

Abstract
Background
Emerging evidence indicates that impaired angiogenesis may contribute to hypertension‐induced cardiac remodeling. The nicotinamide adenine dinucleotide–dependent deacetylase Sirtuin 3 (SIRT3) has the potential to modulate angiogenesis, but this has not been confirmed. As such, the aim of this study was to examine the relationship between SIRT3‐mediated angiogenesis and cardiac remodeling.
Methods and Results
Our experiments were performed on SIRT3 knockout and age‐matched wild‐type mice infused with angiotensin II (1400 ng/kg per minute) or saline for 14 days. After angiotensin II infusion, SIRT3 knockout mice developed more severe microvascular rarefaction and functional hypoxia in cardiac tissues compared with wild‐type mice. These events were concomitant with mitochondrial dysfunction and enhanced collagen I and collagen III expression, leading to cardiac fibrosis. Silencing SIRT3 facilitated angiotensin II–induced aberrant Pink/Parkin acetylation and impaired mitophagy, while excessive mitochondrial reactive oxygen species generation limited angiogenic capacity in primary mouse cardiac microvascular endothelial cells. Moreover, SIRT3 overexpression in cardiac microvascular endothelial cells enhanced Pink/Parkin‐mediated mitophagy, attenuated mitochondrial reactive oxygen species generation, and restored vessel sprouting and tube formation. In parallel, endothelial cell–specific SIRT3 transgenic mice showed decreased fibrosis, as well as improved cardiac function and microvascular network, compared with wild‐type mice with similar stimuli.
Conclusions
Collectively, these findings suggest that SIRT3 could promote angiogenesis through attenuating mitochondrial dysfunction caused by defective mitophagy.
Keywords: angiogenesis, cardiac remodeling, mitochondria, mitophagy, oxidative stress, Sirtuin 3
Subject Categories: Hypertension, Oxidant Stress, Angiogenesis
Clinical Perspective
What Is New?
Sirtuin 3 (SIRT3) deficiency accelerates angiotensin II–induced microvascular rarefaction.
SIRT3‐mediated angiogenesis participates in hypertensive cardiac remodeling.
SIRT3‐mediated angiogenesis contributes to the induction of mitophagy.
SIRT3 induces mitophagy via direct deacetylation of Pink1/Parkin.
What Are the Clinical Implications?
Our data add to the findings of a few other studies suggesting that the angiogenic potential of cardiac endothelial cells may play a crucial role in modulating cardiac myocyte dysfunction.
Our findings suggest that SIRT3 could promote angiogenesis through attenuating mitochondrial dysfunction caused by defective mitophagy.
Introduction
Hypertension is causally related to cardiac remodeling, including left ventricular (LV) hypertrophy, myocardial fibrosis, and LV systolic and diastolic dysfunction.1, 2 Findings from numerous studies demonstrate that heart size and cardiac function are tightly coupled to myocardial angiogenesis. Impaired angiogenesis may result in myocardial microvascular rarefaction, leading to a reduction in oxygen and nutrient supply that can contribute to the progression of heart diseases.3, 4, 5 Thus, the angiogenic potential of cardiac endothelial cells may play a crucial role in modulating cardiac myocyte dysfunction, but this has not yet been confirmed.
Sirtuins (SIRTs) are a family of nicotinamide adenine dinucleotide–dependent deacetylases that consists of 7 isoforms (SIRT1–SIRT7) involved in glucose and fatty acid metabolism, apoptosis, and DNA repair.6, 7 SIRT3 localizes to the mitochondrial matrix, where it may function as a primary stress‐responsive protein deacetylase. Findings from recent studies reveal that SIRT3 has beneficial effects on hypertensive heart diseases. Mechanistically, SIRT3 blocks the cardiac hypertrophic response through activation of FOXO3‐dependent antioxidants, manganese superoxide dismutase (MnSOD), and catalase, as well as suppressing reactive oxygen species (ROS)–mediated Ras activation and downstream mitogen‐activated protein kinase/extracellular signal‐regulated kinase and phosphoinositide 3‐kinase/protein kinase B signaling pathways.8, 9 From these studies, the direct protective effect of SIRT3 on cardiomyocytes is apparent. In parallel, several reports have implicated SIRT3 as an apelin‐induced angiogenic growth factor expression and angiogenesis, while SIRT3 inactivation limits bone marrow cell–mediated angiogenesis and cardiac repair in post‐myocardial infarction.10 Conversely, SIRT3 overexpression inhibits glucose‐induced angiogenic gene expression in human retinal endothelial cells.11 Therefore, SIRT3 expression may play a dual role in angiogenesis based on the inducing stimuli. However, the role of SIRT3 in myocardial angiogenesis and its effect on hypertensive cardiac remodeling has not been defined.
Mitophagy, selective mitochondrial degradation through autophagy, is a conserved cellular process used to eliminate damaged mitochondria.12, 13 Defective mitophagy leads to an accumulation of damaged mitochondria and exacerbates mitochondrial oxidative stress, which may shift the cell's metabolic phenotype to adapt to different pathological stimuli.14, 15, 16 Our studies and others demonstrate that mitophagy/autophagy may participate in endothelial senescence, migration, and angiogenesis.17, 18 Thus, this autophagic response may be a novel target to enhance endothelial angiogenesis. A recent study also reported that SIRT3‐mediated mitophagy not only protects tumor cells against hypoxia‐induced apoptosis,19 but also suppresses age‐related cardiac hypertrophy.20 However, it remains uncertain whether SIRT3‐mediated angiogenesis contributes to the induction of mitophagy.
In the present study, we speculated that the potential beneficial effects of SIRT3 on hypertensive cardiac remodeling may be mediated through enhanced angiogenesis. To address this question, the present study examined the functional significance of SIRT3 in angiogenesis and mitophagy using SIRT3‐deficient and transgenic (Tg) mice, as well as isolated cardiac microvascular endothelial cells (CMVECs) stimulated with angiotensin II (Ang II). Notably, we show that SIRT3 is a negative regulator of cardiac hypertrophy in mice with Ang II–induced hypertension. Similarly, conditional SIRT3 expression in the endothelium of hypertensive mice restored vascularity and ameliorated cardiac dysfunction. Further, we demonstrate that SIRT3 is a stress‐responsive deacetylase that promotes myocardial angiogenesis via Pink1/Parkin‐dependent mitophagy, thereby decreasing mitochondrial ROS production.
Methods
Materials
Ang II and β‐actin antibodies were purchased from Sigma Chemicals. Microfil was obtained from Flow Tech, Inc. Collagen type I, collagen type III, aldehyde dehydrogenase 2, vascular endothelial growth factor (VEGF), Pink, and Parkin antibodies were obtained from Santa Cruz Biotechnology. MnSOD and anti‐4‐hydroxy‐nonenal antibodies were purchased from Millipore and Calbiochem, respectively. Antibodies against SIRT3, LC3B, and α‐smooth muscle actin were obtained from Cell Signaling Technology. Secondary antibodies, including peroxidase‐conjugated rabbit anti‐goat IgG, rabbit anti‐mouse IgG, and goat anti‐rabbit IgG, were supplied by Life Technologies. Matrigel and collagen I rat tail protein were obtained from Corning. Lectin was purchased from Vector Laboratories. SIRT3‐short hairpin RNA (shRNA) and SIRT3‐overexpressing lentivirus were synthesized by Genechem Corporation. Alexa Fluor 555‐labeled anti‐mouse IgG, Alexa Fluor 594‐labeled anti‐rabbit IgG, Alexa Fluor 488‐labeled anti‐rabbit IgG, MitoTracker Red, MitoTracker Green, and wheat germ agglutinin were from Life Technologies. Cell culture reagents were purchased from Hyclone.
