles were as follows: Multiplate using AA as agonist showed (in aggregation units×minute) 60 to 100 in quartile 1 (q1), 101 to 164 in q2, 165 to 238 in q3, and 240 to 659 in q4; Multiplate using collagen as agonist showed 0 to 170 in q1, 172 to 266 in q2, 267 to 397 in q3, and 398 to 867 in q4; and the VerifyNow Aspirin Assay showed (in aspirin reaction units) 315 to 409 in q1, 410 to 426 in q2, 427 to 452 in q3, and 453 to 654 in q4. The ranges of TXB2 (in ng/mL) within the quartiles were 0.02 to 0.52 in q1, 0.53 to 0.97 in q2, 0.98 to 1.82 in q3, and 1.83 to 26.44 in q4. Adherence to aspirin was confirmed in 883 patients (98%) by serum TXB2 levels <10 ng/mL (median: 0.94 ng/mL [interquartile range: 0.52–1.77]) corresponding to 95% COX‐1 inhibition.21 Sixteen patients had serum TXB2 levels >10 ng/mL (median: 15.09 ng/mL [interquartile range: 12.47–16.10]; range: 10.37–26.44 ng/mL).
Table 1.
Baseline Characteristics of the Study Population (n=900 as Presented in Larsen et al9)
| All Patients (n=900) | Patients With Eventa (n=78) | |
|---|---|---|
| Age, y | 65±4 | 67±9 |
| Body mass index, kg/m2 | 27±8 | 30±6 |
| Men, n (%) | 704 (78) | 61 (79) |
| Current smokers, n (%) | 199 (22) | 19 (25) |
| Blood pressure, systolic, mm Hg | 142±20 | 139±19 |
| Blood pressure, diastolic, mm Hg | 83±12 | 83±12 |
| Biochemistry | ||
| B‐leukocyte count, 109/Lb | 7.1±1.9 | 7.7±2.2 |
| B‐hemoglobin, mmol/L | 8.8±0.8 | 8.7±0.7 |
| B‐red blood cell count, 1012/Lc | 4.7±0.4 | 4.7±0.4 |
| B‐reticulocyte count, 109/L | 49±16 | 48±16 |
| B‐platelet count, 109/L | 233±58 | 242±58 |
| B‐mean platelet volume, fL | 10.9±0.9 | 10.8±0.9 |
| P‐creatinine, μmol/L | 82 (71–96) | 87 (76–106) |
| B‐eGFR, mL/min | 78 (65–90) | 72 (59–87) |
| S‐thromboxane B2, ng/mL | 1.0 (0.5–1.8) | 0.8 (0.4–1.4) |
| Cardiovascular morbidity, n (%) | ||
| Prior percutaneous coronary intervention | 849 (94) | 71 (92) |
| Prior myocardial infarction | 795 (88) | 66 (86) |
| Prior coronary artery bypass grafting | 122 (14) | 4 (5) |
| Prior stroke | 53 (6) | 8 (10) |
| Type 2 diabetes mellitus | 250 (28) | 27 (35) |
| Medication, n (%) | ||
| Aspirin | 900 (100) | 77 (100) |
| Statins | 813 (90) | 68 (88) |
| Beta‐blockers | 682 (76) | 53 (69) |
| Angiotensin‐converting enzyme inhibitors | 424 (47) | 34 (44) |
| Angiotensin II receptor blockers | 139 (15) | 14 (18) |
| Calcium antagonists | 194 (22) | 17 (22) |
| Diuretics | 272 (30) | 30 (39) |
| Proton pump inhibitors | 105 (12) | 14 (18) |
| Insulin | 76 (30) | 21 (78) |
| Oral antidiabetic medication | 205 (82) | 21 (78) |
Data are presented as mean±SD, n (%), or median (interquartile range). B indicates blood; eGFR, estimated glomerular filtration rate; P, plasma; S, serum.
Patients with an event included in the primary end point.
Leukocyte count available for only 714 patients.
Red blood cell count available for only 751 patients.
Primary End Point
Patients were followed for a median of 3.1 years (minimum: 2.0; maximum: 5.1). Based on calculations on data from the Western Denmark Heart Registry, we estimated 1% to 2% loss to follow‐up due to patients moving out of western Denmark. The total number of events included in the primary end point was 78 (8.7%), indicating the first event of acute nonfatal myocardial infarction (n=48 [5.3%]), ischemic stroke (n=12 [1.3%]), or cardiovascular death (n=18 [2.0%]). The number of events in the fourth quartile of platelet aggregation evaluated with VerifyNow was 13 (5.9%) versus 23 (9.7%) in the first quartile. The primary end point did not occur more frequently in patients with high platelet‐aggregation levels (first versus fourth quartile) when evaluated with VerifyNow or Multiplate using AA or collagen as agonists (Figure 1). Associations between high platelet‐aggregation levels and cardiovascular events are shown in Table 2. Renal insufficiency (estimated glomerular filtration rate ≤60 mL/min) was the only clinical risk factor independently predicting the primary end point (hazard ratio [HR]: 1.7 [95% confidence interval [CI], 1.0–2.7]; P=0.04). There was no significant effect of age, sex, type 2 diabetes mellitus, prior myocardial infarction, smoking, or body mass index on the occurrence of the primary end point.
Figure 1.
