

Abstract
Background
Idiopathic ventricular tachycardia (VT) is a type of cardiac arrhythmia occurring in structurally normal hearts. The heritability of idiopathic VT remains to be clarified, and numerous genetic factors responsible for development of idiopathic VT are as yet unclear. Variations in FGF12 (fibroblast growth factor 12), which is expressed in the human ventricle and modulates the cardiac Na+ channel NaV1.5, may play an important role in the genetic pathogenesis of VT.
Methods and Results
We tested the hypothesis that genetic variations in FGF12 are associated with VT in 2 independent Chinese cohorts and resequenced all the exons and exon–intron boundaries and the 5′ and 3′ untranslated regions of FGF12 in 320 unrelated participants with idiopathic VT. For population‐based case–control association studies, we chose 3 single‐nucleotide polymorphisms—rs1460922, rs4687326, and rs2686464—which included all the exons of FGF12. The results showed that the single‐nucleotide polymorphism rs1460922 in FGF12 was significantly associated with VT after adjusting for covariates of sex and age in 2 independent Chinese populations: adjusted P=0.015 (odds ratio: 1.54 [95% CI, 1.09–2.19]) in the discovery sample, adjusted P=0.018 (odds ratio: 1.64 [95% CI, 1.09–2.48]) in the replication sample, and adjusted P=2.52E‐04 (odds ratio: 1.59 [95% CI, 1.24–2.03]) in the combined sample. After resequencing all amino acid coding regions and untranslated regions of FGF12, 5 rare variations were identified. The result of western blotting revealed that a de novo functional variation, p.P211Q (1.84% of 163 patients with right ventricular outflow tract VT), could downregulate FGF12 expression significantly.
Conclusions
In this study, we observed that rs1460922 of FGF12 was significantly associated with VT and identified that a de novo variation of FGF12 may be an important genetic risk factor for the pathogenesis of VT.
Keywords: fibroblast growth factor 12, genetic risk factor, variations, ventricular tachycardia
Subject Categories: Arrhythmias; Genetic, Association Studies
Clinical Perspective
What Is New?
Our studies are the first analysis of the genetic association between FGF12 (fibroblast growth factor 12) and ventricular tachycardia. The results revealed that a functional variation (p.P211Q) significantly reduced FGF12 expression, which may affect the interaction of FGF12 and Na+ channels and confer risk of right ventricular outflow tract ventricular tachycardia.
What Are the Clinical Implications?
The findings suggest that known of variations of FGF12 may help identify patients at risk of right ventricular outflow tract ventricular tachycardia in the population, stratify idiopathic ventricular tachycardia, and develop a novel potential treatment.
Introduction
Idiopathic ventricular tachycardia (VT) is a distinct type of monomorphic VT that occurs commonly without structural heart disease.1 The morbidity of idopathic VT is ≈10% in the United States and 20% in Japan.2 Genetic factors play an important role in the pathogenesis of idopathic VT.3 Mutations in genes such as RYR2 (Ryanodine receptor 2), DPP6 (Dipeptidyl aminopeptidase‐like protein 6), CASQ2 (Calsequestrin‐2), TRDN (Triadin), CALM1 (Calmodulin‐1), SCN5A (Sodium channel protein type 5 subunit alpha), SCN4B (Sodium channel subunit beta‐4), KCNQ1 (Potassium voltage‐gated channel subfamily KQT member 1), KCNE1 (Potassium voltage‐gated channel subfamily E member 1), KCNJ2 (Inward rectifier potassium channel 2), KCNH2 (Potassium voltage‐gated channel subfamily H member 2), KCNJ5 (Inward rectifier potassium channel 2), KCNJ8 (ATP‐sensitive inward rectifier potassium channel 8), KCNE2 (Potassium voltage‐gated channel subfamily E member 2), CACNB2 (Voltage‐dependent L‐type calcium channel subunit beta‐2), CACNA1C (Voltage‐dependent L‐type calcium channel subunit alpha‐1C), and CACNA2D1 (Voltage‐dependent calcium channel subunit alpha‐2)3, 4, 5 have been identified as causes of inherited arrhythmogenic disorders, including idopathic VT.
Fibroblast growth factor (FGF) homologous factors (FHFs; FGF11–14) are members of FGFs. Different from other secretory FGFs (FGF1–10 and FGF15–23), FHFs belong to intracellular nonsecretory forms.6 FHFs lack signal sequence, cannot release from cells,7 and activate FGF receptors.8 FHFs can modulate both Na+ and Ca2+ channels, and genes encoding FHFs are responsible for the development of Brugada syndrome (BrS), characterized by VT or ventricular fibrillation without cardiac abnormality.9, 10, 11 Recent studies also demonstrated that a missense mutation (p.Q7R) in FGF12 (encoding a member of FHFs that expresses abundantly in the human ventricle) is a disease‐associated functional variation of BrS. The p.Q7R mutation reduced binding to the NaV1.5 C‐terminus and Na+ channel current density, leading to Na+ channel loss‐of‐function phenotype consistent with that in BrS.12, 13 Furthermore, Musa et al reported that a variation in SCN5A (p.H1849R) blocks the regulation of FGF12 and causes human arrhythmia.14 This evidence highlighted that the variations in FGF12 may affect the interaction between FGF12 and Na+ channel, leading to arrhythmia.
Because changes in sodium channel function are important in the pathogenesis of idopathic VT and other inherited arrhythmias, we supposed that variations in FGF12 may be associated with VT/idopathic VT. To test the potential association between FGF12 and VT/idopathic VT, we performed a 3‐stage study. In the first stage, we chose 3 single‐nucleotide polymorphisms (SNPs)—rs1460922, rs4687326, and rs2686464—that included all exons of FGF12 to observe the association between FGF12 and VT/idopathic VT in a Chinese population (case:control of 255:289). In the second stage, we replicated the result of the first stage in an independent sample (case:control of 180:288). In the third stage, we resequenced all the exons and exon–intron boundaries and the 5′ and 3′ untranslated regions (UTRs) of FGF12 in 320 unrelated participants with idopathic VT to identify functional variations with risk effect on disease.15, 16
Methods
Study Samples
All participants were of Chinese descent and were chosen from GeneID.17, 18, 19, 20 The study was approved by appropriate local institutional review boards on human subject research and conformed to the guidelines set forth by the Declaration of Helsinki. Written informed consent was obtained from all participants.
A total of 255 participants with VT and 289 controls were enrolled in the first stage (discovery sample); 180 participants with VT and 288 controls were enrolled in the second stage (replication sample); 320 unrelated participants with idopathic VT (including 31 patients with idopathic VT from discovery and replication samples) were resequenced in the third stage.