Animal Model
SIRT3‐knockout (KO) (SIRT3 −/−) mice on a 129/Sv background were purchased from Jackson Laboratories. SIRT3flox/flox Tg mice were a gift from Professor Weiliang Xia (Shanghai Jiaotong University). Endothelial‐specific SIRT3 overexpression (SIRT3‐TgEC) mice were constructed by crossing SIRT3‐Tg mice with Tek‐Cre Tg mice (Shanghai Research Center for Model Organisms). Offspring were identified by tail‐snip polymerase chain reaction with the following primers: forward 5′‐ACTGCTCATCAACCGGGAC‐3′; reverse 5′‐CGCACACCGGCCTTATTCCAA‐3′; forward 5′‐ACTCCAAGGCCACTTATCACC‐3′; reverse 5′‐ATTGTTACCAACTGGGACGACA‐3′. Eight‐week‐old male wild‐type (WT), SIRT3‐KO, and SIRT3‐TgEC mice were infused with 1400 ng/kg per minute of Ang II or saline for 2 weeks (n=12/group) via subcutaneously implanted Alzet miniosmotic pumps.21, 22 Systolic blood pressure was measured by tail cuff plethysmography (BP‐2000; Visitech Systems) every 2 days. All procedures were approved by the Institutional Animal Care and Use Committee at Shanghai Jiaotong University and performed in conformance with the Guide for the Care and Use of Laboratory Animals (8th edition).
Echocardiography
Mice were anaesthetized via continuous inhalation of a mixture of isoflurane (1.5%) and oxygen. Cardiac ultrasound was performed using a Visual Sonics Vevo 770 Imaging System with a 30‐MHz high frequency transducer. M‐mode echocardiographic images were recorded. LV end‐systolic diameter (LVESD), LV end‐diastolic diameter (LVEDD), LV posterior wall thickness, and LV anterior wall thickness were recorded. LV fractional shortening (LVFS) is calculated according to the rule: LVFS=(LVEDD‐LVESD)/LVEDD×100%.
Histology and Immunohistochemistry
Hearts were collected from euthanized mice, fixed in 4% paraformaldehyde, and embedded in paraffin. Serial 5‐μm sections were stained with hematoxylin and eosin for general morphology. Masson's trichrome and Picrosirius red staining was performed to exhibit interstitial fibrosis. The collagen volume fraction was calculated by the proportion of blue areas in myocardium sections with Masson's staining using Image‐Pro plus software (Media Cybernetics, Inc). For immunohistochemistry, cross sections were blocked with 5% bovine serum albumin in phosphate‐buffered saline for 30 minutes, followed by incubation with collagen I in 5% bovine serum albumin overnight at 4°C. Images were captured with an Olympus BX51 microscope (OLYMPUS CORPORATION). Cardiomyocyte membranes were visualized using wheat germ agglutinin (5 μg/mL) followed by determination of the single cardiomyocyte cross‐sectional area. Quantitative data from at least 100 cells were determined per group. Hypoxia was visualized with a Hypoxyprobe‐1 Plus Kit (Hypoxyprobe). To evaluate microvascular density, slides were incubated with lectin (5 μg/mL) in the dark. Fluorescence images were obtained with a Zeiss Axiovert A1 microscope (Carl Zeiss) and then analyzed with ImageJ software.
Microfil Perfusion
Perfusions were performed as previously described.23 Briefly, mice were anesthetized with ketamine/xylazine intraperitoneally (100 mg/10 mg per kg). The chest and right heart atrium were infused with heparinized saline via the left ventricle, followed by 4% paraformaldehyde. The Microfil solution containing 42% MV‐122, 53% diluent, and 5% curing agent was injected into the hearts at 0.3 mL/min. Mice were placed at 4°C for 2 hours, and then hearts were collected and fixed in 4% paraformaldehyde before microCT (Skyscan 1176; Bruker) imaging.24 Images were obtained using 50‐kV, 500‐μA, and 1170‐ms exposure at a resolution of 9 μm voxel size. Three‐dimensional morphometric quantification was performed with CT‐Analyser (Bruker). Due to the cubic voxel side dimension was 9 μm, the coronary arterial lumen diameters were derived in 9‐μm increments. Coronary vessel volumes by luminal diameter from 18 to 540 μm were assessed. Because the transition point of which the epicardial vessels penetrated the myocardium and became intramyocardial vessels was within the range of 126 to 162 μm in luminal diameters, intramyocardial vessels were defined as luminal diameters from 18 to 162 μm. To avoid tomographic image blurring errors, vessels <18 μm were excluded from our quantitative analysis.25
Ex Vivo Aortic Sprout Assay
Mouse endothelial cell proliferation was monitored by aortic sprout assays using type I collagen as previously described.18, 26, 27 The whole thoracic aorta was removed from WT, SIRT3‐KO, and SIRT3‐TgEC mice and placed in sterile phosphate‐buffered saline. The isolated vessels were cleaned off perivascular tissue and cut into 1‐mm segments. The aortic strips were individually imbedded in a 48‐well plate coated with 150 μL type I collagen at 37°C for 30 minutes. MCDB131 medium containing 2.5% fetal bovine serum and Ang II (1 μmol/L) was added and then vessel sprouting was analyzed 8 days later with a Zeiss inverted microscope.
Transmission Electron Microscopy
Hearts were cut into 1‐mm3 sections, fixed in 2% glutaraldehyde at 4°C overnight, and then post‐fixed with 1% OsO4 before embedding in Epon. Ultra‐thin sections were stained with uranyl acetate and counterstained with lead citrate.28 Ultrastructure images were acquired with a JEM‐1200EX transmission electron microscope (JEOL Ltd).29
Cell Culture
CMVECs were obtained from Chi Scientific, Ltd, and cultured in DMEM supplemented with 10% fetal bovine serum and 100 μg/mL endothelial cell growth factor. The cells were infected with lentivirus harboring murine SIRT3 shRNA, the SIRT3 coding sequence, or GFP‐expressing control vector at a multiplicity of infection of 25. The transduced CMVECs were then treated with or without Ang II (1 μmol/L) for 12 hours.
Determination of ROS Generation
Cells were treated with Ang II for 4 hours before labeling with MitoTracker Red CMXRos for 30 minutes at 37°C, and then stained with Hochest 33 258. ROS staining was then analyzed with ImageJ software.
Quantification of LC3 Puncta, Mitophagy, and Mitochondrial Morphology
CMVECs were treated with Ang II and/or chloroquine (6 μmol/L) for 12 hours and then incubated with MitoTracker Green at 37°C for 30 minutes. To examine autophagic flux, cells were fixed and stained with anti‐LC3B (1:500) at 4°C overnight followed by Alexa Flour 594 goat anti‐rabbit IgG. The autophagic flux alterations and mitochondrial morphology was detected using a Zeiss LSM 710 microscope.
Tube Formation Assay
Matrigel was thawed on ice at 4°C overnight. Pipette tips and 48‐well plates were kept at −20°C for 2 hours before coating each well with 100 μL Matrigel per well at 37°C for 30 minutes. CMVECs infected with SIRT3‐shRNA, lentivirus‐mediated SIRT3, or vector control were seeded at 5×104 cells in 200 μL medium with or without Ang II for 8 hours. Tube formation was then assessed by microscopy and the images analyzed for tube length with ImageJ software.18
Immunoprecipitation
For immunoprecipitation studies, cells were lysed in buffer supplemented with protease inhibitor. Protein lysates (500 μg) were incubated with lysine antibody (ImmuneChem) containing protein A/G agarose beads overnight at 4°C with orbital rotation. After washing 5 times, proteins were eluted with SDS‐PAGE sample buffer and analyzed with Western blotting. A Pierce Co‐Immunoprecipitation Kit (Thermo Scientific) was used to study the interaction between SIRT3 and Pink1/Parkin according to the manufacturer's instructions. Briefly, affinity‐purified SIRT3 antibody (25 μg) was immobilized onto resin for 90 minutes at room temperature and washed with 1x coupling buffer. Equal amounts of protein (500 μg) was added to the resin and incubated with gentle mixing overnight at 4°C. After washing 3 times with immunoprecipitation lysis buffer, the protein was eluted and examined with Western blotting. To analyze Pink1/Parkin acetylation, 1 mg of protein lysate was directly immunoprecipitated with an acetyl‐lysine antibody and then subjected to Western blotting for Pink1 and Parkin.
Western Blot Analysis
Protein lysate concentrations were assessed with a BCA Protein Assay Kit (Thermo Scientific). Equal amounts of protein (10 μg) were separated by SDS‐PAGE and transferred onto polyvinylidene difluoride membranes (Bio‐Rad Laboratories). Blots were blocked with 5% nonfat milk in tris‐buffered saline with Tween 20 for 1 hour at room temperature and incubated with primary antibodies against SIRT3 (1:1000), collagen I and III (1:1000), α‐smooth muscle actin (1:8000), anti‐4‐hydroxy‐nonenal (1:1000), MnSOD (1:5000), aldehyde dehydrogenase 2 (1:4000), VEGF (1:1000), LC3 (1:2000), Pink1 (1:1000), and Parkin (1:1000) at 4°C overnight. After washing 4 times, the membranes were incubated with horseradish peroxidase–conjugated secondary antibodies for 1 hour. Immunoreactive bands were detected with ECL reagent (Thermo Scientific) and quantified in Quantity One.