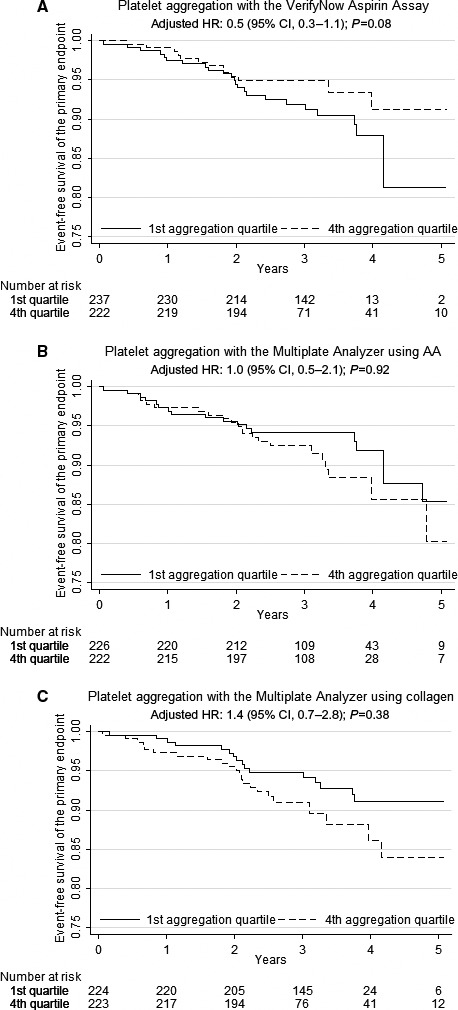
Platelet aggregation as predictor of the primary outcome (cardiovascular death, nonfatal myocardial infarction, and ischemic stroke) with (A) the VerifyNow Aspirin Assay, (B) the Multiplate Analyzer using 1.0 mmol/L AA as agonist, and (C) the Multiplate Analyzer using 1.0 μg/mL collagen as agonist. AA indicates arachidonic acid; CI, confidence interval; HR, hazard ratio.
Table 2.
Clinical End Points According to High and Low Platelet‐Aggregation Levels Evaluated by the VerifyNow and the Multiplate Analyzer
| End points | No. (%) | VerifyNow Aspirin Assay | Multiplate Analyzer Using AA | Multiplate Analyzer Using Collagen | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q4 | Q1 | Crude (95% CI) | P Value | Adjusted HRa (95% CI) | P Value | Q4 | Q1 | Crude (95% CI) | P Value | Adjusted HRa (95% CI) | P Value | Q4 | Q1 | Crude (95% CI) | P Value | Adjusted HRa (95% CI) | P Value | ||
| Primary composite | 78 (8.7) | 13 (1.4) | 23 (2.6) | 0.6 (0.3–1.2) | 0.13 | 0.5 (0.3–1.1) | 0.08 | 22 (2.4) | 18 (2.0) | 1.3 (0.7–2.5) | 0.37 | 1.0 (0.5–2.1) | 0.92 | 22 (2.4) | 16 (1.8) | 1.6 (0.8–3.0) | 0.18 | 1.4 (0.7–2.8) | 0.38 |
| Secondary composite | 104 (11.6) | 20 (2.2) | 27 (3.0) | 0.8 (0.5–1.4) | 0.46 | 0.7 (0.4–1.3) | 0.26 | 28 (3.1) | 20 (2.2) | 1.5 (0.9–2.7) | 0.17 | 1.0 (0.5–1.9) | 0.99 | 26 (2.9) | 27 (3.0) | 1.1 (0.6–1.9) | 0.73 | 0.8 (0.4–1.5) | 0.48 |
| Myocardial infarction | 49 (5.4) | 7 (0.8) | 16 (1.8) | 0.5 (0.2–1.2) | 0.08 | 0.4 (0.2–1.0) | 0.05 | 15 (1.7) | 9 (1.0) | 1.8 (0.8–4.2) | 0.16 | 1.5 (0.6–3.6) | 0.36 | 15 (1.7) | 11 (1.2) | 1.5 (0.7–3.3) | 0.31 | 1.2 (0.5–2.7) | 0.7 |
| Ischemic stroke | 13 (1.4) | 3 (0.3) | 3 (0.3) | 1.1 (0.2–5.5) | 0.90 | 1.2 (0.2–5.9) | 0.85 | 4 (0.4) | 4 (0.4) | 1.0 (0.3–4.2) | 0.96 | 1.3 (0.3–5.8) | 0.73 | 3 (0.3) (0.3) | 3 (0.3) | 1.1 (0.2–5.6) | 0.88 | 1.4 (0.3–8.0) | 0.68 |
| Stent thrombosis | 15 (1.7) | 1 (0.1) | 6 (0.7) | 0.2 (0.0–1.5) | 0.11 | 0.2 (0.0–1.4) | 0.10 | 4 (0.4) | 3 (0.3) | 1.4 (0.3–6.4) | 0.64 | 1.3 (0.3–6.4) | 0.75 | 4 (0.4) | 4 (0.4) | 1.2 (0.3–4.8) | 0.82 | 1.1 (0.2–4.9) | 0.94 |
| Cardiovascular death | 23 (2.6) | 5 (0.6) | 7 (0.8) | 0.7 (0.2–2.4) | 0.62 | 0.7 (0.2–2.2) | 0.54 | 6 (0.7) | 6 (0.7) | 1.1 (0.3–3.3) | 0.91 | 0.8 (0.2–2.7) | 0.75 | 7 (0.8) | 5 (0.6) | 1.5 (0.5–4.8) | 0.48 | 1.2 (0.3–4.2) | 0.78 |
| All‐cause death | 53 (5.9) | 13 (1.4) | 13 (1.4) | 1.1 (0.5–2.4) | 0.77 | 1.0 (0.5–2.3) | 0.92 | 13 (1.4) | 8 (0.9) | 1.7 (0.7–4.1) | 0.24 | 1.3 (0.5–3.2) | 0.57 | 12 (1.3) | 17 (1.9) | 0.8 (0.4–1.7) | 0.56 | 0.6 (0.3–1.3) | 0.16 |
Data are presented as number of patients (%). Primary composite end point: first event of nonfatal myocardial infarction, ischemic stroke, or cardiovascular death. Secondary composite end point: first event of nonfatal myocardial infarction, ischemic stroke, stent thrombosis, or all‐cause death. AA indicates arachidonic acid; CI, confidence interval; HR, hazard ratio; Q, quartile.