All participants were precisely diagnosed with VT by ECG and/or Holter ECG recordings. VT was diagnosed according to the standards mentioned in the American College of Cardiology, American Heart Association, and European Society of Cardiology ventricular arrhythmia guidelines.21 Briefly, a patient showing wide‐QRS complex tachycardia on ECG was diagnosed as a patient with VT. Idopathic VT was defined as VT with structurally normal heart, and participants with coronary artery disease, ischemic stroke, congestive heart failure, essential hypertension, or diabetes mellitus were excluded.2 Those without a history of arrhythmia or detectable abnormal ECG were defined as controls. Demographic and other relevant clinical information, if present, was obtained from the medical records.
SNP Selection and Genotyping
A total of 157 SNPs flanked the 266.3 kb genomic region of FGF12 on chromosome 3 (International HapMap Project showed from 193 342 424 to 193 608 706 bp). Three SNPs—rs1460922, rs4687326, and rs2686464—were selected from the genotyped SNPs in the Han Chinese population of the HapMap project (the phase 2 database) using Haploview 4.2 for the study. The 3 SNPs were located in different linkage disequilibrium blocks and covered all exons and regulatory regions of FGF12 (D′=1, r 2 between 0.048 and 0.5; Figures S1 and S2).
Human genomic DNA was extracted from the peripheral white blood cells using the Wizard Genomic DNA Purification Kit (Promega). The primer sequences are given in Table 1. All SNPs were genotyped by a Rotor‐Gene 6000 high‐resolution melt system (Corbett Life Science) using standard protocols with minor modifications. Reaction mixture and genotyping procedures were described previously.22 Three positive controls with genotypes of 3 SNPs and a negative control of ddH2O were included during each high‐resolution melt run. Twenty samples were randomly selected for direct Sanger sequencing to confirm the accuracy of genotyping.
Table 1.
The Primers of Genotyping and Mutational Analysis for FGF12B
| Exon | Forward | Reverse |
|---|---|---|
| FGF12‐exon01‐HRM | gccctgattaaaatgaaaattga | tgcaaacatttattaaccttttcct |
| FGF12‐exon02‐HRM | ccggcgtttatttttagcag | cgtgcctgtcagcaattcta |
| FGF12‐exon03‐HRM | ttttatggatgtgggcaattt | aggcaagacacacttggaaa |
| FGF12‐exon04‐1‐HRM | caagcggaaagagaaagagc | tgcgaagtagacgtttgcac |
| FGF12‐exon04‐2‐HRM | ttcttccccttccacttggt | cactctccgggcttctactg |
| FGF12‐exon05‐HRM | tttgcagaaccccagctca | ctgggccctacatttgatttg |
| FGF12‐exon06‐HRM | ggattatttattcaaaaggtcactg | gcctaacatgatggttactccat |
| FGF12‐exon07‐HRM | gacaatagttttgatcggctca | cctgcattgctcctgatttt |
| FGF12‐exon08‐1‐HRM | cagaggacatggatttcaagc | ggcggtacagtgtggaagaa |
| FGF12‐exon08‐2‐HRM | gtaccgccagcaagaatcag | gggtccaacaaagacagtcag |
| FGF12‐exon09‐1‐HRM | tgaaggaaatttatgtccactg | agggaagaaggggagagttc |
| FGF12‐exon09‐2‐HRM | tgagaactctccccttcttcc | ccactaggtcttgcgttgtc |
| rs1460922‐HRM | cacgtgcacaaagattagcac | ttcaattctccaaatcctttcc |
| rs4687326‐HRM | tgtatggtgccatattgtttcc | tgcagtttggtagattatcagc |
| rs2686464‐HRM | gggccagactctcttaacca | atcccactccgaagtccag |
| FGF12‐exon01‐SEQ | gggatgtgggctagctagatt | ggaaagtatatctccccttttgg |
FGF12 Variation Analysis by Direct DNA Sequencing
All exons of FGF12 were screened to find functional variations or alleles by polymerase chain reaction (PCR) and DNA Sanger sequencing in 320 unrelated patients with idopathic VT with the clear subtype of VT (including 31 patients with idopathic VT from the discovery and replication samples). The information on primers for PCR and sequencing are also shown in Table 1. Variations observed in FGF12 were verified in 1000 control individuals chosen from our GeneID database. Those without a history of arrhythmia or detectable abnormal ECG were selected and randomly picked as controls.
Bioinformatics Analysis for Variations in FGF12
To observe the possible function of rare variations in FGF12, conservative analysis of mutational amino acid was performed by the Center for Integrative Bioinformatics VU (http://www.ibi.vu.nl/programs/pralinewww/). ExAC Browser (http://exac.broadinstitute.org/) and MutationTaster (http://www.mutationtaster.org/) were used to test the frequency of the detected variants. The extent of injury of variations was predicted by Variant Effect Predictor online (http://www.ensembl.org/info/docs/tools/vep/index.html). SIFT (http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and Condel (https://omictools.com/consensus-deleteriousness-score-of-missense-snvs-tool) were used to predict the extent of injury. The possible transcriptional factor binding regions of noncoding variations discovered in the 5′ UTR of FGF12 were predicted by using TFSEARCH (http://diyhpl.us/~bryan/irc/protocol-online/protocol-cache/TFSEARCH.html) and the JASPAR database (http://jaspar.genereg.net/cgi-bin/jaspar_db.pl?rm=browse&db=core&tax_group=vertebrates).
Cell Lines and Plasmids
Rat myocardial H9C2 cells and Hela cells (human epitheloid cervix carcinoma cell) were purchased from the American Type Culture Collection. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum in a humidified incubator with 5% CO2 at 37°C.
We amplified the full‐length coding region of FGF12 using human genomic cDNA, and it was subcloned into the p3xFLAG‐CMV‐10 by Tianyi Huiyuan. The plasmid of variant p.P211Q was constructed using PCR‐based site‐directed mutagenesis, and the construct was referred to as p3xFLAG‐CMV‐10‐FGF12‐Mut. Primers used for constructing p3xFLAG‐CMV‐10‐FGF12‐Mut were 5′‐GTATGTACAGAGAACAATCGCTACATGAAAT‐3′ (forward) and 5′‐ATTTCATGTAGCGATTGTTCTCTGTACATAC‐3′ (reverse). The promoter including the 5′ UTR of FGF12B was amplified by PCR using human genomic DNA as the template. The PCR product was digested with NheI and HindIII, and subcloned into the pGL3‐Basic luciferase plasmid, resulting in pGL3‐Basic‐5′‐UTR‐Wt. Two pairs of primers were used for constructing pGL3‐Basic‐5′‐UTR‐Wt. The first pair was 5′‐GGG ACT GAG TGA TCG GCC TTG CGT CCG GCG GGT AA‐3′ (forward) and 5′‐TGT TGG ACT CCC TCG CCT GCC GCT TCT G‐3′ (reverse), and the second pair was 5′‐GCC GCT AGC GCG GGT CAC TTC CTT CCT CGG CCG GGA TGG GCG GCG CGG G‐3′ (forward) and 5′‐GGC AAG CTT AGC TGC TCA GCG AGG GCC TCA GGC‐3′ (reverse). The plasmid of variant c.G723A was constructed as described, and the primers used were 5′‐TAG CAC TGC CTC CCC ACG ACT GCC CTT TCC C‐3′ (forward) and 5′‐GGG AAA GGG CAG TCG TGG GGA GGC AGT GCT A‐3′ (reverse).