Statistical Analysis
Data represent mean±SEM. Statistical significance between 2 and multiple groups were evaluated with Student t test and 1‐way ANOVA, respectively, using GraphPad Prism 5.0. P<0.05 was considered statistically significant.
Results
SIRT3 Deficiency Accelerates Ang II–Induced Cardiac Remodeling
To determine the role of SIRT3 in regulating Ang II–induced cardiac remodeling, SIRT3‐KO and WT mice were infused with Ang II for 2 weeks. As expected, treatment induced a significant increase in systolic blood pressure that was similar in magnitude between WT and SIRT3‐KO mice compared with their saline‐treated controls (Figure S1). Heart/body weight (HW/BW) ratios in SIRT3‐KO mice were higher than the WT counterparts (Figure 1A). As exemplified by the M‐mode tracings in Figure 1B through 1F, SIRT3‐KO mice did not show any aberrant cardiac phenotype; however, there was greater concentric remodeling, with a greater increase in LV posterior wall thickness, LV anterior wall thickness, and LVFS and a decrease in LVESD after Ang II infusion. Consistently, histological analysis showed increased fibrosis and collagen volume in SIRT3‐KO (Figure 1G and 1H), accompanied by increased α‐smooth muscle actin, collagen Iα, and collagen IIIα with Ang II treatment (Figure 1I through 1L). Thus, these results indicated that SIRT3 deficiency accelerated Ang II–induced cardiac remodeling.
Figure 1.
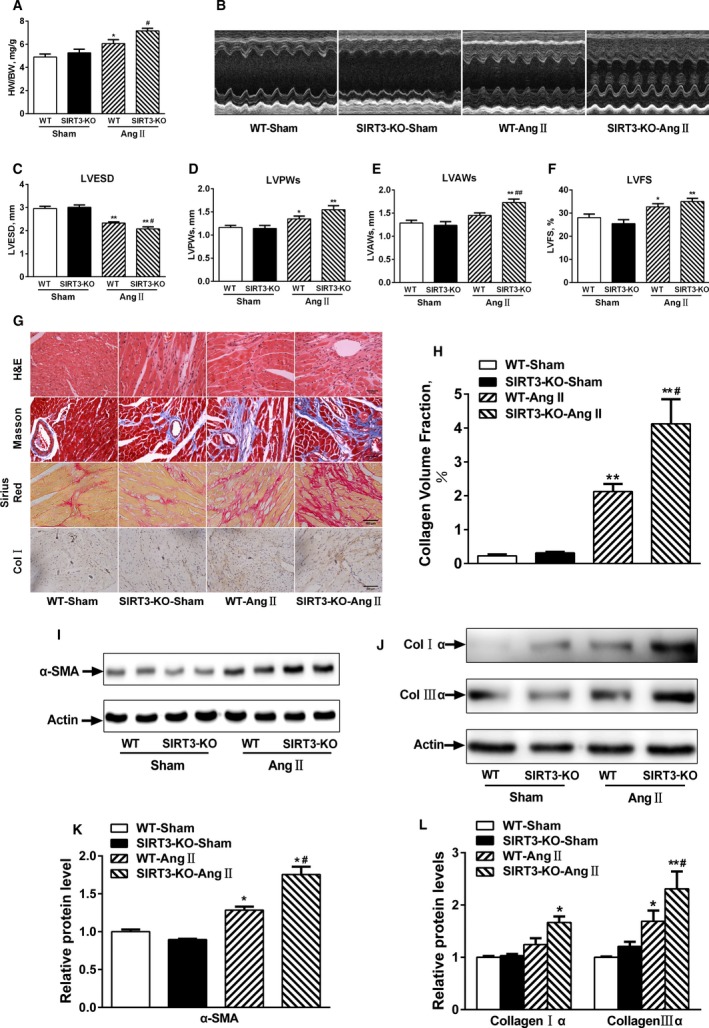
Sirtuin 3 (SIRT3) deficiency accelerates angiotensin II (Ang II)–induced cardiac remodeling. A, Heart to body weight (HW/BW ratios, n=10). B, Representative echocardiography images of wild‐type (WT) and SIRT3‐knockout (KO) hearts. C through F, Quantitative analysis of left ventricular end‐systolic diameter (LVESD), left ventricular posterior wall thickness (LVPWs), left ventricular anterior wall thickness (LVAWs), and left ventricular fractional shortening (LVFS) (n=10/group). G, Representative hematoxylin and eosin (H&E), Masson trichrome, Picrosirius red, and collagen I staining of WT and SIRT3‐KO hearts. H, Quantitative analysis of collagen volume fraction. I and J, Representative Western blot and quantitation for (K) α‐smooth muscle actin (α‐SMA) and (L) collagen I and collagen III. Data are presented as mean±SEM. *P<0.05, **P<0.01 vs genotype‐matched sham mice; # P<0.05, ## P<0.01 vs WT‐Ang II mice. Col indicates collagen.
SIRT3 Deficiency Exacerbates Microvascular Rarefaction
Ang II–dependent hypertension led to a reduction in the cardiac vascular network as determined by micro‐computed tomography analysis with a spatial resolution of ≈9 μm voxel side length (Figure 2A). The total and intramyocardial vessel volumes in WT mice were comparable to SIRT3‐KO mice but decreased significantly after Ang II infusion in SIRT3‐KO mice (Figure 2B and 2C).
Figure 2.
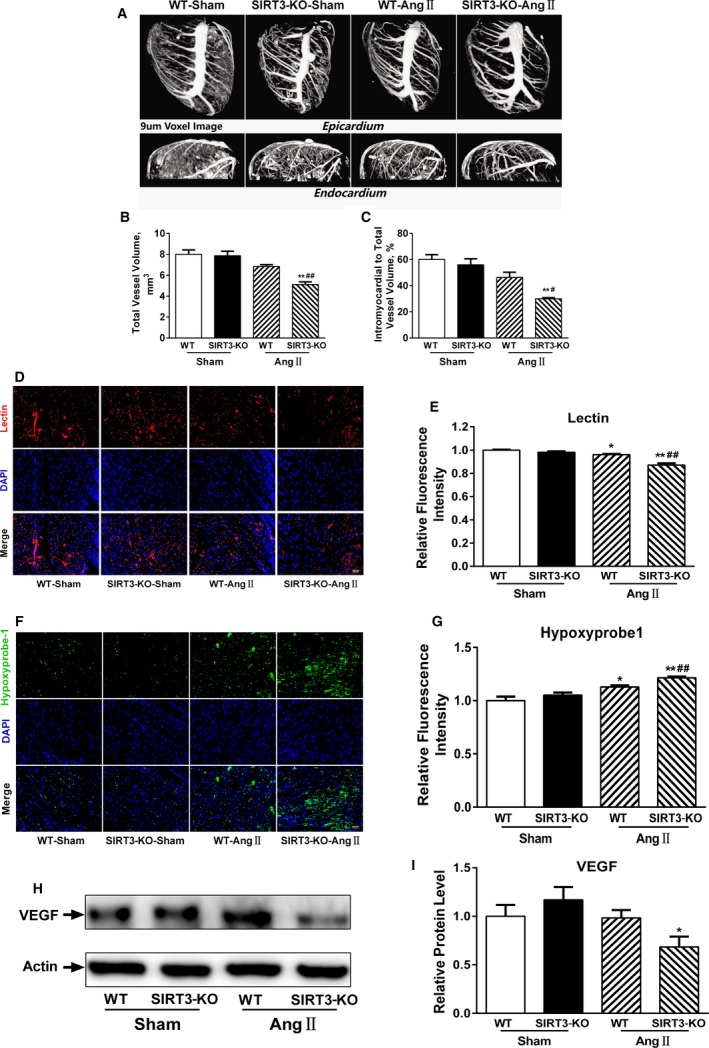
Sirtuin 3 (SIRT3) deficiency exacerbates microvascular rarefaction after angiotensin II (Ang II) challenge. A, Representative micro‐computed tomography images of myocardial vasculatures after Microfil perfusion. B and C, Quantitative assessment of total vessel volume, and intramyocardial vessel volume normalized to total vessel volume (n=3/group). Data are presented as mean±SEM. D, Representative images of cardiac sections stained with lectin (red) and 4’,6‐diamidino‐2‐phenylindole (DAPI; blue). Scale, 200 μm. E, Quantification of lectin intensity. F, Representative images of cardiac sections stained with hypoxyprobe‐1 (green) and DAPI (blue). Scale, 200 μm. G, Quantification of hypoxyprobe‐1 intensity. H through I, Representative vascular endothelial growth factor (VEGF) Western blot and densitometry. Data are relative to that of wild‐type (WT)‐sham mice (mean±SEM). *P<0.05, **P<0.01 vs genotype‐matched sham mice; # P<0.05, ## P<0.01 vs WT‐Ang II mice. KO indicates knockout.