Composite end points adjusted for age, sex, diabetes mellitus, prior myocardial infarction, active smoking, body mass index, platelet count, and renal function. Single end points adjusted for platelet count.
The number of events in the fourth quartile of TXB2 was 15 (6.7%) versus 26 (11.6%) in the first quartile. The primary end point did not occur more frequently in patients with high TXB2 levels (first versus fourth quartile, HR: 0.49 [95% CI, 0.24–1.00]; P=0.05). Of 900 patients, 16 had serum TXB2 levels >10 ng/mL (median: 15.09 ng/mL [interquartile range: 12.47–16.10]; range: 10.37–26.44 ng/mL). Of these 16 patients, 2 had events included in the primary end point (TXB2 levels: 10.37 and 17.41 ng/mL, respectively). Serum TXB2 analysis failed in 1 of 900 patients.
There were no proportional differences between patients with and without an event included in the primary end point regarding the use of nonantiplatelet medications (statins, beta blockers, angiotensin‐converting enzyme inhibitors, angiotensin receptor blockers, calcium channel blockers, diuretics, or proton pump inhibitors).
Secondary End Point
The total number of events included in the secondary end point was 104 (11.6%), indicating the first event of acute nonfatal myocardial infarction (n=47 [5.2%]), ischemic stroke (n=12 [1.3%]), stent thrombosis (n=0 [0%]), or all‐cause death (n=45 [5.0%]). The secondary end point did not occur more frequently in patients with high platelet‐aggregation levels (first versus fourth quartile) when evaluated by VerifyNow or Multiplate using AA (Figure 2A and 2B). Similar results were found with Multiplate using collagen (HR: 0.8 [95% CI, 0.4–1.5]; P=0.48; Figure 2C). As for the primary end point, renal insufficiency independently predicted the secondary end point (HR: 1.9 [95% CI, 1.2–2.8]; P=0.01.)
Figure 2.
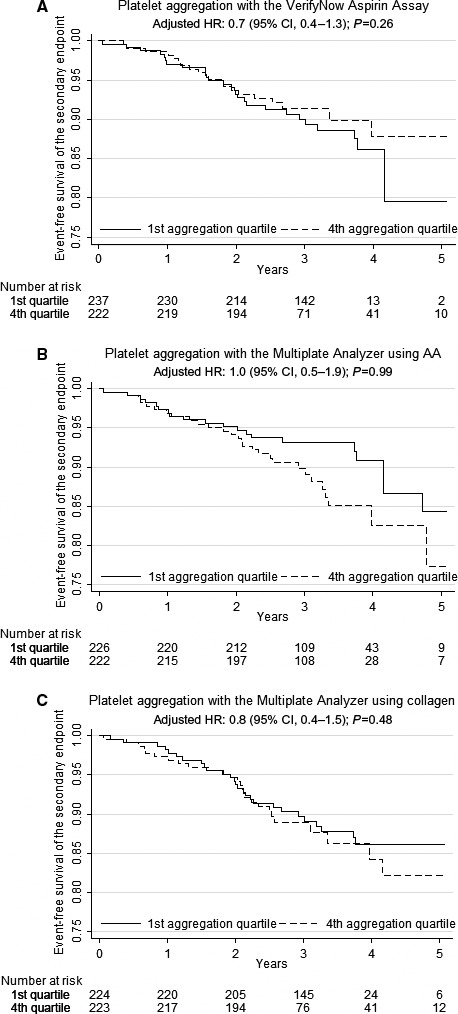
Platelet aggregation as predictor of the secondary outcome (all‐cause death, nonfatal myocardial infarction, ischemic stroke, and stent thrombosis) with (A) the VerifyNow Aspirin Assay, (B) the Multiplate Analyzer using 1.0 mmol/L AA as agonist, and (C) the Multiplate Analyzer using 1.0 μg/mL collagen as agonist. AA indicates arachidonic acid; CI, confidence interval; HR, hazard ratio.
The number of events in the fourth quartile of TXB2 was 19 (8.4%) versus 29 (12.9%) in the first quartile. The secondary end point did not occur more frequently in patients with high TXB2 levels (first versus fourth quartile, HR: 0.56 [95% CI, 0.29–1.07]); P=0.08).
Previous studies have used assay‐specific cutoff values to identify patients with reduced antiplatelet effect of aspirin.15, 22 Using these cutoff values (Multiplate >300 aggregation units×minute, VerifyNow >550 aspirin reaction units), we found that 124 patients had aggregation levels >300 aggregation units×minute using Multiplate with AA as agonist, and 16 of these had events included in the primary end point. Overall, 13 patients had aggregation levels >550 aspirin reaction units using VerifyNow; however, none of these patients had events included in the combined primary or secondary end points.
Single Secondary End Points
The occurrence of single end points did not occur more frequently in the fourth versus the first aggregation quartile with any of the platelet‐aggregation tests (Table 2). Renal insufficiency independently predicted all‐cause death (HR: 3.0 [95% CI, 1.7–5.2]; P<0.0001) and cardiovascular death (HR: 2.6 [95% CI, 1.1–6.0]; P=0.03) but not myocardial infarction (HR: 1.5 [95% CI, 0.8–2.8]; P=0.26), stent thrombosis (HR: 0.3 [95% CI, 0.0–2.0]; P=0.19), or ischemic stroke (HR: 1.1 [95% CI, 0.3–4.0]; P=0.88). Finally, TXB2 values in the fourth quartile did not predict any of the single end points.