Dual Luciferase Reporter Assays
Hela cells were cultured in 24‐well plates for 24 hours and transfected with 250 ng pGL3‐Basic‐FGF12B‐5′UTR‐Wt or pGL3‐Basic‐FGF12B‐5′UTR‐Mut (c.723G>A), and 250 ng transcription factors (pCDNA3.1[+]‐MZF1 and p3xFLAG‐CMV‐10‐ZNF354C) or empty vector (pCDNA3.1[+] and p3xFLAG‐CMV‐10), along with 20 ng of the pRL‐TK vector containing the Renilla luciferase gene. Transfection was carried out using 1 μL lipofectamine 2000 and 500 μL Opti‐MEM (Gibco Life Technologies) reduced serum medium, according to the manufacturer's protocol. Cells were harvested 48 hours after transfection and lysed using 1× passive lysis buffer. Luciferase assays were performed as described in our previous study23 and using the Dual‐Glo luciferase assay kit (Gibco Life Technologies). The ratio of firefly over Renilla luciferase activities was calculated and considered as the final luciferase activity value. Each assay was performed in triplicate and repeated at least 3 times.
Western Blot Analysis
H9C2 cells were cultured in 12‐well plates for 24 hours and transfected with 2 μg either p3xFLAG‐CMV‐10‐FGF12‐Wt or p3xFLAG‐CMV‐10‐FGF12‐p.P211Q, while empty vector (p3xFLAG‐CMV‐10) was used as a negative control. After 48 hours, transfected cells were collected and incubated in ice‐cold TNEN lysis buffer (50 mmol/L Tris/HCl, pH 7.5, 150 mmol/L NaCl, 2.0 mmol/L EDTA, 1.0% Nonidet P‐40) with 1 mini tab of EDTA‐free protease inhibitors and 1 mmol/L phenylmethylsulfonyl fluoride for 30 minutes at 4°C. The insoluble fraction was pelleted by centrifugation at 12 000g for 15 minutes at 4°C. Supernatant (100 μL) was mixed with 20 μL 6× Laemmli buffer (0.3 mol/L Tris‐HCl, 6% SDS, 60% glycerol, 120 mmol/L dithiothreitol, and proprietary pink tracking dye), and heated at 100°C for 10 minutes. Then, 40 μL samples were subjected to SDS‐PAGE (10%). After electrophoresis, proteins were transferred onto a 0.45‐μm polyvinylidene fluoride membrane. The membrane was probed with an anti‐DDDDK‐tag mouse monoclonal antibody (1:3000), followed by incubation with horseradish peroxidase–conjugated secondary goat antimouse antibody (1:5000). The protein signal was visualized by a Super Signal West Pico Chemiluminescent substrate (Pierce Chemical Co), according to the manufacturer's instructions. Human α‐tubulin (1:3000) was used as loading control. Each assay was performed in triplicate and repeated at least 3 times.
Statistical Analysis
Power analysis of each study sample was conducted using the Power and Sample Size Calculations program (PS version 3.0.43, by William D. Dupont and Walton D. Plummer, Jr. http://ps-power-and-sample-size-calculation.software.informer.com/). The genotyping results of SNPs were screened for deviations from Hardy‐Weinberg equilibrium using PLINK version 1.07 (http://zzz.bwh.harvard.edu/plink/index.shtml), and no SNPs showed significant deviation (P>0.05). An independent t test was used to analyze the difference of sex and age in case and control groups by SPSS version 17.0 (IBM Corp). Association analysis for 3 SNPs before adjusting for covariates of age and sex were performed by 2×2 contingency tables using PLINK version 1.07. Association analysis for the 3 SNPs after adjusting for covariates of age and sex was performed using logistic regression analysis with SPSS version 17.0. Odds ratios (ORs) and corresponding 95% confidential intervals (CIs) were also calculated. In addition, we performed multiple logistic regression analysis to adjust significant covariates of sex and age for VT.
Results
Clinical Characteristics
The clinical characteristics of subjects with VT and controls are summarized in Table 2. Age and sex were also observed between cases and controls.
Table 2.
Clinical Characteristics of Participants in This Study
| Items | VT/VF Cases | Comparison Controls | P, t test |
|---|---|---|---|
| Discovery sample | |||
| Sample size, n | 255 | 289 | |
| Sex, male, n (%) | 157 (62) | 194 (67) | P<0.001 |
| Age, y, mean±SD | 61±15 | 58±11 | 2.00E‐03 |
| Replication sample | |||
| Sample size, n | 180 | 288 | |
| Sex, male, n (%) | 113 (73) | 159 (55) | 0.06 |
| Age, y, mean±SD | 47±18 | 62±8 | P<0.001 |
VF indicates ventricular fibrillation; VT, ventricular tachycardia.
A total of 255 cases and 289 controls were enrolled in the discovery study. Among the cases, 62% were male, and the mean age was 61±15 years; among the controls, 67% were male, and the mean age was 58±11 years. A total of 180 cases and 288 controls were enrolled in the replication study. Among the study participants, 73% were male, and the mean age was 47±18 years; among the controls, 55% were male, and the mean age was 62±8 years. Statistical power analysis showed power >80% to detect the association between SNPs and VT in 2 samples and in the combined sample.
Among 320 unrelated patients with idopathic VT, 81 (25%) were male, and 239 (75%) were female. The mean age at diagnosis was 37±15 years. Among 163 (51%) cases with right ventricular outflow tract VT (RVOT), 113 (35%) cases showed left ventricular idopathic VT, 18 (6%) showed left ventricular outflow tract VT, and 26 (8%) showed miscellaneous VT.
Significant Allelic Association Between SNP rs1460922 of FGF12 and Risk of VT
The genotyping data of all 3 SNPs did not deviate from the Hardy–Weinberg equilibrium in the control group (P>0.05). In the discovery sample, only rs1460922G was significantly associated with the risk of VT (P adj=0.015; OR: 1.54 [95% CI, 1.09–2.19]; Table 3) after adjusting for covariates of sex and age. Genotypic association analysis was then performed under different inheritance models (additive, dominant, or recessive). SNP rs1460922G was significantly associated with the risk of VT in the additive model after adjusting for covariates of sex and age (P adj=0.010; OR: 1.64 [95% CI, 1.12–2.39]; Table 4) in genotypic association. SNPs rs4687326 and rs2686464 in FGF12, which failed to show significant association with risk of VT in the discovery sample (the adjusted P value is 0.390 for rs2686464 and 0.414 for rs4687326; Table 3), were excluded from the replication stage of the study.
Table 3.