Cardiac tissue sections were stained with fluorescein‐conjugated tomato lectin to identify blood vessels. Immunofluorescence analysis revealed a reduction in microvascular density in WT and SIRT3‐KO mice infused with Ang II, while the level tended to be lower in SIRT3‐KO mice (Figure 2D and 2E). Consistent with these observations, tissue oxygen levels—indicated by pimonidazole staining—declined in SIRT3‐KO heart but only modestly in WT controls in response to Ang II (Figure 2F and 2G). Previous studies indicate that VEGF‐A plays a crucial role in controlling vascular network formation in cardiac tissues. Ang II–induced cardiac remodeling was also associated with decreased VEGF‐A protein, which was greater in SIRT3‐KO mice compared with controls—consistent with the reductions in cardiac vasculature (Figure 2H and 2I). These results suggested that SIRT3 deficiency exacerbates microvascular rarefaction and secondary to induce myocardial hypoxia.
SIRT3 Deficiency Aggravates Cardiac Mitochondrial Dysfunction and Oxidative Stress
SIRT3 exists in 44‐kDa long and 28‐kDa short form. The long form is found throughout the cell, whereas the short form is localized exclusively in the mitochondria. Notably, we found that the short form of SIRT3 was decreased in the hearts of mice subjected to Ang II infusion (Figure 3A and 3B).
Figure 3.
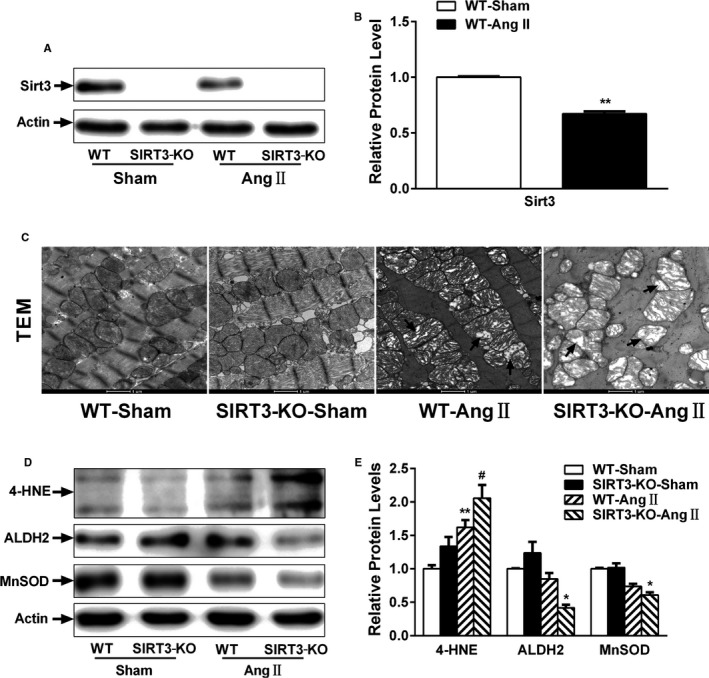
Loss of Sirtuin 3 (SIRT3) aggravates mitochondrial dysfunction and oxidative stress. A and B, Representative SIRT3 Western blot and desnitometry. C, Representative transmission electron microscopy (TEM) images of mitochondria in wild‐type (WT) and SIRT3‐knockout (KO) hearts. D and E, Anti‐4‐hydroxy‐nonenal (4‐HNE), aldehyde dehydrogenase 2 (ALDH2), and manganese superoxide dismutase (MnSOD) Western blot and densitometry. Data are relative to that of WT‐sham control mice (mean±SEM). *P<0.05, **P<0.01 vs genotype‐matched sham mice; # P<0.05 vs WT‐Ang II mice.
Previous studies demonstrate that SIRT3 resides in the mitochondria and regulates many aspects of mitochondrial function. As shown in Figure 3C, electron microscopy of WT cardiac sections showed mitochondria regular in shape, compact, and with a high electron dense matrix. Conversely, SIRT3‐KO hearts displayed defective mitochondrial organization. Moreover, following Ang II infusion, mitochondria in WT hearts were swollen with distorted cristae. Similar ultrastructural changes were observed in SIRT3‐KO mice but more severe.
Anti‐4‐hydroxy‐nonenal is a major aldehyde produced during lipid peroxidation. As shown in Figure 3D and 3E, anti‐4‐hydroxy‐nonenal adducts increased in cardiac tissue from SIRT3‐KO mice compared with WT controls following Ang II infusion. Moreover, aldehyde dehydrogenase 2 and MnSOD levels were significantly lower in SIRT3‐KO mice. Collectively, these results suggest that the mitochondrial defects observed in Ang II–induced cardiac remodeling result from reduced SIRT3 function.
Role of SIRT3 in Modulating CMVEC Angiogenic Capacity After Ang II Challenge
To examine the effects of SIRT3 on angiogenesis, we performed endothelial‐sprouting assay and microvascular tube formation assay to examine angiogenic capacity in CMVECs ex vivo. Aortic rings from WT, SIRT3‐KO, and SIRT3‐TgEC mice were cultured with or without Ang II (1 μmol/L) for 8 days. As shown in Figure 4A through 4C, aortic rings from SIRT3‐KO mice show a trend in reduced microvascular sprout outgrowth. Moreover, Ang II decreased sprouting in aortic rings from WT mice and to a greater extent in SIRT3‐KO mice, whereas SIRT3 overexpression in the endothelium restored outgrowth.
Figure 4.
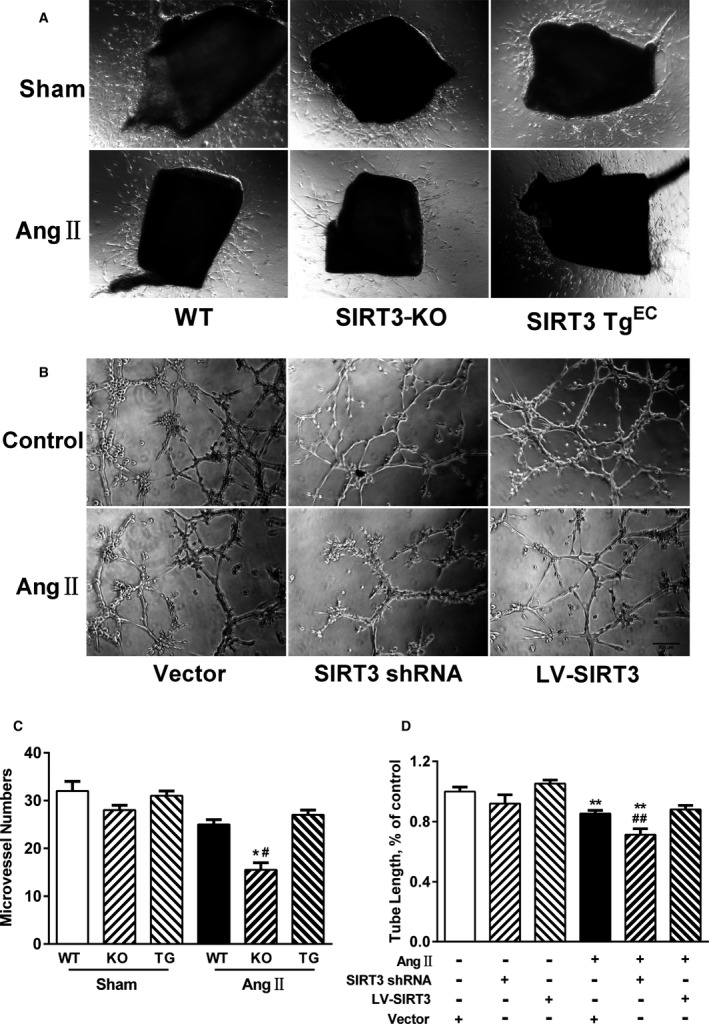
Sirtuin 3 (SIRT3) deficiency impairs angiogenic capacity. A, Representative micrographs of microvessel sprouting in aortic rings from wild‐type (WT), SIRT3‐knockout (KO), and endothelium‐specific SIRT3‐transgenic (SIRT3 TgEC) mice. B, Representative tube formation images of cardiac microvascular endothelial cells infected with SIRT3‐short hairpin RNA (shRNA), lentivirus‐mediated SIRT3 (LV‐SIRT3), or negative controls treated with or without angiotensin II (Ang II; 1 μmol/L) for 12 hours. Scale, 500 μm. C, Quantitative analysis of microvessel sprouting in aortic rings. Data are presented as mean±SEM. *P<0.05 vs genotype‐matched sham mice; # P<0.05 vs WT‐Ang II mice. D, Quantitative analysis of tube length in each well. Five images per well were randomly chosen to be measured. Results are expressed as fold‐change over untreated vector‐infected cells. **P<0.01 vs untreated genotype‐matched cells; ## P<0.01 vs Ang II–treated vector‐infected cells.