Nested Case–Control Analyses
For nested case–control analyses, platelet‐aggregation results were dichotomized according to median values. There was no difference in the proportion of patients with high versus low platelet‐aggregation levels in terms of reaching the primary end point (Table 3). Similarly, cases did not have an increased risk of events included in the primary end point compared with matched controls when evaluated with VerifyNow or Multiplate using AA or collagen as agonists (Table 3). Results were comparable when analyzing the secondary end point as well as all single end points.
Table 3.
Nested Case–Control Analyses According to Platelet Aggregation Levels Evaluated by the VerifyNow and the Multiplate
| End points | No. (%) | VerifyNow Aspirin Assay | Multiplate Analyzer With AA | Multiplate Analyzer With Collagen | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Below Median, No. (%) | Median or Above, No. (%) | OR (95% CI) | P Value | Below Median, No. (%) | Median or Above, No. (%) | OR (95% CI) | P Value | Below Median, No. (%) | Median or Above, No. (%) | OR (95% CI) | P Value | ||
| Primary composite | 78 (8.7) | 41 (53) | 37 (47) | 0.8 (0.5–1.4) | 0.53 | 37 (47) | 41 (53) | 1.1 (0.7–2.0) | 0.64 | 35 (45) | 43 (55) | 1.4 (0.8–2.5) | 0.22 |
| Secondary composite | 104 (11.6) | 53 (51) | 51 (49) | 1.0 (0.6–1.6) | 0.94 | 51 (49) | 53 (51) | 1.1 (0.6–1.8) | 0.80 | 53 (51) | 51 (49) | 0.9 (0.6–1.5) | 0.82 |
| Myocardial infarction | 49 (5.4) | 26 (53) | 23 (47) | 0.8 (0.4–1.6) | 0.57 | 20 (41) | 29 (59) | 1.8 (0.9–3.8) | 0.12 | 23 (47) | 26 (53) | 1.2 (0.6–2.4) | 0.57 |
| Ischemic stroke | 13 (1.4) | 8 (62) | 5 (38) | 0.6 (0.1–2.6) | 0.46 | 8 (62) | 5 (38) | 0.6 (0.1–2.6) | 0.48 | 7 (54) | 6 (46) | 0.9 (0.3–3.0) | 0.84 |
| Stent thrombosis | 15 (1.7) | 10 (67) | 5 (33) | 0.3 (0.1–1.3) | 0.09 | 7 (47) | 8 (53) | 1.3 (0.4–4.5) | 0.68 | 8 (53) | 7 (47) | 0.9 (0.3–2.9) | 0.84 |
| Cardiovascular death | 23 (2.6) | 15 (65) | 8 (35) | 0.6 (0.2–1.5) | 0.28 | 13 (57) | 10 (43) | 0.7 (0.3:1.9) | 0.51 | 13 (57) | 10 (43) | 0.7 (0.3–2.0) | 0.49 |
| All‐cause death | 53 (5.9) | 27 (51) | 26 (49) | 1.0 (0.5–1.8) | 0.92 | 29 (55) | 24 (45) | 0.8 (0.4–1.5) | 0.49 | 30 (57) | 23 (43) | 0.7 (0.3–1.3) | 0.22 |
Data are presented as number of patients (%). Primary composite end point: first event of nonfatal myocardial infarction, ischemic stroke, or cardiovascular death. Secondary composite end point: first event of nonfatal myocardial infarction, ischemic stroke, stent thrombosis, or all‐cause death. AA indicates arachidonic acid; CI, confidence interval; OR, odds ratio.
Discussion
This work is the largest prospective study so far investigating the association between platelet‐aggregation levels and cardiovascular events in stable CAD patients treated with aspirin as single antithrombotic therapy. Our main finding was that high platelet‐aggregation levels measured 1 hour after aspirin intake did not predict cardiovascular events.
Reduced antiplatelet effect of aspirin has been associated previously with an increased risk of ischemic events.7, 8 However, studies have been heterogeneous in terms of cohort size, cardiovascular disease manifestation, treatment regimen (monotherapy versus dual‐antiplatelet therapy), and platelet‐function testing (COX‐1–specific versus COX‐1–nonspecific). Furthermore, the majority of previous studies have included patients with acute coronary syndromes or patients undergoing percutaneous coronary intervention.