Analysis of Allelic Association of SNPs in FGF12B With VT/VF
| SNP | Sample Size, n (Case/Control) | R.A | Frequency (Case/Control) | Without Adjustment | With Adjustment | ||
|---|---|---|---|---|---|---|---|
| P obs | OR (95% CI) | P adj | OR (95% CI) | ||||
| rs2686464, Discovery sample | 255/289 | C | 0.763/0.750 | 0.701 | 1.07 (0.76–1.51) | 0.390 | 1.18 (0.81–1.71) |
| rs4687326, Discovery sample | 255/289 | T | 0.230/0.217 | 0.637 | 1.08 (0.79–1.47) | 0.414 | 1.14 (0.83–1.58) |
| rs1460922, Discovery sample | 255/289 | G | 0.350/0.252 | 5.77E‐03 | 1.60 (1.14–2.23) | 0.015 | 1.54 (1.09–2.19) |
| rs1460922, Replication sample | 180/288 | G | 0.298/0.225 | 0.029 | 1.46 (1.04–2.05) | 0.018 | 1.64 (1.09–2.48) |
| rs1460922, Combined sample | 435/577 | G | 0.327/0.242 | 2.12E‐04 | 1.56 (1.23–1.98) | 2.52E‐04 | 1.59 (1.24–2.03) |
CI indicates confidential interval; OR, odds ratio; P adj, P value for association after adjusting for covariates of sex and age by multiple logistic regression analysis using SPSS version 17.0; P obs, P value for association before adjusting for covariates of age and sex by 2×2 contingence tables using PLINK version 1.07; R.A, risk allele; SNP, single‐nucleotide polymorphism; VF indicates ventricular fibrillation; VT, ventricular tachycardia.
Table 4.
Analysis of Genotypic Association of SNPs in FGF12B Under 3 Genetic Models
| SNP | Model | Without Adjustment | With Adjustment | ||
|---|---|---|---|---|---|
| P obs | OR (95% CI) | P adj | OR (95% CI) | ||
| rs2686464, Discovery sample | Dominant (C) | 0.073 | 2.36 (0.90–6.18) | 0.082 | 2.47 (0.89–6.84) |
| Recessive (C) | 0.682 | 0.92 (0.60–1.40) | 0.848 | 1.05 (0.66–1.65) | |
| Additive (C) | 0.122 | ··· | 0.392 | 1.18 (0.81–1.71) | |
| rs4687326, Discovery sample | Dominant (T) | 0.364 | 1.19 (0.82–1.73) | 0.224 | 1.28 (0.86–1.89) |
| Recessive (T) | 0.447 | 0.72 (0.30–1.69) | 0.606 | 0.79 (0.33–1.91) | |
| Additive (T) | 0.377 | ··· | 0.414 | 1.14 (0.83–1.58) | |
| rs1460922, Discovery sample | Dominant (G) | 0.018 | 1.68 (1.09–2.58) | 0.042 | 1.60 (1.02–2.51) |
| Recessive (G) | 0.017 | 3.18 (1.17–8.64) | 0.030 | 3.17 (1.12–9.01) | |
| Additive (G) | 0.011 | ··· | 0.010 | 1.64 (1.12–2.39) | |
| rs1460922, Replication sample | Dominant (G) | 0.190 | 1.33 (0.87–2.03) | 0.104 | 1.53 (0.92–2.56) |
| Recessive (G) | 0.007 | 3.23 (1.32–7.90) | 0.008 | 4.60 (1.48–14.29) | |
| Additive (G) | 0.024 | ··· | 0.017 | 1.67 (1.10–2.56) | |
| rs1460922, Combined sample | Dominant (G) | 4.00E‐03 | 1.55 (1.15–2.08) | 4.00E‐03 | 1.59 (1.12–2.17) |
| Recessive (G) | 3.70E‐04 | 3.16 (1.63–6.14) | 1.00E‐03 | 3.31 (1.65–6.64) | |
| Additive (G) | 2.79E‐04 | ··· | 1.79E‐04 | 1.64 (1.27–2.12) | |
CI indicates confidential interval; OR, odds ratio; P adj, P value for association after adjusting for covariates of sex and age by multiple logistic regression analysis using SPSS version 17.0; P obs, P value for association before adjusting for covariates of age and sex by 2×2 contingence tables using PLINK version 1.07.
We verified the association between rs1460922 and VT in an independent sample. The results showed that rs1460922G was still significantly associated with VT (P adj=0.018; OR: 1.64 [95% CI, 1.09–2.48]; Table 3) after adjusting for covariates of sex and age. In genotypic association, rs1460922 was significantly associated with VT in recessive and additive models (recessive: P adj=0.008; OR: 4.60 [95% CI, 1.48–14.29]; additive: P adj=0.017; OR: 1.67 [95% CI, 1.10–2.56]; Table 4). In the combined sample of the 2 Chinese cohorts, the VT association remained significant for rs1460922G (P adj=2.52E‐04; OR: 1.59 [95% CI, 1.24–2.03]). Significant genotypic association was also found assuming an additive model (dominant: P adj=4.00E‐03; OR: 1.59 [95% CI, 1.12–2.17]; recessive: P adj=1.00E‐03; OR: 3.31 [95% CI, 1.65–6.64]; additive: P adj=1.79E‐04; OR: 1.64 [95% CI, 1.27–2.12]; Table 4).
Functional Variations of FGF12 Identified in Patients With idopathic VT
The SNP rs1460922 was noted to be associated with VT in 2 independent case–control studies. Consequently, to further verify the new mutation of FGF12 associated with the risk of VT, all exons of FGF12 in 320 unrelated samples with idopathic VT showing a clear subtype of VT (including 31 patients with idopathic VT from discovery and replication samples) were resequenced.
A nonsynonymous variation, rs17852067 (p.P211Q), in exon 5 of FGF12 was identified in 3 (1%) participants with RVOT. Because no minor allele frequency (MAF) of rs17852067 was observed in National Center for Biotechnology Information (NCBI), ExAC, and 1000 Genomes databases, and the variation did not exist in 1000 controls in the present study, it is supposed that rs17852067 is a rare variation (MAF <0.01)24 associated with the disease. Two other rare variations (c.742C>T and c.723G>A in 2 different patients, respectively) in the 5′ UTR of FGF12 were identified in 320 individuals with idopathic VT (Figure 1A and 1B, Table 5). Information of patients who carried these variations is shown in Table 5. Three common SNPs (MAF >0.01)24—rs3109189 (MAF of 0.0797 in our study and 0.1860 in NCBI), rs75224764 (MAF of 0.0010 in our study and 0.0016 in NCBI), and rs13088552 (MAF of 0.2953 in our study and 0.3372 in NCBI)—were identified in patients. In addition, the MAFs of these 3 SNPs identified in 320 patients with idopathic VT in this study appeared to be slightly different from the NCBI MAFs. It could be assumed that the NCBI MAFs are for the general population; if so, these SNPs may be associated with VT in the general population.