SIRT3‐shRNA expression in CMVECs resulted in an 80% reduction in protein expression (Figure S2). Notably, SIRT3‐shRNA cells showed decreased tube length in CMVECs, while no effect was observed in empty vector controls (Figure 4B). Conversely, cells with lentivirus‐mediated SIRT3 overexpression, which increased SIRT3 expression by 1.5‐fold, rescued the Ang II–induced decrease in tube formation (Figure 4D), suggesting that SIRT3 overexpression could ameliorate impaired angiogenesis in response to Ang II challenge.
Inhibition of Endogenous SIRT3 Suppresses Pink/Parkin‐Mediated Mitophagy
Defects in mitophagy may exacerbate mitochondrial oxidative stress. To investigate the impact of Ang II on mitophagy in CMVECs, cells were stained with LC3B antibody. Although Ang II–induced LC3 foci formation was comparable to control, and using lysosomal inhibitor chloroquine alone resulted to an increase in LC3 foci that largely colocalized with MitoTracker Green–labeled mitochondria, the increase was much smaller in combination with Ang II. These data indicated that Ang II was effective in inhibiting autophagic flux in CMVECs (Figure 5A). Moreover, SIRT3‐shRNA CMVECs displayed an accelerated decrease in LC3‐II protein expression, whereas SIRT3 overexpression rescued this effect (Figure 5B and 5C). We subsequently performed co‐immunoprecipitation studies to determine whether SIRT3‐mediated mitophagy occurred in a Pink1/Parkin‐dependent manner. As shown in Figure 5D, endogenous Pink1 and Parkin co‐immunoprecipitated with SIRT3 in CMVEC lysates. Based on these observations, we next sought to understand whether Pink1/Parkin was a potential SIRT3 substrate. As seen in Figure 5E and 5F, Pink1/Parkin was acetylated under basal conditions and the acetylation increased in response to Ang II; however, CMVECs with SIRT3 overexpression displayed a decrease in Pink1/Parkin acetylation (Figure 5G and 5H), which were increased in SIRT3‐shRNA cells. These results indicated that SIRT3 could induce Pink/Parkin pathway activation to potentiate mitophagy.
Figure 5.
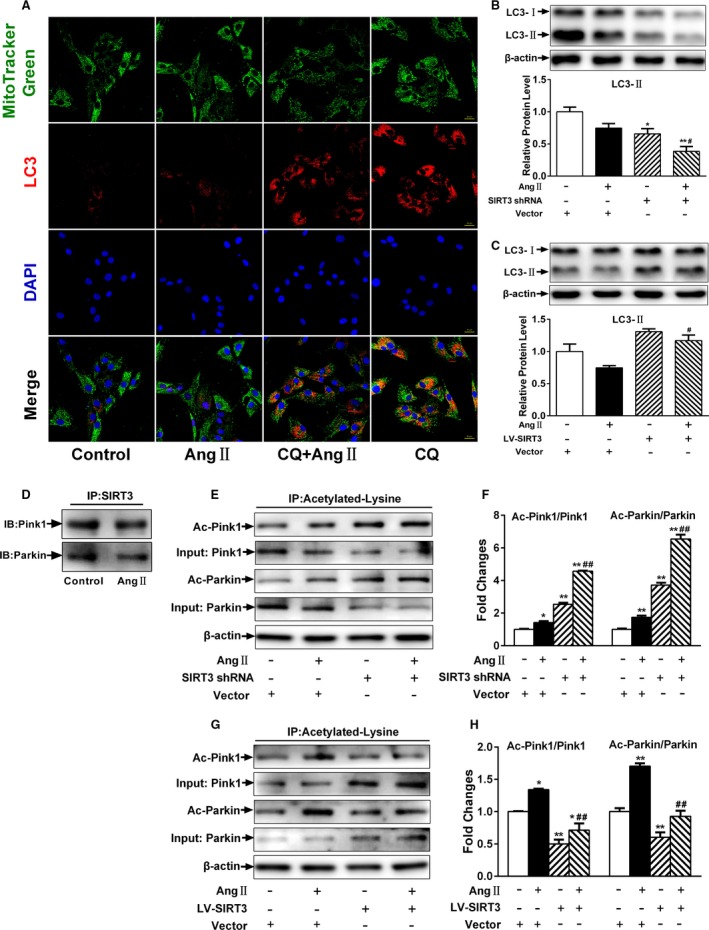
Loss of Sirtuin 3 (SIRT3) suppresses Pink/Parkin‐mediated mitophagy. A, Representative confocal images of autophagic flux stained with LC3B (red), MitoTracker Green, and 4′,6‐diamidino‐2‐phenylindole (blue). Scale, 200 μm. B, LC3‐II expression was detected by Western blotting with quantitative analysis in SIRT3‐short hairpin RNA (shRNA) or negative control cells. C, LC3‐II expression was detected by Western blotting with quantitative analysis in lentivirus‐mediated SIRT3 (LV‐SIRT3) or negative control infected cells. D, Representative Western blot analysis of Pink1 and parkin following immunoprecipitation with SIRT3 antibody. E, Representative Western blot and (F) quantitative analysis of acetylated Pink1 and acetylated Parkin in SIRT3‐shRNA or negative control cells. G, Representative Western blot and (H) quantitative analysis of acetylated Pink1 and acetylated Parkin in LV‐SIRT3 or negative control cells. Results are expressed as fold‐change over untreated vector‐infected cells. Data are presented as means±SEM. *P<0.05, **P<0.01 vs untreated vector‐infected cells; # P<0.05, ## P<0.01 vs angiotensin II (Ang II)–treated vector‐infected cells. CQ indicates chloroquine.
Restoration of Angiogenic Capacity by Attenuating Mitochondrial Oxidative Stress
Excessive mitochondrial‐derived oxidative stress can impair angiogenesis.30 Ang II–treated cells showed enhanced mitochondrial ROS generation compared with control cells. In addition, SIRT3‐shRNA cells showed an increase in Ang II–induced ROS production (Figure 6A and 6B), whereas these effects were ameliorated in lentivirus‐mediated SIRT3 counterparts (Figure 6C and 6D). Moreover, Ang II also decreased MnSOD expression, which was potentiated in SIRT3‐shRNA cells but partly rescued in those with SIRT3 overexpression (Figure 6E and 6F).
Figure 6.