The ASCET (Aspirin Nonresponsiveness and Clopidogrel Endpoint Trial) study was the first prospective randomized trial relating platelet aggregation to clinical outcome in a large cohort of stable CAD patients (n=1001) treated with aspirin as single‐antithrombotic therapy.23 Patients were randomized to continue with aspirin or to switch to clopidogrel. The main finding was that high on‐aspirin platelet reactivity did not predict clinical outcome after 2‐year follow‐up. The observed end point rate in the study (n=106 [10.6%]) was lower than expected, which may partly explain the results. Platelet reactivity was evaluated by the PFA‐100 system using COX‐1–nonspecific collagen/epinephrine cartridges. Similar results were reported in a recent cohort study including 592 stable cardiovascular patients treated with aspirin monotherapy for secondary prevention.24 Platelet aggregation was determined by light transmission aggregometry using AA and collagen as agonists. After 2 years of follow‐up, cardiovascular events occurred independently of high platelet aggregation.24
In the ADRIE (Results of the Antiplatelet Drug resistance and Ischemic Events) study, a large prospective multicenter study of 771 stable CAD patients treated with aspirin and/or clopidogrel, the predictive values for major adverse cardiovascular events of both specific and nonspecific platelet function assays were investigated.25 After 3‐year follow‐up, the primary end point of recurrent major adverse cardiovascular events occurred in 120 patients (15.6%). The main finding was that none of the platelet‐function assays added incremental predictive value to conventional risk factors for the occurrence of ischemic events. This is in line with our results and other recent studies including stable CAD patients undergoing elective percutaneous coronary intervention or coronary artery bypass grafting26, 27 as well as a recent comprehensive report from the National Institute for Health Research.28
Other studies reported that high on‐aspirin platelet aggregation measured with COX‐1–specific and –nonspecific assays was associated with the occurrence of ischemic cardiovascular events6, 29, 30; however, patients received dual antiplatelet therapy, and platelet aggregation was primarily investigated in the acute phase of acute coronary syndromes and/or assessed <1 month after antiplatelet therapy initiation. Comparison of results between studies including patients receiving monotherapy versus dual‐antiplatelet therapy is inexpedient because these patient groups differ in terms of cardiovascular risk profile. Moreover, the influence of a second antiplatelet agent on an aspirin‐specific platelet‐function test is unclear, and the second antiplatelet agent likely influences the occurrence of adverse cardiovascular events.28
Previous studies have demonstrated that aspirin does not provide consistent 24‐hour platelet inhibition in a significant proportion of CAD patients31, 32 and that residual platelet aggregation is 5‐fold more frequent 24 versus 2 hours after aspirin ingestion.33 Few previous studies have accounted for these pharmacokinetic conditions, which in part may explain the large proportion of patients with reduced antiplatelet effect of aspirin.7, 8 Furthermore, the absorption of acetylsalicylic acid depends on whether enteric‐coated or immediate‐release aspirin is used.34 Enteric‐coated aspirin is reported to delay and reduce drug absorption, causing “pseudoresistance,” a phenomenon that is not present when using non–enteric‐coated aspirin.35 This may partly explain the large difference in “aspirin resistance” reported in previous studies in which both enteric‐ and non–enteric‐coated aspirin were used. In our study, all patients were treated with 75 mg non–enteric‐coated aspirin. Blood sampling was standardized as blood for platelet aggregometry, and serum TXB2 measurement was drawn exactly 1 hour after aspirin intake. The peak plasma level of acetylsalicylic acid occurs 30 to 40 minutes after aspirin intake.34 Because blood samples were drawn 60 minutes after intake of non–enteric‐coated aspirin, high plasma levels of acetylsalicylic acid may have influenced our results, inducing high levels of platelet inhibition due to a short time interval from aspirin intake to blood sampling.
Surprisingly, renal insufficiency was the only conventional risk factor independently predicting the primary and secondary end points in our study. We reported previously that prior myocardial infarction, type 2 diabetes mellitus, high body mass index, and high platelet count predicted increased platelet aggregation in stable CAD patients treated with aspirin monotherapy.9 In the present study, however, we found no effect of age, sex, type 2 diabetes mellitus, prior myocardial infarction, smoking, or body mass index on any end points. This result conflicts with recent articles showing the influence of clinical risk factors on major adverse cardiovascular events in patients on dual‐antiplatelet therapy with aspirin and clopidogrel.36, 37
The low event rate may partly explain our results because only 5.9% of patients in the fourth aggregation quartile reached the primary end point, which was considerably lower than the expected 15% used in our sample size calculation. We used classical clinical end points, and despite studying a group of stable CAD patients with a high‐risk clinical profile, our study had lower event rates than comparable studies.23, 25 In our study, 88% of the patients had a history of myocardial infarction, 28% had type 2 diabetes mellitus, and 94% had previously undergone percutaneous coronary intervention. The relatively low number of events likely reflects both improved secondary prevention and improved stent technology within the past decade.38
In our study, the risk of the primary end point was increased by 40% in patients with high platelet‐aggregation levels (first versus fourth quartile) when evaluated with Multiplate using collagen as agonist (HR: 1.4 [95% CI, 0.7–2.8]; P=0.38; Figure 1C). This finding, however, was statistically nonsignificant, which may reflect that relatively small groups were compared. Of note, the numerical differences regarding the primary end point actually differed between the 2 platelet‐function tests showing opposite results. In our opinion, these results should be interpreted as neutral overall.
The optimal method to evaluate the antiplatelet effect of aspirin in a clinical setting remains unclear. AA‐induced platelet aggregation and measurement of thromboxane metabolites reflect COX‐1–specific pathways, whereas nonspecific platelet‐aggregation methods reflect multiple signaling pathways.3 The predictive value for clinical events of 6 different assays was investigated in the POPULAR (Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation) trial, including clopidogrel‐pretreated patients undergoing elective percutaneous coronary intervention.5 Only VerifyNow, light transmittance aggregometry, and PlateletWorks predicted the occurrence of the primary end point (myocardial infarction, stent thrombosis, stroke, and death), whereas shear‐based methods (IMPACT‐R and PFA‐100) did not. Multiplate was not included in the study, yet this assay predicted stent thrombosis in another study.4 Both VerifyNow and Multiplate are currently recommended for platelet‐function testing.39 Finally, serum TXB2 measurement reflects the pharmacodynamics of low‐dose aspirin, and in CAD patients treated with aspirin, increased serum TXB2 levels were related to adverse cardiovascular outcomes.6 Similar results were reported with urinary 11‐dehydro‐TXB2.40, 41
Some limitations of the present study must be acknowledged. Platelet‐aggregation levels over time were not explored because platelet function was measured only once. Blood for platelet aggregation was drawn between 8 am and 3 pm, and circadian variation of platelet function during aspirin therapy might have influenced the results.42 Not all patients were fasting before blood sampling, and that may have influenced platelet‐aggregation results because absorption and efficacy of aspirin can vary depending on food intake.43 The time interval from aspirin intake to blood sampling was standardized to 1 hour in all patients; however, this short interval may have resulted in more consistent and strong platelet inhibition due to high plasma levels of acetylsalicylic acid than after for example, 24 hours.