Figure 1.
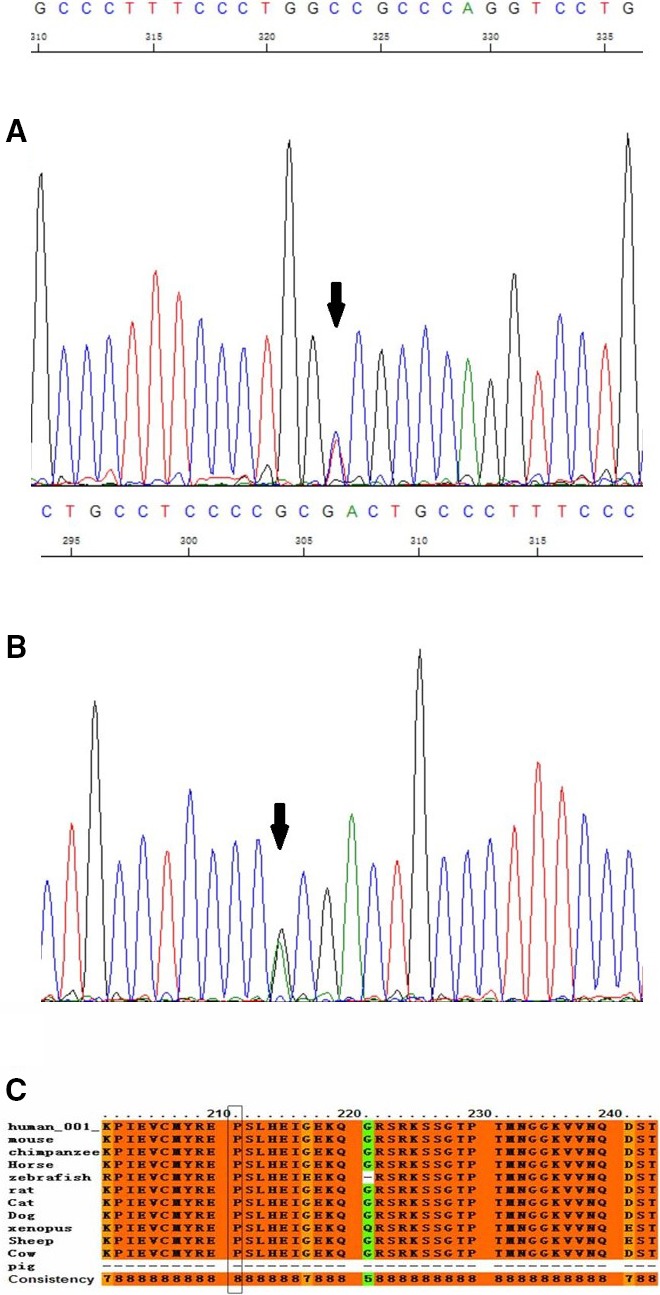
The sequencing results for variations and the conservation of 1 nonsynonymous variant and 1 functional SNP. A, The sequencing results for noncoding variation c.742C>T. B, The sequencing results for noncoding variation c.723G>A. C, The conservation of 1 nonsynonymous SNP rs17852067 (p.P211Q).
Table 5.
The Clinical Information of VT Patients With Variations
| GeneID | Mutation | Sex | Age, y | Diagnosis |
|---|---|---|---|---|
| 634987 | c.742C>T | Female | 39 | LOVT |
| 529139 | c.723G>A | Female | 53 | RVOT |
| 614250 | P211Q | Female | 47 | RVOT |
| 633085 | P211Q | Male | 14 | RVOT |
| 662250 | P211Q | Male | 64 | ROVT |
LOVT indicates idiopathic ventricular tachycardia from the left ventricular outflow tract; RVOT, right ventricular outflow tract ventricular tachycardia; VT, ventricular tachycardia.
The p.P211Q mutation was highly conservative in most species (score: 8; Figure 1C). The prediction analysis results showed that p.P211Q was deleterious (Table 6). The score of 3 online programs predicting the injury of mutations was from 1 to 10. Lower scores showed higher injury in SIFT and Condel, and higher scores showed higher injury in PolyPhen2. Moreover, p.P211Q was noted to be highly deleterious in the 3 scoring programs (SIFT: 0.04 [deleterious]; PolyPhen2: 1 [probably damaging]; Condel: 0.849 [deleterious]). The regulation of p.P211Q at the protein level was also examined by western blotting. Protein extracts were isolated from rat myocardial H9C2 cells transfected with p3xFLAG‐CMV‐10‐FGF12‐Wt or p3xflag‐cmv‐10‐FGF12‐p.P211Q. The results of western blotting showed that the mutation p.P211Q significantly reduced 52% of the FGF12 protein expression (P<0.0001; Figure 2A and 2B).
Table 6.
Critical Analysis Information of Variable Locus in FGF12B
| Gene | Location | Type | Transcript | Mutation | SIFT | PolyPhen2 | Condel |
|---|---|---|---|---|---|---|---|
| FGF12B | 5′ UTR | Noncoding | ENSP00000413496 | c.742C>T | ··· | ··· | ··· |
| 5′ UTR | Noncoding | ENSP00000413496 | c.723G>A | ··· | ··· | ··· | |
| Exon | SNP | ENSP00000413496 | rs17852067 (p.P211Q) | 0.04 (deleterious) | 1 (probably damaging) | 0.849 (deleterious) |
SIFT predicts whether an amino acid substitution affects protein function. SIFT can be applied to naturally occurring nonsynonymous polymorphisms or laboratory‐induced missense mutations. PolyPhen2 predicts the effect of an amino acid substitution on the structure and function of a protein. Condel is a general method for calculating a consensus prediction from the output of tools designed to predict the effect of an amino acid substitution. The Condel score is the consensus probability that a substitution is deleterious, so values nearer 1 are predicted with greater confidence to affect protein function. Chr indicates chromosome; SNP, single‐nucleotide polymorphism; UTR, untranslated region.
Figure 2.