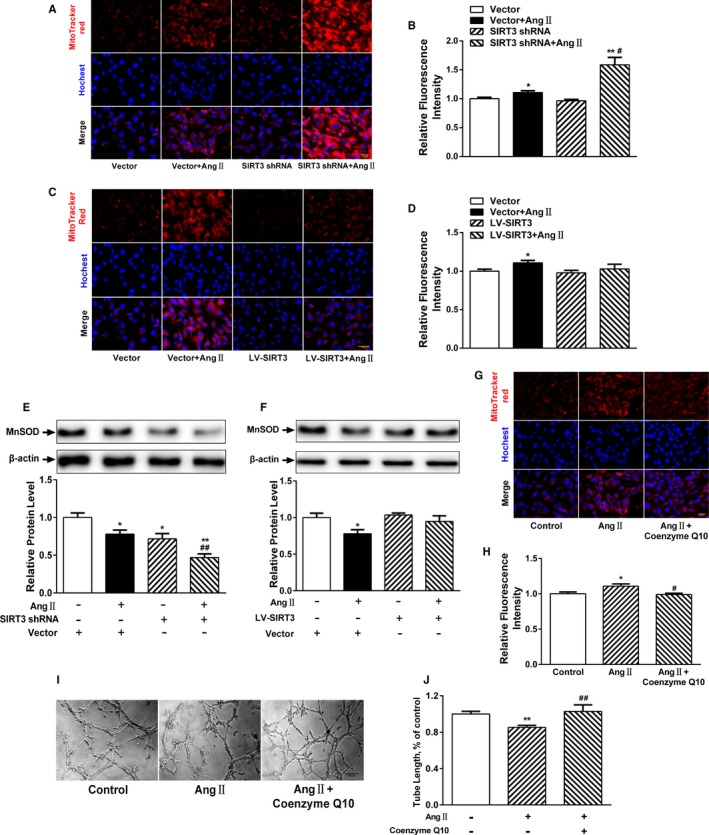
Attenuated mitochondrial oxidative stress restores angiogenic capacity. A, Representative images of reactive oxygen species (ROS) and MitoTracker Red staining in Sirtuin 3 (SIRT3)‐short hairpin RNA (shRNA) or negative control cardiac microvascular endothelial cells (CMVECs). Scale, 200 μm. B, MitoTracker Red quantification. C, Representative images and (D) quantification of ROS with MitoTracker Red staining in lentivirus‐mediated SIRT3 (LV‐SIRT3) or negative control CMVECs. Scale, 200 μm. E, Representative Western blot analysis and quantification of manganese superoxide dismutase (MnSOD) expression in SIRT3‐shRNA or negative control cells. F, Representative Western blot analysis and (H) quantification of MnSOD expression in LV‐SIRT3 or negative control cells. Results are expressed as fold‐change over untreated vector‐infected cells. *P<0.05, **P<0.01 vs untreated vector‐infected cells; # P<0.05, ## P<0.01 vs angiotensin II (Ang II)–treated vector‐infected cells. G, Representative fluorescence images and (H) quantification of ROS with MitoTracker Red staining in CMVECs pretreated with or without coenzyme Q10. Scale, 200 μm. I, Representative tube formation images of CMVECs pretreated with or without coenzyme Q10. J, Quantitative analysis of tube length in each well. Five images per well were randomly chosen to be measured. *P<0.05, **P<0.01 vs control cells; # P<0.05, ## P<0.01 vs Ang II treatment.
Coenzyme Q10 is an essential cofactor of the electron transport chain and a potent antioxidant that acts as a mitochondrial ROS scavenger. Interestingly, coenzyme Q10 treatment significantly suppressed Ang II–mediated ROS production (Figure 6G and 6H). Further, to assess whether Ang II–induced ROS production induced antiangiogenic effects, CMVECs were pretreated with coenzyme Q10 before Ang II treatment, which resulted in enhanced microvascular tube formation (Figure 6I and 6J). Thus, these results suggest that attenuating mitochondrial oxidative stress may restore the angiogenic capacity of CMVECs.
SIRT3 Overexpression Prevents Hypertension‐Induced Cardiac Remodeling by Restoring Angiogenesis
To test whether restored cardiac vascularity attenuated hypertension‐induced cardiac remodeling, we generated homozygous SIRT3‐TgEC mice by crossing SIRT3flox/flox with Tie2‐CreTg mice. Notably, Ang II infusion for 14 days resulted in the development of 20% cardiac hypertrophy in WT mice, whereas SIRT3‐Tg mice showed a decrease in heart/body weight ratio compared with WT mice (Figure 7A). Then, we performed echocardiography to evaluate the prevalence of hypertrophy and found that WT mice exhibited a greater increase in LV thickness and LVFS and a decrease in LVESD, which remained unchanged in SIRT3‐Tg mice after Ang II infusion (Figure 7B through 7F). Similarly, SIRT3‐TgEC mice were also resistant to Ang II–induced cardiac fibrosis and hypertrophy, which was robust in WT controls (Figure 7G through 7I). Ang II treatment also significantly induced a reduction in microvessel density that was associated with severe hypoxia in the cardiac tissue of WT mice. This also was not detected in SIRT3‐TgEC mice (Figure 7J through 7M), suggesting that LV functions of SIRT3‐TgEC mice were preserved.
Figure 7.
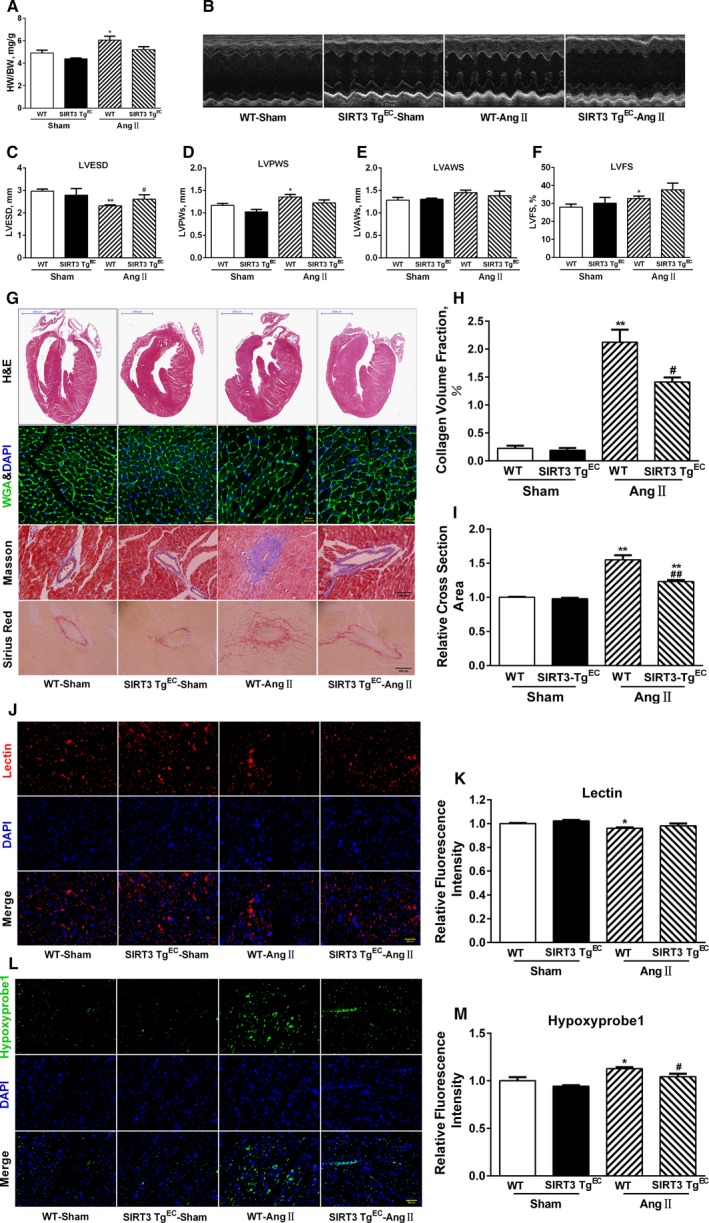
Sirtuin 3 (SIRT3) overexpression reverses hypertensive cardiac remodeling. A, Heart/body weight (HW/BW) ratios in wild‐type (WT) and endothelium‐specific SIRT3‐transgenic (SIRT3‐TgEC) mice (n=10). B, Representative echocardiography images of WT and SIRT3‐TgEC mice hearts. C through F, Quantitative analysis of left ventricular end‐systolic diameter (LVESD), left ventricular posterior wall thickness (LVPW), left ventricular anterior wall thickness (LVAW), and left ventricular fractional shortening (LVFS) (n=10/group). G, Representative hematoxylin and eosin (H&E), wheat germ agglutinin (WGA), Masson trichrome, and Picrosirius red staining of WT and SIRT3‐TgEC mice hearts. H, Quantitative analysis of collagen volume fraction. I, Quantitative analyses of cardiomyocyte cross‐sectional area. J, Representative images of cardiac sections stained with lectin (red) and 4′,6‐diamidino‐2‐phenylindole (DAPI; blue). Scale, 200 μm. K, Quantification of lectin intensity. L, Representative images of cardiac sections stained with hypoxyprobe‐1 (green) and DAPI (blue). Scale, 200 μm. M, Quantification of hypoxyprobe‐1 intensity. Data are presented as mean±SEM. *P<0.05, **P<0.01 vs genotype‐matched sham mice; # P<0.05, ## P<0.01 vs WT‐angiotensin II (Ang II) mice.