Cardiac events (acute nonfatal myocardial infarction and stent thrombosis) treated without intervention or treated in the eastern part of Denmark were not included in our study; however, this loss to follow‐up is unlikely to have been skewed. Finally, the end point event rate was lower than expected, which reduced the study power.
Conclusion
This study is the largest so far to investigate platelet aggregation in stable CAD patients receiving aspirin as single‐antithrombotic therapy. Neither high levels of platelet aggregation nor high serum TXB2 levels predicted cardiovascular events. Consequently, our results do not support routine risk stratification based on platelet aggregation in stable CAD patients treated with aspirin only.
Sources of Funding
This work was supported by the Danish Agency for Science Technology and Innovation (grant no. 2101‐05‐0052 to Kristensen); Novo‐Nordic Foundation (grant no. NNF14OC0008817 to Kristensen); Faculty of Health Sciences, Aarhus University; Denmark, The Danish Heart Foundation; A.P. Møller Foundation; Department of Clinical Medicine, Aarhus University; The Eva and Henry Fraenkel Foundation; The Aase and Ejnar Danielsen Foundation; The Korning Foundation; The Physician's assurance association anno 1891, and The Sophus Jacobsen and Spouse Astrid Jacobsen Foundation. The funding sources had no influence on the study design, collection, analysis or interpretation of the data, writing of the article, or the decision to submit the article for publication.
Disclosures
None of the authors have conflicts of interest regarding the present article. Kristensen, Grove, and Hvas report the following general conflicts of interest: Kristensen: speaker honoraria from Aspen and AstraZeneca. Grove: speaker honoraria from AstraZeneca, Baxter, Bayer, Boehringer Ingelheim, Pfizer, Sysmex. Advisory board meetings for Boehringer Ingelheim, AstraZeneca, Bayer, and Bristol‐Myers Squibb. Hvas: speaker honoraria from CSL Behring, Bayer, Boehringer‐Ingelheim, Bristol‐Myers Squibb and Leo Pharma and unrestricted research support from Octapharma, CSL Behring and Leo Pharma. Larsen, Würtz, and Petersen have no conflicts of interest to declare.
Acknowledgments
We gratefully acknowledge the help from Jens Flensted Lassen, MD, PhD, in planning the end point evaluation; Morten Madsen, PhD, for his support with statistical analyses; Lise Nielsen Wulff, MD, for helping with patient inclusion, and finally, technician Vivi Bo Mogensen for laboratory assistance.
(J Am Heart Assoc. 2017;6:e006050 DOI: 10.1161/JAHA.117.006050.)28780510
References
- 1. Juul‐Moller S, Edvardsson N, Jahnmatz B, Rosen A, Sorensen S, Omblus R. Double‐blind trial of aspirin in primary prevention of myocardial infarction in patients with stable chronic angina pectoris. The Swedish Angina Pectoris Aspirin Trial (SAPAT) Group. Lancet. 1992;340:1421–1425. [DOI] [PubMed] [Google Scholar]
- 2. Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, Buring J, Hennekens C, Kearney P, Meade T, Patrono C, Roncaglioni MC, Zanchetti A. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta‐analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Santilli F, Rocca B, De Cristofaro R, Lattanzio S, Pietrangelo L, Habib A, Pettinella C, Recchiuti A, Ferrante E, Ciabattoni G, Davi G, Patrono C. Platelet cyclooxygenase inhibition by low‐dose aspirin is not reflected consistently by platelet function assays: implications for aspirin “resistance”. J Am Coll Cardiol. 2009;53:667–677. [DOI] [PubMed] [Google Scholar]
- 4. Siller‐Matula JM, Christ G, Lang IM, Delle‐Karth G, Huber K, Jilma B. Multiple electrode aggregometry predicts stent thrombosis better than the vasodilator‐stimulated phosphoprotein phosphorylation assay. J Thromb Haemost. 2010;8:351–359. [DOI] [PubMed] [Google Scholar]
- 5. Breet NJ, van Werkum JW, Bouman HJ, Kelder JC, Ruven HJ, Bal ET, Deneer VH, Harmsze AM, van der Heyden JA, Rensing BJ, Suttorp MJ, Hackeng CM, ten Berg JM. Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation. JAMA. 2010;303:754–762. [DOI] [PubMed] [Google Scholar]
- 6. Frelinger AL III, Li Y, Linden MD, Barnard MR, Fox ML, Christie DJ, Furman MI, Michelson AD. Association of cyclooxygenase‐1‐dependent and ‐independent platelet function assays with adverse clinical outcomes in aspirin‐treated patients presenting for cardiac catheterization. Circulation. 2009;120:2586–2596. [DOI] [PubMed] [Google Scholar]
- 7. Krasopoulos G, Brister SJ, Beattie WS, Buchanan MR. Aspirin “resistance” and risk of cardiovascular morbidity: systematic review and meta‐analysis. BMJ. 2008;336:195–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Snoep JD, Hovens MM, Eikenboom JC, van der Bom JG, Huisman MV. Association of laboratory‐defined aspirin resistance with a higher risk of recurrent cardiovascular events: a systematic review and meta‐analysis. Arch Intern Med. 2007;167:1593–1599. [DOI] [PubMed] [Google Scholar]