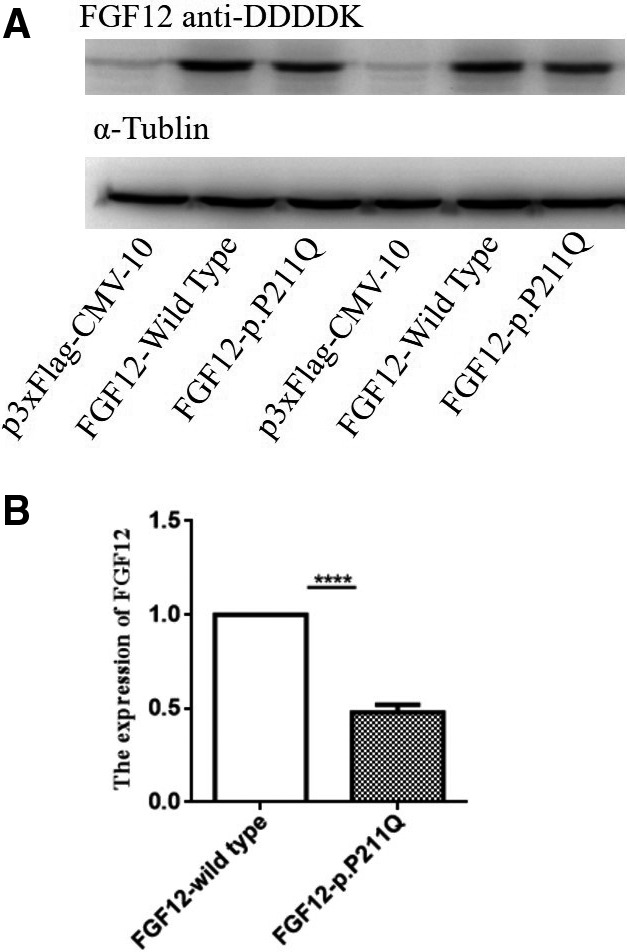
P211Q negatively regulates expression of FGF12 protein. A, P211Q significantly decreased the expression level of FGF12 protein in rat cardiac myocyte H9C2 cells compared with wild type by western blot analysis. α‐Tublin was used as loading control. B, The images of western blot analysis shown in (A) were scanned, quantified, and plotted. ****P <0.0001.
The prediction of binding domains for the transcription factors in the 2 mutation regions was noted. In case of c.723G>A, 2 transcription factors—MZF1 (myeloid zinc finger 1) and ZNF354C (zinc finger protein 354C)—were predicted to interact with the binding domain by TFSEARCH and JASPAR, respectively (Figure 3). In case of C.742C>T, no transcription factor was predicted to bind to the domain (data not shown). Based on the results of bioinformatics analysis, we cloned the region that contained the predicted MZF1 and ZNF354C binding sites and flanking sequences at the 5′ UTR of FGF12 into the pGL3‐Basic‐REPORT luciferase vector, resulting in a reporter gene pGL3‐Basic‐FGF12‐WT with the G allele and pGL3‐Basic‐FGF12‐5′UTR‐Mut with the A allele. Each reporter was cotransfected with MZF1 or ZNF354c, and negative NC‐control (pcDNA3.1(+) or p3x flag‐CMV‐10 plasmid) into cells and luciferase assays was carried out. The results showed that the luciferase activities were not regulated by MZF1 and ZNF354c (Figure S3A and S3B).
Figure 3.

The prediction of binding domains for the transcription factors in the noncoding variation c.723G>A. A, The binding domains for the transcription factors before the gene point change predicted by TFSEARCH. B, The binding domains for the transcription factors after the gene point change predicted by TFSEARCH. C, The binding domains for the transcription factors before the gene point change by predicted JASPAR. D, The binding domains for the transcription factors after the gene point change predicted by JASPAR.
Discussion
In the present study, we observed that SNP rs1460922 of FGF12 was associated with the risk of VT in the Chinese population. Furthermore, we identified a de novo functional variation (p.P211Q) that affects the expression of FGF12 in idopathic VT (RVOT). To the best of our knowledge, this is the first time that the genetic association between FGF12 and VT was observed.
We used 2 different approaches to test the association between FGF12 and VT. First, we carried out a case–control association study with SNPs in FGF12. Significant association was demonstrated for the minor allele G of rs1460922 and VT after adjusting for covariates of sex and age (P adj=2.52E‐04; OR: 1.59 [95% CI, 1.24–2.03]; Table 3). The genotypic association was also significantly associated with VT in dominant, recessive, and additive models (dominant: P adj=0.004; OR: 1.59 [95% CI, 1.12–2.17]; recessive: P adj=0.001; OR: 3.31 [95% CI, 1.65–6.64]; additive: P adj=1.79E‐04, OR: 1.64 [95% CI, 1.27–2.12]; Table 4). These results indicated that common SNPs in FGF12 can increase the genetic risk of VT.
Second, identification of de novo functional variations in patients with VT is important for confirming the association between FGF12 and VT. Consequently, we resequenced all exons and exon–intron boundaries and 5′ and 3′ UTRs of FGF12 in 320 patients with idopathic VT (idopathic VT is a special phenotype of VT in which patients represent only VT or premature ventricular contraction with normal structure of the heart1). According to the origin of idopathic VT, it is commonly classified into 3 types—idopathic VT from left ventricular outflow tract, idopathic VT from RVOT, and fascicular idopathic VT.25 RVOT is the most common form, accounting for 70% of all cases26 and 51% (163 cases with RVOT in 320 cases with idopathic VT) in our study. Five rare variations were identified after resequencing. A nonsynonymous variation, rs17852067 (p.P211Q), in exon 5 of FGF12 was identified in 3 participants with RVOT (1% of all patients with idopathic VT, 2% of 163 patients with RVOT). The results of western blotting revealed that p.P211Q significantly reduced FGF12 expression (52%, P<0.0001). Recent studies showed that some patients with BrS experienced RVOT leading to sudden death.27 idopathic VT is also a clinical symptom of BrS with ECG patterns similar to those of a left bundle‐branch block. Dysfunction of SCN5A is a major cause of both BrS and idopathic VT.5, 28, 29 These results are consistent with our study. A previous study14 reported that a variation in SCN5A (p.H1849R) could block the regulation of FGF12 and cause human arrhythmia. In the present study, p.P211Q downregulated the expression of FGF12 and might reduce binding to the NaV1.5 C‐terminus and Na+ channel current density, leading to an Na+ channel loss‐of‐function phenotype. The exact mechanism should be confirmed by further studies. In the present study, we could not detect more common variations (MAF >0.0001) because the sample size was limited.
It is interesting to note that FGF12 protein can interact with ion channels in the nervous and cardiac systems, bind to and modulate the cardiac NaV1.5 Na+ channel, and play a role in various arrhythmias, including VT.6, 30, 31, 32, 33 After binding to FGF12, recombinant NaV1.5 in human embryonic kidney 293 (HEK293) cells was observed to be a significant hyperpolarizing shift in the channel inactivation.34 Mutations of FGF12, such as p.P149Q, which decreased the binding affinity to the C‐terminus of specific voltage‐gated Na+ channels, affected the function of NaV1.533 and induced cardiac arrhythmias. Another mutation of FGF12, p.Q7R, reduced FGF12 expression and Na+ channel density and availability, leading to the development of BrS.12, 13 In contrast, abnormal mutations of genes encoding the Na+ channel, such as p.H1849R in SCN5A, can block the interaction and regulation of FGF12 and cause human arrhythmia.14 These studies suggested that the interaction of FGF12 and Na+ channels may play an important role in causing arrhythmia.