Discussion
In the present study, we demonstrated that SIRT3‐KO resulted in more severe microvascular rarefaction and functional hypoxia in cardiac tissue compared with WT mice. These events were concomitant with cardiac mitochondrial dysfunction and enhanced collagen I and collagen III expression, which led to cardiac fibrosis in Ang II infusion mice. This was further accompanied by an impairment of Pink1/Parkin‐mediated mitophagy in CMVECs. SIRT3 overexpression restored angiogenic capacity as well as improved cardiac function and decreased fibrosis, thereby suggesting that SIRT3 could promote angiogenesis and contribute to attenuated mitochondrial dysfunction and increased Pink/Parkin‐mediated mitophagy.
Multiple lines of evidence suggest vulnerability to hypoxia is greater in the endocardium of the left ventricle than in the epicardial layers. This phenomenon has been proven in the hearts of spontaneously hypertensive rats.31 Similarly, several reports have indicated that Ang II—the main effector peptide of the renin‐angiotensin system—induced hypertensive cardiac hypertrophy that was also associated with a reduction in microvascular density.32 Meanwhile, angiotensin‐converting enzyme inhibitors could increase microvascular density independently of its antihypertensive and antihypertrophic actions.33 The underlying mechanism is caused, in part, by defects in the angiogenic capacity of cardiac endothelial cells, resulting in microvascular rarefaction in the endocardium. Inhibition of new blood vessel formation accelerates the development of LV dysfunction, whereas stimulation of angiogenesis improves cardiac function and delays the onset of heart failure.34, 35, 36 Thus, therapeutic myocardial angiogenesis is a promising approach for the treatment of hypertension‐induced cardiac damage.4
Mitochondrial SIRT3 is highly expressed in metabolically active tissues, such as the heart, liver, kidney, and brown adipose tissue.37 Although mitochondrial content in endothelial cells is modest compared with other cell types, SIRT3 exerts protective effects in response to various damage factors involved in endothelial dysfunction, such as Ang II, transforming growth factor‐β, and high glucose.38, 39, 40 Therefore, we conclude that the reduced microvascular formation and VEGF expression in cardiac tissue was accompanied by a loss of mitochondrial SIRT3 during Ang II–induced cardiac remodeling. In parallel, SIRT3 depletion reveals no compensatory effect existing from other mitochondrial Sirtuins (SIRT4 and SIRT 5) (data not shown). In addition, Ang II–induced mitochondrial dysfunction in CMVECs was characterized by elevated mitochondrial ROS production and decreased mitophagy.
To our knowledge, the majority of research regarding Pink/Parkin‐mediated mitophagy has been related to neurodegenerative diseases, such as Parkinson's disease.41 Here, we found that Pink/Parkin is also involved in endothelial dysfunction. Further, we found that SIRT3 interacted with Pink1 and Parkin, resulting in their deacetylation by SIRT3 in vitro. Posttranslational modifications are critical for regulating Pink1 and Parkin activity and function. A review of the literature suggests that Pink1/Parkin‐mediated mitophagy is mainly dependent on Pink1 kinase activity, which is required to induce translocation of the E3 ligase Parkin to depolarized mitochondria. However, a recent study showed that Pink1 can bind Beclin1 and enhance autophagy, but this is not dependent on Pink1 kinase activity.42 Other reports demonstrate that SIRT3‐mediated Pink1/Parkin pathway activation is mainly dependent on FOXO3A deacetylation.16 Nevertheless, few studies have explored gain‐of‐function effects with SIRT3 on the Pink1/Parkin pathway. In the current study, we found that SIRT3 reduces ROS levels not only by enhancing MnSOD and catalase expression, but also by inducing mitophagy via direct Pink1/Parkin deacetylation.
Knockdown or overexpression of SIRT3 in CMVECs altered Ang II–induced ROS generation and mitophagy. Further, mitochondrial ROS caused by defects in mitophagy was attenuated by coenzyme Q10, which also restored CMVEC angiogenesis. Collectively, these data indicated that Ang II induced mitochondrial oxidative stress partly by suppressing SIRT3‐mediated mitophagy. In agreement with our results, recent studies report that SIRT3‐mediated mitophagy may be involved in protecting cardiomyocytes from diabetic cardiomyopathys.
Conclusions
SIRT3‐mediated attenuations in cardiac hypertrophy have been previously documented; however, our data show that the effect of SIRT3 on angiogenesis may contribute to the progression of hypertensive cardiac remodeling. Moreover, endothelial‐specific SIRT3 expression in hypertensive mice restored vascularity and ameliorated cardiac dysfunction. As per the scheme presented in Figure S3, we propose that SIRT3 drives Pink1/Parkin activity to increase rates of mitophagy in response to oxidative stress, and thereby limits the production of damaging ROS that would further promote angiogenesis.
Sources of Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 91439113, 81370255, 81272010, and 81472099).
Disclosures
None.
Supporting information
Figure S1. Systolic blood pressure in angiotensin II (Ang II)–induced mice. A, Systolic blood pressure profile of wild‐type (WT) and SIRT3−/− mice measured every 2 days for 2 weeks. B, Systolic blood pressure of WT and SIRT3−/− mice infused with Ang II in day 14. The values are presented as means±SEM. ** P<0.01 vs genotype‐matched sham mice.
Figure S2. Sirtuin 3 (SIRT3) expression in cardiac microvascular endothelial cells (CMVECs). A, SIRT3 expression was detected by Western blotting with quantitative analysis in SIRT3‐short hairpin RNA (shRNA) or negative control cells. B, SIRT3 expression was detected by Western blotting with quantitative analysis in lentivirus‐mediated SIRT3 (LV‐SIRT3) or negative control infected cells. Results are expressed as fold‐change over untreated vector‐infected cells. Data are presented as means±SEM. *P<0.05, **P<0.01 vs untreated vector‐infected cells; # P<0.05, ## P<0.01 vs angiotensin II (Ang II)–treated vector‐infected cells.
Figure S3. Graphical summery. Sirtuin 3 (SIRT3) drives Pink1/Parkin activity to increase rates of mitophagy in response to oxidative stress, and thereby limits the production of damaging reactive oxygen species (ROS) that would further promote angiogenesis.
Acknowledgments
We thank Professor Weiliang Xia (Shanghai Jiaotong University) for providing SIRT3flox/flox Tg mice.
(J Am Heart Assoc. 2017;6:e006114 DOI: 10.1161/JAHA.117.006114.)