- 9. Larsen SB, Grove EL, Neergaard‐Petersen S, Wurtz M, Hvas AM, Kristensen SD. Determinants of reduced antiplatelet effect of aspirin in patients with stable coronary artery disease. PLoS One. 2015;10:e0126767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Larsen SB, Neergaard‐Petersen S, Grove EL, Kristensen SD, Hvas AM. Increased platelet aggregation and serum thromboxane levels in aspirin‐treated patients with prior myocardial infarction. Thromb Haemost. 2012;108:140–147. [DOI] [PubMed] [Google Scholar]
- 11. Larsen SB, Grove EL, Kristensen SD, Hvas AM. Reduced antiplatelet effect of aspirin is associated with low‐grade inflammation in patients with coronary artery disease. Thromb Haemost. 2013;109:920–929. [DOI] [PubMed] [Google Scholar]
- 12. Wurtz M, Grove EL, Wulff LN, Kaltoft AK, Tilsted HH, Jensen LO, Hvas AM, Kristensen SD. Patients with previous definite stent thrombosis have a reduced antiplatelet effect of aspirin and a larger fraction of immature platelets. JACC Cardiovasc Interv. 2010;3:828–835. [DOI] [PubMed] [Google Scholar]
- 13. Wurtz M, Wulff LN, Grove EL, Kristensen SD, Hvas AM. Influence of renal function and platelet turnover on the antiplatelet effect of aspirin. Thromb Res. 2012;129:434–440. [DOI] [PubMed] [Google Scholar]
- 14. Schmidt M, Maeng M, Jakobsen CJ, Madsen M, Thuesen L, Nielsen PH, Botker HE, Sorensen HT. Existing data sources for clinical epidemiology: the Western Denmark Heart Registry. Clin Epidemiol. 2010;2:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grove EL, Hvas AM, Johnsen HL, Hedegaard SS, Pedersen SB, Mortensen J, Kristensen SD. A comparison of platelet function tests and thromboxane metabolites to evaluate aspirin response in healthy individuals and patients with coronary artery disease. Thromb Haemost. 2010;103:1245–1253. [DOI] [PubMed] [Google Scholar]
- 16. Patrono C, Ciabattoni G, Pinca E, Pugliese F, Castrucci G, De Salvo A, Satta MA, Peskar BA. Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb Res. 1980;17:317–327. [DOI] [PubMed] [Google Scholar]
- 17. Wildenschild C, Mehnert F, Thomsen RW, Iversen HK, Vestergaard K, Ingeman A, Johnsen SP. Registration of acute stroke: validity in the Danish Stroke Registry and the Danish National Registry of Patients. Clin Epidemiol. 2014;6:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Juel K, Helweg‐Larsen K. The Danish registers of causes of death. Dan Med Bull. 1999;46:354–357. [PubMed] [Google Scholar]
- 19. Thygesen K, Alpert JS, White HD. Universal definition of myocardial infarction. J Am Coll Cardiol. 2007;50:2173–2195. [DOI] [PubMed] [Google Scholar]
- 20. Cutlip DE, Windecker S, Mehran R, Boam A, Cohen DJ, van Es GA, Steg PG, Morel MA, Mauri L, Vranckx P, McFadden E, Lansky A, Hamon M, Krucoff MW, Serruys PW. Clinical end points in coronary stent trials: a case for standardized definitions. Circulation. 2007;115:2344–2351. [DOI] [PubMed] [Google Scholar]
- 21. Patrono C, Rocca B. Drug insight: aspirin resistance—fact or fashion? Nat Clin Pract Cardiovasc Med. 2007;4:42–50. [DOI] [PubMed] [Google Scholar]
- 22. Lordkipanidze M, Pharand C, Schampaert E, Turgeon J, Palisaitis DA, Diodati JG. A comparison of six major platelet function tests to determine the prevalence of aspirin resistance in patients with stable coronary artery disease. Eur Heart J. 2007;28:1702–1708. [DOI] [PubMed] [Google Scholar]
- 23. Pettersen AA, Seljeflot I, Abdelnoor M, Arnesen H. High on‐aspirin platelet reactivity and clinical outcome in patients with stable coronary artery disease: results from ASCET (Aspirin Nonresponsiveness and Clopidogrel Endpoint Trial). J Am Heart Assoc. 2012;1:e000703 DOI: 10.1161/JAHA.112.000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagatsuka K, Miyata S, Kada A, Kawamura A, Nakagawara J, Furui E, Takiuchi S, Taomoto K, Kario K, Uchiyama S, Saito K, Nagao T, Kitagawa K, Hosomi N, Tanaka K, Kaikita K, Katayama Y, Abumiya T, Nakane H, Wada H, Hattori A, Kimura K, Isshiki T, Nishikawa M, Yamawaki T, Yonemoto N, Okada H, Ogawa H, Minematsu K, Miyata T. Cardiovascular events occur independently of high on‐aspirin platelet reactivity and residual COX‐1 activity in stable cardiovascular patients. Thromb Haemost. 2016;116:356–368. [DOI] [PubMed] [Google Scholar]
- 25. Reny JL, Berdague P, Poncet A, Barazer I, Nolli S, Fabbro‐Peray P, Schved JF, Bounameaux H, Mach F, de Moerloose P, Fontana P. Antiplatelet drug response status does not predict recurrent ischemic events in stable cardiovascular patients: results of the Antiplatelet Drug Resistances and Ischemic Events study. Circulation. 2012;125:3201–3210. [DOI] [PubMed] [Google Scholar]
- 26. Park DW, Ahn JM, Song HG, Lee JY, Kim WJ, Kang SJ, Yun SC, Lee SW, Kim YH, Lee CW, Park SW, Park SJ. Differential prognostic impact of high on‐treatment platelet reactivity among patients with acute coronary syndromes versus stable coronary artery disease undergoing percutaneous coronary intervention. Am Heart J. 2013;165:34–42. [DOI] [PubMed] [Google Scholar]
- 27. Bolliger D, Filipovic M, Matt P, Tanaka KA, Gregor M, Zenklusen U, Seeberger MD, Lurati Buse G. Reduced aspirin responsiveness as assessed by impedance aggregometry is not associated with adverse outcome after cardiac surgery in a small low‐risk cohort. Platelets. 2016;27:254–261. [DOI] [PubMed] [Google Scholar]