In conclusion, for the first time, we demonstrated a significant association between FGF12 and VT and identified a de novo functional variation, p.P211Q (2% of 163 patients with RVOT), that can significantly downregulate FGF12 expression. The exact mechanism underlying the development of VT/idopathic VT due to FGF12 needs further validation and functional study.
Sources of Funding
This work was supported by grants from National Basic Research Program of China (973 Program: 2013CB531103 and 2013CB531101), the National Natural Science Foundation of China (No. 91439109, 81270163, 81670363, 81630002, 31430047, and 91439129), NIH/NHLBI (USA) grants R01 HL121358 and R01 HL126729, Hubei Province Natural Science Programs (2016CFB224 and 2014CFA074), and the Program for New Century Excellent Talents at Chinese Universities (NCET‐11‐0181).
Disclosures
None.
Supporting information
Figure S1. Linkage disequilibrium structure and haplotype block in FGF12B in a Chinese Han population. Linkage disequilibrium (D′) for single‐nucleotide polymorphisms spanning a 266.3‐kb genomic region in FGF12B on chromosome 3 is generated by Haploview 4.0.
Figure S2. A schematic drawing giving the standard, stylized intron/exon gene map of FGF12. Three‐tag single‐nucleotide polymorphisms (SNPs) and area covered by the disequilibrium blocks they tag, the 3 new rare mutations, and the 3 common SNPs identified in patients by resequencing were indicated in the schematic drawing.
Figure S3. Transcription factors MZF1 and ZNF354C do not regulate the expression of the FGF12 at the mutant site c.G723A. A, Effect of MZF1 on the pGL3‐Basic‐FGF12B‐5′UTRMut luciferase reporters compared with pGL3‐Basic‐FGF12B‐5′UTR‐Wt transfected into Hela cells. B, Effects of ZNF354c on the pGL3‐Basic‐FGF12B‐5′UTR‐Mut luciferase reporters compared with pGL3‐Basic‐FGF12B‐5′UTR‐Wt. Luciferase activities were calculated as the ratio of firefly/Renilla activities and normalized to the negative control (empty vectors) group. Results were obtained from 3 independent experiments. Data are shown as mean±SD.
Acknowledgments
The authors thank the study subjects for their participation and support in this study and all members of the GeneID team for their help and assistance.
(J Am Heart Assoc. 2017;6:e006130 DOI: 10.1161/JAHA.117.006130.)28775062
Contributor Information
Qing Kenneth Wang, Email: qkwang@hust.edu.cn, Email: wangq2@ccf.org.
Xin Tu, Email: xtu@hust.edu.cn.
References
- 1. Yang SG, Mlcek M, Kittnar O. Gender differences in electrophysiological characteristics of idiopathic ventricular tachycardia originating from right ventricular outflow tract. Physiol Res. 2014;63(suppl 4):S451–S458. [DOI] [PubMed] [Google Scholar]
- 2. Badhwar N, Scheinman MM. Idiopathic ventricular tachycardia: diagnosis and management. Curr Probl Cardiol. 2007;32:7–43. [DOI] [PubMed] [Google Scholar]
- 3. Chopra N, Knollmann BC. Genetics of sudden cardiac death syndromes. Curr Opin Cardiol. 2011;26:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Albert CM, MacRae CA, Chasman DI, VanDenburgh M, Buring JE, Manson JE, Cook NR, Newton‐Cheh C. Common variants in cardiac ion channel genes are associated with sudden cardiac death. Circ Arrhythm Electrophysiol. 2010;3:222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Son MK, Ki CS, Park SJ, Huh J, Kim JS, On YK. Genetic mutation in Korean patients of sudden cardiac arrest as a surrogating marker of idiopathic ventricular arrhythmia. J Korean Med Sci. 2013;28:1021–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang X, Bao L, Yang L, Wu Q, Li S. Roles of intracellular fibroblast growth factors in neural development and functions. Sci China Life Sci. 2012;55:1038–1044. [DOI] [PubMed] [Google Scholar]
- 7. Pablo JL, Pitt GS. Fibroblast growth factor homologous factors: new roles in neuronal health and disease. Neuroscientist. 2016;22:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olsen SK, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M, Mohammadi M. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem. 2003;278:34226–34236. [DOI] [PubMed] [Google Scholar]
- 9. Hennessey JA, Wei EQ, Pitt GS. Fibroblast growth factor homologous factors modulate cardiac calcium channels. Circ Res. 2013;113:381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang C, Hennessey JA, Kirkton RD, Wang C, Graham V, Puranam RS, Rosenberg PB, Bursac N, Pitt GS. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ Res. 2011;109:775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan H, Pablo JL, Pitt GS. FGF14 regulates presynaptic Ca2+ channels and synaptic transmission. Cell Rep. 2013;4:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. [DOI] [PubMed] [Google Scholar]
- 13. Hennessey JA, Marcou CA, Wang C, Wei EQ, Wang C, Tester DJ, Torchio M, Dagradi F, Crotti L, Schwartz PJ, Ackerman MJ, Pitt GS. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm. 2013;10:1886–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Musa H, Kline CF, Sturm AC, Murphy N, Adelman S, Wang C, Yan H, Johnson BL, Csepe TA, Kilic A, Higgins RS, Janssen PM, Fedorov VV, Weiss R, Salazar C, Hund TJ, Pitt GS, Mohler PJ. SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci USA. 2015;112:12528–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, Voight BF, Bonnycastle LL, Jackson AU, Crawford G, Surti A, Guiducci C, Burtt NP, Parish S, Clarke R, Zelenika D, Kubalanza KA, Morken MA, Scott LJ, Stringham HM, Galan P, Swift AJ, Kuusisto J, Bergman RN, Sundvall J, Laakso M, Ferrucci L, Scheet P, Sanna S, Uda M, Yang Q, Lunetta KL, Dupuis J, de Bakker PI, O'Donnell CJ, Chambers JC, Kooner JS, Hercberg S, Meneton P, Lakatta EG, Scuteri A, Schlessinger D, Tuomilehto J, Collins FS, Groop L, Altshuler D, Collins R, Lathrop GM, Melander O, Salomaa V, Peltonen L, Orho‐Melander M, Ordovas JM, Boehnke M, Abecasis GR, Mohlke KL, Cupples LA. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lusis AJ, Pajukanta P. A treasure trove for lipoprotein biology. Nat Genet. 2008;40:129–130. [DOI] [PubMed] [Google Scholar]