References
- 1. Deschepper CF, Llamas B. Hypertensive cardiac remodeling in males and females: From the bench to the bedside. Hypertension. 2007;49:401–407. [DOI] [PubMed] [Google Scholar]
- 2. Ishizu T, Seo Y, Kameda Y, Kawamura R, Kimura T, Shimojo N, Xu D, Murakoshi N, Aonuma K. Left ventricular strain and transmural distribution of structural remodeling in hypertensive heart disease. Hypertension. 2014;63:500–506. [DOI] [PubMed] [Google Scholar]
- 3. Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114:565–571. [DOI] [PubMed] [Google Scholar]
- 4. Humar R, Zimmerli L, Battegay E. Angiogenesis and hypertension: an update. J Hum Hypertens. 2009;23:773–782. [DOI] [PubMed] [Google Scholar]
- 5. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park S, Mori R, Shimokawa I. Do sirtuins promote mammalian longevity? A critical review on its relevance to the longevity effect induced by calorie restriction. Mol Cells. 2013;35:474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV Jr, Weissman S, Verdin E, Schwer B. Mammalian SIR2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. SIRT3 blocks the cardiac hypertrophic response by augmenting Foxo3a‐dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, Meyer‐Steenbuck M, Cenkerova K, Hoffmann MM, Jaeger C, Odening KE, Kammerer B, Hein L, Bode C, Bugger H. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol. 2015;110:36 DOI: 10.1007/s00395‐015‐0493‐6. [DOI] [PubMed] [Google Scholar]
- 10. Hou X, Zeng H, He X, Chen JX. SIRT3 is essential for apelin‐induced angiogenesis in post‐myocardial infarction of diabetes. J Cell Mol Med. 2015;19:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mao XB, You ZP, Wu C, Huang J. Potential suppression of the high glucose and insulin‐induced retinal neovascularization by Sirtuin 3 in the human retinal endothelial cells. Biochem Biophys Res Commun. 2017;482:341–345. [DOI] [PubMed] [Google Scholar]
- 12. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Springer MZ, Macleod KF. In brief: mitophagy: mechanisms and role in human disease. J Pathol. 2016;240:253–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Requejo‐Aguilar R, Lopez‐Fabuel I, Fernandez E, Martins LM, Almeida A, Bolanos JP. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat Commun. 2014;5:4514. [DOI] [PubMed] [Google Scholar]
- 15. Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW II. Parkin‐mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu W, Gao B, Li N, Wang J, Qiu C, Zhang G, Liu M, Zhang R, Li C, Ji G, Zhang Y. SIRT3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A‐Parkin‐mediated mitophagy. Biochim Biophys Acta. 2017;1863:1973–1983. [DOI] [PubMed] [Google Scholar]
- 17. Shen W, Tian C, Chen H, Yang Y, Zhu D, Gao P, Liu J. Oxidative stress mediates chemerin‐induced autophagy in endothelial cells. Free Radic Biol Med. 2013;55:73–82. [DOI] [PubMed] [Google Scholar]
- 18. Lin JR, Shen WL, Yan C, Gao PJ. Downregulation of dynamin‐related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler Thromb Vasc Biol. 2015;35:1413–1422. [DOI] [PubMed] [Google Scholar]
- 19. Qiao A, Wang K, Yuan Y, Guan Y, Ren X, Li L, Chen X, Li F, Chen AF, Zhou J, Yang JM, Cheng Y. SIRT3‐mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget. 2016;7:43390–43400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3‐mediated deacetylation of CypD at lysine 166 suppresses age‐related cardiac hypertrophy. Aging. 2010;2:914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jia LX, Qi GM, Liu O, Li TT, Yang M, Cui W, Zhang WM, Qi YF, Du J. Inhibition of platelet activation by clopidogrel prevents hypertension‐induced cardiac inflammation and fibrosis. Cardiovasc Drugs Ther. 2013;27:521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siddesha JM, Valente AJ, Sakamuri SS, Yoshida T, Gardner JD, Somanna N, Takahashi C, Noda M, Chandrasekar B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and reck. J Mol Cell Cardiol. 2013;65:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie B, Miao P, Sun Y, Wang Y, Yang GY. Micro‐computed tomography for hemorrhage disruption of mouse brain vasculature. Transl Stroke Res. 2012;3:174–179. [DOI] [PubMed] [Google Scholar]
- 24. Ananda S, Marsden V, Vekemans K, Korkmaz E, Tsafnat N, Soon L, Jones A, Braet F. The visualization of hepatic vasculature by X‐ray micro‐computed tomography. J Electron Microsc. 2006;55:151–155. [DOI] [PubMed] [Google Scholar]
- 25. Sangaralingham SJ, Ritman EL, McKie PM, Ichiki T, Lerman A, Scott CG, Martin FL, Harders GE, Bellavia D, Burnett JC Jr. Cardiac micro‐computed tomography imaging of the aging coronary vasculature. Circ Cardiovasc Imaging. 2012;5:518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lluri G, Huang V, Touma M, Liu X, Harmon AW, Nakano A. Hematopoietic progenitors are required for proper development of coronary vasculature. J Mol Cell Cardiol. 2015;86:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aplin AC, Gelati M, Fogel E, Carnevale E, Nicosia RF. Angiopoietin‐1 and vascular endothelial growth factor induce expression of inflammatory cytokines before angiogenesis. Physiol Genomics. 2006;27:20–28. [DOI] [PubMed] [Google Scholar]
- 28. Condello M, Caraglia M, Castellano M, Arancia G, Meschini S. Structural and functional alterations of cellular components as revealed by electron microscopy. Microsc Res Tech. 2013;76:1057–1069. [DOI] [PubMed] [Google Scholar]
- 29. Cury DP, Dias FJ, Sosthenes MC, Dos Santos Haemmerle CA, Ogawa K, Da Silva MC, Mardegan Issa JP, Iyomasa MM, Watanabe IS. Morphometric, quantitative, and three‐dimensional analysis of the heart muscle fibers of old rats: transmission electron microscopy and high‐resolution scanning electron microscopy methods. Microsc Res Tech. 2013;76:184–195. [DOI] [PubMed] [Google Scholar]
- 30. Bae ON, Wang JM, Baek SH, Wang Q, Yuan H, Chen AF. Oxidative stress‐mediated thrombospondin‐2 upregulation impairs bone marrow‐derived angiogenic cell function in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2013;33:1920–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murfee WL, Schmid‐Schonbein GW. Chapter 12. Structure of microvascular networks in genetic hypertension. Methods Enzymol. 2008;444:271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Belabbas H, Zalvidea S, Casellas D, Moles JP, Galbes O, Mercier J, Jover B. Contrasting effect of exercise and angiotensin ii hypertension on in vivo and in vitro cardiac angiogenesis in rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1512–R1518. [DOI] [PubMed] [Google Scholar]
- 33. Rusai K, Jianxing C, Schneider R, Struijker‐Boudier H, Lutz J, Heemann U, Baumann M. Renin inhibition mitigates anti‐angiogenesis in spontaneously hypertensive rats. J Hypertens. 2011;29:266–272. [DOI] [PubMed] [Google Scholar]
- 34. Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz‐Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J Jr, Chien KR, Ferrara N. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA. 2001;98:5780–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gogiraju R, Xu X, Bochenek ML, Steinbrecher JH, Lehnart SE, Wenzel P, Kessel M, Zeisberg EM, Dobbelstein M, Schafer K. Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J Am Heart Assoc. 2015; 4:e001770 DOI: 10.1161/JAHA.115.001770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, Horvath TL, Sinclair DA, Pfluger PT, Tschop MH. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev. 2012;92:1479–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Akamata K, Wei J, Bhattacharyya M, Cheresh P, Bonner MY, Arbiser JL, Raparia K, Gupta MP, Kamp DW, Varga J. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget. 2016;7:69321–69336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang L, Zhang J, Xing W, Zhang X, Xu J, Zhang H, Chen L, Ning X, Ji G, Li J, Zhao Q, Gao F. SIRT3 deficiency induces endothelial insulin resistance and blunts endothelial‐dependent vasorelaxation in mice and human with obesity. Sci Rep. 2016;6:23366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu H, Chen T, Li N, Wang S, Bu P. Role of SIRT3 in angiotensin II‐induced human umbilical vein endothelial cells dysfunction. BMC Cardiovasc Disord. 2015;15:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini‐Molteni S, Sale P, Vago R, Arena G, Torosantucci L, Cassina L, Russo MA, Dallapiccola B, Valente EM, Casari G. The Parkinson‐associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Systolic blood pressure in angiotensin II (Ang II)–induced mice. A, Systolic blood pressure profile of wild‐type (WT) and SIRT3−/− mice measured every 2 days for 2 weeks. B, Systolic blood pressure of WT and SIRT3−/− mice infused with Ang II in day 14. The values are presented as means±SEM. ** P<0.01 vs genotype‐matched sham mice.
Figure S2. Sirtuin 3 (SIRT3) expression in cardiac microvascular endothelial cells (CMVECs). A, SIRT3 expression was detected by Western blotting with quantitative analysis in SIRT3‐short hairpin RNA (shRNA) or negative control cells. B, SIRT3 expression was detected by Western blotting with quantitative analysis in lentivirus‐mediated SIRT3 (LV‐SIRT3) or negative control infected cells. Results are expressed as fold‐change over untreated vector‐infected cells. Data are presented as means±SEM. *P<0.05, **P<0.01 vs untreated vector‐infected cells; # P<0.05, ## P<0.01 vs angiotensin II (Ang II)–treated vector‐infected cells.
Figure S3. Graphical summery. Sirtuin 3 (SIRT3) drives Pink1/Parkin activity to increase rates of mitophagy in response to oxidative stress, and thereby limits the production of damaging reactive oxygen species (ROS) that would further promote angiogenesis.