- 28. Dretzke J, Riley RD, Lordkipanidze M, Jowett S, O'Donnell J, Ensor J, Moloney E, Price M, Raichand S, Hodgkinson J, Bayliss S, Fitzmaurice D, Moore D. The prognostic utility of tests of platelet function for the detection of “aspirin resistance” in patients with established cardiovascular or cerebrovascular disease: a systematic review and economic evaluation. Health Technol Assess. 2015;19:1–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Breet NJ, van Werkum JW, Bouman HJ, Kelder JC, ten Berg JM, Hackeng CM. High on‐aspirin platelet reactivity as measured with aggregation‐based, cyclooxygenase‐1 inhibition sensitive platelet function tests is associated with the occurrence of atherothrombotic events. J Thromb Haemost. 2010;8:2140–2148. [DOI] [PubMed] [Google Scholar]
- 30. Mayer K, Bernlochner I, Braun S, Schulz S, Orban M, Morath T, Cala L, Hoppmann P, Schunkert H, Laugwitz KL, Kastrati A, Sibbing D. Aspirin treatment and outcomes after percutaneous coronary intervention: results of the ISAR‐ASPI Registry. J Am Coll Cardiol. 2014;64:863–871. [DOI] [PubMed] [Google Scholar]
- 31. Rocca B, Santilli F, Pitocco D, Mucci L, Petrucci G, Vitacolonna E, Lattanzio S, Mattoscio D, Zaccardi F, Liani R, Vazzana N, Del Ponte A, Ferrante E, Martini F, Cardillo C, Morosetti R, Mirabella M, Ghirlanda G, Davi G, Patrono C. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low‐dose aspirin in patients with and without diabetes. J Thromb Haemost. 2012;10:1220–1230. [DOI] [PubMed] [Google Scholar]
- 32. Wurtz M, Hvas AM, Jensen LO, Kaltoft AK, Tilsted HH, Kristensen SD, Grove EL. 24‐hour antiplatelet effect of aspirin in patients with previous definite stent thrombosis. Int J Cardiol. 2014;175:274–279. [DOI] [PubMed] [Google Scholar]
- 33. Henry P, Vermillet A, Boval B, Guyetand C, Petroni T, Dillinger JG, Sideris G, Sollier CB, Drouet L. 24‐hour time‐dependent aspirin efficacy in patients with stable coronary artery disease. Thromb Haemost. 2011;105:336–344. [DOI] [PubMed] [Google Scholar]
- 34. Brantmark B, Wahlin‐Boll E, Melander A. Bioavailability of acetylsalicylic acid and salicylic acid from rapid‐and slow‐release formulations, and in combination with dipyridamol. Eur J Clin Pharmacol. 1982;22:309–314. [DOI] [PubMed] [Google Scholar]
- 35. Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation. 2013;127:377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Droppa M, Tschernow D, Muller KA, Tavlaki E, Karathanos A, Stimpfle F, Schaeffeler E, Schwab M, Tolios A, Siller‐Matula JM, Gawaz M, Geisler T. Evaluation of clinical risk factors to predict high on‐treatment platelet reactivity and outcome in patients with stable coronary artery disease (PREDICT‐STABLE). PLoS One. 2015;10:e0121620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reny JL, Fontana P, Hochholzer W, Neumann FJ, Ten BJ, Janssen PW, Geisler T, Gawaz M, Marcucci R, Gori AM, Cuisset T, Alessi MC, Berdague P, Gurbel PA, Yong G, Angiolillo DJ, Aradi D, Beigel R, Campo G, Combescure C. Vascular risk levels affect the predictive value of platelet reactivity for the occurrence of MACE in patients on clopidogrel. Systematic review and meta‐analysis of individual patient data. Thromb Haemost. 2016;115:844–855. [DOI] [PubMed] [Google Scholar]
- 38. Fokkema ML, James SK, Albertsson P, Aasa M, Akerblom A, Calais F, Eriksson P, Jensen J, Schersten F, de Smet BJ, Sjogren I, Tornvall P, Lagerqvist B. Outcome after percutaneous coronary intervention for different indications: long‐term results from the Swedish Coronary Angiography and Angioplasty Registry (SCAAR). EuroIntervention. 2016;12:303–311. [DOI] [PubMed] [Google Scholar]
- 39. Aradi D, Storey RF, Komocsi A, Trenk D, Gulba D, Kiss RG, Husted S, Bonello L, Sibbing D, Collet JP, Huber K. Expert position paper on the role of platelet function testing in patients undergoing percutaneous coronary intervention. Eur Heart J. 2014;35:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szczeklik W, Stodolkiewicz E, Rzeszutko M, Tomala M, Chrustowicz A, Zmudka K, Sanak M. Urinary 11‐dehydro‐thromboxane B2 as a predictor of acute myocardial infarction outcomes: results of Leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) study. J Am Heart Assoc. 2016;5:e003702 DOI: 10.1161/jaha.116.003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin‐resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–1655. [DOI] [PubMed] [Google Scholar]
- 42. Tofler GH, Brezinski D, Schafer AI, Czeisler CA, Rutherford JD, Willich SN, Gleason RE, Williams GH, Muller JE. Concurrent morning increase in platelet aggregability and the risk of myocardial infarction and sudden cardiac death. N Engl J Med. 1987;316:1514–1518. [DOI] [PubMed] [Google Scholar]
- 43. Moore RA, Derry S, Wiffen PJ, Straube S. Effects of food on pharmacokinetics of immediate release oral formulations of aspirin, dipyrone, paracetamol and NSAIDs—a systematic review. Br J Clin Pharmacol. 2015;80:381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]