- 17. Cheng X, Shi L, Nie S, Wang F, Li X, Xu C, Wang P, Yang B, Li Q, Pan Z, Li Y, Xia H, Zheng C, Ke Y, Wu Y, Tang T, Yan X, Yang Y, Xia N, Yao R, Wang B, Ma X, Zeng Q, Tu X, Liao Y, Wang QK. The same chromosome 9p21.3 locus is associated with type 2 diabetes and coronary artery disease in a Chinese Han population. Diabetes. 2011;60:680–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li C, Wang F, Yang Y, Fu F, Xu C, Shi L, Li S, Xia Y, Wu G, Cheng X, Liu H, Wang C, Wang P, Hao J, Ke Y, Zhao Y, Liu M, Zhang R, Gao L, Yu B, Zeng Q, Liao Y, Yang B, Tu X, Wang QK. Significant association of SNP rs2106261 in the ZFHX3 gene with atrial fibrillation in a Chinese Han GeneID population. Hum Genet. 2011;129:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang F, Xu CQ, He Q, Cai JP, Li XC, Wang D, Xiong X, Liao YH, Zeng QT, Yang YZ, Cheng X, Li C, Yang R, Wang CC, Wu G, Lu QL, Bai Y, Huang YF, Yin D, Yang Q, Wang XJ, Dai DP, Zhang RF, Wan J, Ren JH, Li SS, Zhao YY, Fu FF, Huang Y, Li QX, Shi SW, Lin N, Pan ZW, Li Y, Yu B, Wu YX, Ke YH, Lei J, Wang N, Luo CY, Ji LY, Gao LJ, Li L, Liu H, Huang EW, Cui J, Jia N, Ren X, Li H, Ke T, Zhang XQ, Liu JY, Liu MG, Xia H, Yang B, Shi LS, Xia YL, Tu X, Wang QK. Genome‐wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat Genet. 2011;43:345–349. [DOI] [PubMed] [Google Scholar]
- 20. Xiong X, Xu C, Zhang Y, Li X, Wang B, Wang F, Yang Q, Wang D, Wang X, Li S, Chen S, Zhao Y, Yin D, Huang Y, Zhu X, Wang L, Wang L, Chang L, Xu C, Li H, Ke T, Ren X, Wu Y, Zhang R, Wu T, Xia Y, Yang Y, Ma X, Tu X, Wang QK. BRG1 variant rs1122608 on chromosome 19p13.2 confers protection against stroke and regulates expression of pre‐mRNA‐splicing factor SFRS3. Hum Genet. 2014;133:499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, Priori SG, Quinones MA, Roden DM, Silka MJ, Tracy C, Smith SC Jr, Jacobs AK, Adams CD, Antman EM, Anderson JL, Hunt SA, Halperin JL, Nishimura R, Ornato JP, Page RL, Riegel B, Priori SG, Blanc JJ, Budaj A, Camm AJ, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Tamargo JL, Zamorano JL. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death). J Am Coll Cardiol. 2006;48:e247–e346. [DOI] [PubMed] [Google Scholar]
- 22. Ding H, Tu X, Xu Y, Xu C, Wang X, Cui G, Bao X, Hui R, Wang QK, Wang DW. No evidence for association of 12p13 SNPs rs11833579 and rs12425791 within NINJ2 gene with ischemic stroke in Chinese Han population. Atherosclerosis. 2011;216:381–382. [DOI] [PubMed] [Google Scholar]
- 23. Fan C, Ouyang P, Timur AA, He P, You SA, Hu Y, Ke T, Driscoll DJ, Chen Q, Wang QK. Novel roles of GATA1 in regulation of angiogenic factor AGGF1 and endothelial cell function. J Biol Chem. 2009;284:23331–23343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Panoutsopoulou K, Tachmazidou I, Zeggini E. In search of low‐frequency and rare variants affecting complex traits. Hum Mol Genet. 2013;22:R16–R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marchlinski FE, Deely MP, Zado ES. Sex‐specific triggers for right ventricular outflow tract tachycardia. Am Heart J. 2000;139:1009–1013. [DOI] [PubMed] [Google Scholar]
- 26. Pellegrini CN, Scheinman MM. Clinical management of ventricular tachycardia. Curr Probl Cardiol. 2010;35:453–504. [DOI] [PubMed] [Google Scholar]
- 27. Nademanee K, Veerakul G, Chandanamattha P, Chaothawee L, Ariyachaipanich A, Jirasirirojanakorn K, Likittanasombat K, Bhuripanyo K, Ngarmukos T. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123:1270–1279. [DOI] [PubMed] [Google Scholar]
- 28. Gaborit N, Wichter T, Varro A, Szuts V, Lamirault G, Eckardt L, Paul M, Breithardt G, Schulze‐Bahr E, Escande D, Nattel S, Demolombe S. Transcriptional profiling of ion channel genes in Brugada syndrome and other right ventricular arrhythmogenic diseases. Eur Heart J. 2009;30:487–496. [DOI] [PubMed] [Google Scholar]
- 29. King JH, Huang CL, Fraser JA. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front Physiol. 2013;4:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X, Giza J, Tchetchik D, Kelley K, Vega A, Matthews G, Rossi P, Ornitz DM, D'Angelo E. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage‐gated sodium channels. Neuron. 2007;55:449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dover K, Solinas S, D'Angelo E, Goldfarb M. Long‐term inactivation particle for voltage‐gated sodium channels. J Physiol. 2010;588:3695–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wildburger NC, Ali SR, Hsu WC, Shavkunov AS, Nenov MN, Lichti CF, LeDuc RD, Mostovenko E, Panova‐Elektronova NI, Emmett MR, Nilsson CL, Laezza F. Quantitative proteomics reveals protein‐protein interactions with fibroblast growth factor 12 as a component of the voltage‐gated sodium channel 1.2 (nav1.2) macromolecular complex in Mammalian brain. Mol Cell Proteomics. 2015;14:1288–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang C, Wang C, Hoch EG, Pitt GS. Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage‐gated sodium channels. J Biol Chem. 2011;286:24253–24263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu CJ, Dib‐Hajj SD, Renganathan M, Cummins TR, Waxman SG. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J Biol Chem. 2003;278:1029–1036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Linkage disequilibrium structure and haplotype block in FGF12B in a Chinese Han population. Linkage disequilibrium (D′) for single‐nucleotide polymorphisms spanning a 266.3‐kb genomic region in FGF12B on chromosome 3 is generated by Haploview 4.0.
Figure S2. A schematic drawing giving the standard, stylized intron/exon gene map of FGF12. Three‐tag single‐nucleotide polymorphisms (SNPs) and area covered by the disequilibrium blocks they tag, the 3 new rare mutations, and the 3 common SNPs identified in patients by resequencing were indicated in the schematic drawing.
Figure S3. Transcription factors MZF1 and ZNF354C do not regulate the expression of the FGF12 at the mutant site c.G723A. A, Effect of MZF1 on the pGL3‐Basic‐FGF12B‐5′UTRMut luciferase reporters compared with pGL3‐Basic‐FGF12B‐5′UTR‐Wt transfected into Hela cells. B, Effects of ZNF354c on the pGL3‐Basic‐FGF12B‐5′UTR‐Mut luciferase reporters compared with pGL3‐Basic‐FGF12B‐5′UTR‐Wt. Luciferase activities were calculated as the ratio of firefly/Renilla activities and normalized to the negative control (empty vectors) group. Results were obtained from 3 independent experiments. Data are shown as mean±SD.